

Background: Sulfatases are emerging as key adaptive tools of commensal bacteria to their host.
Results: The first bacterial endo-O-sulfatase and three exo-O-sulfatases from the human commensal Bacteroides thetaiotaomicron, specific for glycosaminoglycans, have been discovered and characterized.
Conclusion: Commensal bacteria possess a unique array of highly specific sulfatases to metabolize host glycans.
Significance: Bacterial sulfatases are much more diverse than anticipated.
Keywords: Bacterial Metabolism, Chondroitin Sulfate, Enzyme Mechanism, Glycosaminoglycan, Heparan Sulfate, Bacteroides thetaiotaomicron, Bacterial Metabolism, Enzyme Mechanism, Microbiota, Sulfatase
Abstract
Despite the importance of the microbiota in human physiology, the molecular bases that govern the interactions between these commensal bacteria and their host remain poorly understood. We recently reported that sulfatases play a key role in the adaptation of a major human commensal bacterium, Bacteroides thetaiotaomicron, to its host (Benjdia, A., Martens, E. C., Gordon, J. I., and Berteau, O. (2011) J. Biol. Chem. 286, 25973–25982). We hypothesized that sulfatases are instrumental for this bacterium, and related Bacteroides species, to metabolize highly sulfated glycans (i.e. mucins and glycosaminoglycans (GAGs)) and to colonize the intestinal mucosal layer. Based on our previous study, we investigated 10 sulfatase genes induced in the presence of host glycans. Biochemical characterization of these potential sulfatases allowed the identification of GAG-specific sulfatases selective for the type of saccharide residue and the attachment position of the sulfate group. Although some GAG-specific bacterial sulfatase activities have been described in the literature, we report here for the first time the identity and the biochemical characterization of four GAG-specific sulfatases. Furthermore, contrary to the current paradigm, we discovered that B. thetaiotaomicron possesses an authentic GAG endosulfatase that is active at the polymer level. This type of sulfatase is the first one to be identified in a bacterium. Our study thus demonstrates that bacteria have evolved more sophisticated and diverse GAG sulfatases than anticipated and establishes how B. thetaiotaomicron, and other major human commensal bacteria, can metabolize and potentially tailor complex host glycans.
Introduction
Bacteria are by far the most abundant and diverse microorganisms inside the human gastrointestinal tract with Firmicutes and Bacteroidetes being the major phyla (1–3). How these bacteria efficiently compete in this fierce ecosystem is still poorly understood despite their major role in human physiology (4). Although the human gut provides a constant source of nutrients and tightly controlled conditions, only a few phyla are abundant, suggesting a selective adaptation of these bacteria to their host (1). Key studies have highlighted that the prominent gut bacterium Bacteroides thetaiotaomicron has evolved complex relationships with its host (5, 6). Of particular interest, it has been established that B. thetaiotaomicron relies on host glycan foraging to enhance the fitness to its host (7). Recently, we have shown that in this process sulfatases play a pivotal role to allow B. thetaiotaomicron persistence in the gastrointestinal tract (8).
Sulfatases catalyze hydrolysis of sulfate groups from a broad range of substrate molecules, including small organic compounds to large macromolecules such as mucins or glycosaminoglycans (GAGs).5 In humans, sulfatases have been studied in great detail and are shown to be involved in genetic disorders or cancer (9). Most of them are lysosomal, and at least six are exo-enzymes responsible for GAG degradation (10). In addition, one GAG endosulfatase has been identified and shown to be critical for heparan sulfate (HS) remodeling (11). Opposite to the mammalian sulfatases, few bacterial GAG sulfatases have been characterized with three heparin sulfatases identified in the bacterium Flavobacterium heparinum (now called Pedobacter heparinus) (12–14). In addition, several GAG sulfatase activities, acting on chondroitin sulfate (CS), have been reported in B. thetaiotaomicron (15) and Proteus vulgaris (16).
Among hydrolases from bacteria to human, sulfatases are unique in requiring a critical amino acid post-translational modification on a key active site residue. This modification leads to the formation of the so-called “formylglycine” (3-oxoalanine), which is the only naturally occurring amino acid having an aldehyde function (17). Although the role of formylglycine in catalysis is still not completely understood, we have shown this modification to be dependent on three distinct enzymatic systems in bacteria (17–20). In B. thetaiotaomicron only one of these systems, the anaerobic sulfatase-maturating enzyme (anSME), which is part of the large and diverse family of radical S-adenosyl-l-methionine enzymes (21, 22), is involved in sulfatase activation (20). By inactivating anSME, we have demonstrated that the ability of B. thetaiotaomicron to survive in the gastrointestinal tract is strongly impaired (8). Transcriptomic analyses further revealed that, in the absence of a functional anSME, the expression profile of genes related to host glycan metabolism, especially encoding putative sulfatase genes, was markedly altered suggesting their involvement in host glycan metabolism.
To decipher the function of these potential sulfatases, we cloned 10 B. thetaiotaomicron genes, nine of which are induced by host glycans and five belong to polysaccharide utilization loci (Table 1) (7, 8). These genes were expressed in Escherichia coli in the presence of the B. thetaiotaomicron anSME (anSMEbt) that we have previously characterized (20, 23). The activity of the soluble purified proteins was then assayed with synthetic and natural GAG substrates. We especially focused on heparin/HS and chondroitin sulfate/dermatan sulfate (CS/DS), two major classes of sulfated GAGs, to determine the activity and specificity of these enzymes. Both classes of GAGs are characterized by alternating units of a hexuronate and an amino sugar: N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc), respectively, with heparan polymer containing GlcUAβ1–4GlcNAcα1–4 and CS GlcUAβ1–3GalNAcβ1–4 as basic repeat units.
TABLE 1.
B. thetaiotaomicron sulfatases expressed in this study
Genes induced by host glycans (7, 8) are shown in boldface type.
| Namea | Activity on pNP-Sb | Activity on the GlcNAc6S-O-pNPc | CS disaccharidesd |
|---|---|---|---|
| BT_0756 | None | None | None |
| BT_1596 | Yes | None | None |
| BT_1624 | Yes | None | None |
| BT_1628 | Yes | Low | None |
| BT_1918 | Yes | None | None |
| BT_3095 | Yes | None | None |
| BT_3101 | None | None | None |
| BT_3333 | None | None | None |
| BT_3349 | Yes | None | Yes |
| BT_4656 | None | Yes | None |
a These are the sulfatase gene numbers.
b Sulfatase activity was assayed with p-nitrophenyl sulfate (see “Experimental Procedures”).
c Sulfatase activity assayed with GlcNAc6S-O-pNP. Assay was performed in the presence of the indicated sulfatase with 10 mm GlcNAc6S-O-pNP and 1 unit of β-N-acetylglucosaminidase (Canavalia ensiformis). The released pNP was measured spectrophotometrically at 405 nm.
d Sulfatase activity was assayed on synthetic CS libraries as described (see “Experimental Procedures” and Ref. 27) for synthesis and composition).
During biosynthesis, these structures are further modified by extensive sulfation and epimerization. In HS, part of the GlcNAc are N-deacetylated and N-sulfated during biosynthesis resulting in an N-sulfated GlcN unit (GlcNS). The hexuronate, either glucuronate (GlcUA) or its C5-epimer iduronate (IdoUA), may be 2-O-sulfated, whereas the GlcNAc and GlcNS units can further be 6-O- and, rarely, 3-O-sulfated. CS/DS can be sulfated at position 2 on IdoUA and, more rarely on GlcUA, at positions 4 and 6 of the GalNAc unit. These sulfate groups are not only critical for the functional properties of the GAGs but also confer resistance to the hydrolytic activity of bacterial glycosidases (24).
In B. thetaiotaomicron, we identified two different 6-O-sulfatases with a strict specificity for either gluco- or galactosaminoglycans. We also report a 2-O-sulfatase that removes sulfates from a hexuronate unit independent of the parent GAG and whose structure is reported. Whereas these three enzymes are exolytic hydrolases, like all bacterial sulfatases reported to date, we also identified, in a totally unanticipated manner, a unique GAG endosulfatase. This novel enzyme very efficiently removes sulfate groups in the 4-O-position from CS/DS disaccharides to large polymeric chains and is thus the first bacterial GAG endosulfatase reported to date active at the polymer level.
We not only discovered novel bacterial sulfatases, we also demonstrate the complex interplay between these enzymes for host glycan degradation. Finally, we show that these enzymes are widespread in gut-associated bacteria underlining the importance of such metabolic pathways for a dominant phylum of the human microbiota, the Bacteroides. These novel sulfatases constitute attractive targets to manipulate the human microbiota and could potentially influence gut epithelial integrity by their unique capacity to selectively modify human GAGs.
EXPERIMENTAL PROCEDURES
Materials
CSA from bovine trachea, CSB from porcine intestinal mucosa, and CSC from shark cartilage were from Sigma. CSD from shark cartilage and CSE from squid cartilage were purchased from Seikagaku. HS from porcine intestine was a gift from G. van Dedem (Diosynth, Oss, The Netherlands), and heparin from bovine lung was purified as described previously (see Table 2 for composition) (16). CS-derived Δ4,5-unsaturated disaccharides were purchased from Iduron, and HS-derived Δ4,5-unsaturated disaccharides were obtained from Calbiochem. Monosaccharide substrates N-acetyl glucosamine 6-O-sulfate (GlcNAc6S), N-acetyl glucosamine 3,6-O-sulfate (GlcNAc3S6S), N-sulfate glucosamine (GlcNS), N-sulfate glucosamine 3-O-sulfate (GlcNS3S), N-sulfate glucosamine 6-O-sulfate (GlcNS6S) and N-sulfate glucosamine 3,6-O-sulfate (GlcNS3S6S) were purchased from Dextra. All other synthetic or semisynthetic HS derivatives were from Sanofi. Chondroitinase ABC was bought from Seikagaku, heparin lyases I–III from IBEX Pharmaceuticals, Inc. (Montreal, Canada), and Δ4,5-glycosidase from Grampian enzymes (Orkney, UK). A Luna 5-μ C18 reversed phase column (4.6 × 150 mm) was from Phenomenex.
TABLE 2.
Composition of polysaccharide chains used as sulfatase substrates
Disaccharides obtained by exhaustive enzymatic cleavage of chains were quantified, and the relative proportions of different disaccharide units were calculated for each type of chain.
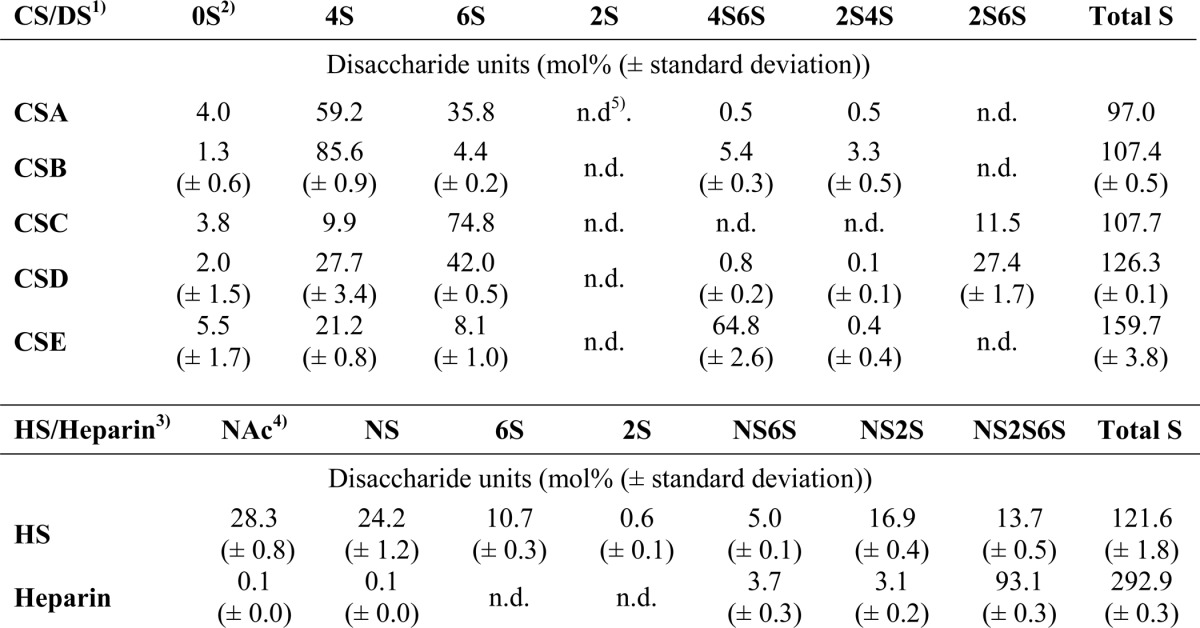
1) CS/DS disaccharides were generated by chondroitinase ABC lyase, and all units therefore contain a 4.5-unsaturated hexuronate unit (ΔHexA) at the nonreducing end.
2) Disaccharide abbreviations reflect the positions in ΔHexA-GalNAc modified with a sulfate group (where the uronate unit can be modified at position C2 and the N-acetylgalactosamine at positions 4 and 6).
3) HS/heparin disaccharides were generated by a mixture of heparin lyases I–III.
4) Disaccharide abbreviations reflect sulfate positions in the basic ΔHexA-GlcNR unit, where R can be either an N-acetyl (NAc) or an N-sulfate (NS) group. O-Sulfation in HS and heparin are indicated for positions C2 of HexA and C6 of GlcN.
5) n.d. means not detected.
Cloning of Sulfatase Enzymes
Sulfatases were cloned into the pRSF-Duet1 expression vector (Novagen) at the MCS1 cloning site with an N-terminal His6 tag that allowed purification on the Ni-NTA column. The sulfatase-maturating enzyme from B. thetaiotaomicron (anSMEbt (20)) was cloned into the same vector at the MCS2-cloning site that allowed sulfatase to be activated during expression in E. coli. The expression vectors were transformed into One Shot BL21 StarTM (DE3) E. coli (Invitrogen). The following primers were used for the corresponding sulfatase genes: BT_0756, 5′-GGA TCC AAT GGC CTC TGC TGT GCA-3′ and 5′-CTG CAG TTA TTT TTT TAA TGA TGA TTT CTT CAC TT-3′; BT_1596, 5′-GGA TCC AAT GGG ATT AGC CCT TTG TGG-3′ and 5′-CTG CAG TTA TTT TCT TTT GAG GAT CTC CCG-3′; BT_1624, 5′-GGA TCC AAT GAG AAA AGA ATT TTA TGG TAT ATT ACC-3′ and 5′-CTG CAG TTA TAG TGG CAG ACC GTA GCG-3′; BT_1628, 5′-GGA TCC AAT GCC GGA AGG CCA TC-3′ and 5′-CTG CAG TTA TTC CTT GTC CCT TTC CG-3′; BT_1918, 5′-GGA TCC AAT GAT TAA CCT GAA ATG TAC ATT TGC-3′ and 5′-CTG CAG TTA TCG CTT TTC TTT CGG ATA GTT-3′; BT_3095, 5′-GCA TGA CTG AAT TCA ATG CAG CGT TTT GTA TTA CGG-3′ and 5′-GAG TCT ACG TCG ACT TAT TGT TCG GGT TGG AGA TAA TT-3′; BT_3101, 5′-GGA TCC AAT GAA TAG ACT ATT TTT GAG TGT TTC TGT-3′ and 5′-CTC CAG TCA CTT TTG TTT GGA AGG CA-3′; BT_3333, 5′-GGA TCC AAT GAA GAA TGT CTC ACG TTT ACT ACC-3′ and 5′-CTG CAG TTA TCT TGT CTT TAC CGA TTC AAG C-3′; BT_3349, 5′-GGA TCC AAT GGG AGG CTT GAC CCT C-3′ and 5′-CTG CAG TCA GTA AGG TAT ACG GTC CGA A-3′; and BT_4656, 5′-GGA TCC AAT GCC CGC ACG GAA AAG-3′ and 5′-CTG CAG TTA TCG ATT TTC CAT CAG TCT TCT G-3′.
Expression and Purification of Sulfatase Enzymes
E. coli was used for cloning and expression of sulfatase genes using previously reported procedures (20). Briefly, plasmid coexpressing sulfatase genes with anSMEbt were selected on LB-agar plates containing 50 μg/ml kanamycin and grown in LB medium supplemented with the same antibiotic at 37 °C under agitation at 200 rpm. When the A600 reached 0.7, incubation temperature was lowered at 21 °C, and sulfatase expression was induced by adding isopropyl β-d-1-galactopyranoside (500 μm final concentration). The culture was continued overnight, and bacteria were harvested by centrifugation at 5000 × g for 20 min at 4 °C. After resuspension in buffer A (50 mm Tris/HCl, pH 7.0, 100 mm KCl, 10 mm MgCl2), the cells were disrupted by sonication and centrifuged at 100,000 × g for 1 h at 4 °C. The cell extract was then loaded onto a Ni-NTA-Sepharose column equilibrated with buffer A. The column was washed extensively with the same buffer. Weakly adsorbed proteins were washed off by applying 3 column volumes of 25 mm imidazole in buffer A, followed by 2 column volumes of 100 mm imidazole in the same buffer. Sulfatases were then eluted by applying 1 column volume of 500 mm imidazole in buffer A. Imidazole was removed using PD-10 desalting columns (GE Healthcare) equilibrated in buffer A. The sulfatase-containing fractions were immediately concentrated in Ultrafree cells (Millipore) with a molecular cutoff of 10 kDa. Sulfatase purity was assessed by SDS-PAGE, and their identity was confirmed by mass spectrometry analyses.
Synthesis of the Chromogenic Substrate 4-Nitrophenyl 2-Acetamido-2-deoxy-6-O-sodium Sulfonato-β-d-glucopyranoside (GlcNAc6S-O-pNP)
Triethylamine·SO3 (21 mg, 1 eq) was added to a solution of 4-nitrophenyl 2-acetamido-2-deoxy-β-d-glucopyranoside (40 mg, 0.12 mmol) in dimethylformamide (5.4 ml) under argon atmosphere at 0 °C. After stirring at 0 °C for 0.5 h and at room temperature for 1 h, a 0.15 m aqueous solution of sodium hydrogen carbonate (2.6 ml) was added at 0 °C and stirred for 16 h. The reaction mixture was then concentrated under reduced pressure and purified by reverse phase column chromatography (H2O/MeOH) to finally give pure GlcNAc6S-O-pNP (21 mg, 34%) after lyophilization. 1H and 13C NMR data were in agreement with previously reported data (25).
Synthesis of Methyl (2-O-Sulfonato-α-l-idopyranosyluronate)-(1–4)-2-acetamido-2-deoxy-6-O-sulfonato-α-d-glucopyranoside, Sodium Salt (IdoUA2S-GlcNAc6S-O-Me)
Sodium hydrogen carbonate (45 mg, 0.54 mmol) and acetic anhydride (25 μl, 0.27 mmol) were successively added at 0 °C to a saturated sodium hydrogen carbonate solution (1.8 ml) of methyl (2-O-sulfonato-α-l-idopyranosyluronate)-(1–4)-2-amino-2-deoxy-6-O-sulfonato-α-d-glucopyranoside (7.0 mg, 13.3 μmol) (26). After stirring 3 h at 0 °C and then 17 h at room temperature, the reaction mixture was diluted with 0.2 m aqueous NaCl (2 ml) and layered on top of a Sephadex G-25 column (in 0.2 m NaCl). Effluent fractions were pooled, concentrated, and desalted on the same gel filtration column, equilibrated with H2O. Lyophilization gave the sodium salt of methyl (2-O-sulfonato-α-l-idopyranosyluronate)-(1–4)-2-acetamido-2-deoxy-6-O-sulfonato-α-d-gluco-pyranoside (6.2 mg, 82%). [α]D + 0.063° (c 0.18, H2O); 1H NMR (500 MHz, D2O) δ = 5.18 (d, J1,2 = 2.4 Hz, 1 H, H-1II), 4.79 (d, J1,2 = 3.7 Hz, 1 H, H-1I), 4.74 (br. s, 1 H, H-5II), 4.39–4.29 (m, 3 H, H-6aI, H-6bI, H-2II), 4.07–4.00 (m, 4 H, H-3II, H-5I, H-4II, H-2I), 3.85 (dd, J = 8.8 Hz, J = 10.3 Hz, 1 H, H-3I), 3.77 (dd, J = 8.8 Hz, J = 9.6 Hz, 1 H, H-4I), 3.43 (s, 3 H, CH3O), 2.06 (s, 3 H, CH3CO). 13C NMR (125.76 MHz, D2O) δ = 101.0 (C-1II), 99.5 (C-1I), 79.5 (C-4I), 76.8 (C-2II), 71.6 (C-3I), 71.3 (C-4II), 71.2 (2 C, C-5II, C-3II), 70.4 (C-5I), 68.7 (C-6I), 57.0 (CH3O), 55.4 (C-2I), 23.7 (CH3CO).
Screening for Sulfatase Activity
Sulfatase activity was routinely assayed using the chromogenic reagent p-nitrophenyl sulfate (pNP-S) as a substrate. Standard assays were performed with 10 μm enzyme incubated at 30 °C for 10 min with 50 mm substrate in 100 mm Tris/HCl buffer, pH 7.25, containing 10 mm MgCl2, as described previously (17, 18). The pNP released was measured spectrophotometrically at 405 nm (ϵ = 9 000 mol−1·cm−1 at pH 7.25).
Sulfatase Assays with Synthetic and Natural Sulfated Glycans
Synthetic libraries of sulfated glycans, previously synthesized (27), were tested with enzymes as follows.: One μl (10 mm) of enzyme was added to 68 μl of 34 mm Tris/HCl buffer, pH 7.5, containing 6 μl of library solution (1 mg/ml). Incubations were performed overnight at 25 °C before analysis by capillary electrophoresis. Sulfated mono- and disaccharides (3 mm) were assayed at identical conditions.
To measure sulfatase activity on natural GAGs, sulfatases were incubated at a final concentration of 10 μm in 20 μl of 50 mm Tris/HCl, pH 7.5, 100 mm KCl, 10 mm MgCl2, containing 1 μg of GAG at 30 °C for 8 h. Sulfatase activity was stopped by boiling the sample at 96 °C for 10 min. For specificity analyses, variable enzyme concentrations were used as indicated in the figure legends. Sulfated saturated mono- and unsaturated disaccharides were used at a 20 μm final concentration and enzymatically digested as indicated, followed by analysis by reversed-phase ion pairing-high performance liquid chromatography (RPIP-HPLC).
Analysis of Sulfatase Products by Capillary Electrophoresis
Analyses by capillary electrophoresis (CE) were carried out on an Agilent CE apparatus, using a bare fused-silica capillary (64 cm × 50 μm). New capillaries were conditioned by successive flushes with NaOH (1 and 0.1 m) and water for 10, 5, and 10 min, respectively. Borate buffer (Agilent Technologies) (23 mm, pH 9.0, filtered through 0.2-μm filtered before use) was used as CE background electrolyte. The capillary was thermostatically controlled at 25 °C, and CE experiments were performed by applying a positive voltage of 30 kV at the capillary inlet. Prior to each sample injection, the capillary was flushed with the separation electrolyte for 10 min. Samples were loaded hydrodynamically by applying 50 millibars at the capillary inlet for 4 s. Detection was performed at 192 nm.
Analysis of Sulfatase Products by Reversed-phase Ion Pairing Chromatography
Sulfatase-treated GAGs (1 μg) were prepared for RPIP-HPLC analysis by exhaustive digestion to disaccharides with either 20 milliunits chondroitinase ABC (for CS/DS) or a mixture of 0.4 milliunit each of heparin lyase I–III (for heparin or HS) at 37 °C overnight as described (28) before heat inactivation of the enzymes. Products were separated by RPIP-HPLC and monitored by post-column fluorescence detection as described earlier (28).
Structure of the BT_1596 Sulfatase
The BT_1596 structure was solved by the Joint Center for Structural Genomics (29) and deposited in the PDB under code 3B5Q. Manual docking of the enzyme substrate was made using the disaccharide ΔHexA2S-GalNAc4,6S derived from a structurally characterized heparin tetrasaccharide (PDB code 1BFB). The human cerebroside-3-sulfate 3-sulfohydrolyase (ArsA) in interaction with a synthetic substrate (PDB code 1E2S) was superimposed with the BT_1596 structure using Coot and the SSM program. The heparin disaccharide was manually docked using ArsA substrate as reference.
NMR Analysis
NMR experiments were performed on a Bruker 600 MHz Avance III spectrometer using a 2.5-mm 1H/13C inverse detection probe, a 5-mm 1H/13C/15N/31P inverse detection QCI probe, or a 5-mm 1H/13C/15N/31P cryoprobe all equipped with z-gradient and controlled by Topspin 2.1 and 3.0 software. The incubated samples were repeatedly exchanged with D2O with intermediate lyophilization. The residual HOD signal was suppressed by saturation of the water peak during the recycle delay or using the WATERGATE pulse sequence. For the one-dimensional NMR experiments with the substrates incubated with the enzymes, the diffusion-edited NMR experiment (ledbpgp2s1d) was used for effective suppression of the residual water and buffer signals while retaining the signals from the substrates. The diffusion delay Δ and the gradient pulse length δ were 100 and 1.9 ms, respectively. The gradient strength was set to 95%. Assignments of 1H and 13C resonances were achieved using COSY, TOCSY, NOESY, HSQC, HMBC, and HSQC-TOCSY experiments from the Bruker pulse sequence library.
RESULTS
Expression and Functional Assay of B. thetaiotaomicron Sulfatases
Based on our and other transcriptomic studies (7, 8), we selected 10 potential sulfatase genes in the B. thetaiotaomicron genome, induced by host glycans (Table 1). These potential sulfatases were cloned and expressed in E. coli in the presence of the sulfatase-activating enzyme (anSMEbt) from B. thetaiotaomicron (20). Indeed, we have shown earlier that anSMEbt is critical for the post-translational modification and thus the activation of sulfatases from B. thetaiotaomicron (20). Purified sulfatases were first assayed with the simple chromogenic substrate pNP-S (Table 1). Half of these enzymes proved to be active on this synthetic substrate identifying them as authentic sulfatases.
We further synthesized a more relevant substrate in which the sulfate group was on a carbohydrate moiety rather than on an aromatic ring. This substrate (GlcNAc6S-O-pNP; see Scheme 1a and “Experimental Procedures”) (25) also contained a chromogenic group allowing the spectroscopic monitoring of the reaction by a coupled enzyme assay (Table 1). All the purified sulfatases were tested on this substrate; however, only two sulfatases, BT_4656 and to a much lesser extent BT_1628, hydrolyzed this reagent. Interestingly, despite its strong activity on this substrate, the BT_4656 sulfatase was inactive on pNP-S (Table 1).
SCHEME 1.

Structure of the synthetic and semisynthetic oligosaccharides used in this study.
Sulfatase Activity on Synthetic GAG-like Substrates
The B. thetaiotaomicron sulfatases were then tested against a library of synthetic saturated CS disaccharides with sulfate groups on the 2-O-position of the uronate unit and 4-O- and 6-O-positions of the amino sugar as described previously (27, 30).
Only the sulfatase BT_3349 (Table 1) proved to be active on these disaccharides. This enzyme led to the hydrolysis of all the disaccharides containing a sulfate group in the 4-O-position, whether or not a sulfate group is present in 6-O-position. To confirm BT_3349's specificity, we incubated this enzyme with the synthetic di-O-sulfated disaccharide GlcUA-GalNAc4,6S (Scheme 1b). Comparison of the NMR spectra of the disaccharide in the absence or presence of BT_3349 (Fig. 1A) showed that the signals at ∼4.8 and 4.9 ppm corresponding to the H4 of the GalNAc4,6S (α, β) unit disappeared in agreement with desulfation at the 4-O-position of the GalNAc residue.
FIGURE 1.

Identification of BT_3349 as a 4-O-sulfatase and of BT_4656 and BT_3333 as exosulfatases acting on the 6-O-sulfated nonreducing end hexosamine unit. A, comparison of 1H 1D NMR spectra of GlcUA-GalNAc4,6S before (upper trace) and after (lower trace) incubation with BT_3349. The characteristic signals of H4 (α/β) of GalNAc4,6S at 4.9 and 4.8 ppm are upfield shifted upon incubation with the enzyme. The asterisks denote signals from the glucuronic acid residue. B, mixtures of HS and CS disaccharides were incubated for 1 h at 30 °C with the indicated putative sulfatases. Disaccharides pretreated with glycuronidase to release the nonreducing end ΔHexA unit were incubated with BT_3333 and BT_4656. All samples were analyzed by RPIP-HPLC and quantified by comparison against known amount of standards. The average number of sulfate groups per unit (either disaccharide mixture or monosaccharide) is indicated for substrates without (Ø) or with pretreatment by the corresponding enzyme as marked. C, unsaturated CS disaccharide mixture was treated with BT_3333 for 1 h either with or without previous digestion by Δ4.5-glycuronidase that converted Δ4.5 HexA-GalNAc6S (6S) to GalNAc6S, which was the only structure further digested by BT_3333, whereas all the other unsaturated CS disaccharides present were neither substrates for glycuronidase nor BT_3333. D, 1H NMR spectra of GlcNAc6S-O-pNP before (upper trace) and after (lower trace) incubation with BT_4656. Addition of the enzyme results in an upfield shift of the H6 signals indicative of desulfation at position 6 of GlcNAc. The asterisks indicate signals from the buffer. E, to assess the influence of substitution for BT_4656 in either position 3 or position 2, GlcNAc6S, GlcNS6S, N-acetylglucosamine 3,6-O-sulfate, and GlcNS3S6S were incubated at a constant concentration with increasing concentrations of BT_4656 for 1 h, before analysis by RPIP-HPLC. Remaining substrate molecules were quantified as a proportion of originally added monosaccharides.
These results were consistent with transcriptomic analysis showing that two putative sulfatases genes, BT_3349 and BT_3333, are induced when B. thetaiotaomicron grows with CS as sole carbon source (7). B. thetaiotaomicron is known to possess two distinct CS sulfatase activities, one specific for the 4-O-sulfate group and the other for the 6-O-sulfate group (15). Because we established BT_3349 as a 4-O-sulfatase, BT_3333 was the logical candidate for the 6-O-sulfatase activity reported for that bacterium. Nevertheless, none of the synthetic substrates tested (CS disaccharide libraries or GlcNAc6S-O-pNP) proved to be a suitable substrate for the BT_3333 enzyme.
B. thetaiotaomicron GAG-specific 6-O-Sulfatases Require an Unsubstituted, Nonreducing End Hexosamine as Substrate
Because the synthetic CS disaccharides only partially mimic products formed during bacterial CS degradation, we assayed our library of enzymes on disaccharide mixtures obtained after treatment of HS and CS with bacterial eliminases. The disaccharides produced, in contrast to the synthetic ones, contain a 4,5-unsaturated hexuronate ring at their nonreducing end. Nevertheless, neither CS- nor HS-derived disaccharides (Fig. 1B) proved to be suitable substrates for BT_3333 or the 6-O-sulfatase BT_4656.
As exoenzymes would not be expected to attack the sulfate groups on the sugar in reducing-end position, we treated these disaccharides with a bacterial Δ4,5-glycuronidase that catalyzes the hydrolysis of the unsaturated hexuronate ring (31). The resulting monosaccharide mixtures were assayed with BT_3333 and BT_4656 sulfatases. As shown, the monosaccharides GalNAc6S and GlcNAc6S were hydrolyzed by BT_3333 and BT_4656, respectively (Fig. 1B). Using a mixture of differentially sulfated CS disaccharides as substrates, we demonstrated that only the 6-O-sulfate group on the GalNAc unit was hydrolyzed by BT_3333 after removal of the ΔHexA unit (Fig. 1C) (where Δ stands for the 4,5-unsaturated ring structure and HexA for the uronate unit).
1H NMR analysis of BT_4656 in the presence of GlcNAc6S-O-pNP showed that signals at ∼4.4 and 4.2 ppm, corresponding to the two H6 protons of the substrate, disappeared in the NMR spectrum (Fig. 1D), consistent with its identity as an N-acetylglucosamine-6-O-sulfatase. On the contrary, BT_4656 was inactive on a synthetic disaccharide with the GlcNAc6S unit in reducing-end position (i.e. IdoUA2S-GlcNAc6S-O-Me, Scheme 1c and “Experimental Procedures”), in agreement with the experiments performed on the enzymatically produced disaccharides.
In HS not only the GlcNAc can be 6-O-sulfated but also the GlcN-sulfated unit. Furthermore, the rare 3-O-sulfation of the GlcN unit may hinder 6-O-sulfatase activity. We therefore tested the impact of additional sulfation on the GlcNAc unit for 6-O-desulfation. Although the substitution with either an N-acetyl- or an N-sulfate group did not affect sulfatase activity, 3-O-sulfation totally hampered enzyme activity, demonstrating tight substrate specificity (Fig. 1E). Taken together, these data established that the sulfatases BT_3333 and BT_4656 are strict 6-O-sulfatases with an exclusive exolytic mode of action.
Link between the CS and HS Degradation Pathways in B. thetaiotaomicron
Further investigation with B. thetaiotaomicron sulfatases led to the identification of BT_1596 as a sulfatase active on HS-derived disaccharides but also on CS-derived disaccharides (Fig. 1B). Analysis of the different disaccharides present in solution showed that all types of 2-O-sulfated disaccharides, independent of the type of hexosamine unit and the presence of additional sulfate groups, were hydrolyzed by this enzyme. Notably, ΔHexA2S-GlcNAc, ΔHexA2S-GlcNAc6S, ΔHexA2S-GlcNS, and ΔHexA2S-GlcNS6S were completely desulfated (Fig. 2A), as well as ΔHexA2S-GalNAc4S, ΔHexA2S-GalNAc6S, and ΔHexA2S-GalNAc4,6S (Fig. 2B). Comparison of BT_1596 on ΔHexA2S-GalNAc and ΔHexA2S-GlcNAc indicated similar hydrolytic activities on both CS- and HS-derived disaccharides (data not shown). However, BT_1596 had no effect on polymeric CS or HS indicating that this enzyme is an exolytic sulfatase (Fig. 2C).
FIGURE 2.

Common 2-O-sulfatase (BT_1596) for HS and CS degradation products. A, incubation of a mixture of lyase-produced HS disaccharides with BT_1596. Recovery of disaccharides incubated with heat-inactivated enzyme (filled black boxes) or active BT_1596 enzyme (open white boxes) after separation by RPIP-HPLC and quantification are indicated. The standard HS disaccharide units are abbreviated as follows: NAc, ΔHexA-GlcNAc; NS, ΔHexA-GlcNS; 6S, ΔHexA-GlcNAc6S; 2S, ΔHexA2S-GlcNAc; NS2S, ΔHexA2S-GlcNS; NS6S, ΔHexA-GlcNS6S; NS2S6S, ΔHexA2S-GlcNS6S. B, analogous experiment with lyase-produced CS disaccharides incubated with BT_1596 and analyzed. C, HS and heparin were incubated without or with BT_1596 for 8 h under conditions described under “Experimental Procedures” and thereafter exhaustively digested by a mixture of heparin lyases (I–III) before analysis by RPIP-HPLC and quantification as described in Fig. 1C. The average sulfation degree was calculated and plotted on the y axis. CS-derived disaccharides are abbreviated as follows: 0S, ΔHexA-GalNAc; 4S, ΔHexA-GalNAc4S; 6S, ΔHexA-GalNAc6S; 2S, ΔHexA2S-GalNAc; 4S6S, ΔHexA-GalNAc4,6S; 2S4S, ΔHexA2S-GalNAc4S; 2S4S6S, ΔHexA2S-GalNAc4,6S. D, 1H-1D NMR spectra of a size-defined, lyase-produced heparin-derived hexasaccharide before (lower trace) and after (upper trace) incubation with BT_1596. The large upfield shift of H2 and smaller upfield shifts of the other signals from the nonreducing end unsaturated uronate residue (Δ) are indicative of desulfation at the C2 position. E, superimposition of 1H-13C HSQC spectra of AVE5026 alone (red, CH and CH3 groups, and blue, CH2 groups) and incubated with BT_1596 (black, CH and CH3 groups, and green, CH2 groups) show the large shifts experienced by the carbon and proton signals of the nonreducing end residue upon desulfation at the C2 position, although signals for internal IdoUA2S remain unaltered. The upper spectrum displays the anomeric region and the lower spectrum the C2/H2 to C6/H6 region. F, overall structure of the BT_1596 sulfatase (PDB code 3B5Q) with the disaccharide ΔHexA2S-GalNAc46S (PDB code 1BFB) manually docked into the active site. G, zoom of the BT_1596 sulfatase showing conserved critical residues. Residues involved in cation coordination are shown in salmon, and residues involved in substrate coordination are shown in pink. Serine 64 is likely a Cα-formylglycine in the active enzyme. The green sphere represents a modeled cation.
To confirm its exolytic nature and specificity, BT_1596 was incubated with a heparin-derived hexasaccharide (Scheme 1d) (32), and the reaction was analyzed by NMR (Fig. 2D). Comparison of the 1H and 13C signals in the hexasaccharide before and after incubation with BT_1596 showed chemical shift differences for the signals H1/C1 to H4/C4 at the nonreducing end. The large upfield shift of H2 by 0.84 ppm from 4.64 to 3.80 ppm together with the upfield shift of C2 by 3 ppm proved desulfation at this position. The changes in chemical shifts for the other signals were also characteristic for a change from a Δ4,5-HexA2S to the nonsulfated Δ4,5-HexA (33, 34). BT_1596 is thus a 2-O-sulfatase active on unsaturated nonreducing end hexuronate units.
To establish whether the 4,5-unsaturated ring is critical for enzyme recognition, we synthesized a heparin-like octasaccharide containing internal 2-O-sulfate groups and an IdoUA2S unit at its nonreducing end, IdoUA2S-GlcNAc-IdoUA2S-GlcNAc6S-IdoUA2S-GlcNAc6S-IdoUA2S-GlcNAc6S-OMe(Scheme 1e) (35). Despite extensive incubation with BT_1596, no modification of the NMR spectrum could be monitored. Notably, no changes were measured regarding both internal 2-O-sulfated units and the saturated 2-O-sulfated nonreducing end unit.
The activity of the BT_1596 sulfatase was further investigated by incubating the enzyme with the semisynthetic ultra-low molecular weight heparin AVE5026 (Scheme 1f) (36). AVE5026 consists of a mixture of different oligosaccharides with an average Mr of ∼2400 g/mol (i.e. ∼8 units) and the internal IdoUA residues partially sulfated on position 2. NMR analysis showed AVE5026 to be an enzyme substrate (Fig. 2E). It also demonstrated that BT_1596 exclusively hydrolyzes the ester sulfate at the 2-O-position of the Δ4,5HexA2S nonreducing end of all oligosaccharides present in AVE5026. Furthermore, no change for the C2 chemical shifts and the H1 signal intensity of the internal IdoUA2S units was observed, confirming that they are not targets of the enzyme (33). Our study thus establishes BT_1596 as a Δ4,5-hexuronate-2-O-sulfatase connecting HS and CS metabolism in B. thetaiotaomicron, consistent with transcriptomic analysis showing its induction in presence of host glycans (7).
Structure of the Δ4,5-Hexuronate-2-O-sulfatase BT_1596
Investigation of public databases revealed that the structure of the BT_1596 enzyme has been solved by the Joint Center for Structural Genomics (29) (PDB code 3B5Q) and identified as a putative sulfatase. Its structure shows an α/β topology, similar to other solved arylsulfatase structures (9). It is composed of six β-sheets in the larger N-terminal domain and of the canonical four anti-parallel β-sheets in the C-terminal domain, both surrounded by α-helices (Fig. 2F). In the highly charged active site, the critical residue (i.e. Ser-64) is located, as expected, at the beginning of an α-helix (9). Only one structure of a sulfatase in interaction with a substrate analog is available. It was obtained using an alanine mutant of the human cerebroside 3-sulfate 3-sulfohydrolyase (ArsA) and the synthetic substrate p-nitrocatechol sulfate (37). Interestingly, the superposition of ArsA and BT_1596 sulfatase shows that both enzyme structures share high structural similarities (root mean square deviation of 2.1) and the conservation of several amino acids involved in catalysis and substrate binding. Inside the BT_1596-active site, in addition to the catalytic residue Ser-64 (formylglycine 64 in the post-translationally activated enzyme), His-119 and Arg-68 are potentially involved in the elimination of the sulfate ester intermediate, whereas His-180, Lys-117 and Lys-296 may assist the binding of the anionic sulfate of the substrate (Fig. 2G). Moreover, although no cation has been modeled in the BT_1596 crystal structure, a putative conserved metal-binding region is found, composed of Asp-24, His-25, Asp-283, and His-284, likely coordinating a metal cation in the active enzyme. Incubation of BT_1596 in the presence of EDTA abolished its activity, further supporting the requirement of a cation, in the enzyme-active site.
Based on this analysis and using the ArsA substrate as a reference, we manually docked a disaccharide containing a Δ4,5-HexA2S (PDB code 1BFB) inside the BT_1596-active site (Fig. 2G). Interestingly, validations of our model are provided by the fact that the docked disaccharide superimposed with a sulfated piperazine crystallized within the enzyme-active site and that the four sulfate oxygen atoms coincide perfectly with four water molecules located inside the enzyme-active site. In this model, two charged amino acids (i.e. Lys-296 and Lys-117) are ideally positioned to interact with the sulfate group, although Glu-378 and Asn-95 seem to be responsible for the correct orientation of the unsaturated unit inside the enzyme-active site.
BT_3349 Is a Bacterial Endo-4-O-sulfatase Active from Di- to Polysaccharides
As demonstrated, the 2-O-sulfatase and the two 6-O-sulfatases isolated from B. thetaiotaomicron proved to be strict exolytic hydrolases, active only on the nonreducing end of oligosaccharides, in agreement with what is known for bacterial sulfatases. On the contrary, BT_3349 was active on the reducing-end unit of saturated (Fig. 1A) or unsaturated CS disaccharides (Fig. 1B) but not on monosaccharide units (30) suggesting a possible endolytic mode of activity.
To probe this hypothesis, we assayed BT_3349 on various polymeric CS with different degrees and locations of sulfate modifications (Table 2). With all CS used, we monitored extensive polymer desulfation related to the chain content of 4-O-sulfate groups (Fig. 3, A and B). Even with excess of enzyme, only 4-O-sulfate groups were removed from either substrate indicating a high degree of specificity of this enzyme.
FIGURE 3.

BT_3349 is an endo-4-O-sulfatase. A, two preparations of CS (B and D) were incubated for 8 h with or without BT_3349 as indicated, before exhaustive degradation of the chains with chondroitinase ABC followed by RPIP-HPLC. The average degree of sulfation was calculated for the recovered disaccharides and plotted. B, four different types of CS chains containing different degree and distribution of sulfate groups (see Table 2 for composition) were treated by BT_3349 before analysis as described in A. Degree of sulfation is plotted for each of the three different positions, C4 and C6 in GalNAc and C2 in ΔHexA. C, 1H-1H COSY of CS before (red) and after (black) addition of BT_3349. D, superimposition of the 1H-13C HSQC of CS before (red) and after (black) addition of BT_3349. The arrows show the change in chemical shifts of H4/C4, H3/C3, and H5/C5 of the GalNAc residues upon desulfation at the C4 position.
To univocally demonstrate 4-O-desulfation, we incubated the BT_3349 sulfatase with CS from shark cartilage (CSD) and followed the reaction by NMR. Analysis of the two-dimensional COSY, TOCSY, and HSQC NMR spectra showed shifts of all GalNAc4S H4/C4 signals by −0.61/−8.6 ppm, H3/C3 signal by −0.21/+5 ppm, and H5/C5 signal by −0.13/+0.4 ppm upon enzyme addition (Fig. 3, C and D). These changes in 1H and 13C chemical shifts indicate desulfation on the 4-O-position of GalNAc4S (38–40). Taken together these data demonstrated that the vast majority of the 4-O-sulfate groups present on the GalNAc units in the polymer chain were hydrolyzed. Quantitative analysis confirmed that, depending on the CS used, between 90 and 100% of 4-O-sulfate groups were removed (Fig. 3B).
Nevertheless, the substitution of CS units with sulfate groups in other positions and the presence of epimerized hexuronate unit affected the sulfatase activity (Fig. 4A). Mono-4-O-sulfated disaccharides were essentially completely hydrolyzed by <0.5 μm enzyme at a constant substrate level, whereas the presence of iduronate or sulfation of the uronate residue (more often on IdoUA than GlcUA) in the chain reduced the efficiency of the enzyme (Fig. 4, cf. CSB versus CSA, -D, or -E). Substitution of the 4-O-sulfated GalNAc with an additional sulfate group in position C6 reduced the efficiency slightly with all 4-O-sulfate groups hydrolyzed by ∼1 μm enzyme (Fig. 4A, CSE).
FIGURE 4.

Endo-4-O-sulfatase (BT_3349) recognizes 4-O-sulfation in different contexts. A, CS chains A, B, D, and E, as indicated (1 μg), were incubated for 8 h with increasing concentrations of enzyme before exhaustive digestion of chains by chondroitinase. Analysis of resulting disaccharides was performed by RPIP-HPLC. Relative proportions of the different disaccharide units recovered were plotted against enzyme concentrations. B, BT_3349 is affected by surrounding sulfation. Constant concentrations of CS-derived disaccharides were incubated for 1 h with increasing concentrations of sulfatase BT_3349 before analysis of products by RPIP-HPLC. Recovered disaccharides were plotted as a proportion of the original starting material.
As distribution of sulfate groups in different chain types is heterogeneous, analysis at chain level gives only a semi-quantitative indication of the impact of modification surrounding the target sulfate group. We therefore employed lyase-generated disaccharides to determine the influence of sulfate groups in position C2 of the hexuronate and C6 of the hexosamine unit on 4-O-sulfatase efficiency. Although 6-O-sulfation increased the enzyme concentration required to hydrolyze a disaccharide substrate from ∼0.05 μm (for mono-4-O-sulfated disaccharides) to ∼0.5 μm (for di-4,6-O-sulfated disaccharides), an additional 10-fold more enzyme (∼5 μm) was necessary when a sulfate group was present in position C2 of the hexuronate (Fig. 4B). Sulfation in all three positions further hindered sulfatase activity.
DISCUSSION
Despite their broad distribution in living organisms, only scarce information is available on sulfatases. Notably, because no sulfatase has been crystallized with its physiological substrate, almost no information about their specificity and selectivity is available. Furthermore, even if GAG-active sulfatases have been reported in some bacterial species, the identity of many of them remains unknown limiting biochemical characterization and sequence/structure comparisons. Finally, because of the lack of knowledge on these enzymes, their role in bacteria is usually confined to sulfate scavenging, although we have recently demonstrated their critical involvement for bacteria/host relationships in the context of the human microbiota (8).
We thus undertook a comprehensive analysis of potential GAG-specific sulfatases from the major human gut commensal B. thetaiotaomicron. In our study, we identified four sulfatases (BT_1596, BT_3333, BT_3349, and BT_4656) specific for GAG degradation. Three of these sulfatases are exolytic enzymes, in line with what is known about bacterial sulfatases, although one is an authentic endolytic O-sulfatase.
The BT_4656 sulfatase proved to be an N-acetylglucosamine 6-O-sulfatase sharing 57.5% identity with the heparin/HS 6-O-sulfatase from P. heparinus (14). Both enzymes exhibit similar properties, notably their specificity toward glucosamine over galactosamine residues. Using a high homology threshold (E-value 0, identity >50%), we identified more than 200 related potential sulfatases genes in bacterial genomes. This group contains two other sulfatase genes from B. thetaiotaomicron, BT_3177 and BT_1628 (52.5 and 51.7% identity, respectively). We have shown here that the BT_4656 sulfatase has a different specificity compared with the BT_1628 enzyme, suggesting that these 200 genes are unlikely to be GAG-specific glucosamine 6-O-sulfatases, although the BT_1628 exhibited weak 6-O-sulfatase activity. Phylogenetic analysis revealed that the glucosamine 6-O-sulfatase from B. thetaiotaomicron and P. heparinus defined a gene cluster containing 61 sulfatase genes. Interestingly, this cluster is composed mostly of genes originating from 38 major gut Bacteroides species, including Bacteroides cellulosilyticus, Bacteroides eggerthii, Bacteroides finegoldi, Bacteroides intestinalis, Bacteroides ovatus, Bacteroides pyogenes, Bacteroides stercoris, B. thetaiotaomicron, Bacteroides uniformis, Bacteroides xylanisolvens and several uncharacterized Bacteroides species.
BT_3333 is the other 6-O-sulfatase we have identified. It represents the first bacterial N-acetylgalactosamine-6-O-sulfatase gene identified so far, although this type of activity has been reported in P. vulgaris (16). This enzyme does not present significant sequence homologies with the human N-acetylgalactosamine-6-sulfatase (UniProt P34059, identity <24%) and is thus currently only annotated as an arylsulfatase in bacterial genomes. We were able to identify 64 homologs (E-value 0, identity >50%) with a distribution mirroring essentially the one of BT_4656 among gut bacteria.
Although both of these 6-O-sulfatases are highly specific for either CS or HS units and cannot cross-react, we identified the BT_1596 sulfatase as a Δ4,5-hexuronate-2-O-sulfatase able to efficiently hydrolyze both types of substrates with slight preference for HS-derived structures. BT_1596 exhibits only 29% sequence identity with the analogous enzyme identified in P. heparinus (12). The latter enzyme, contrary to BT_1596, has been described as a 2-O-sulfatase kinetically preferentially acting on HS-derived unsaturated disaccharides, yet capable to cleave 2-O-sulfated unsaturated CS-derived units at saturating conditions. Whereas both enzymes hydrolyze sulfate groups on 4,5-unsaturated units derived from HS and CS disaccharides, we demonstrate here that the BT_1596 sulfatase is efficient on longer oligosaccharide and strictly active on the unsaturated units. Indeed, even after an extended period of time, BT_1596 cannot hydrolyze saturated units.
The solved structure of this enzyme shows the typical α/β-topology of sulfatases and a narrow active site consistent with its strict exolytic activity. Our structural analysis allowed us to identify characteristic residues likely involved in catalysis and cation coordination, although no metal ion was found in the electron density map. A model of P. heparinus sulfatase was previously built, and several residues were predicted to be essential for substrate interaction (41). None of them were conserved in the structure of the BT_1596 sulfatase.
Structural analysis revealed the presence of two charged amino acids, likely involved in the interaction with the sulfate group. Interestingly, both residues (i.e. Lys-296 and Lys-117) are conserved in the P. heparinus sulfatase. Based on our model, we also identified Glu-378 and Asn-95 as likely candidates for the selectivity of BT_1596 toward Δ4,5HexA2S over IdoUA2S units.
Interestingly, the BT_1596 sulfatase has a more narrow distribution with only 32 homologs (E-value 0, identity >85%) found exclusively in a few Bacteroides species including B. xylanisolvens, B. pyogenes, B. ovatus, B. finegoldi, and B. uniformis. Because of its lack of homology with previously known sulfatases, all these genes are annotated as putative sulfatase (similar to the yidJ gene from E. coli (18)) or as hypothetical proteins. The P. heparinus 2-O-sulfatase (31) has no homolog at such a threshold indicating the existence of at least two distinct groups of Δ4,5-hexuronate-2-O-sulfatases among bacteria.
Finally, our study allowed us to identify a fourth GAG-specific sulfatase (BT_3349) in B. thetaiotaomicron, which we have demonstrated to be a CS/DS-specific 4-O-sulfatase. We have established that this enzyme efficiently removes sulfate groups from a broad substrate range of saturated disaccharides to high molecular weight polymers. As expected, this enzyme shares no significant homologies with the human N-acetylgalactosamine-4-O-sulfatase, ArsB (UniProt ID: P15848), which is a strict exo-enzyme active only on the sulfate group present on the GAG nonreducing end (42).
The BT_3349 enzyme exhibits similar properties to an enzyme from P. vulgaris (16). Although the P. vulgaris sulfatases were never further characterized, two sulfatase activities (a CS 4-O- and 6-O-sulfatase) were found in this bacterium like in B. thetaiotaomicron. Few major differences are apparent between the activities identified in P. vulgaris and the enzymes we characterized from B. thetaiotaomicron. First, we have unequivocally demonstrated that the CS 6-O-sulfatase (N-acetylgalactosamine-6-O-sulfatase) from B. thetaiotaomicron (i.e. BT_3333) is an exo-enzyme active only on the nonreducing end of CS oligosaccharides, whereas the P. vulgaris CS 6-O-sulfatase was reported to be specific for the sulfate groups present at the reducing end of hexasaccharides (16). Second, the P. vulgaris CS 4-O-sulfatase was shown to be active at the reducing end of oligosaccharides up to hexasaccharides and, under some conditions, able to act on monosulfated internal units. Here, we demonstrate that the BT_3349 efficiently hydrolyzes sulfate groups from a broad range of substrate size, including disaccharide to high molecular weight CS and DS polymers. We have shown that this enzyme is active from mono- to tri-O-substituted units with a strict specificity for the 4-O-sulfate groups of galactosamine. Furthermore, a noteworthy feature of this enzyme is its ability to extensively and efficiently remove sulfate groups on the intact polymer and, as such, is the first reported bacterial GAG endosulfatase active at polymer level. To date, only one other GAG endosulfatase, the HS 6-O-endosulfatase, present in eukaryotes from Drosophila to humans, is able to hydrolyze 6-O-sulfate groups on the chain level without requiring prior polymer degradation. This latter enzyme has attracted considerable interest during the last 10 years because of its major roles in metazoan development and homeostasis (11). Although no structural data are available, this enzyme has been reported to be able to act as an endosulfatase because of the unique presence of an additional positively charged domain. This domain has been speculated to be critical for the interaction with the highly negatively charged GAG chains. The BT_3349 sulfatase does not possess such an additional domain and has a predicted topology similar to other bacterial sulfatases. This enzyme thus represents a novel group of GAG-specific endosulfatases and is the first identified CS-endosulfatase and the first bacterial GAG-specific endosulfatase ever reported.
Interestingly, whereas genes, encoding for homologs of BT_3349, are also present in several Bacteroides species (e.g. B. xylanisolvens, B. pyogenes, B. ovatus, B. finegoldi, and B. uniformis), no homologs were found in the P. heparinus genome. Conversely, the sequenced Proteus species are apparently deprived of HS-related sulfatases. The gut Bacteroides are thus uniquely equipped to extensively modify and degrade both types of host GAGs.
Based on our biochemical studies, we can tentatively propose a metabolic pathway for GAG degradation in B. thetaiotaomicron (Fig. 5). The CS and HS degradation pathways might differ in the first step because CS can be extensively desulfated at the 4-O-position in the extracellular environment by the BT_3349 sulfatase. Then CS and HS are cleaved by lyases producing unsaturated di- to oligosaccharides. Because most of these enzymes belong to polysaccharide utilization loci, these enzymes are likely to exert their activity in the bacterial environment and the resulting oligosaccharides to be imported into the bacterium as described for other types of glycans (43).
FIGURE 5.

Proposed functions of B. thetaiotaomicron sulfatases in the bacterial degradation pathways of glycosaminoglycans. Chondroitin and dermatan sulfate can be first desulfated by the unique endo-4-O-sulfatase (BT_3349) before lyases act, whereas heparin/HS are degraded directly into oligosaccharides and disaccharides. Independently of their origin, GAG oligosaccharides and disaccharides are further processed by the Δ4,5-hexuronate-2-O-sulfatase (BT_1596) and hydrolyzed into shorter oligosaccharides or monosaccharides. Oligosaccharides with a nonreducing end hexosamine and monosaccharides then become substrates for the two specific 6-O-sulfatases (BT_3333 and BT_4656). Further action of an N-sulfamidase and 3-O-sulfatase is required to obtain totally desulfated units from HS and heparin.
The 2-O-sulfate groups present on the unsaturated hexuronate units of CS and HS oligosaccharides are cleaved off by the BT_1596 sulfatase. These products are then further hydrolyzed by glycosidases releasing oligosaccharides containing 6-O-sulfatated galactosamine or glucosamine at their nonreducing end. The BT_4656 and BT_3333 sulfatases are involved in the final steps reducing sulfation of oligo- or monosaccharides. Despite our attempts, we failed to identify in B. thetaiotaomicron an N- and a 3-O-sulfatase whose activities are required for completion of GAG desulfation even though we expressed and assayed BT_3095 and BT_3101, the closest homologs of P. heparinus N-sulfoglucosamine sulfohydrolase. Although we cannot exclude that these activities would require different assay conditions, it is likely that genes coding for these enzymes are present among the 14 other putative sulfatase genes of this organism. Interestingly, although many bacteria and Bacteroides species have either the CS or HS degradation pathway, only a few Bacteroides species possess both complete degradation pathways. These species are microbiota dominant species such as B. thetaiotaomicron, B. ovatus, or B. uniformis.
Although the role of the enzymes we have discovered is likely to metabolize host or food-derived GAGs, the unexpected discovery of a bacterial GAG endosulfatase raises the possibility that, as we previously hypothesized (8), Bacteroides use sulfatases to shape their glycosidic landscape. Indeed, these enzymes provide B. thetaiotaomicron (and other major human commensal Bacteroides) with the unique ability not only to metabolize host glycans but possibly also to modify host macromolecules with a potential impact on host physiology and pathology. This hypothesis will nevertheless require further investigations. Finally, these enzymes represent unique tools for the selective modification and characterization of GAGs, notably the novel endosulfatase identified here could be employed to produce novel CS-derived polymers.
Acknowledgments
We thank the Joint Center for Structural Genomics (jcsg.org) for the use of the BT_1596 coordinates (PDB code 3B5Q). Dr. Sandrine Dhénin is acknowledged for chemical synthesis of heparin derivatives.
This work was supported in part by European Community within a consortium PolyModE KBBE-2007-3-3-07.
- GAG
- glycosaminoglycan
- anSME
- anaerobic sulfatase-maturating enzyme
- anSMEbt
- B. thetaiotaomicron anSME
- HS
- heparan sulfate
- CS
- chondroitin sulfate
- DS
- dermatan sulfate
- GlcNAc
- N-acetyl-glucosamine
- GalNAc
- N-acetyl-galactosamine
- GlcN
- glucosamine
- GlcNS
- N-sulfated glucosamine
- GlcUA
- glucuronate
- IdoUA
- iduronate
- GlcNAc6S
- N-acetylglucosamine-6-O-sulfate
- GlcNS6S
- N-sulfated glucosamine-6-O-sulfate
- GlcNS3S6S
- N-sulfated glucosamine-3,6-O-sulfate
- GlcNAc6S-O-pNP
- p-nitrophenyl GlcNAc6S
- CE
- capillary electrophoresis
- RPIP-HPLC
- reversed-phase ion pairing-high performance liquid chromatography
- ΔHexA
- Δ4,5 unsaturated hexuronate
- PDB
- Protein Data Bank
- Ni-NTA
- nickel-nitrilotriacetic acid.
REFERENCES
- 1. Qin J., Li R., Raes J., Arumugam M., Burgdorf K. S., Manichanh C., Nielsen T., Pons N., Levenez F., Yamada T., Mende D. R., Li J., Xu J., Li S., Li D., Cao J., Wang B., Liang H., Zheng H., Xie Y., Tap J., Lepage P., Bertalan M., Batto J. M., Hansen T., Le Paslier D., Linneberg A., Nielsen H. B., Pelletier E., Renault P., Sicheritz-Ponten T., Turner K., Zhu H., Yu C., Jian M., Zhou Y., Li Y., Zhang X., Qin N., Yang H., Wang J., Brunak S., Doré J., Guarner F., Kristiansen K., Pedersen O., Parkhill J., Weissenbach J., MetaHIT Consortium, Bork P., Ehrlich S. D., Wang J. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Turnbaugh P. J., Hamady M., Yatsunenko T., Cantarel B. L., Duncan A., Ley R. E., Sogin M. L., Jones W. J., Roe B. A., Affourtit J. P., Egholm M., Henrissat B., Heath A. C., Knight R., Gordon J. I. (2009) A core gut microbiome in obese and lean twins. Nature 457, 480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Turnbaugh P. J., Quince C., Faith J. J., McHardy A. C., Yatsunenko T., Niazi F., Affourtit J., Egholm M., Henrissat B., Knight R., Gordon J. I. (2010) Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc. Natl. Acad. Sci. U.S.A. 107, 7503–7508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lozupone C. A., Stombaugh J. I., Gordon J. I., Jansson J. K., Knight R. (2012) Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hooper L. V., Midtvedt T., Gordon J. I. (2002) How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 22, 283–307 [DOI] [PubMed] [Google Scholar]
- 6. Hooper L. V., Xu J., Falk P. G., Midtvedt T., Gordon J. I. (1999) A molecular sensor that allows a gut commensal to control its nutrient foundation in a competitive ecosystem. Proc. Natl. Acad. Sci. U.S.A. 96, 9833–9838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martens E. C., Chiang H. C., Gordon J. I. (2008) Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4, 447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Benjdia A., Martens E. C., Gordon J. I., Berteau O. (2011) Sulfatases and a radical S-adenosyl-l-methionine (AdoMet) enzyme are key for mucosal foraging and fitness of the prominent human gut symbiont, Bacteroides thetaiotaomicron. J. Biol. Chem. 286, 25973–25982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hanson S. R., Best M. D., Wong C. H. (2004) Sulfatases: structure, mechanism, biological activity, inhibition, and synthetic utility. Angew. Chem. Int. Ed. Engl. 43, 5736–5763 [DOI] [PubMed] [Google Scholar]
- 10. Diez-Roux G., Ballabio A. (2005) Sulfatases and human disease. Annu. Rev. Genomics Hum. Genet. 6, 355–379 [DOI] [PubMed] [Google Scholar]
- 11. Dhoot G. K., Gustafsson M. K., Ai X., Sun W., Standiford D. M., Emerson C. P., Jr. (2001) Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science 293, 1663–1666 [DOI] [PubMed] [Google Scholar]
- 12. Myette J. R., Shriver Z., Claycamp C., McLean M. W., Venkataraman G., Sasisekharan R. (2003) The heparin/heparan sulfate 2-O-sulfatase from Flavobacterium heparinum. Molecular cloning, recombinant expression, and biochemical characterization. J. Biol. Chem. 278, 12157–12166 [DOI] [PubMed] [Google Scholar]
- 13. Myette J. R., Soundararajan V., Behr J., Shriver Z., Raman R., Sasisekharan R. (2009) Heparin/heparan sulfate N-sulfamidase from Flavobacterium heparinum: structural and biochemical investigation of catalytic nitrogen-sulfur bond cleavage. J. Biol. Chem. 284, 35189–35200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Myette J. R., Soundararajan V., Shriver Z., Raman R., Sasisekharan R. (2009) Heparin/heparan sulfate 6-O-sulfatase from Flavobacterium heparinum: integrated structural and biochemical investigation of enzyme active site and substrate specificity. J. Biol. Chem. 284, 35177–35188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hwa V., Salyers A. A. (1992) Analysis of two chondroitin sulfate utilization mutants of Bacteroides thetaiotaomicron that differ in their abilities to compete with the wild type in the gastrointestinal tracts of germfree mice. Appl. Environ. Microbiol. 58, 869–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sugahara K., Kojima T. (1996) Specificity studies of bacterial sulfatases by means of structurally defined sulfated oligosaccharides isolated from shark cartilage chondroitin sulfate D. Eur. J. Biochem. 239, 865–870 [DOI] [PubMed] [Google Scholar]
- 17. Berteau O., Guillot A., Benjdia A., Rabot S. (2006) A new type of bacterial sulfatase reveals a novel maturation pathway in prokaryotes. J. Biol. Chem. 281, 22464–22470 [DOI] [PubMed] [Google Scholar]
- 18. Benjdia A., Dehò G., Rabot S., Berteau O. (2007) First evidences for a third sulfatase maturation system in prokaryotes from E. coli aslB and ydeM deletion mutants. FEBS Lett. 581, 1009–1014 [DOI] [PubMed] [Google Scholar]
- 19. Benjdia A., Leprince J., Guillot A., Vaudry H., Rabot S., Berteau O. (2007) Anaerobic sulfatase-maturating enzymes: radical SAM enzymes able to catalyze in vitro sulfatase post-translational modification. J. Am. Chem. Soc. 129, 3462–3463 [DOI] [PubMed] [Google Scholar]
- 20. Benjdia A., Subramanian S., Leprince J., Vaudry H., Johnson M. K., Berteau O. (2008) Anaerobic sulfatase-maturating enzymes–first dual substrate radical S-adenosylmethionine enzymes. J. Biol. Chem. 283, 17815–17826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pierre S., Guillot A., Benjdia A., Sandström C., Langella P., Berteau O. (2012) Thiostrepton tryptophan methyltransferase expands the chemistry of radical SAM enzymes. Nat. Chem. Biol. 8, 957–959 [DOI] [PubMed] [Google Scholar]
- 22. Decamps L., Philmus B., Benjdia A., White R., Begley T. P., Berteau O. (2012) Biosynthesis of F0, precursor of the F420 cofactor, requires a unique two radical-SAM domain enzyme and tyrosine as substrate. J. Am. Chem. Soc. 134, 18173–18176 [DOI] [PubMed] [Google Scholar]
- 23. Benjdia A., Subramanian S., Leprince J., Vaudry H., Johnson M. K., Berteau O. (2010) Anaerobic sulfatase-maturating enzyme–a mechanistic link with glycyl radical-activating enzymes? FEBS J. 277, 1906–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karamanos N. K., Linhardt R. J. (2013) Special issue: Proteoglycans: signaling, targeting and therapeutics: introduction. FEBS J. 280, 2119. [DOI] [PubMed] [Google Scholar]
- 25. Loft K. J., Bojarová P., Slámová K., Kren V., Williams S. J. (2009) Synthesis of sulfated glucosaminides for profiling substrate specificities of sulfatases and fungal β-N-acetylhexosaminidases. Chembiochem 10, 565–576 [DOI] [PubMed] [Google Scholar]
- 26. Jaurand G., Tabeur C., Petitou M. (1994) Synthesis of the basic disaccharide unit of heparin. Carbohydr. Res. 255, 295–301 [DOI] [PubMed] [Google Scholar]
- 27. Lubineau A., Bonnaffe D. (1999) Access to molecular diversity in glycosaminoglycans: combinatorial synthesis of eight chondroitin sulfate disaccharides. Eur. J. Org. Chem. 2523–2532 [Google Scholar]
- 28. Ledin J., Staatz W., Li J. P., Götte M., Selleck S., Kjellén L., Spillmann D. (2004) Heparan sulfate structure in mice with genetically modified heparan sulfate production. J. Biol. Chem. 279, 42732–42741 [DOI] [PubMed] [Google Scholar]
- 29. Elsliger M. A., Deacon A. M., Godzik A., Lesley S. A., Wooley J., Wüthrich K., Wilson I. A. (2010) The JCSG high-throughput structural biology pipeline. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 1137–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Malleron A., Benjdia A., Berteau O., Le Narvor C. (2012) Chondroitin-4-O-sulfatase from Bacteroides thetaiotaomicron: exploration of the substrate specificity. Carbohydr. Res. 353, 96–99 [DOI] [PubMed] [Google Scholar]
- 31. Myette J. R., Shriver Z., Kiziltepe T., McLean M. W., Venkataraman G., Sasisekharan R. (2002) Molecular cloning of the heparin/heparan sulfate Δ4,5-unsaturated glycuronidase from Flavobacterium heparinum, its recombinant expression in Escherichia coli, and biochemical determination of its unique substrate specificity. Biochemistry 41, 7424–7434 [DOI] [PubMed] [Google Scholar]
- 32. Petitou M., Lormeau J. C., Perly B., Berthault P., Bossennec V., Sié P., Choay J. (1988) Is there a unique sequence in heparin for interaction with heparin cofactor II? Structural and biological studies of heparin-derived oligosaccharides. J. Biol. Chem. 263, 8685–8690 [PubMed] [Google Scholar]
- 33. Pervin A., Gallo C., Jandik K. A., Han X. J., Linhardt R. J. (1995) Preparation and structural characterization of large heparin-derived oligosaccharides. Glycobiology 5, 83–95 [DOI] [PubMed] [Google Scholar]
- 34. Guerrini M., Raman R., Venkataraman G., Torri G., Sasisekharan R., Casu B. (2002) A novel computational approach to integrate NMR spectroscopy and capillary electrophoresis for structure assignment of heparin and heparan sulfate oligosaccharides. Glycobiology 12, 713–719 [DOI] [PubMed] [Google Scholar]
- 35. Driguez P. A. (2013) Synthesis of natural and non-natural heparin fragments–from monosaccharides to complex glycoconjugates. Modern Synthetic Methods Carbohydr. Chem. 191–220 [Google Scholar]
- 36. Viskov C., Just M., Laux V., Mourier P., Lorenz M. (2009) Description of the chemical and pharmacological characteristics of a new hemisynthetic ultra-low-molecular-weight heparin, AVE5026. J. Thromb. Haemost. 7, 1143–1151 [DOI] [PubMed] [Google Scholar]
- 37. von Bülow R., Schmidt B., Dierks T., von Figura K., Usón I. (2001) Crystal structure of an enzyme-substrate complex provides insight into the interaction between human arylsulfatase A and its substrates during catalysis. J. Mol. Biol. 305, 269–277 [DOI] [PubMed] [Google Scholar]
- 38. de Beer T., Inui A., Tsuda H., Sugahara K., Vliegenthart J. F. (1996) Polydispersity in sulfation profile of oligosaccharide alditols isolated from the protein-linkage region and the repeating disaccharide region of chondroitin 4-sulfate of bovine nasal septal cartilage. Eur. J. Biochem. 240, 789–797 [DOI] [PubMed] [Google Scholar]
- 39. Maruyama T., Toida T., Imanari T., Yu G., Linhardt R. J. (1998) Conformational changes and anticoagulant activity of chondroitin sulfate following its O-sulfonation. Carbohydr. Res. 306, 35–43 [DOI] [PubMed] [Google Scholar]
- 40. Gargiulo V., Lanzetta R., Parrilli M., De Castro C. (2009) Structural analysis of chondroitin sulfate from Scyliorhinus canicula: a useful source of this polysaccharide. Glycobiology 19, 1485–1491 [DOI] [PubMed] [Google Scholar]
- 41. Raman R., Myette J. R., Shriver Z., Pojasek K., Venkataraman G., Sasisekharan R. (2003) The heparin/heparan sulfate 2-O-sulfatase from Flavobacterium heparinum. A structural and biochemical study of the enzyme active site and saccharide substrate specificity. J. Biol. Chem. 278, 12167–12174 [DOI] [PubMed] [Google Scholar]
- 42. Bond C. S., Clements P. R., Ashby S. J., Collyer C. A., Harrop S. J., Hopwood J. J., Guss J. M. (1997) Structure of a human lysosomal sulfatase. Structure 5, 277–289 [DOI] [PubMed] [Google Scholar]
- 43. Martens E. C., Koropatkin N. M., Smith T. J., Gordon J. I. (2009) Complex glycan catabolism by the human gut microbiota: The bacteroidetes Sus-like paradigm. J. Biol. Chem. 284, 24673–24677 [DOI] [PMC free article] [PubMed] [Google Scholar]