

Background: Mismatch repair is involved in the cellular response to DNA-alkylating agents.
Results: Alkylation damage causes mismatch repair-dependent apoptosis in human pluripotent stem cells (PSCs) in the first S phase after damage.
Conclusion: Human PSCs utilize a unique MMR-dependent damage response to alkylation damage.
Significance: Understanding how PSCs respond to DNA damage is crucial for their potential use in regenerative medicine.
Keywords: Apoptosis, DNA Damage Response, DNA Mismatch Repair, DNA Repair, Embryonic Stem Cell, Induced Pluripotent Stem Cells, Pluripotent Stem Cells
Abstract
Human pluripotent stem cells (PSCs) are presumed to have robust DNA repair pathways to ensure genome stability. PSCs likely need to protect against mutations that would otherwise be propagated throughout all tissues of the developing embryo. How these cells respond to genotoxic stress has only recently begun to be investigated. Although PSCs appear to respond to certain forms of damage more efficiently than somatic cells, some DNA damage response pathways such as the replication stress response may be lacking. Not all DNA repair pathways, including the DNA mismatch repair (MMR) pathway, have been well characterized in PSCs to date. MMR maintains genomic stability by repairing DNA polymerase errors. MMR is also involved in the induction of cell cycle arrest and apoptosis in response to certain exogenous DNA-damaging agents. Here, we examined MMR function in PSCs. We have demonstrated that PSCs contain a robust MMR pathway and are highly sensitive to DNA alkylation damage in an MMR-dependent manner. Interestingly, the nature of this alkylation response differs from that previously reported in somatic cell types. In somatic cells, a permanent G2/M cell cycle arrest is induced in the second cell cycle after DNA damage. The PSCs, however, directly undergo apoptosis in the first cell cycle. This response reveals that PSCs rely on apoptotic cell death as an important defense to avoid mutation accumulation. Our results also suggest an alternative molecular mechanism by which the MMR pathway can induce a response to DNA damage that may have implications for tumorigenesis.
Introduction
Human pluripotent stem cells (PSCs),2 including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), can replicate indefinitely in culture and give rise to all the different somatic cell lineages. These features make PSCs attractive for their potential use in regenerative therapy and as a useful model system for drug screening, genotoxicity testing, and general mechanistic studies of development. The role of these cells in the early stages of human development likely requires a strict maintenance of genome stability to protect the developing embryo from the damaging effects of mutations. Not surprisingly, some of the initial studies examining DNA repair pathways in PSCs indicate that they are highly efficient at removing DNA damage compared with somatic cells (1–4). DNA damage caused by γ irradiation, ultraviolet (UV) irradiation, H2O2, or the cross-linking reagent psoralen is repaired more rapidly in human ESC lines than in primary human fibroblasts (4).
However, in addition to damage repair, cells can respond to genotoxic stress through the induction of protective cell cycle checkpoints. As an example, impeded replication forks result in activation of an S phase checkpoint that leads to stabilization of the replication fork and coordination of DNA repair with the resumption of DNA synthesis (5). This important damage response protects the viability of the cell while at the same time reduces the incidence of broken chromosomes that can lead to genomic rearrangements. Interestingly, PSCs have been reported to lack this S phase checkpoint in response to replication stress (6). Rather PSCs upon encountering replication stress are much more prone to apoptosis. This same increased propensity to undergo apoptosis is also observed in PSCs treated with UV and γ irradiation (3, 7–10). Understanding the response of PSCs to different sources of genotoxic stress and the molecular mechanisms involved becomes crucial if these cells are to ever realize their potential for therapeutic purposes.
An important repair pathway that needs to be examined in PSCs is the DNA mismatch repair (MMR) pathway. MMR increases the fidelity of DNA replication by up to 3 orders of magnitude to maintain genome integrity through correcting DNA polymerase errors that escape proofreading (11–13). Loss of MMR function has been proposed to create a mutator phenotype in cells that increases the risk of tumorigenesis (14). Consistent with this hypothesis, germ line mutations in the major MMR genes are associated with the inherited cancer predisposition disease Lynch syndrome (15). Defects in MMR, mostly due to epigenetic inactivation of the MMR gene MLH1, have also been associated with 10–40% of sporadic colorectal and other cancer types (16, 17).
In addition to repairing DNA polymerase mistakes, the MMR pathway is also required for activation of cell cycle checkpoints and apoptosis in response to certain DNA-damaging agents (18). For example, MMR-deficient cells are up to 100-fold more resistant to the Sn1 alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) than isogenic MMR-proficient cells (19–21). Studies in multiple cell lines have revealed a consistent observation that treatment with MNNG induces an MMR-dependent G2 arrest in the second cell cycle after treatment (20, 22, 23). It is not clear why it takes two cell cycles to induce the G2 arrest. The primary cytotoxic lesion generated by MNNG is O6-methylguanine (MeG), which is commonly mispaired with T during replication. The MeG-T mispair is recognized by the MMR heterodimer MSH2-MSH6, which activates the MMR response (24). Two major models have been proposed to explain the molecular mechanism of this damage response. The “futile cycle” model suggests that the MeG-T mispair generated during the first S phase after treatment with MNNG initiates the MMR process. Successful MMR is executed, leading to excision of the mispaired T in the daughter strand. However, as the modified MeG remains in the template strand, the polymerase will regenerate a MeG-T mispair again during repair synthesis. The MMR process will be triggered repeatedly, resulting in an unreplicated gap opposite the lesion. In the next S phase, the new replication fork encounters this gap and converts it to a double strand break. It is this double strand break that initiates a DNA damage response that ultimately leads to cell cycle arrest and eventual apoptosis. The second model, the “direct signaling” model, suggests that following binding of the MeG-T mismatches by the MMR proteins a damage signal is transmitted directly to the checkpoint machinery without the need for DNA processing. Evidence supporting the direct signaling model includes findings that overexpression of MSH2 or MLH1 induces apoptosis in either MMR-proficient or -deficient cells (25) and that checkpoint kinases Chk1, Chk2, ataxia telangiectasia mutated and Rad3-related (ATR), and ataxia telangiectasia mutated (ATM) co-immunoprecipitate with MSH2 in cell extracts after MNNG treatment (26–29).
In this study, we examined the activity of the DNA MMR pathway in human PSCs. We were particularly interested in determining whether PSCs are capable of eliciting the MMR-dependent damage response to alkylation damage as observed in human cancer cell lines and other somatic cell types. Our results reveal that iPSCs and ESCs are hypersensitive to the alkylating agent MNNG, although the mechanism by which they respond to the DNA damage is different. Our results demonstrate that the MMR pathway is an important repair pathway for maintaining genome stability in human PSCs. These results also reveal the need for further studies to fully understand the mechanisms by which the MMR pathway can elicit a DNA damage response.
EXPERIMENTAL PROCEDURES
Cell Culture
Human ESCs (H1 and CT-2) were obtained from the University of Connecticut Stem Cell Core. Human YK26 iPSCs were reprogrammed from human dermal fibroblast (HDFa) cells using retroviral vectors as described (30), and Rx13 iPSCs were reprogrammed from BJ human foreskin fibroblasts (HFFs) using a single excisable polycistronic lentiviral stem cell cassette (STEMCCA) encoding the Yamanaka factors at the University of Connecticut Stem Cell Core facility. Both ESCs and iPSCs were cultured on BD Matrigel (BD Biosciences) with irradiated mouse embryonic fibroblast-conditioned ESC medium (GlobalStem) containing DMEM/F-12, 20% knock-out serum replacer (Invitrogen), non-essential amino acids (Invitrogen), 1 mm l-glutamine (Invitrogen), 0.1 mm β-mercaptoethanol (Sigma), and 4 ng/ml basic fibroblast growth factor (Invitrogen). HDFa cells (ATCC) and HFFs (ATCC) were cultured in DMEM containing 10% fetal bovine serum (FBS; Invitrogen) and non-essential amino acids. Hec59 cells (a kind gift from Drs. Thomas Kunkel and Alan Clark) were grown in DMEM/F-12 containing 10% FBS. HeLa cells (ATCC) were grown in DMEM containing 10% FBS.
Western Blotting
An equal number of H1, CT-2, YK26, Rx13, HDFa, HFF, HeLa, or Hec59 cells were harvested and lysed with radioimmune precipitation assay buffer supplemented with protease inhibitors. The cell lysates were separated by electrophoresis on a 6% SDS-polyacrylamide gel. The primary antibodies used included: anti-MSH2 (BD Biosciences 556349), anti-MSH6 (Bethyl Laboratories A300-023A), anti-MLH1 (BD Biosciences 550838), anti-PMS2 (BD Biosciences 556415), anti-proliferating cell nuclear antigen (Santa Cruz Biotechnology sc-56), anti-polymerase δ (Santa Cruz Biotechnology sc-10784), anti-RFC4 (Santa Cruz Biotechnology sc-20996), anti-replication protein A (Calbiochem RPA34-20), anti-phospho-Chk1 (Ser-345) (Cell Signaling Technology 2341), anti-phospho-Chk2 (Thr-68) (Cell Signaling Technology 2661), anti-Chk1 (Cell Signaling Technology 2345), anti-Chk2 (Cell Signaling Technology 2662), anti-γ H2AX, (Ser-139) (Millipore 05-636), anti-phospho-p53 (Ser-15) (Cell Signaling Technology 9284), anti-p53 (Cell Signaling Technology 9282), anti-phospho-ATM (Ser-1981) (Cell Signaling Technology 5883), anti-phospho-ATR (Ser-428) (Cell Signaling Technology 2853), anti-ATM (Cell Signaling Technology 2873), anti-ATR (Cell Signaling Technology 2790) and anti-actin (Sigma A5060). Where indicated, cells were treated with a 10 μm concentration of the ATM-specific inhibitor KU5593 (Selleck Chemicals) and/or the ATR-specific inhibitor VE-821 (Selleck Chemicals) for 24 h prior to harvesting.
MNNG Treatment and Cell Cycle Analysis
MNNG (obtained from the National Cancer Institute Chemical Carcinogen Reference Standard Repository; Chemical Abstracts Registry number 70-25-7) was dissolved in DMSO to a concentration of 10 mm and stored at −20 °C until use. O6-Benzylguanine (O6-BG; Chemical Abstracts Registry number 19916-73-5) was purchased from Sigma, dissolved in DMSO to a concentration of 25 mm, and stored at −80 °C until use. Cells were treated with 25 μm O6-BG for 2 h, and then medium was replaced with fresh medium containing 25 μm O6-BG and 2 μm MNNG for 48 h. Cell cycle analyses were performed using propidium iodide (PI) staining for DNA content and subsequent detection by flow cytometry. Briefly, cells were harvested and fixed in 70% ethanol at −20 °C. Cells were then treated with 20 μg/ml PI and 200 μg/ml RNase A and incubated at 37 °C for 1 h, filtered, and analyzed with a FACSCalibur flow cytometer (BD Biosciences). The resulting data were analyzed by Modfit analysis software.
MMR Knockdown
MMR knockdown YK26 cells were generated with lentiviral vectors containing shRNAs targeting either MSH2 or MLH1. sh-MSH2 and sh-MLH1 lentiviruses were a kind gift from Drs. Kareem Mohni and Sandra Weller. Briefly, YK26 cells were incubated with lentivirus containing sh-MSH2 or sh-MLH1 for 1 h, and then fresh medium was added to continue incubation overnight. Stable expression of the shRNAs was maintained by adding 0.8 μg/ml puromycin to the normal medium.
Annexin V Staining and Apoptosis Analysis
YK26 cells were treated with 2 μm MNNG for 24 h, then harvested, and stained with anti-Annexin V and PI using the Annexin V apoptosis kit (Molecular Probes v13241). The cells were analyzed with an LSRII flow cytometer (BD Biosciences). The resulting data were analyzed by FlowJo analysis software.
Cell Synchronization
Synchronization in mitosis was performed by treating YK26 cells with 0.2 μm nocodazole for 18 h. Cells were released in fresh medium containing 25 μm O6-BG. At 4 h postrelease, cells were treated with 2 μm MNNG for an additional 4 h. Cells were harvested at different time points as indicated and subjected to cell cycle analysis.
MMR Assay
The heteroduplex MMR substrate was prepared according to Zhou et al. (31). The p111 and p189 plasmids were a kind gift from Dr. Lu-Zhe Sun. p189 encodes for a premature stop codon in the enhanced GFP gene. To generate single-stranded DNA circles, p111 was nicked with Nb.Bpu10I (Thermo Scientific) and further digested with ExoIII (New England Biolabs). The heteroduplex substrate was prepared by annealing the single-stranded DNA circles to linearized, denatured p189 DNA. Excess linear DNA and single-stranded DNA were removed by plasmid-safe DNase (Epicenter Biotechnologies). To assess MMR activity, PSCs were transfected with 2.5 μg of the heteroduplex plasmid and 2.5 μg of pDsRed2-N1 (Clontech), which encodes the red fluorescent protein, using the Amaxa Human Stem Cell Nucleofector kit 2 (Lonza VPH-5022). HeLa cells were transfected using Lipofectamine2000 (Invitrogen), and HDFa cells were transfected using GeneIn transfection reagent (GlobalStem). After incubation for 48 h, the cells were harvested and analyzed for fluorescence intensity with an LSRII flow cytometer using BD FACSDiva software. The ratio of GFP-positive cells to red fluorescent protein-positive cells was determined to account for differences in transfection efficiency.
Immunofluorescent Staining
H1 cells with or without MNNG treatment were fixed with 4% paraformaldehyde for 10 min and permeabilized with cold acetone for 2 min. After blocking in 1% BSA in PBS for 1 h at room temperature, cells were incubated with the diluted primary antibodies anti-cleaved caspase-3 (BD Biosciences 559565) and anti-cleaved caspase-9 (Pierce PA5-17913) for 1 h at room temperature and then incubated with diluted Alexa Fluor 488 secondary antibody (Molecular Probes) for 45 min at room temperature. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and cells were analyzed on a Nikon Eclipse inverted fluorescence microscope.
RESULTS
The MMR Proteins Are Highly Expressed in PSCs Compared with Parental Fibroblasts
To begin characterizing the MMR pathway in iPSCs, we first examined the expression of the four major MMR proteins, MSH2, MSH6, MLH1, and PMS2. Whole cell extracts were prepared from an equal number of HDFa cells, HFFs, human ESCs (H1 and CT-2), and human iPSCs (YK26 reprogrammed from HDFa cells (30) and Rx13 reprogrammed from BJ foreskin fibroblasts). Consistent with previous reports of increased MMR gene expression in iPSCs (9), we showed that the expression of all four MMR proteins increased 5–8-fold in YK26 cells compared with the parental HDFa cells (Fig. 1A) and similarly increased in H1 and Rx13 cells compared with HFF cells (Fig. 1B). The expression between the different iPSCs and ESCs was similar (Fig. 1, A and B). PSCs undergo rapid cell division compared with both fibroblast lines, so we compared expression of the MMR proteins in the PSC lines with a more proliferative cell type. We found that the levels of MSH2, MSH6, and MLH1 in YK26 cells were 1.5–2-fold higher than in the MMR-proficient HeLa cervical cancer cells, whereas PMS2 levels were similar (Fig. 1C). These results suggest that PSCs may have a robust MMR system to protect their genome. We also found that essential replication proteins such as proliferating cell nuclear antigen, polymerase δ, and RFC4 were expressed at higher levels in PSCs compared with fibroblasts, although levels of replication protein A were similar between the cell types (Fig. 1D).
FIGURE 1.
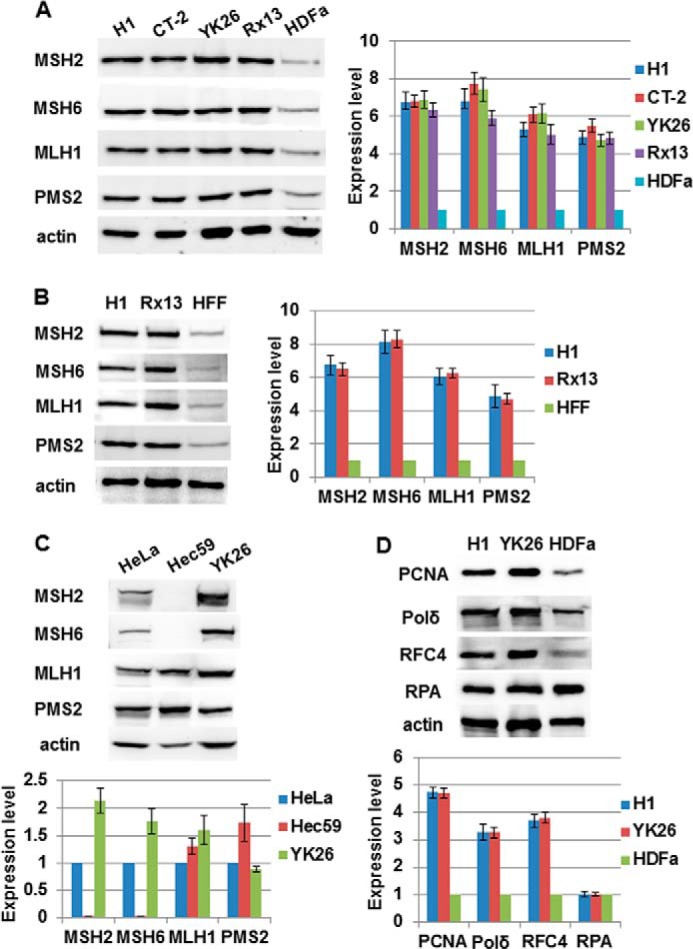
Human pluripotent stem cells express higher levels of MMR proteins than parental fibroblasts. A, Western blot analysis of MMR proteins in an equal number of human embryonic stem cells (H1 and CT-2), human induced pluripotent stem cells (YK26 and Rx13), and parental fibroblasts (HDFa). B, Western blot analysis of MMR proteins in an equal number of H1, Rx13, and HFF cells. C, Western blot analysis and quantitation of MMR proteins in HeLa, Hec59, and YK26 cells. D, Western blot analysis of various replication proteins in H1, YK26, and HDFa cells. The values represent the means of three independent experiments. Error bars represent S.E. Pol, polymerase; RPA, replication protein A; PCNA, proliferating cell nuclear antigen.
PSCs Repair Mismatches More Efficiently than Parental Fibroblasts
Considering the increased expression of MMR proteins in PSCs, we asked whether their single base pair mismatch repair capacity is enhanced compared with the parental fibroblasts. To test repair activity, we introduced a plasmid into cells that encodes GFP containing a single G-T mispair that disrupts protein translation (31). In vivo repair of the mismatch leads to restored GFP expression that can be quantitated using flow cytometry. As a control for transfection efficiency, cells were co-transfected with a red fluorescent protein-expressing plasmid. We found that the majority of transfected ESCs and iPSCs expressed GFP, indicating robust repair of the heteroduplex substrate (Fig. 2, A and B). This repair efficiency was significantly enhanced over that in parental HDFa cells. The repair rate in PSCs was similar to that in MMR-competent HeLa cells (Fig. 2, A and B). To confirm that restoration of GFP expression is MMR-dependent, we used lentiviral vectors encoding shRNAs to knock down levels of MSH2 or MLH1 in the YK26 cells (Fig. 2C). Knockdown of MSH2 or MLH1 also resulted in loss of stability of their obligate heterodimer partners MSH6 and PMS2, respectively. Knockdown of MSH2 did not affect levels of MLH1-PMS2, nor did MLH1 knockdown alter levels of MSH2-MSH6 (Fig. 2C). We found that the levels of repair in either MSH2 knockdown or MLH1 knockdown YK26 cells were 2–2.5-fold reduced compared with YK26 cells infected with a luciferase shRNA-expressing lentivirus (Fig. 2D). These results reveal that PSCs have robust MMR repair function compared with differentiated cell types that is similar to that observed in highly proliferative cancer cells.
FIGURE 2.
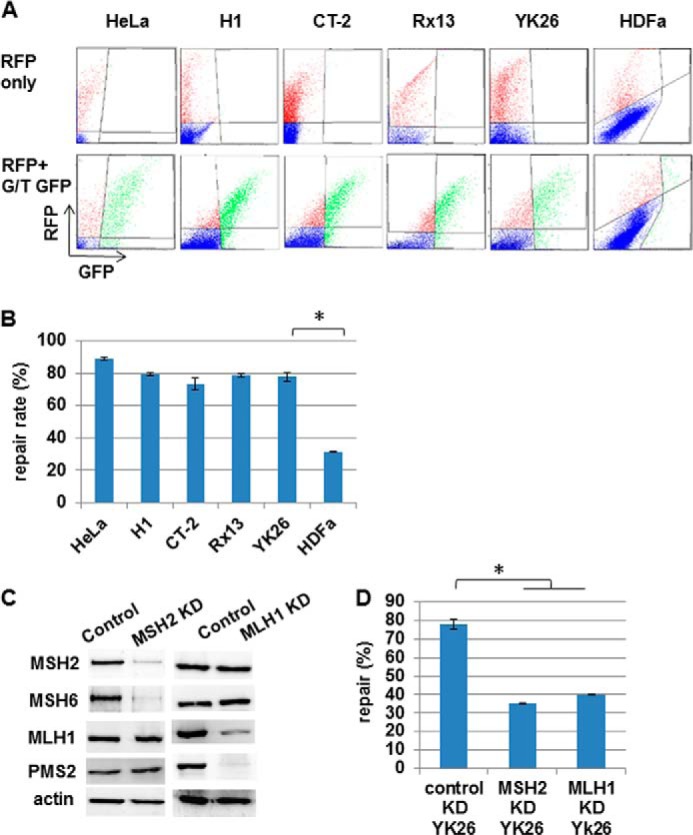
Human pluripotent stem cells repair mismatches more efficiently than parental fibroblasts. Repair of a transfected heteroduplex plasmid encoding GFP with a premature stop codon and red fluorescent protein (RFP) as a transfection control was measured in HeLa, H1, CT-2, YK26, Rx13, and HDFa cells. A, representative flow cytometry images. B, quantitation of repair rates in transfected cells. * represents p < 0.01. C, Western blot analysis confirming the knockdown of MSH2 or MLH1 in YK26. Actin is included as a loading control. D, the percentage of heteroduplex repair in control and MSH2 or MLH1 knockdown (KD) YK26 cells. The values represent the means of three independent experiments. * represents p < 0.01. Error bars represent S.E.
PSCs Are Hypersensitive to the Alkylating Agent MNNG
To test whether PSCs have the protective MMR-dependent response to alkylation damage, we treated ESCs and iPSCs along with HeLa cells (MMR-proficient), Hec59 endometrial cancer cells (MMR-deficient; see Fig. 1C), and HDFa cells with 2 μm MNNG for 48 h. Cells were pretreated with the methylguanine methyltransferase inhibitor O6-BG for 2 h to enhance the effects of the alkylation damage. The cells were examined by flow cytometry to determine whether MNNG induces a cell cycle arrest. Consistent with our previous studies (21, 23), HeLa cells were permanently arrested at G2/M after MNNG treatment, whereas no cell cycle arrest was observed in the MMR-deficient Hec59 cells (Fig. 3A). Surprisingly, although we did not see any evidence for a G2/M arrest in either the iPSC or ESC line, we observed large sub-G1 peaks consistent with the cells undergoing apoptosis (Fig. 3, A and B). The fibroblasts from which the YK26 iPSCs were derived did not show any apoptosis and only a modest G2/M arrest after MNNG treatment. As the HDFa cells replicate more slowly than PSCs and HeLa cells, we incubated them for an additional 72 h following treatment to ensure that the cells could finish the two cell cycles necessary to undergo a G2/M arrest consistent with the futile cycle model. We also tested a higher dose of MNNG. These changes led to slightly increased populations of cells in G2/M but still not the dramatic response observed in HeLa cells, suggesting that the MMR-dependent response to alkylation damage is not very strong in HDFa cells (Fig. 3C). Conversely, treatment of iPSCs with a 10-fold lower concentration of MNNG for 48 h still resulted in a substantial sub-G1 population (Fig. 3D). These results highlight the extent to which the iPSCs reactivate the alkylation damage response during reprogramming.
FIGURE 3.

DNA alkylation damage induces apoptosis in human pluripotent stem cells. A, representative cell cycle profiles of HeLa, Hec59, CT-2, YK26, and HDFa cell lines with or without 2 μm MNNG for 48 h as measured by flow cytometry. The arrows indicate the presence of sub-G1 populations associated with apoptotic cells. B, representative cell cycle profiles of H1 and Rx13 cells with or without 2 μm MNNG for 48 h. C, representative cell cycle profiles of HDFa cells with or without 2 or 5 μm MNNG for 5 days. D, representative cell cycle profiles of YK26 cells mock-treated or treated with 2 or 0.2 μm MNNG.
The Alkylation Damage Response Is MMR-dependent
We next tested whether the response to MNNG in PSCs is MMR-dependent by comparing the damage response between control and MMR knockdown iPSCs. We treated the control and MMR knockdown iPSCs with 2 μm MNNG and analyzed their cell cycle profiles. The MNNG-induced apoptotic response was entirely abrogated in the MSH2 or MLH1 knockdown line, suggesting that the hypersensitive response of PSCs to alkylation damage is MMR-dependent (Fig. 4A). To confirm that the sub-G1 populations observed in the cell cycle profiles are apoptotic PSCs, we used an apoptotic marker, Annexin V, to detect apoptotic cells. We found that after MNNG treatment most of the control YK26 cells were dually positive for Annexin V and PI, whereas a majority of the MSH2 or MLH1 knockdown YK26 cells were negative for Annexin V and PI staining (Fig. 4B). We also observed activation of caspase-9 and caspase-3 in MNNG-treated YK26 cells by immunofluorescence (Fig. 4C). These results demonstrate that PSCs respond to MNNG by inducing an intrinsic apoptotic pathway that is MMR-dependent.
FIGURE 4.

The apoptotic response to alkylation damage in induced pluripotent stem cells is mismatch repair-dependent. A, representative cell cycle profiles of control, MSH2 knockdown (KD), or MLH1 knockdown YK26 cells with or without 2 μm MNNG for 48 h by flow cytometry. B, Annexin V and PI staining of control, MSH2 knockdown, or MLH1 knockdown YK26 cells with or without 2 μm MNNG treatment for 24 h as analyzed by flow cytometry. C, immunofluorescence imaging of cleaved caspase-9 and cleaved caspase-3 in H1 cells with or without 2 μm MNNG treatment for 24 h.
MNNG-induced Apoptosis Occurs in the First S Phase after Damage without Undergoing G2 Arrest
Previous studies have shown that MNNG induces an MMR-dependent G2/M arrest in the second cell cycle after treatment in multiple somatic cell types, and this permanent G2/M arrest eventually leads to apoptosis (20, 22, 23). In both the ESCs and iPSCs, we observed apoptosis after MNNG treatment without any apparent G2/M arrest. We speculated that due to the rapid proliferation rate of PSCs it was possible the cells underwent a G2/M arrest prior to apoptosis that we failed to observe due to the timing of our experiment. To assess the timing of the response, we synchronized YK26 cells in mitosis with the microtubule inhibitor nocodazole. The cells were then released back into the cell cycle in normal growth medium. At 4 h postrelease when most cells were in G1 phase, we treated them with 2 μm MNNG for an additional 4 h. We returned the cells to normal medium again and harvested them at different time points for cell cycle profile analysis (Fig. 5). We observed that our mock-treated cells were beginning to enter S phase 8 h after release from the nocodazole block, and by 16 h, they had all cycled through to G2/M. By 24 h postrelease, the cells were continuing through the cell cycle in an asynchronous fashion. Similarly, our MNNG-treated cells were also entering S phase at the 8-h time point; however, we observed a fraction of the cells in a sub-G1 population. By 16 h, the treated cells remained mostly in S phase, suggesting a delay in progression through S phase compared with the untreated cells. A sub-G1 peak was also evident at 16 h. By 24 h, the sub-G1 peak had diminished, and the surviving cells continued through the cell cycle. To determine whether the cells incur a G2/M arrest after the second cell cycle following MNNG treatment, we harvested treated cells at 48 and 72 h postrelease but did not observe any cell cycle arrest. We confirmed that the observed apoptotic response to MNNG was MMR-dependent by repeating the synchronization experiments in our MMR knockdown YK26 cells. As observed in our asynchronous populations, the sub-G1 peak following MNNG treatment is absent in the MSH2 and MLH1 knockdown iPSCs (Fig. 5).
FIGURE 5.
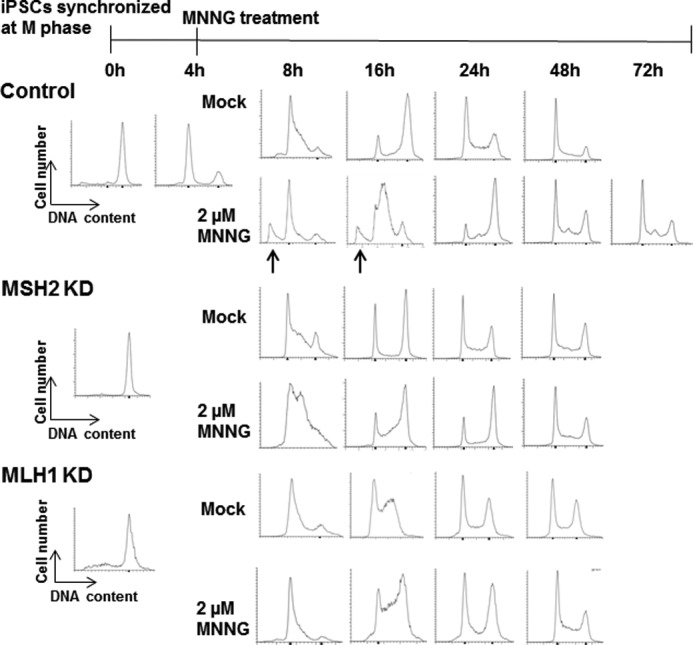
MNNG-induced apoptosis occurs in a mismatch repair-dependent manner in the first S phase after damage. Representative cell cycle profiles of control, MSH2 knockdown (KD), or MLH1 knockdown YK26 cells originally synchronized in mitosis by nocodazole were measured by flow cytometry. Cells were released into normal growth medium and then, with or without a 4 h treatment with 2 μm MNNG, harvested at different time points after release. The arrows indicate sub-G1 populations of cells. The absence of sub-G1 populations observed in MSH2 knockdown cells indicates a loss of the apoptotic response observed in control cells.
Unlike the 48-h MNNG treatment of PSCs that resulted in nearly 85% of the cells undergoing apoptosis (Fig. 4B), a 4-h treatment resulted in the death of only a fraction of the cells. To test whether we had selected for a population of cells that can tolerate this treatment level, we allowed the surviving cells to recover in normal growth medium for 24 h before subjecting them to a second round of MNNG treatment for 4 h. If the initial treatment led to a selection of resistant cells, we would not expect any response to the second round of MNNG. However, we once again observed that a similar fraction of cells displayed a sub-G1 peak indicative of apoptosis (Fig. 6). Taken together, these results suggest that PSCs undergo an immediate apoptotic response to MNNG in the first S phase after treatment without undergoing a G2/M arrest first. This result is very different from the response observed in HeLa and other somatic cell types.
FIGURE 6.
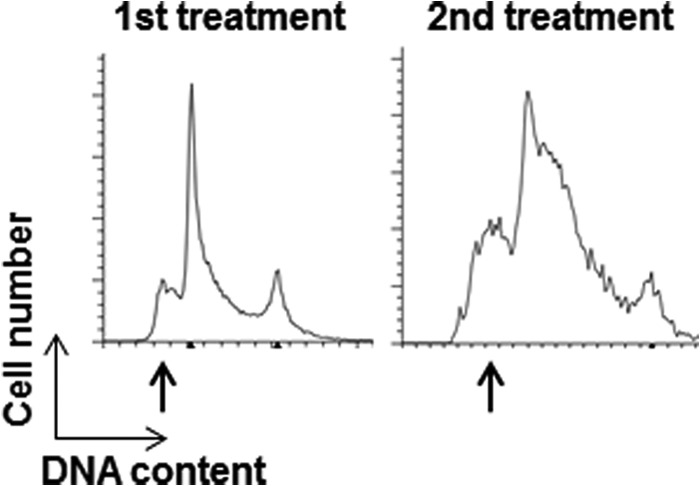
Pluripotent stem cells that survive MNNG treatment retain sensitivity to MNNG. Synchronized YK26 cells were treated with 2 μm MNNG for 4 h (1st treatment). Cells were allowed to recover in fresh medium for 24 h and then treated again with 2 μm MNNG for 4 h (2nd treatment). Cell cycle profiles of synchronized YK26 after the first and second treatments are displayed.
DNA Damage Checkpoint Kinases Are Not Activated in iPSCs in Response to MNNG
Previous studies of the MMR damage response indicate activation of the checkpoint kinases Chk1 and Chk2 following MNNG treatment that may be responsible for the cell cycle arrest and cell death observed (23, 32, 33). We wanted to test whether the checkpoint kinases are activated in PSCs after MNNG treatment. Asynchronous iPSCs or HeLa cells were mock-treated or treated with MNNG for 24 h and then harvested 1 day later. We found that treatment of HeLa cells with MNNG resulted in robust activation of Chk1 and Chk2 as indicated by phosphorylation of Ser-345 and Thr-68, respectively (Fig. 7A). No activation of either Chk1 or Chk2 was observed in iPSCs after MNNG treatment (Fig. 7A). To confirm that this response was consistent across multiple PSC lines, we performed similar experiments in Rx13 iPSCs and the two ESC lines. As in YK26 cells, MNNG treatment failed to activate Chk1 and Chk2 in this panel of PSCs (Fig. 7B). To rule out a transient activation of Chk1 or Chk2 early in the response to damage, we repeated the synchronization experiments described previously and analyzed cell extracts at different time points. We found no significant activation of Chk1 and Chk2 at any point during the cell cycle following MNNG treatment (Fig. 7, C and D). However, we did observe a strong induction of γH2AX that is indicative of double strand breaks and/or replicative stress in the first S phase coinciding with the sub-G1 peaks (Fig. 7, C and D). These results indicate that the typical checkpoint kinases activated during the MMR-dependent damage response in somatic cells are not involved in the PSC response to alkylation damage, again suggesting that a different damage response mechanism is utilized in these cells.
FIGURE 7.
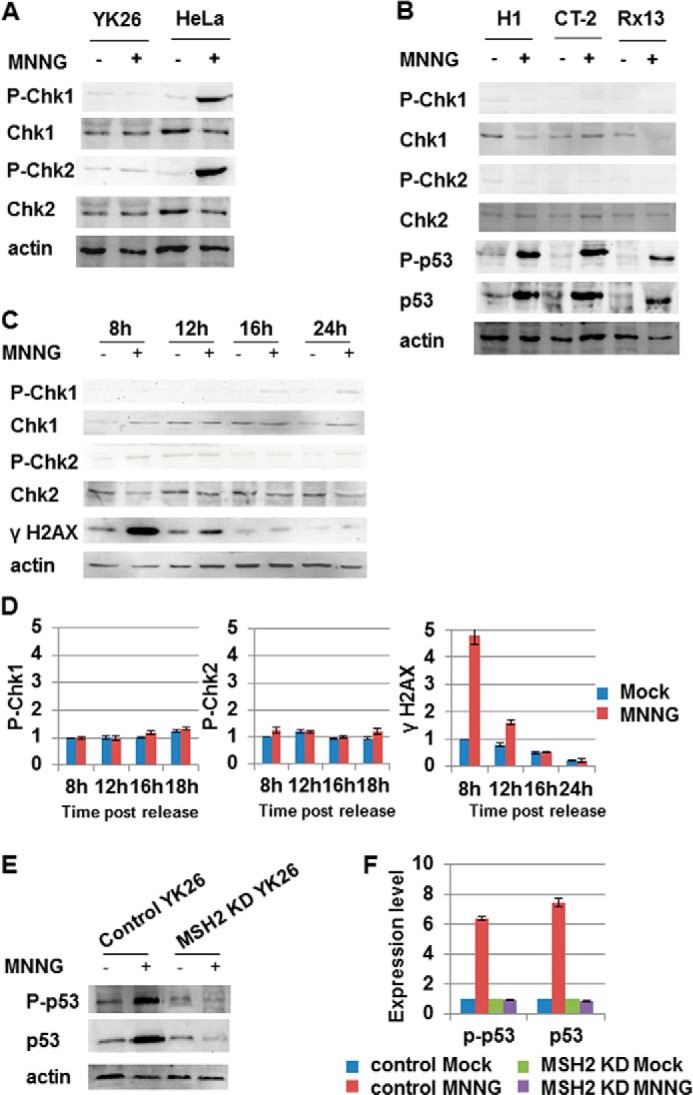
Chk1 and Chk2 are not activated in induced pluripotent stem cells in response to MNNG, but p53 is. A, Western blot analysis of phospho-Chk1, phospho-Chk2, total Chk1, and total Chk2 in HeLa and YK26 cells with or without 2 μm MNNG treatment for 24 h. B, Western blot analysis of phospho-Chk1, phospho-Chk2, total Chk1, total Chk2, phospho-p53, and total p53 in H1, CT-2, and Rx13 cells with or without 2 μm MNNG treatment for 24 h. C, Western blot analysis of phospho-Chk1, phospho-Chk2, total Chk1, total Chk2, and γH2AX in YK26 cells originally synchronized in mitosis with nocodazole and then, with or without a 4-h treatment with 2 μm MNNG, harvested at different time points after release. D, quantitation of Western blots represented in C. E, Western blot analysis of phospho-p53 (Ser-15) and total p53 in control or MSH2 knockdown (KD) YK26 cells with or without 2 μm MNNG treatment for 24 h. Actin is included as a loading control. F, quantitation of Western blots represented in E. The values represent the means of three independent experiments. Error bars represent S.E.
p53 Is Induced and Activated in iPSCs after MNNG Treatment
PSCs treated with the DNA-damaging agent etoposide undergo a rapid and extensive induction of apoptosis that is abrogated by knocking down p53 (34). To test whether p53 is activated in PSCs after MNNG treatment, we examined MNNG-treated iPSC lysates for increased levels of total p53 protein and increased phospho-p53 (Ser-15) levels. We found that both p53 and phospho-p53 levels were increased in iPSCs after a 24-h MNNG treatment; however, there was no induction or activation of p53 in MSH2 knockdown YK26 cells (Fig. 7, E and F). We observed a similar MNNG-induced activation of p53 in the other iPSC and ESC lines tested (Fig. 7B). These results indicate that MNNG treatment causes an MMR-dependent activation of p53 in PSCs that may be responsible for the apoptosis observed.
ATM and ATR Are Involved in the MNNG-induced Phosphorylation of p53
Activation of p53 following DNA damage can result from direct phosphorylation by the phosphatidylinositol 3-kinase-related kinases ATM (35, 36) and ATR (37). To test whether these phosphatidylinositol 3-kinase-related kinases were involved in the response to alkylation damage, we first examined MNNG-treated YK26 and H1 cells for the presence of Ser-1981 phosphorylation of ATM (38) and Ser-428 phosphorylation of ATR (39) as markers for DNA damage-dependent activation. We found that both ATM and ATR are phosphorylated following MNNG treatment of PSCs (Fig. 8A). ATR phosphorylation was similar to that observed in HeLa cells; however, ATM phosphorylation was not as enhanced in PSCs as in HeLa cells. We next asked whether inhibiting the activity of ATM or ATR affected p53 induction and phosphorylation. We treated H1 cells with the ATM-specific inhibitor KU55933 or the ATR-specific inhibitor VE-821 along with MNNG for 24 h and found that treatment with both inhibitors led to a partial reduction in the levels of damaged-induced total and phosphorylated p53 (Fig. 8B). When combining both inhibitors, we saw an additive effect as the levels of p53 activation were reduced even further than with either single agent alone. However, complete inhibition of p53 induction or phosphorylation was never observed, which may suggest that other kinases such as DNA-dependent protein kinase may be involved.
FIGURE 8.
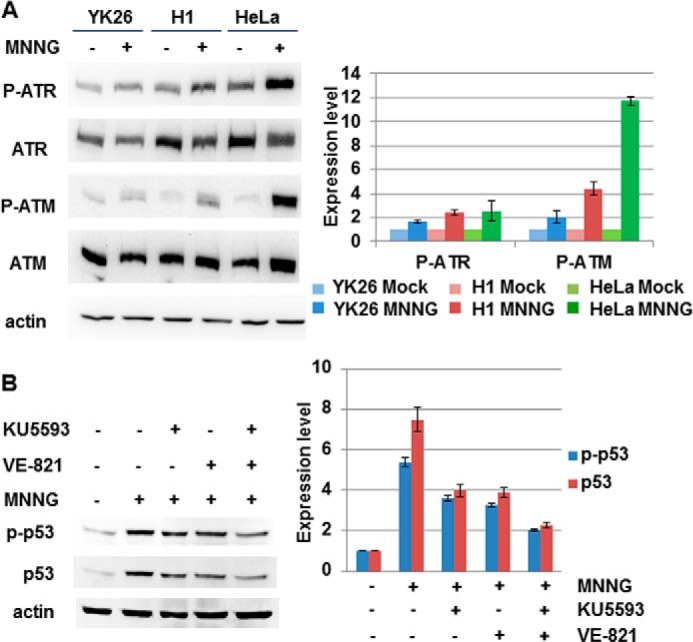
MNNG treatment leads to phosphorylation of ATR and ATM in pluripotent stem cells. A, Western blot analysis of phospho-ATM (Ser-1981), phospho-ATR (Ser-428), total ATM, and total ATR in H1, YK26, and HeLa cells with or without 2 μm MNNG treatment for 24 h. B, Western blot analysis of phospho-p53 and total p53 in H1 cells treated with 2 μm MNNG and the ATM-specific inhibitor KU5593, the ATR-specific inhibitor VE-821, or both for 24 h. The values represent the means of three independent experiments. Error bars represent S.E.
DISCUSSION
Our results show that PSCs, including both iPSCs and ESCs, have a robust MMR pathway to protect the stability of their genome. Expression of the four major MMR proteins is greatly enhanced compared with primary fibroblasts from which our iPSCs were derived and slightly enhanced compared with rapidly dividing HeLa cancer cells. This is consistent with the up-regulation of other DNA repair factors observed in PSCs compared with more differentiated cell types. Increased expression of factors involved in homologous recombination repair such as RAD51 and BRCA1, non-homologous end joining such as Ku70, and base excision repair such as uracil-DNA glycosylase and FEN1 has been reported in PSCs (4, 9, 40). The enhanced expression of DNA repair proteins in PSCs may underlie the increased repair efficiency observed in these cells. PSCs display accelerated repair of cyclobutane pyrimidine dimers caused by UV radiation, suggesting an enhanced nucleotide excision repair pathway (3, 4). Repair of modified bases caused by treatment with hydrogen peroxide or dimethyl sulfate is improved in PSCs compared with somatic cell types, suggesting enhanced base excision repair (3, 4). Double strand breaks caused by hydrogen peroxide or γ irradiation are repaired more efficiently in PSCs than in somatic cells (1, 3, 4, 40). Similarly, we detected high levels of single base pair mismatch correction by the MMR pathway in iPSCs and ESCs. Thus, the enhancement of DNA repair pathways appears to be an important strategy that PSCs utilize to protect their genome.
However, increased expression of repair factors may not always promote increased DNA repair. The levels of MMR proteins expressed in PSCs were slightly higher than in HeLa cells, but the repair activity in PSCs was slightly reduced compared with the cancer cells. These results may suggest that at the high expression levels observed in both PSCs and HeLa cells the amounts of the four major MMR proteins are no longer rate-limiting in the repair process. Localization of the MMR proteins to the mismatched template or the availability of other proteins involved in repairing the mismatch may be limiting. Alternatively, as 80% or more of the transfected mismatched template is repaired in PSCs, we may be reaching the limitations of the assay to discern repair efficiency.
Another important strategy used by PSCs to prevent mutation is an increased hypersensitivity to DNA damage. Increased apoptosis has been observed in PSCs treated with a variety of DNA-damaging agents, including UV irradiation (3, 7), γ irradiation (8–10, 40), cisplatin (6), and thymidine (6). Our results show that PSCs undergo massive apoptosis in response to the alkylating agent MNNG. Interestingly, Fig. 4B also reveals an increased level of background apoptosis in untreated iPSCs compared with MMR knockdown iPSCs. These results may suggest sensitivity to even endogenously generated DNA damage in an MMR-dependent manner. Alternatively, the high proliferation rate of PSCs may increase mismatch formation, which, if a certain threshold is reached, may result in replication stress due to the excessive MMR activity. As PSCs are particularly sensitive to replicative stress (6), this may lead to increased cell death. Although such a mechanism has not been described in somatic cells, yeast displaying a mutator phenotype due to mutations in polymerase δ have been shown to have prolonged S phase and evidence of a G2/M arrest consistent with replication stress signals due to increased mutation generation (41).
Whereas somatic cell types have also been shown to be sensitive to MNNG in an MMR-dependent fashion, we show here that the commitment to cell death occurs much more quickly in PSCs. Somatic cells treated with MNNG require two rounds of S phase following treatment, resulting in a G2/M arrest and eventually cell death. Why two cell cycles are necessary is not entirely clear; however, the futile cycle model suggests that MMR processing of MeG-T mismatches in the first S phase results in persistent unreplicated gaps that are converted to lethal double strand breaks in the second S phase (Fig. 9). One potential implication is that the two cell cycles provide an increased opportunity to resolve the primary MeG lesion. For example, cells suffering low levels of MeG damage may be protected against its mutagenic effects by the MMR pathway until methylguanine methyltransferase is able to remove the MeG lesion. Repair of the unreplicated region caused by MMR processing at a later time point would allow the cell to ultimately survive. Accumulating evidence suggests that somatic cells are capable of exiting S phase with incompletely replicated chromosomes and may be able to repair these regions during the subsequent cell cycle (42). Cells undergoing replicative stress are marked in the following G1 phase by large, 53BP1 foci that may play a role in shielding unreplicated gaps until they can be repaired during the next S phase (43, 44). Even if the unreplicated regions go unrepaired and are converted to double strand breaks, the breaks could be a substrate for homologous recombination repair, again leading to cell survival. Consistent with this model, depletion of the homologous recombination repair protein RAD51D in mouse embryonic fibroblasts resulted in a 5-fold-increased sensitivity to MNNG compared with wild-type cells (45). This increased sensitivity was alleviated when MLH1 was also depleted, indicative of homologous recombination repair playing a role in resolving secondary damage generated by MMR lesion processing. The immediate apoptotic response observed in PSCs suggests that these cells do not have any extra time to resolve the alkylation damage.
FIGURE 9.
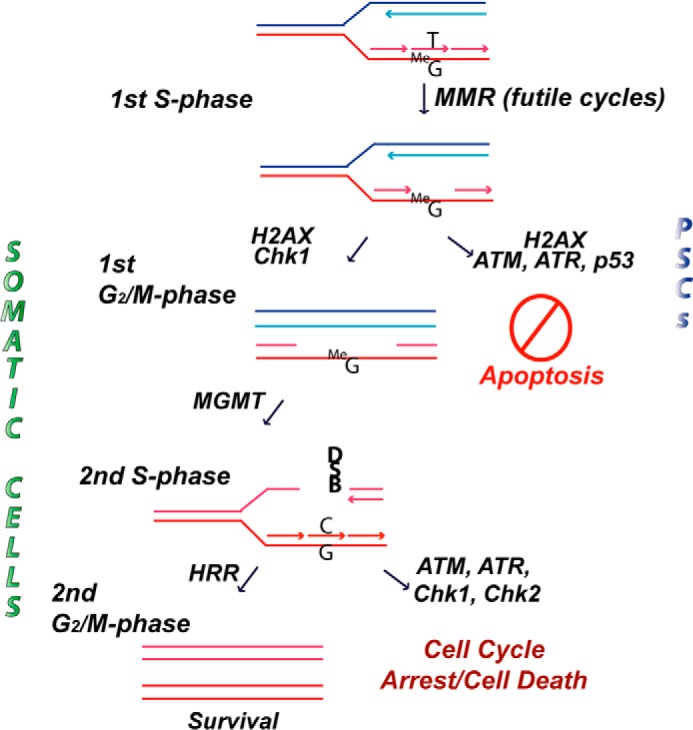
Model of the mismatch repair-dependent damage response to alkylation damage in somatic cells versus human pluripotent stem cells. Details are described in the text. HRR, homologous recombination repair; MGMT, methylguanine methyltransferase; DSB, double strand break.
One possibility that will require further investigation is that PSCs, unlike somatic cells, cannot tolerate perturbed S phase progression such as might occur during futile cycles of MMR (Fig. 9). PSCs have already been shown to lack intra-S phase checkpoints in response to replication stress that normally function to stabilize replication forks and allow for replication restart (6). In the current study, we observed a similar failure of PSCs to activate Chk1 in response to MNNG, thus missing a possibly important signaling pathway by which somatic cells survive MMR processing of MeG-T mismatches in the first S phase (32). Therefore, the futile MMR cycling at MeG-T mismatches may result in replication stress through either stalled proliferating cell nuclear antigen forks or the generation of unreplicated single-stranded gaps that lead to immediate apoptosis induction. Interestingly, we did see an increase in γH2AX activation in the first S phase after damage that has been associated with increased replication stress (46). Alternatively, the MMR proteins may be functioning through a direct signaling mechanism to recruit stress response proteins to the sites of damage. We have shown that MNNG results in the phosphorylation of ATM and ATR as well as the MMR-dependent stabilization and activation of p53, which, unlike in somatic cells (32), leads to cell death during the first S phase in PSCs. The explanation for this differing outcome between cell types may come from recent data showing that ESCs have an enhanced mitochondrial readiness for apoptosis compared with more differentiated cell types (47). Although p53 activation is similar between ESCs and differentiated cells following treatment with the radiomimetic drug neocarzinostatin, the balance of pro- and antiapoptotic proteins in ESCs is such that they are more prone to undergo apoptosis.
Our results raise interesting questions about the molecular pathways involved in the apoptotic response to MNNG in PSCs. Both ATM and ATR are involved in activating p53. Inhibiting both kinases reduced the level of p53 activation, although it did not eliminate p53 activation entirely. Whether the increased activation of p53 by multiple kinases is required to induce an apoptotic response or whether the overlap provides a fail-safe mechanism to ensure p53 activation and apoptosis upon damage is not clear. In addition, it is not clear why ATM and ATR activation is not accompanied by activation of Chk1 and Chk2. Determining whether Chk1 and Chk2 are prevented from being phosphorylated by the phosphatidylinositol 3-kinase-related kinases or whether they are phosphorylated and rapidly turned over is an important mechanistic question for determining how PSCs respond to replicative stress.
Understanding the mechanisms by which PSCs handle genotoxic stress will be extremely important if these cells are to realize their full potential as therapeutic agents in regenerative medicine. In addition, our results may provide insight into the role of the MMR pathway in preventing tumorigenesis. An important question that our studies raise is whether adult stem cells and cancer stem cells behave more like PSCs with regard to their MMR damage response or more like differentiated cells in culture. If they are similar to PSCs, the increased sensitivity to DNA damage may result in a strong selection pressure for loss of MMR function. We have previously proposed that colonic stem cells from Lynch syndrome patients, which are heterozygous for a given MMR gene, may be under selection pressure for loss of the remaining wild-type allele when exposed to DNA-damaging agents in the colonic environment, thus enhancing tumorigenesis (48). The damage response mechanism may also have implications for tumor response to therapy. If cancer stem cells do not share the same rapid apoptotic response to damage with PSCs, it is possible that their response may be made more similar to PSCs by priming the cells for apoptosis through the use of antiapoptotic protein inhibitors (47). More studies will be required to better understand the MMR damage response in multiple cell types; however, our results reveal the utility of using PSCs for drug response testing.
Acknowledgment
We thank Dr. Stormy J. Chamberlain for technical assistance with pluripotent stem cells.
This work was supported, in whole or in part, by National Institutes of Health Grant CA115783. This work was also supported by State of Connecticut Stem Cell Grant 13SCB-UCHC-06.
- PSC
- pluripotent stem cell
- MMR
- mismatch repair
- ESC
- embryonic stem cell
- iPSC
- induced pluripotent stem cell
- MNNG
- N-methyl-N′-nitro-N-nitrosoguanidine
- MeG
- O6-methylguanine
- ATR
- ataxia telangiectasia mutated and Rad3-related
- ATM
- ataxia telangiectasia mutated
- HDFa
- human dermal fibroblast
- HFF
- human foreskin fibroblast
- O6-BG
- O6-benzylguanine
- PI
- propidium iodide.
REFERENCES
- 1. Adams B. R., Golding S. E., Rao R. R., Valerie K. (2010) Dynamic dependence on ATR and ATM for double-strand break repair in human embryonic stem cells and neural descendants. PLoS One 5, e10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fung H., Weinstock D. M. (2011) Repair at single targeted DNA double-strand breaks in pluripotent and differentiated human cells. PLoS One 6, e20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Luo L. Z., Gopalakrishna-Pillai S., Nay S. L., Park S.-W., Bates S. E., Zeng X., Iverson L. E., O'Connor T. R. (2012) DNA repair in human pluripotent stem cells is distinct from that in non-pluripotent human cells. PLoS One 7, e30541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maynard S., Swistowska A. M., Lee J. W., Liu Y., Liu S.-T., Da Cruz A. B., Rao M., de Souza-Pinto N. C., Zeng X., Bohr V. A. (2008) Human embryonic stem cells have enhanced repair of multiple forms of DNA damage. Stem Cells 26, 2266–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Branzei D., Foiani M. (2009) The checkpoint response to replication stress. DNA Repair 8, 1038–1046 [DOI] [PubMed] [Google Scholar]
- 6. Desmarais J. A., Hoffmann M. J., Bingham G., Gagou M. E., Meuth M., Andrews P. W. (2012) Human embryonic stem cells fail to activate CHK1 and commit to apoptosis in response to DNA replication stress. Stem Cells 30, 1385–1393 [DOI] [PubMed] [Google Scholar]
- 7. Qin H., Yu T., Qing T., Liu Y., Zhao Y., Cai J., Li J., Song Z., Qu X., Zhou P., Wu J., Ding M., Deng H. (2007) Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J. Biol. Chem. 282, 5842–5852 [DOI] [PubMed] [Google Scholar]
- 8. Wilson K. D., Sun N., Huang M., Zhang W. Y., Lee A. S., Li Z., Wang S. X., Wu J. C. (2010) Effects of ionizing radiation on self-renewal and pluripotency of human embryonic stem cells. Cancer Res. 70, 5539–5548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Momcilovic O., Knobloch L., Fornsaglio J., Varum S., Easley C., Schatten G. (2010) DNA damage responses in human induced pluripotent stem cells and embryonic stem cells. PLoS One 5, e13410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Filion T. M., Qiao M., Ghule P. N., Mandeville M., van Wijnen A. J., Stein J. L., Lian J. B., Altieri D. C., Stein G. S. (2009) Survival responses of human embryonic stem cells to DNA damage. J. Cell Physiol. 220, 586–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kunkel T. A., Erie D. A. (2005) DNA mismatch repair. Annu. Rev. Biochem. 74, 681–710 [DOI] [PubMed] [Google Scholar]
- 12. Kolodner R. D., Marsischky G. T. (1999) Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 9, 89–96 [DOI] [PubMed] [Google Scholar]
- 13. Modrich P. (2006) Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 281, 30305–30309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fishel R., Kolodner R. D. (1995) Identification of mismatch repair genes and their role in the development of cancer. Curr. Opin. Genet. Dev. 5, 382–395 [DOI] [PubMed] [Google Scholar]
- 15. Lynch H. T., Lynch P. M., Lanspa S. J., Snyder C. L., Lynch J. F., Boland C. R. (2009) Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin. Genet. 76, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dietmaier W., Wallinger S., Bocker T., Kullmann F., Fishel R., Rüschoff J. (1997) Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res. 57, 4749–4756 [PubMed] [Google Scholar]
- 17. Kane M. F., Loda M., Gaida G. M., Lipman J., Mishra R., Goldman H., Jessup J. M., Kolodner R. (1997) Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 57, 808–811 [PubMed] [Google Scholar]
- 18. Stojic L., Brun R., Jiricny J. (2004) Mismatch repair and DNA damage signalling. DNA Repair 3, 1091–1101 [DOI] [PubMed] [Google Scholar]
- 19. Cejka P., Marra G., Hemmerle C., Cannavó E., Storchova Z., Jiricny J. (2003) Differential killing of mismatch repair-deficient and -proficient cells: towards the therapy of tumors with microsatellite instability. Cancer Res. 63, 8113–8117 [PubMed] [Google Scholar]
- 20. Kaina B., Ziouta A., Ochs K., Coquerelle T. (1997) Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex−, Mex+ and methylation-tolerant mismatch repair compromised cells: facts and models. Mutat. Res. 381, 227–241 [DOI] [PubMed] [Google Scholar]
- 21. Mastrocola A. S., Heinen C. D. (2010) Lynch syndrome-associated mutations in MSH2 alter DNA repair and checkpoint response functions in vivo. Hum. Mutat. 31, E1699–E1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cejka P., Stojic L., Mojas N., Russell A. M., Heinimann K., Cannavó E., di Pietro M., Marra G., Jiricny J. (2003) Methylation-induced G2/M arrest requires a full complement of the mismatch repair protein hMLH1. EMBO J. 22, 2245–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mastrocola A. S., Heinen C. D. (2010) Nuclear reorganization of DNA mismatch repair proteins in response to DNA damage. DNA Repair 9, 120–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Duckett D. R., Drummond J. T., Murchie A. I., Reardon J. T., Sancar A., Lilley D. M., Modrich P. (1996) Human MutSα recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc. Natl. Acad. Sci. U.S.A. 93, 6443–6447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang H., Richards B., Wilson T., Lloyd M., Cranston A., Thorburn A., Fishel R., Meuth M. (1999) Apoptosis induced by overexpression of hMSH2 or hMLH1. Cancer Res. 59, 3021–3027 [PubMed] [Google Scholar]
- 26. Adamson A. W., Beardsley D. I., Kim W. J., Gao Y., Baskaran R., Brown K. D. (2005) Methylator-induced, mismatch repair-dependent G2 arrest is activated through Chk1 and Chk2. Mol. Biol. Cell 16, 1513–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Y., Fang Y., Shao H., Lindsey-Boltz L., Sancar A., Modrich P. (2010) Interactions of human mismatch repair proteins MutSα and MutLα with proteins of the ATR-Chk1 pathway. J. Biol. Chem. 285, 5974–5982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Y., Qin J. (2003) MSH2 and ATR form a signaling module and regulate two branches of the damage response to DNA methylation. Proc. Natl. Acad. Sci. U.S.A. 100, 15387–15392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoshioka K., Yoshioka Y., Hsieh P. (2006) ATR kinase activation mediated by MutSα and MutLα in response to cytotoxic O6-methylguanine adducts. Mol. Cell 22, 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zeng H., Guo M., Martins-Taylor K., Wang X., Zhang Z., Park J. W., Zhan S., Kronenberg M. S., Lichtler A., Liu H.-X., Chen F.-P., Yue L., Li X.-J., Xu R.-H. (2010) Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS One 5, e11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou B., Huang C., Yang J., Lu J., Dong Q., Sun L. Z. (2009) Preparation of heteroduplex EGFP plasmid for in vivo mismatch repair activity assay. Anal. Biochem. 388, 167–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Noonan E. M., Shah D., Yaffe M. B., Lauffenburger D. A., Samson L. D. (2012) O-6-Methylguanine DNA lesions induce an intra-S-phase arrest from which cells exit into apoptosis governed by early and late multi-pathway signaling network activation. Integr. Biol. 4, 1237–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stojic L., Mojas N., Cejka P., Di Pietro M., Ferrari S., Marra G., Jiricny J. (2004) Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 18, 1331–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grandela C., Pera M. F., Grimmond S. M., Kolle G., Wolvetang E. J. (2007) p53 is required for etoposide-induced apoptosis of human embryonic stem cells. Stem Cell Res. 1, 116–128 [DOI] [PubMed] [Google Scholar]
- 35. Banin S., Moyal L., Shieh S.-Y., Taya Y., Anderson C. W., Chessa L., Smorodinsky N. I., Prives C., Reiss Y., Shiloh Y., Ziv Y. (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674–1677 [DOI] [PubMed] [Google Scholar]
- 36. Canman C. E., Lim D.-S., Cimprich K. A., Taya Y., Tamai K., Sakaguchi K., Appella E., Kastan M. B., Siliciano J. D. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677–1679 [DOI] [PubMed] [Google Scholar]
- 37. Tibbetts R. S., Brumbaugh K. M., Williams J. M., Sarkaria J. N., Cliby W. A., Shieh S. Y., Taya Y., Prives C., Abraham R. T. (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 13, 152–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bakkenist C. J., Kastan M. B. (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 [DOI] [PubMed] [Google Scholar]
- 39. Liu S., Shiotani B., Lahiri M., Maréchal A., Tse A., Leung C. C., Glover J. N., Yang X. H., Zou L. (2011) ATR autophosphorylation as a molecular switch for checkpoint activation. Mol. Cell 43, 192–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fan J., Robert C., Jang Y. Y., Liu H., Sharkis S., Baylin S. B., Rassool F. V. (2011) Human induced pluripotent cells resemble embryonic stem cells demonstrating enhanced levels of DNA repair and efficacy of nonhomologous end-joining. Mutat Res. 713, 8–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Venkatesan R. N., Hsu J. J., Lawrence N. A., Preston B. D., Loeb L. A. (2006) Mutator phenotypes caused by substitution at a conserved motif A residue in eukaryotic DNA polymerase delta. J. Biol. Chem. 281, 4486–4494 [DOI] [PubMed] [Google Scholar]
- 42. Mankouri H. W., Huttner D., Hickson I. D. (2013) How unfinished business from S-phase affects mitosis and beyond. EMBO J. 32, 2661–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Harrigan J. A., Belotserkovskaya R., Coates J., Dimitrova D. S., Polo S. E., Bradshaw C. R., Fraser P., Jackson S. P. (2011) Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 193, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lukas C., Savic V., Bekker-Jensen S., Doil C., Neumann B., Pedersen R. S., Grøfte M., Chan K. L., Hickson I. D., Bartek J., Lukas J. (2011) 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 13, 243–253 [DOI] [PubMed] [Google Scholar]
- 45. Rajesh P., Rajesh C., Wyatt M. D., Pittman D. L. (2010) RAD51D protects against MLH1-dependent cytotoxic responses to O6-methylguanine. DNA Repair 9, 458–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sirbu B. M., Couch F. B., Feigerle J. T., Bhaskara S., Hiebert S. W., Cortez D. (2011) Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 25, 1320–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu J. C., Guan X., Ryan J. A., Rivera A. G., Mock C., Agrawal V., Letai A., Lerou P. H., Lahav G. (2013) High mitochondrial priming sensitizes hESCs to DNA-damage-induced apoptosis. Cell Stem Cell 13, 483–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Heinen C. D., Schmutte C., Fishel R. (2002) DNA repair and tumorigenesis: lessons from hereditary cancer syndromes. Cancer Biol. Ther. 1, 477–485 [DOI] [PubMed] [Google Scholar]