

Background: How can HCV require only 10 proteins for decades-long evasion of the immune system?
Results: Phosphorylation of the intrinsically disordered domain (IDD) of NS5A changes its dynamics, inducing unique structure and function.
Conclusion: IDD phosphorylation expands the HCV proteome.
Significance: Post-translational modification of a viral IDD represents a strategy to expand a viral proteome when coding capacity is limited.
Keywords: Hepatitis C Virus (HCV), Intrinsically Disordered Protein, Phosphorylation, RNA Virus, Viral Replication, NMR, Non-structural Protein 5A
Abstract
The human proteome contains myriad intrinsically disordered proteins. Within intrinsically disordered proteins, polyproline-II motifs are often located near sites of phosphorylation. We have used an unconventional experimental paradigm to discover that phosphorylation by protein kinase A (PKA) occurs in the intrinsically disordered domain of hepatitis C virus non-structural protein 5A (NS5A) on Thr-2332 near one of its polyproline-II motifs. Phosphorylation shifts the conformational ensemble of the NS5A intrinsically disordered domain to a state that permits detection of the polyproline motif by using 15N-, 13C-based multidimensional NMR spectroscopy. PKA-dependent proline resonances were lost in the presence of the Src homology 3 domain of c-Src, consistent with formation of a complex. Changing Thr-2332 to alanine in hepatitis C virus genotype 1b reduced the steady-state level of RNA by 10-fold; this change was lethal for genotype 2a. The lethal phenotype could be rescued by changing Thr-2332 to glutamic acid, a phosphomimetic substitution. Immunofluorescence and transmission electron microscopy showed that the inability to produce Thr(P)-2332-NS5A caused loss of integrity of the virus-induced membranous web/replication organelle. An even more extreme phenotype was observed in the presence of small molecule inhibitors of PKA. We conclude that the PKA-phosphorylated form of NS5A exhibits unique structure and function relative to the unphosphorylated protein. We suggest that post-translational modification of viral proteins containing intrinsic disorder may be a general mechanism to expand the viral proteome without a corresponding expansion of the genome.
Introduction
Hepatitis C virus (HCV)2 establishes chronic infections in humans that can persist for decades before causing any clinical manifestations. The ability of a positive-strand RNA virus with such limited coding capacity to so effectively evade host defenses is extraordinary. A unique feature of HCV, relative to most acute RNA viruses of similar genetic size and encoded functions, is nonstructural protein 5A (NS5A). NS5A is a two-domain protein. The amino-terminal domain can form at least two structurally distinct homodimers (1, 2). One of these dimers binds to RNA, with a preference for GU-rich RNA (3, 4). The carboxyl-terminal domain is an intrinsically disordered domain (IDD) (5, 6) that contains numerous putative sites of phosphorylation and has been reported to bind dozens of cellular proteins (7). The ability of NS5A to antagonize and/or to hijack numerous cellular pathways may be a key determinant of HCV persistence.
Specific combinations of post-translational modifications, including phosphorylation, in the IDD of p53 confer the ability to interface with myriad cellular pathways (8). Conformational sampling of an IDD leads to multiple, unique structures capable of unique interactions (8). Although empirical examples are quite limited, the thought is that the addition of a post-translational modification to a specific location will restrict conformations sampled, thus channeling the IDD toward a single structure or subset of structures. Therefore, the interaction of NS5A with so many proteins may rely on a similar mechanism, phosphorylation-dependent acquisition of structure.
Several observations suggest an important role for NS5A phosphorylation in HCV multiplication and/or pathogenesis. First, persistent replication of the prototypical genotype 1b subgenomic replicon RNA in Huh-7 cells and sublines thereof requires an adaptive mutation that changes Ser-2204 of NS5A to Ile (9). The S2204I substitution prevents formation of the hyperphosphorylated/p58 form of NS5A (9). It is becoming increasingly clear that full-length genomes encoding adaptive mutations fail to cause disease in chimpanzees (10, 11), consistent with NS5A phosphorylation contributing to pathogenesis. Inhibitors of host cell kinases have been shown to impact HCV replication, presumably due to inhibition of NS5A phosphorylation (12, 13). In addition, the most potent direct acting anti-HCV drug reported to date, daclatasvir, targets NS5A and alters the phosphorylation state of the protein (14, 15). Finally, a clear connection has been made between casein kinase II phosphorylation in the carboxyl-terminal region of the IDD and viral assembly (16, 17), although a molecular explanation for the gain of function caused by phosphorylation remains a mystery. Therefore, knowledge of the kinases responsible for NS5A phosphorylation and the corresponding sites of phosphorylation (the NS5A phosphoproteome) is essential to understand the role of individual phosphorylation states during the HCV life cycle and to test the hypothesis that inhibitors of NS5A may function by perturbing one or more phosphorylated states of NS5A.
The inability to directly reveal the NS5A phosphoproteome (e.g. by using mass spectrometry to analyze NS5A isolated from HCV-infected hepatocytes) demands an alternative experimental approach. Here we define a new experimental paradigm to study phosphorylation of NS5A. We show that protein kinase A (PKA) phosphorylation occurs near the poly-proline-II (PPII) motif of NS5A, leading to changes in the conformational sampling of this SH3 binding determinant. This phosphorylation-dependent change in NS5A dynamics is important for replication because it contributes to the integrity of the replication organelle. We suggest that phosphorylation and other post-translational modifications of viral IDPs represent an important strategy for expanding the functional proteome of viruses with limited coding capacity.
EXPERIMENTAL PROCEDURES
Construction of Recombinant NS5A Plasmids
NS5A 2005–2419 was generated as described previously (18). Oligonucleotides 1 and 2 (Table 1) were used with pET26-Ub-Δ32-NS5A-C(His) (encoding residues 2005–2419 of the polyprotein) to amplify residues 2194–2419. The NS5A deletion mutant 2005–2306 was created using oligonucleotides 3 and 4 (Table 1). NS5A containing ΔP3, T2332A, or T2332E was amplified using oligonucleotides 2 and 3 (Table 1) on pHCVbart.rep1/Ava-II-ΔP3 (3), pHCVbart.rep1/Ava-II-T2332A, or pHCVbart.rep1/Ava-II-T2332E. The PCR fragments were digested and ligated into pET26Ub-CHIS vector. All plasmid constructs were verified by restriction enzyme digestion and DNA sequencing.
TABLE 1.
Oligonucleotides used in this study
| Number | Name | Sequence |
|---|---|---|
| 1 | HCV-NS5A-2194-NcoI-for | 5′-CGC GCC ATG GAT CCT CTG GTT CTC CCC CCT CCT TGG CC-3′ |
| 2 | HCV-NS5A-HindIII-rev | 5′-GGT ACC AAG CTT CTA TTA GCA GCA GAC GAC GTC CTC ACT-3′ |
| 3 | HCV-Δ32-NS5A-NcoI- for | 5′-GCG GGT ACC CCA TGG ATC CTC TGG TGG AGT CCC CTT CTT-3′ |
| 4 | HCV-NS5A- 2307-HindIII-rev | 5′-GGC AAG CTT CTA TTA GTA GTC CGG GTC CTT CCA-3′ |
| 5 | HCV-NS5A-T2332A-for | 5′-CCA CGG AGG AAG AGG GCG GTT GTC CTG TCA GAA-3′ |
| 6 | HCV-NS5A-T2332A-rev | 5′-TTC TGA CAG GAC AAC CGC CCT CTT CCT CCG TGG-3′ |
| 7 | HCV-NS5A-XhoI-for | 5′-GCG AAA TTC CCT CGA GCG ATG CCC ATA-3′ |
| 8 | HCV-NS5B-MfeI-rev | 5′-GCG GGT GGT GTC AAT TGG TGT CTC-3′ |
| 9 | HCV-NS5A-T2332E-for | 5′-CCA CGG AGG AAG AGG GAG GTT GTC CTG TCA GAA-3′ |
| 10 | HCV-NS5A-T2332E-rev | 5′-TTC TGA CAG GAC AAC CTC CCT CTT CCT CCG TGG-3′ |
| 11 | HCV-rep-RT-PCR-for | 5′-GGA AGC GGT CAG CCC AT-3′ |
| 12 | HCV-rep-RT-PCR-rev | 5′-GCG TTG GCT ACC CGT GAT-3′ |
| 13 | HCV-Northern-for | 5′-GGG TGC TTG CGA GTG CC-3′ |
| 14 | HCV-Northern-rev | 5′-GGC CAG TAA CGT TAG GGG-3′ |
Construction of NS5A Subgenomic Replicon Plasmids
Oligonucleotides 5–8 (Table 1) were used to perform overlap extension PCR on pHCVbart.rep1/Ava-II to create the T2332A derivative. The overlap PCR fragment was digested and ligated into vector to generate pHCVbart.rep1/Ava-II-T2332A (T2332A). The T2332E derivative, pHCVbart.rep1/Ava-II-T2332E, was constructed with oligonucleotides 7–10 (Table 1) in the same manner as the T2332A derivative. Both Thr-2332 derivatives were also cloned in the context of the NS5A cell culture adaptive mutation S2204I (SI), to generate SI/T2332A and SI/T2332E, using pHCVbart.rep1/Ava-II-SI (3), as described above.
JC1 Reporter Plasmid Constructs
The plasmids pJC1/GLuc2A and pJC1/GLuc2A(GNN) were described previously (19). To construct the pJC1/GLuc2A(T2332A) and (T2332E) replicons, a 3,676-bp fragment from pYSGR-JFH(T2332A) and T2332E was subcloned into pJC1/GLuc2A using common SpeI and BsrGI restriction sites, generating pJC1/GLuc2A(T2332A) and (T2332E) replicons.
Expression and Purification of NS5A, NS3, and NS5B Proteins
All NS5A, NS3, and NS5B proteins (genotype 1b) used in this study were expressed and purified as described previously (4, 18, 20, 21).
In Vitro Phosphorylation Assays
PKA- or CK2-mediated phosphorylation of HCV non-structural proteins was performed in 50 mm HEPES, pH 7.5, 0.5 mm tris(2-carboxyethyl)phosphine, 20 mm MgCl2, 100 mm NaCl, 125 μm ATP, 0.5 μCi/μl [γ-32P]ATP (MP Biomedicals), and 1 μm NS3, NS5A, NS5A derivative, or NS5B. For reactions that did not require the use of radiolabeled ATP, the [γ-32P]ATP was omitted. Reactions performed in the presence of PKA inhibitors or NS5A inhibitor BMS-790052 were performed by incubating 5 nm PKA with inhibitor at 37 °C for 30 min, after which point NS5A was added to 0.5 μm, and phosphorylation proceeded for the specified time. The phosphorylation reaction was quenched with an equal volume of 2× SDS-PAGE sample buffer. The samples were resolved by 8% SDS-PAGE. For reactions containing radiolabeled ATP, the gels were analyzed with the Typhoon phosphorimager (GE Healthcare) and quantified with ImageQuant software. The amount of phosphorylation was normalized to the amount of radioactivity present in each lane. Gels of reactions not using radiolabeled ATP were analyzed by Western blot. PKA-phosphorylated NS5A IDD used for NMR was prepared as described above but with 100 μm isotopically labeled IDD, 250 μm ATP, and 10 μm PKA. Reactions proceeded for 60 min. Phosphorylation reactions were then desalted using a Zeba column pre-equilibrated with NMR buffer.
Mass Spectrometric Mapping of Phosphorylation Site
Analysis of the PKA-phosphorylated residues on NS5A was performed by mass spectrometry with 8 μg of NS5A phosphorylated by PKA. A sample of the quenched reaction was resolved by 8% SDS-PAGE and stained with Pro-Q Diamond phosphoprotein gel stain (Invitrogen) to verify that NS5A had been phosphorylated by PKA. The remaining reaction was resolved by 8% SDS-PAGE, and the NS5A band was visualized by Coomassie staining. Peptides obtained from in-gel trypsin digestion of the Coomassie-stained band corresponding to in vitro phosphorylated NS5A were analyzed by LC/MS/MS. Tandem MS spectra obtained by collision-induced dissociation were acquired using an LC/MS/MS system that consisted of a Surveyor HPLC pump, a Surveyor Micro AS autosampler, and an LTQ linear ion trap mass spectrometer (ThermoFinnigan). The acquired spectra were processed using Xcalibur version 2.0. The raw tandem MS spectra were also converted to Mascot generic files (.mgf). Detection and mapping of the phosphorylation site were achieved by database searching of tandem mass spectra of the proteolytic peptides against a current Swiss-Prot protein database using the Bioworks version 3.2 and Mascot version 2.2 (Matrix Science) search engines. The database searches were performed with cysteine carbamidomethylation as a fixed modification. The variable modifications were methionine oxidation (+16 Da) and phosphorylation (+80 Da) of Ser, Thr, and Tyr residues. Up to two missed cleavages were allowed for trypsin digestion. Peptide mass tolerance and fragment mass tolerance were set to 3 and 2 Da, respectively. Tandem MS spectra that are matched to phosphorylated peptides were manually evaluated at the raw data level with the consideration of overall data quality, signal/noise ratio of matched peaks, and the presence of dominant peaks that did not match to any theoretical m/z value.
NS5A Alignment
HCV sequence alignments were performed using the Los Alamos HCV Sequence Database (22). Genotype references of the NS5A protein sequence were compared to determine the conservation of the PKA phosphorylation site, residue Thr-2332.
Calculation of IC50 Values of PKA Inhibitors
Phosphorylation of NS5A in the presence of specified PKA inhibitors was performed in 50 mm HEPES, pH 7.5, 0.5 mm tris(2-carboxyethyl)phosphine, 20 mm MgCl2, 100 mm NaCl, 50 μm ATP, 0.5 μCi/μl [γ-32P]ATP, and 0.005 μm PKA. Reactions were incubated with 0–50 μm PKA inhibitor H-7 dihydrochloride, H-89 dihydrochloride, KT-5720, or the myristoylated peptide fragment 14–22 (Sigma), each in a final DMSO concentration of 2.5% in the reaction, at 37 °C for 30 min. After 30 min, NS5A was added to 0.5 μm, and phosphorylation proceeded for 30 min. The phosphorylation reaction was quenched with 2× SDS-PAGE sample buffer. The samples were resolved by 8% SDS-PAGE. Data were fit to a hyperbolic equation to calculate the respective IC50 for each inhibitor.
Antibodies
Rabbit polyclonal anti-NS5A, anti-NS3, and anti-NS5B were generated using purified protein immunogens by Covance, Inc. for our use. Rabbit polyclonal phospho-specific anti-Thr(P)-2332-NS5A was generated using the synthetic peptide PPRRKRpTVVLSESC conjugated to keyhole limpet hemocyanin by Covance Research Products, Inc. for our use. Mouse monoclonal anti-NS5A was purchased from Advanced Immunochemical Services, Inc. Rabbit polyclonal anti-NS4B was a gift from Dr. Kouacou Konan (Albany Medical College). Rabbit polyclonal anti-giantin and mouse monoclonal anti-β-actin were purchased from Abcam. Mouse monoclonal anti-PKAc was purchased from BD Transduction Laboratories. Goat monoclonal anti-calnexin was purchased from Origene. Alexa Fluor 488 goat anti-mouse, Alexa Fluor 488 goat anti-rabbit, Alexa Fluor 594 goat anti-rabbit, and Alexa Fluor 594 donkey anti-mouse IgG were purchased from Invitrogen. The goat anti-rabbit HRP, goat anti-rabbit AP, goat anti-mouse AP, bovine anti-goat HRP, and bovine anti-goat AP were purchased from Santa Cruz Biotechnology, Inc.
Cell Culture
Huh-7.5 cells were maintained in DMEM, as described previously (3). Stable cell lines were maintained in supplemented DMEM containing 0.5 mg/ml G418. Where specified, cells were treated with DMSO; PKA inhibitor H-7, H-89, or KT-5720 or the myristoylated peptide fragment 14–22 (all from Sigma); or NS5A inhibitor Daclatasvir (BMS-790052) for the designated times prior to harvesting.
Transient Transfection of HCV RNA
In vitro transcribed subgenomic RNA was transfected into Huh-7.5 cells using the TransMessenger transfection system (Qiagen). Briefly, 1.6 × 106 cells were mixed with 2 μg of RNA per the manufacturer's protocol. For colony formation assays, 0.5 × 106 cells were plated in 100-mm dishes. Cells were selected under DMEM containing 0.5 mg/ml G418 for 3 weeks, exchanging G418-containing media every 3 days.
Cell Culture and RNA Transfection for JC1/GLuc2A Replication and Infectivity Experiments
Huh-7.5 cells were maintained in DMEM supplemented with 10% fetal calf serum (HyClone, Logan, UT) and 1 mm nonessential amino acids (Invitrogen). Cells were seeded at 0.2 × 106 cells/ml/well in 12-well plates and transfected with HCV RNA transcripts by using the TransIT-mRNA transfection kit (Mirus Bio, Madison, WI).
Replication and Infectivity Measurements Using JC1/GLuc2A Reporter Viruses
To measure the relative replication of JC1/GLuc2A reporter viruses, cell culture medium was collected at various time points post-transfection, clarified by centrifugation (16,000 × g for 5 min), and mixed with ¼ volume of Renilla 5× lysis buffer (Promega, Madison, WI) to kill infectious HCV. GLuc activity was measured, as described previously (19), on a Berthold Centro LB 960 luminescent plate reader with 20 μl of sample injected with 50 μl of Gaussia luciferase assay reagent (New England Biolabs), integrated over 10 s. To measure relative infectivity, cell culture media containing infectious virus were collected at various times post-transfection, clarified by centrifugation, and stored at −80 °C. The virus supernatants were used to infect naive Huh-7.5 cells seeded at 0.2 × 106 cells/ml/well in 12-well plates. After 6 h of virus adsorption, cells were washed three times with Dulbecco's PBS and incubated with complete medium for an additional 72 h. Cell culture media were collected, clarified, and assayed for GLuc activity as described above.
RT-qPCR
Total RNA was extracted from subgenomic replicon transfected Huh-7.5 cells using the RNeasy Plus RNA extraction system (Qiagen). The RT-qPCR was performed at the Nucleic Acid Facility at Penn State. Oligonucleotides 11 and 12 were used for reverse transcription. The Taqman primer 5′-CGC CGC CAA GCT CTT CAG CAA-3′ was used on an Applied Biosystems 7300 system. In vitro transcribed RNA was used to quantify the copy number in cells.
Western Blot Analysis
Recombinant proteins or cell extracts from transiently transfected cells or stable cell lines were separated on 8% SDS-polyacrylamide gels and transferred to nitrocellulose membrane. Membranes were probed with the appropriate antibodies. Proteins were detected by ECL (Millipore) or ECF (GE Healthcare) Western blot detection reagents. ECF-detected blots were visualized by TyphoonImager (GE Healthcare) and quantified using NS5A or PKA-phosphorylated NS5A standards via ImageQuant software.
Northern Blot Analysis
For Northern blotting, 0.5 and 2.0 μg of total RNA from stable cell lines was separated on a 0.6% agarose gel of 0.8 m formaldehyde in 20 mm MOPS buffer containing 5 mm sodium acetate and 1 mm EDTA and transferred to a nylon membrane in 150 mm sodium chloride, 15 mm sodium citrate, pH 7.0. Additionally, 0.5, 1.0, 5.0, and 10.0 ng of in vitro transcribed subgenomic replicon RNA was used as a positive control. The RNA was UV-cross-linked to the membrane and incubated with radiolabeled probes for 12 h. The probes were generated by PCR using oligonucleotides 13 and 14 (Table 1) with pHCVbart.rep1/Ava-II and labeled with [α-32P]dATP (MP Biomedicals). RNA was visualized by the Typhoon imager (GE Healthcare) and quantified using ImageQuant software.
Cycloheximide (CHX) Treatment
Huh-7.5 cells stably replicating the SI, SI/T2332A, and SI/T2332E subgenomic replicons were incubated in the presence of 100 μg/ml CHX for 48 h and harvested every 12 h for analysis via Northern and Western blotting and immunofluorescence.
Immunofluorescence
Cells were seeded on coverslips in 6-well plates. After the designated time and any specified treatment, the cells were washed with PBS and fixed for 15 min in 4% formaldehyde in PBS. Cells were washed with PBS, permeabilized for 5 min in 0.05% Triton X-100 in PBS, and washed with PBS. The cells were blocked with 3% BSA in PBS for 15 min and double-stained by incubation for 1 h each with primary antibody A followed by primary antibody B. Cells were incubated for 1 h each with Alexa-488- or Alexa-594-conjugated secondary antibodies (Invitrogen). After each antibody incubation, PBS was used to wash cells. The coverslips were mounted on glass slides in Vectashield with DAPI (Vectashield Laboratories, Inc., Burlingame, CA) and sealed with nail polish. Samples were analyzed by fluorescence microscopy (Zeiss Axiovert 200 M) with a ×63 lens, and digital images were taken with an Axiocam MRm CCD camera. Optical sections were deconvolved using Axiovision software.
Confocal Microscopy
Slides prepared for immunofluorescence were also used for confocal microscopy. Confocal analysis was performed on an Olympus Fluoview 1000 microscope with an Olympus IX70 inverted microscope with fluorescence burner and four single-line lasers with individual shutters that are software-controlled for sequential acquisition: violet (405 nm, 10 milliwatts), blue argon (488 nm, 10 milliwatts), green HeNe (543 nm, 10 milliwatts), and red HeNe (633 nm, 10 milliwatts). Images were taken using the PlanApo ×60/1.4 oil objective and the two-dimensional X-Y scanning mode. Data were analyzed with the Olympus Fluoview version 3.0a software. Quantification was performed using Pearson's coefficient of colocalization between the green (NS5A) and red (NS4B) channels, and the statistical significance was calculated using a two-tailed paired Student's t test. Colocalization of NS5A and NS4B was calculated using 20 cells per cell line from three independent experiments.
Electron Microscopy
For ultrastructural analysis, cells were trypsinized, resuspended in complete DMEM, and processed as follows. Single cell suspensions were transferred to microcentrifuge tubes and pelleted at 1,000 × g for 10 min. The cell pellets were fixed with an ice-cold fixative containing 2% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, at room temperature for 1 h and then postfixed in 1% OsO4 for 1 h. Samples were dehydrated with a serial treatment in graded ethanol and embedded in Eponite 12. Areas of interest were selected under a dissecting microscope, and 80-nm-thick sections were produced with a Leica UC6 ultramicrotome. Sections were contrasted with uranyl acetate/lead citrate and examined with an FEI Tecnai Spirit transmission electron microscope (FEI, Hillsborough, OR) at 120 kV.
Expression and Purification of 15N-, 13C-Labeled NS5A IDD
NS5A IDD was expressed from the pSUMO-N(His)-NS5A IDD C2215S/C2314S plasmid (encoding residues 2212–2417 of the polyprotein). Rosetta(DE3) cells containing the expression plasmid were grown in NZCYM supplemented with 25 μg/ml kanamycin (K25) and 20 μg/ml chloramphenicol (C20) at 37 °C to A600 = 0.8. The culture was used to inoculate 4 liters of NZCYM with K25 and C20 to A600 = 0.025. The cultures were grown at 37 °C to A600 = 0.8 and then harvested and washed with M9 media without ammonium or dextrose. The cells were resuspended in 1 liter of M9 medium with 15NH4Cl, [13C]glucose, K25, and C20 and grown at 37 °C to A600 = 5. The cultures were then cooled to 20 °C, and 0.8 mm isopropyl 1-thio-β-d-galactopyranoside was added. The cultures were then grown at 20 °C for 12–15 h and then harvested. The cells were lysed in 100 mm potassium phosphate, pH 8, buffer with 500 mm NaCl, 10 mm β-mercaptoethanol, 10% glycerol, and 20 mm imidazole supplemented with protease inhibitors pepstatin A (10.0 μg/ml), leupeptin (10.0 μg/ml), and one protease inhibitor mixture tablet per 4 g of cell paste (Roche Applied Science, catalog no. 1-873-580) by passing through a French Press (SIM-AMINCO) twice at 16,000 p.s.i. Immediately after lysis, PMSF was added to a final concentration of 1 mm, and Nonidet P-40 was added to 0.5%. The extract was clarified by ultracentrifugation for 30 min at 25,000 rpm (75,000 × g) at 4 °C. The clarified lysate was loaded onto a Ni-NTA-agarose column. The column was washed with 5 column volumes of 5 and 50 mm imidazole. The protein was eluted with buffer containing 500 mm imidazole, and Ulp1 protease was used to cleave the SUMO tag. The cleaved NS5A IDD protein was purified from the SUMO by running the sample over another Ni-NTA-agarose column. The sample was then passed over a Q-Sepharose, pH 6, column and an S-200 column. The purified protein was dialyzed into NMR analysis buffer (50 mm HEPES, pH 7.5, 100 mm NaCl, 10 mm β-mercaptoethanol, 10% glycerol) and brought to a final concentration of 400 μm, and 10% (v/v) D2O was added right before analysis.
Expression and Purification of His-c-Src-SH3
His-c-Src-SH3 was expressed from the pET-Ub-based plasmids that are fused to yeast ubiquitin at the amino terminus (23). BL21(DE3)pCG1 cells containing the expression plasmid were grown in 100 ml of NZCYM with K25 and C20 at 37 °C to A600 = 1.0. The culture was used to inoculate 1 liter of autoinducing medium (24) with K25, C20 to A600 = 0.025. The cells were grown at 37 °C to A600 = 0.8 and then cooled to 20 °C, grown overnight (∼12–14 h), and then harvested. Cells were lysed by passing through a French Press. PMSF was added to a final concentration of 1 mm, and Nonidet P-40 was added to 0.5%. The extract was clarified by ultracentrifugation for 30 min at 25,000 rpm (75,000 × g) at 4 °C. The supernatant was loaded onto a Ni-NTA-agarose column and purified using the standard manufacturer's protocol. Protein was eluted with 500 mm imidazole and dialyzed into the NMR analysis buffer and brought to a final concentration of 1 mm and 10% (v/v) D2O.
NMR Spectroscopy
All spectra were acquired on uniformly 15N and 13C isotope-enriched samples at a concentration ranging from 0.4 to 0.7 mm in 50 mm HEPES buffer (pH 7.5), 100 mm NaCl, 10 mm β-mercaptoethanol, 5% glycerol, and 10% D2O. All of the NMR experiments were recorded at 11.7 T on a Bruker AVANCE-3 spectrometer operating at a 1H frequency of 500.13 MHz and equipped with a TCI cryoprobe. All spectra were collected at 298 K. Typical pulse times were 9.79 and 31 μs for hard 1H and 15N 90 pulses, respectively, with some sample-based variation in the 1H pulse time. All pulsed field gradients used in the experiments were applied for 1 ms with a sine shape. In all pulse sequences, unless otherwise noted by Sahu et al. (25), the 90 band-selective 13C pulses have the Q5 shape (or time reversed, Q5tr), and the band-selective 13C 180 pulses use the Q3 shape with durations of 384 and 307 μs, respectively. The standard 1H,15N HSQC, C_CON, HNCO, and HNCACO experiments were used from the TopSpin pulse program library. Chemical shift assignments of residues found in the proline region of the 15N,13C CON were generated as described by Sahu et al. (25). Briefly, several of the proline resonances were uniquely assigned by their presence in the Ala-specific CON, Ser-specific CON, and TAVI-specific CON spectra. Additional resonance assignments were generated from (HN-flip)N(CA)CON and (HN-flip)N(CA)NCO spectra (25). All NMR data were processed in TopSpin version 2.1 and converted to Sparky (26) format for data analysis. For the c-Src SH3 titrations (0–2 molar eq), purified c-Src SH3 was added to the same sample of 15N-, 13C-labeled NS5A IDD from a 1 mm stock, and the sample was concentrated back to its original volume, and spectra were collected. All 15N,13C CON spectra for the c-Src SH3 titrations were collected with 16 scans of signal averaging and 256 increments in the indirect dimension, yielding a total measurement time of 2 h/spectrum. Final data matrix dimensions were 512 × 2,048 points, with sweep widths of 36.00 and 20.00 ppm in the 15N and 13C dimensions, respectively.
RESULTS
Phosphorylation of NS5A on Thr-2332 by PKA in Vitro
HCV NS5A is a phosphoprotein (27). Examples of a link between a specific kinase, a specific site of phosphorylation on NS5A, and a biological function/activity of NS5A remain the exception rather than the rule. This circumstance is further complicated by the fact that mass spectrometry of NS5A isolated from cells replicating HCV RNA has not yielded substantial biological insight (28–32). Therefore, a new paradigm for studying NS5A phosphorylation and the function thereof is needed. We reasoned that as long as a protein is folded, specific phosphorylation might be observed in vitro, especially if our criteria also demanded a stoichiometry of phosphorylation that was greater than or equal to 1. We chose to study PKA because prior biochemical evidence suggested that NS5A is a substrate for this kinase (33). The NetPhosK server predicted one site of PKA phosphorylation at NS5A position Thr-360, corresponding to Thr-2332 of the HCV polyprotein. PKA was also a good test case because the NetPhosK server predicted six sites of phosphorylation in NS3 and three in NS5B. However, for the predicted phosphorylation sites in NS3 and NS5B to be accessible to the kinase active site, tertiary contacts would need to be lost, and, in some cases, secondary structure would need to be lost as well (Fig. 1A).
FIGURE 1.

PKA specifically phosphorylates NS5A in vitro. A, the conserved NetPhosK-predicted PKA phosphorylation sites on HCV NS3 and NS5B are inaccessible to the kinase without the loss of secondary and/or tertiary structure. The five putative PKA phosphorylation sites on NS3 protease (left), the one site on NS3 helicase (middle), and the three sites on NS5B polymerase (right) are shown in red on the crystal structure. Residue numbering corresponds to the HCV sequence of the respective crystal structures. B, PKA does not phosphorylate HCV NS3 or NS5B in vitro. Left, purified recombinant HCV non-structural proteins, with 1 μg loaded per lane. Right top, stoichiometric phosphorylation of HCV non-structural proteins by PKA showed specificity of the kinase for NS5A. Right bottom, quantification of the top panel with numbers corresponding to the amount of phosphate incorporated/mol of viral protein after 60 min. The numbers in parentheses below the labels correspond to the number of predicted PKA-phosphorylated residues based on NetPhosK prediction with a 0.5 threshold setting. C, PKA phosphorylates NS5A within residues 2307–2419. Top, schematic representation of NS5A constructs used to verify the site of PKA phosphorylation in NS5A. Full-length NS5A consists of amino acids 1973–2419 of the HCV polyprotein using Con1 numbering. NS5A contains three clusters of highly conserved serine-rich regions, denoted P1 (residues 2194–2210), P2 (residues 2246–2269), and P3 (residues 2380–2409). The amino-terminal 32-amino acid amphipathic α-helix was removed for purification purposes, yielding Δ32-NS5A (residues 2005–2419) (1). Truncations of this construct included 2194–2419 (2), 2005–2306 (3), and ΔP3 (4). Bottom, stoichiometric phosphorylation of NS5A truncations with PKA indicated that the target residue was located within 2307–2419, but not within P3, based on a lack of phosphate incorporation using NS5A truncation 2005–2306 (lane 3).
We observed stoichiometric phosphorylation of NS5A (Fig. 2A) but less than 0.1 eq of phosphate incorporated into NS3 or NS5B under the same conditions (Fig. 1B). That Thr-2332 was the site of phosphorylation was supported by biochemical analysis of NS5A deletion constructs (Fig. 1C), mass spectrometry (Fig. 2B), and site-directed mutagenesis (Fig. 2C). Thr-2332 is located within a sequence that has been shown to function as a nuclear localization signal, although the biological relevance of the nuclear localization signal remains to be determined (34). Thr-2332 is also adjacent to one of the two PPII motifs known to be important for interactions of NS5A with SH3 domains of several cellular proteins (35, 36). Importantly, Thr-2332 is present in all genotypes that have been shown to replicate in cell culture and those that seem to be the most recalcitrant to the current standard of care in the clinic (Table 2).
FIGURE 2.

PKA phosphorylates NS5A on Thr-2332. A, kinetics of NS5A phosphorylation by PKA in vitro were monitored by using gel electrophoresis (top) to identify phosphorylated NS5A (NS5A-P). Care was taken to retain labeled ATP on gel, thus permitting quantitation (bottom) of phosphorylated NS5A, indicating that 1 eq of phosphate was incorporated/molecule of NS5A. B, spectrum of the shown peptide obtained by tandem mass spectrometry indicating phosphorylation on Thr-2332. C, NS5A derivatives having Thr-2332 changed to alanine (T2332A) or glutamic acid (T2232E) were treated with labeled ATP and PKA for 60 min and analyzed as in A. Loss of Thr-2332 prevented phosphorylation, consistent with this residue being the site of phosphorylation. D, an antibody was produced that specifically recognizes Thr(P)-2332-NS5A. Experimental validation of the specificity is shown, using NS5A wild type and the indicated derivatives. Phosphorylation was performed in vitro with PKA or CK2, resolved by gel electrophoresis, and then processed for Western blotting. Top, anti-Thr(P)-2332-NS5A was used; bottom, anti-NS5A was used.
TABLE 2.
Conservation of PKA phosphorylation site in NS5A across HCV genotypes
Shown is an alignment of HCV genotypes using the Los Alamos HCV Sequence Database, with the PKA phosphorylation site shown in boldface type. The numbers are the genotype 1b reference.
| 1a._.H77.NC_004102 | PP-RKKRTVVLTES |
| 1a.US.99_38.DQ889300 | PP-RRKRTVVLTES |
| 1a.US.H77-H21.AF011753 | PP-RKKRTVVLTES |
| 1a.US.HC-TN.EF621489 | PP-RKKRTVILTES |
| 1b.DE.BID-V502.EU155381 | 2326PP-RRKRTVVLSES2338 |
| 1b.JP.MD1–911.AF165046 | PP-RRKKTVVLTES |
| 1b.TR.HCV-TR1.AF483269 | PP-RRKRTVVLTES |
| 1c.ID.HC-G9.D14853 | PP-RRKRTVVLDES |
| 1c.IN.AY051292.AY051292 | PP-RRKRTVVLDES |
| 2a._.G2AK1.AF169003 | PP-RSRRTVGLRES |
| 2a.JP.AY746460.AY746460 | PP-RRRRTVGLSES |
| 2a.JP.JCH-6.AB047645 | PP-RRRRTVGLSES |
| 2b._.JPUT971017.AB030907 | PP-RRRRARVLTQD |
| 2b._.MD2B-1.AF238486 | PP-RRRRAKVLTQD |
| 2b.JP.MD2b1–2.AY232731 | PP-RRRRAKVLTQD |
| 2c._.BEBE1.D50409 | PP-RRRRAVVLDQS |
| 2i.VN.D54.DQ155561 | PP-RRRRTVALDQS |
| 2k.MD.VAT96.AB031663 | PP-RRRRALVLSQS |
| 3a.CH.452.DQ437509 | PP-RRKRTVQLDGS |
| 3a.DE.HCVCENS1.X76918 | PP-RRKRTIQLDGS |
| 3a.US.TN78–0.DQ430819 | XP-RXKRTIQLDGS |
| 3b.JP.HCV-Tr.D49374 | PP-RRKRTIKLDGS |
| 3k.ID.JK049.D63821 | PP-RRKRTIVLSES |
| 4a._.01–09.DQ418782 | PP-RRKRTVQLTES |
| 4a.EG.Eg7.DQ988076 | PP-RRKRTVQLTES |
| 4a.EG.Eg9.DQ988077 | PP-RRKRTVQLTES |
| 4d._.03–18.DQ418786 | PP-RRKRAVVLSES |
| 4d._.24.DQ516083 | PP-RRKRTVALSES |
| 4f.FR.IFBT84.EF589160 | PPRRKKKTVVLSEA |
| 4f.FR.IFBT88.EF589161 | PPRRRKKTVVLSES |
| 5a.GB.EUH1480.Y13184 | LPRRKRKPMELSDS |
| 5a.ZA.SA13.AF064490 | PPRRKRKPVVLSDS |
| 6a.HK.6a74.DQ480524 | PP-RRKRLVHLDES |
| 6a.HK.EUHK2.Y12083 | PP-RRKRLVHLDES |
| 6b._.Th580.NC_009827 | PP-RRKRLIQLDES |
| 6c.TH.Th846.EF424629 | PP-RRKKVVKLDEA |
| 6d.VN.VN235.D84263 | PP-RKKRVVQLDEG |
| 6e.CN.GX004.DQ314805 | PP-RRKRVVKLDES |
| 6f.TH.C-0044.DQ835760 | PP-RRKRTVLLDSS |
| 6f.TH.C-0046.DQ835764 | PP-RRKRTVLLDSS |
| 6g.HK.HK6554.DQ314806 | PP-RRKKLVQLDDS |
| 6g.ID.JK046.D63822 | PP-RRKKLVQLDDS |
| 6h.VN.VN004.D84265 | PP-RRKKVVQLDSS |
| 6i.TH.C-0159.DQ835762 | PP-RRRKVVQLDAS |
| 6i.TH.Th602.DQ835770 | PP-RRRKVVQLDAS |
| 6j.TH.C-0667.DQ835761 | PP-RRKKMXQLDSS |
| 6j.TH.Th553.DQ835769 | PP-RRKKLXQXDSS |
| 6k.CN.Km41.DQ278893 | PP-RRKKVVRLDES |
| 6k.CN.Km45.DQ278891 | PP-RRKKVVHLDDS |
| 6k.VN.VN405.D84264 | PP-RRKSVVHLDDS |
| 6l.US.537796.EF424628 | PPX-RRKKVVHLDD |
| 6m.TH.C-0185.DQ835765 | PP-RRRKVVQLDQS |
| 6m.TH.C-0192.DQ835766 | PP-RRRKVVQLDRS |
| 6m.TH.C-0208.DQ835763 | PP-RKRKVVQLDQS |
| 6n.CN.Km42.DQ278894 | PP-RRKRTIHLDQS |
| 6n.TH.D86/93.DQ835768 | PP-RRKKTIHLDQS |
| 6o.CA.QC227.EF424627 | PP-RRKKVVRLNES |
| 6p.CA.QC216.EF424626 | PP-RRKKVVQLDES |
| 6q.CA.QC99.EF424625 | PP-RRKKVVQLDES |
| 6t.VN.TV241.EF632069 | PP-RRKKVVQXNDS |
| 6t.VN.TV249.EF632070 | PP-RRKKVVQLDDS |
| 6t.VN.VT21.EF632071 | PP-RRKKVVQLDDS |
| 7a.CA.QC69.EF108306 | PPRRKRAVIQLTES |
We used our knowledge of the PKA phosphorylation site on NS5A to produce an antibody that specifically recognized the PKA-phosphorylated form of NS5A (referred to as Thr(P)-2332-NS5A throughout). This antibody recognized NS5A that had been phosphorylated by PKA (lane 3 of panel pT2332-NS5A in Fig. 2D) but not that which had been phosphorylated by casein kinase 2, CK2 (lane 4 of panel pT2332-NS5A in Fig. 2D). That Thr-2332 is the site of PKA phosphorylation was further supported by the inability of the antibody to recognize the T2332A derivative of NS5A that had been treated with PKA (lane 6 of panel pT2332-NS5A in Fig. 2D). The presence of a negative charge at position 2332 (T2332E derivative of NS5A) was not recognized by the antibody, consistent with the phosphate moiety contributing to the epitope recognized by the antibody (lane 8 of panel pT2332-NS5A in Fig. 2D).
Phosphorylation of NS5A on Thr-2332 in Cells Replicating HCV RNA
It is now well established that sublines of the human hepatoma 7 cell line, in our case Huh-7.5 (37), can be used to efficiently establish a cell culture in which HCV RNA of genotype 1b (consensus 1 sequence) can be replicated persistently as long as an adaptive mutation(s) is present (9). Here we use a subgenomic replicon encoding the S2204I adaptive substitution in NS5A (denoted SI throughout). The replicons also contain coding sequence for neomycin phosphotransferase, thus permitting cells replicating HCV RNA to be selected in the presence of G418 (37).
We engineered the T2332A and T2332E substitutions into the SI replicon. We were able to transduce G418 resistance to Huh-7.5 cells as efficiently as observed for the SI replicon encoding Thr-2332 (Fig. 3A). It is worth noting that substitutions at position 2332 were not themselves adaptive (Fig. 3A) and had no significant impact on production of the p58 form of NS5A or the observed ratio of p58/p56 (Fig. 3B). Cell cultures persistently replicating SI/T2332A or SI/T2332E replicons expressed NS5A (lanes 5 and 6 of panel NS5A in Fig. 3C), although the steady-state level of NS5A was reduced 3-fold (Fig. 3D). Use of the Thr(P)-2332-NS5A antibody clearly showed reactivity in cells replicating the SI replicon (lane 4 of panel pT2332 in Fig. 3C) that was absent when Thr-2332 was changed to alanine or glutamic acid (lanes 5 and 6 of panel pT2332 in Fig. 3C, respectively). Therefore, Thr-2332 of NS5A is phosphorylated in cells replicating HCV RNA. When quantified, we found that only 20% of total NS5A was phosphorylated on Thr-2332 (Fig. 3D). Huh-7.5 cells express a protein that reacted with the Thr(P)-2332-NS5A antibody (lane 3 of panel pT2332-NS5A in Fig. 3C), consistent with the existence of one protein in the human proteome having the exact sequence as that found in NS5A that was used as the antigen (accession number Q9UBX2).
FIGURE 3.

NS5A in cells replicating HCV RNA is phosphorylated on Thr-2332. A, loss of Thr(P)-2332 does not alter replication as measured by colony formation. T2332A and T2232E variants were engineered into the HCV genotype 1b (Con1) subgenomic replicon in the absence (top panels) or presence (bottom panels) of the S2204I adaptive mutation, referred to throughout as the SI replicon. Replicon replication confers resistance to G418, thus permitting replication efficiency to be quantified by determining G418-resistant colonies per microgram of transfected replicon RNA as described (60); these values are shown below each panel. B, loss of Thr(P)-2332 does not have a global effect on NS5A phosphorylation. Wild type replicons produce two forms of NS5A. The p58/p56 ratios were unchanged. Errors represent S.E. n = 3. C, Thr(P)-2332-NS5A is present in cells replicating HCV RNA. Extracts made from populations of cells replicating HCV RNA encoding wild type or mutant NS5A were analyzed by Western blotting for the presence of Thr(P)-2332-NS5A using the Thr(P)-2332-specific antibody (top) or NS5A antibody (bottom) characterized in Fig. 2D. The band indicated by the asterisk is a cellular protein that reacts with the Thr(P)-2332-specific antibody. Recombinant NS5A and Thr(P)-2332-NS5A were used as controls. D, Thr(P)-2332 represents 20% of the total NS5A in cells replicating HCV RNA. Standard curves were prepared using NS5A or PKA-phosphorylated NS5A in order to quantify Western blots as shown in C to determine the number of molecules of NS5A and Thr(P)-2332-NS5A per cell. E, Thr(P)-2332-NS5A colocalizes with NS5A in cells replicating HCV RNA. Huh-7.5 cells without HCV RNA (mock) or expressing the indicated replicon were evaluated by immunofluorescence using antibodies against NS5A and Thr(P)-2332-NS5A. Nuclear staining observed with the antibody to Thr(P)-2332-NS5A is probably the cellular protein observed by Western blotting in C. HCV replicates in the cytoplasm, thus limiting interference.
Given that only a subset of NS5A was phosphorylated on Thr-2332, we used immunofluorescence microscopy to determine whether Thr(P)-2332-NS5A localized to a unique place in the cell relative to other/major form(s) of NS5A. Evaluation of Huh-7.5 cells showed that the Thr(P)-2332-NS5A antibody stained the nucleus most intensely (panel mock in Fig. 3E). Evaluation of cells replicating the SI replicon showed colocalization of Thr(P)-2332-NS5A with bulk NS5A (panel SI in Fig. 3E). As expected, substitution of Thr-2332 with alanine or glutamic acid failed to show any colocalization, thus confirming that the observed colocalization was not between NS5A and the cellular protein recognized by the Thr(P)-2332-NS5A antibody.
Thr(P)-2332-NS5A Influences Steady-state Levels of RNA for Genotype 1b HCV but Is Essential for Replication and Infectious Virus Production by Genotype 2a HCV
Because transduction of G418 resistance to cells provides no information on the levels of RNA produced in the cell, we were motivated to take a closer look at replication in cells in which the Thr(P)-2332/Thr-2332 ratio was perturbed. By using both RT-qPCR (Fig. 4A) and Northern blotting (Fig. 4B), it was clear that loss of the Thr(P)-2332/Thr-2332 ratio resulted in a nearly 10-fold reduction in the steady-state level of RNA. Interestingly, the impact of the perturbed Thr(P)-2332/Thr-2332 ratio was manifested only after multiple passages as if partitioning of replicated RNA during cell division was impacted (Fig. 4C). The lower steady-state level of RNA correlated directly with a lower steady-state level of the non-structural proteins (Fig. 4D) without a change in the stability of these proteins (Figs. 5, A and B) or the replicon RNA (Fig. 5, C and D). Collectively, these data reveal a unique function for a phosphorylated species of NS5A in maintenance of HCV RNA at maximal levels in cells persistently replicating genotype 1b HCV RNA.
FIGURE 4.

Phosphorylation of NS5A Thr-2332 influences steady state levels of RNA for HCV genotype 1b but is essential for replication and infectious virus production for HCV genotype 2a. A and B, cells expressing the indicated genotype 1b replicons were used to measure the steady-state level of RNA by RT-qPCR in A or Northern blotting in B. C, reduced RNA levels lead to reduced protein levels. Extracts were prepared from the same cells used in the previous panels and evaluated by Western blotting using antibodies against NS5A, NS3, and NS5B. D, RNA levels change as a function of passage. We noted variability in the magnitude of the differences observed between wild type and mutants. This variability was traced to very reproducible changes in RNA levels as a function of passage. Total RNA was extracted from G418-resistant colonies at each split, and HCV RNA copy number was determined by RT-qPCR. Errors represent S.E. n = 3. E, requirement of Thr(P)-2332-NS5A evaluated using the HCV genotype 2a infection system, pJC1/GLuc2A (19). This viral genome encodes the secreted Gaussia luciferase, permitting RNA synthesis to be monitored indirectly by luciferase activity. GNN, a viral genome expressing an inactive polymerase. Huh-7.5 cells were transfected as indicated, and media were collected at 6, 24, 48, and 72 h post-transfection. Values on the y axis represent the amount of secreted GLuc activity (shown as relative light units (RLU)) for each transfection. Errors represent S.D. n = 3. F, in this experiment, medium harvested from each time point shown in D was used as an inoculum to infect Huh-7.5 cells. Seventy-two hours later, RLU was measured. Error bars, S.D. n = 3.
FIGURE 5.

Thr-2332 phosphorylation does not affect HCV non-structural protein or RNA decay rates. A and B, T2332A and T2332E substitutions do not affect the decay of HCV non-structural proteins. A, Western blots of HCV non-structural proteins NS5A, NS3, and NS5B from SI, SI/T2332A, and SI/T2332E replicon-containing cells under 100 μg/ml CHX treatment. β-Actin was detected as a loading control. B, plot of NS5A levels under CHX treatment at varying time points (tn) relative to 0 h (t0) showed an equal decay of the viral protein across SI, SI/T2332A, and SI/T2332E replicon-containing cell lines. C and D, T2332A and T2332E substitutions do not affect the decay of HCV RNA. C, top, subgenomic replicon RNA extracted from SI, SI/T2332A, and SI/T2332E replicon-containing Huh-7.5 cells detected by Northern blot showed an increase in replicon levels over time. Bottom, Northern blot of subgenomic replicon RNA extracted from the same replicon-containing Huh-7.5 cells treated with 100 μg/ml CHX. Over 24 h, replicon levels decreased in the presence of CHX treatment. Varying amounts of in vitro transcribed subgenomic replicon RNA were loaded as positive controls in the first four lanes of the top and bottom blots. D, plot of replicon levels under CHX treatment at varying time points (tn) relative to 0 h (t0) showed an equal decay of SI, SI/T2332A, and SI/T2332E replicon levels over time.
Because of the known role of some NS5A phosphorylation events in infectious virus production, we turned to a model that would permit the role of Thr-2332 phosphorylation on virus assembly to be determined. For this, we used the Jc1/GLuc2A reporter virus, which is a genotype 2a HCV strain that expresses a secretable Gaussia luciferase (19). This reporter virus efficiently replicates and secretes luciferase in proportion to virus replication, facilitating rapid measurements of viral replication and infectivity. Furthermore, JC1/GLuc2A infectious virus production can be monitored by measuring the amount of secreted luciferase activity produced by reporter virus in infected cells, and the level of luciferase produced is relative to the amount of input virus (19).
In contrast to our observations with genotype 1b, the genotype 2a replicon encoding the T2332A NS5A variant was incapable of replicating (T2332A in Fig. 4E). In fact, this change was as debilitating as a genome encoding an inactive polymerase (GNN in Fig. 4E). Unexpectedly, the T2332E NS5A variant replicated as well as wild type (T2332E in Fig. 4E). The same phenotypes were observed when infectious virus was monitored (Fig. 4F). Together, these results would suggest that Thr(P)-2332 is essential for replication of genotype 2A HCV and that T2332E phenocopies Thr(P)-2332 in this context.
Thr(P)-2332 Shifts the Conformational Ensemble of the SH3-binding PPII Motif in the IDD of NS5A
As shown in Fig. 6A, the NS5A IDD contains two PPII motifs, which have been termed PPII.1 and PPII.2 (35, 38). Thr(P)-2332 is located adjacent to these motifs (Fig. 6A). Reverse-genetic, biochemical, and biophysical studies have implicated this region of NS5A in binding to a variety of cellular SH3 domains (35, 36, 38, 39). However, no study has monitored the PPII motifs directly. We reasoned that if we could accomplish this goal, then we would be able to determine whether and how Thr(P)-2332 impacts the PPII motifs and how these motifs interact with an SH3 domain. The recent realization that many of the phosphorylation sites in cellular IDPs are located adjacent to a PPII motif expanded the relevance of studying the relationship between PPII motifs and phosphorylation (40).
FIGURE 6.

Phosphorylation of Thr-2332 shifts proline residues of the NS5A polyproline motif that contribute to interaction with the SH3 domain of c-Src. A, schematic of NS5A IDD studied by NMR. Proline residues (black, boldface type) were observed in the unphosphorylated IDD. Residues of the two type-II polyproline motifs, PPII.1 and PPII.2, are indicated. Proline residues (green) were observed only in the Thr(P)-2332- and T2332E IDDs. The location of Thr(P)-2332 is indicated. B, prolines in NS5A IDD can be detected by NMR. Shown is the 15N,13C CON spectrum of unphosphorylated NS5A IDD (proline region). Resonances observed represent a very small subset of the proline residues in the IDD. Three were assigned as indicated; none were in PPII. C, phosphorylation of IDD increases the number of proline resonances detected. Shown is the 15N,13C CON spectrum with PKA-phosphorylated IDD (NS5A IDD pT2332; green). The spectrum was overlaid with that of the unphosphorylated IDD (NS5A IDD; black). Many new resonances were observed. Three could be assigned, and these mapped to PPII. D, NS5A IDD T2332E mimics Thr(P)-2332. The spectrum for NS5A IDD T2332E (purple) superimposes with that of NS5A IDD Thr(P)-2332 (green). E, titration of the SH3 domain into NS5A IDD Thr(P)-2332 leads to loss of resonances induced by Thr-2332 phosphorylation. Shown are 15N,13C CON spectra of NS5A IDD Thr(P)-2332 with 0–2 eq of unlabeled c-Src SH3. The absence of red resonances at positions where green resonances are visible indicates a dynamical response to the presence of c-Src. F, chemical shift perturbation of Thr(P)-2332 in response to the presence of c-Src. 1H,15N HSQC spectra are zoomed in on the Thr(P) region for NS5A IDD Thr(P)-2332 with 0–2 eq of unlabeled c-Src SH3. This resonance is also sensitive to the presence of c-Src. G, molecular model of c-Src SH3 domain in complex with NS5A PPII peptide, APPIPPPR. The model was derived from a structure of FYN-SH3 domain in complex with NS5A PPII peptide (43) and structure of c-Src SH3 domain (61). Proline residues whose resonances appear in the presence of phosphorylation and disappear in the presence of c-Src are clearly capable of binding to the protein. The Thr(P)-2332 would be predicted to be close enough to the protein surface for interaction.
The absence of an amide proton for prolines located in a polypeptide chain precludes the use of an NMR strategy built around 1H,15N HSQC experiments for direct visualization of these prolines. We therefore built our NMR strategy around the 15N,13C CON experiment, using recently developed multidimensional NMR techniques for the analysis and chemical shift assignments of IDPs that do contain resonances corresponding to proline residues (25). We expressed and purified isotopically 15N-, 13C-labeled NS5A IDD and acquired double and triple resonance spectra on both the unphosphorylated and PKA-phosphorylated proteins. The 15N,13C CON spectrum of unphosphorylated HCV NS5A IDD shows nice chemical shift dispersion, with well resolved resonances in the proline region of the spectrum (Fig. 6B). Although NS5A IDD has 25 prolines, far fewer proline resonances were observed in the spectrum. The absence of resonances for many of the prolines is probably due to conformational exchange of the proline-rich region within the IDD between multiple conformations on the millisecond time scale. Additional evidence supporting this conclusion is provided by the diverse line widths (and resulting signal intensities) of the proline resonances that are retained in the spectrum. We were able to assign three resonances in the spectrum of the unphosphorylated protein to Pro-2222, Pro-2369, and Pro-2389 (Fig. 6B); all of these are remote from the polyproline motif (Fig. 6A).
Upon phosphorylation by PKA, resonances for many more prolines were observed (Fig. 6C). With the assistance of amino acid-specific 15N,13C CON spectra (25), we were able to assign three of these new resonances to Pro-2315, Pro-2322, and Pro-2325, suitable probes for both PPII motifs (Fig. 6A). It appears that the average conformations yielding the resonances observed in the Thr(P)-2332 spectrum are not created in response to phosphorylation, because we see them in the unphosphorylated IDD spectrum if the concentration is sufficiently high (Fig. 7). Therefore, it is likely that the ability of phosphorylation to bring resonances of the PPII motif into view originates from a reduction in the conformational sampling of this region of the IDD. Interestingly, an IDD with the T2332E substitution was indistinguishable from the IDD phosphorylated on Thr-2332 (Fig. 6D), suggesting that T2332E-NS5A phenocopies the conformation of Thr(P)-2332-NS5A.
FIGURE 7.
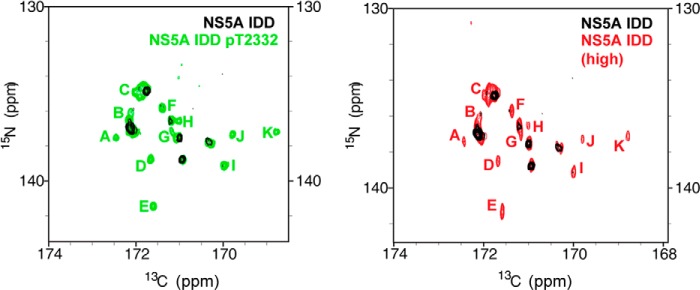
PKA phosphorylation alters the observed resonances for NS5A IDD. Shown is a comparison of NMR spectra for unphosphorylated and PKA-phosphorylated NS5A IDD. In the left panel is the 15N,13C CON spectra of NS5A IDD (black) overlaid with the 15N,13C CON spectra of NS5A IDD Thr(P)-2332 (green) zoomed to the proline region of the spectrum. The proline residue resonances that appear upon phosphorylation are indicated with green letters. In the right panel is the 15N,13C CON spectra of NS5A IDD (black) overlaid with 15N,13C CON spectra of NS5A IDD at high concentration (red). Only at very high concentrations of unphosphorylated NS5A IDD do resonances begin to appear that are similar to resonances observed for PKA-phosphorylated NS5A IDD (resonances labeled with the same letter code as in the left panel but in red).
It is clear that NS5A interacts with several SH3-containing proteins (38). Recently, it has been shown that the NS5A PPII motif is essential for replication of genotype 2a (41). To determine the impact of an SH3 domain on the PPII motifs of NS5A, we expressed and purified the SH3 domain from c-Src. We chose this SH3 domain because there is evidence that the NS5A-c-Src interaction is important for replication (42). Evaluation of Thr(P)-2332·NS5A IDD·SH3 complexes produced at different molar ratios of the SH3 domain showed that the resonances induced by phosphorylation disappear in the presence of the SH3 domain (red resonances are not present for any of the prolines labeled green in Fig. 6E). In contrast, none of the resonances remote from the PPII motifs are impacted by the presence of the SH3 domain (red resonances are present for all of the prolines labeled black in Fig. 6E). Performing this experiment with T2332E-NS5A IDD yielded the same result as observed for Thr(P)-2332-NS5A IDD (Fig. 8A) without any perturbations in the spectrum for the unphosphorylated protein (Fig. 8B). We conclude that both PPII motifs contribute to interactions with SH3 domains and that PKA phosphorylation near these motifs stabilizes a conformation competent for binding to SH3 domains.
FIGURE 8.

Binding of SH3 domain favors interaction with PKA-phosphorylated NS5A IDD. A, the 15N,13C CON spectra of NS5A IDD T2332E with 0 (purple), 1 (pink), and 2 (red) eq of unlabeled SH3 added zoomed to the proline region of the spectrum. B, the 15N,13C CON spectra of unphosphorylated NS5A IDD with 0 (black), 0.5 (cyan), 1 (pink), and 2 (red) eq of unlabeled SH3 domain added zoomed to the proline region of the spectrum.
Because of the unique location of phosphothreonines in spectra produced by performing 1H,15N HSQC experiments, we were able to identify and assign the resonance for Thr(P)-2332 (Fig. 6F). In the presence of the SH3 domain, there was a chemical shift perturbation followed by loss of the resonance. This observation suggests a possible interaction of Thr(P)-2332 with the SH3 domain as well. Given the substantial structural overlap that exists between SH3 domains, we were able to construct a model of the c-Src SH3 domain bound to the PPII.2 motif based on a structure of this motif bound to the Fyn SH3 domain (Fig. 6G) (43). This model places both Pro-2322 and Pro-2325 on c-Src SH3 domain and would suggest that Thr(P)-2332 should be close enough to interact with the SH3 domain as well.
Thr(P)-2332-NS5A Contributes to the Form and Function of the HCV Replication Organelle
At this point, we assumed that loss of Thr(P)-2332 negatively impacted interaction with one or more cellular SH3 domains. In order to further investigate the basis for the phenotype observed in cells producing T2332A- or T2332E-NS5A, we decided to evaluate the colocalization of NS5A with NS4B, a well accepted marker of the HCV membranous web/replication organelle (44). By using immunofluorescence alone, it was obvious that the amount of NS5A-NS4B colocalization was reduced when Thr-2332 was changed to alanine (Fig. 9A). Mutation of position 2332 to glutamic acid yielded an intermediate phenotype (Fig. 9A). In order to perform this experiment more quantitatively, we used confocal microscopy to obtain the Pearson's coefficient for NS5A-NS4B colocalization in single cells (45). In each case, 60 cells were evaluated, and a significant difference was observed in both the range and average colocalization observed when cells producing T2332A- or T2332E-NS5A were compared with those producing Thr(P)/Thr-2332-NS5A (Fig. 9B). We also analyzed the membranous web by using transmission electron microscopy (Fig. 9C). Clustered, tubular-vesicular structures were very apparent in cells producing Thr(P)-2332/Thr-2332-NS5A (SI in Fig. 9C). In the absence of Thr(P)-2332, only vesicular structures were observed; these were not clustered (SI/T2332A in Fig. 9C). T2332E-NS5A led to the production of demonstrably larger vesicular structures with very few tubular structures (SI/T2332E in Fig. 9C). Some vesicles were clustered; others were not (SI/T2332E in Fig. 9C). Collectively, these data suggest that the Thr(P)-2332/Thr-2332-NS5A ratio is an important determinant of the assembly and/or stability of the replication organelle. The outcome associated with changing this ratio or this residue varies depending on the HCV genotype.
FIGURE 9.

Phosphorylation of Thr-2332 contributes to the integrity of the HCV membranous web/replication organelle. A, reduced colocalization of NS5A and NS4B in the absence of Thr(P)-2332-NS5A. Immunofluorescence was used to evaluate the replication organelle. NS4B is used as a marker for the replication organelle, with good colocalization implying integrity of this organelle (44). Cells replicating the indicated HCV 1b replicon were stained for NS5A (green) and NS4B (red), so yellow indicates colocalization. When there is a fourth panel in a row, it shows the region in the merge panel that is in the white box. B, quantitative analysis of NS5A-NS4B colocalization. Visual analysis of images like those shown in A revealed extensive heterogeneity in the extent of NS5A-NS4B colocalization in cells replicating the SI replicon. To remove bias, we used a confocal microscope and determined colocalization (Pearson's coefficient) in entire cells. Three trials were performed; each trial used 20 cells per construct. p values were calculated using a two-tailed paired t test. C, loss of integrity of replication organelle also observed by transmission electron microscopy. In order to demonstrate that the changes in NS5A-NS4B colocalization corresponded to changes in the replication organelle ultrastructure, portions of the cells used in A were processed for transmission electron microscopy. Bar, 1 μm.
PKA Activity Is Required for HCV Replication beyond Phosphorylation of NS5A on Thr-2332
In order to obtain evidence for PKA as the kinase responsible for phosphorylation of Thr-2332 in vivo, we treated cells with the PKA inhibitor, KT-5720 (46), which caused a substantial reduction in the level of Thr(P)-2332-NS5A relative to untreated cells (compare lane 3 with lane 2 of panel pT2332-NS5A in Fig. 10A). In addition to KT-5720, three other inhibitors of PKA have been used widely in the literature: H-7 (47), H-89 (48), and 14–22 (49). All of these inhibitors prevent NS5A phosphorylation by PKA in vitro (Fig. 11A) and in cells replicating HCV RNA (Fig. 11B).
FIGURE 10.

PKA targets other than NS5A are required for HCV replication. A, PKA phosphorylates NS5A at Thr-2332 in cells replicating HCV RNA. In order to demonstrate that PKA was indeed phosphorylating NS5A on Thr-2332 in cells, cells were treated with DMSO (lane 2) or the PKA inhibitor, KT-5720 (lane 3), for 24 h. The effect of treatment on Thr(P)-2332-NS5A and NS5A was determined by Western blotting. Huh-7.5 cells were used as a negative control (mock, lane 1); detection of calnexin served as a loading control. In order to determine whether dephosphorylated NS5A could be rephosphorylated, cycloheximide, a translation inhibitor, was added upon removal of the PKA inhibitor, KT-5720, and probed 12 (lane 4) or 24 h (lane 5) thereafter. Similar observations were made using other PKA inhibitors (Fig. 11). B, PKA inhibitors cause an irreversible loss in integrity of the replication organelle. The impact of the treatments performed in A on the replication organelle was determined by using immunofluorescence to evaluate NS5A-NS4B colocalization as described above. C, inhibition of PKA has a greater impact on HCV RNA levels than loss of pT2332. RT-qPCR was used to determine the copy number of HCV RNA in cells treated with DMSO or KT-5720. Errors represent S.E. n = 3. D, inhibition of PKA further exacerbates loss of integrity of the replication organelle caused by substitutions at Thr-2332. Immunofluorescence was used to evaluate the integrity of the replication organelle for the wild-type replicon or the indicated Thr-2332 variant in the absence (DMSO) or presence (KT-5720) of the PKA inhibitor. The white box in the merge panel is enlarged in the bottom right of each quadrant. This experiment was repeated with other PKA inhibitors (Fig. 11).
FIGURE 11.

Commercially available PKA inhibitors (PKAi) disrupt HCV replication organelles. A, the IC50 values of four commercially available PKA inhibitors were calculated using standard phosphorylation reaction conditions with 50 μm ATP, 0.5 μm NS5A, 0.005 μm PKA, and 2.5% DMSO. Final concentrations of the inhibitors varied from 0 to 50 μm. The IC50 values were determined by plotting the data and fitting to a hyperbolic equation. S.E. values are from non-linear regression fits of the data. B, PKA inhibitors decrease Thr(P)-2332-NS5A, as shown by Western blot. Top, a decrease in Thr(P)-2332-NS5A was observed in SI replicon-containing cells treated for 24 h with 10× IC50 of PKA inhibitors H-7, H-89, KT-5720, and 14-22 relative to cells treated for the same time with 0.5% DMSO. The same blot was also probed with anti-NS5A (middle) and anti-calnexin (bottom). C, immunofluorescence of SI replicon-containing Huh-7.5 cells in the presence of 0.5% DMSO or 10× IC50 PKA inhibitor H-7, H-89, KT-5720, or 14-22 for 24 h. Colocalization between NS5A (green) and NS4B (red) was nearly ubiquitous in DMSO-treated cells. Cells treated with H-7 showed a slight change in colocalization between the viral proteins; however, cells treated with H-89, KT-5720, and 14-22 had more dispersed HCV replication organelles.
Because loss of NS5A phosphorylation on Thr-2332 caused perturbations in the integrity of the replication organelle, as determined by NS5A-NS4B colocalization (Fig. 9), we evaluated the impact of KT-5720 on integrity of the replication organelle (Fig. 10B). As expected, treatment of SI cells with KT-5720 caused disruption of the replication organelle (panel +KT-5720 in Fig. 10B). In order to determine whether loss of Thr(P)-2332-NS5A was reversible and would lead to reestablishment of the replication organelle (i.e. NS5A-NS4B colocalization), we removed the PKA inhibitor but added CHX to ensure that any Thr(P)-2332-NS5A observed would derive from existing pools of NS5A instead of newly translated NS5A. Under these conditions, Thr(P)-2332-NS5A was again produced (lanes 4 and 5 of panel pT2332 in Fig. 10A). Interestingly, reestablishment of the replication organelle was not observed based on NS5A-NS4B colocalization (panels +CHX in Fig. 10B). This suggests that once phosphorylation ceases and the replication organelle is disrupted, polyprotein synthesis may be required for reestablishment of the replication organelle.
If phosphorylation of NS5A on Thr-2332 is the only function of PKA for replication of HCV RNA, then treatment of SI cells with a PKA inhibitor should phenocopy the SI/T2332A mutant. When SI cells were treated for 24 h with 2 μm (10 × IC50) KT-5720, the steady-state level of HCV replicon RNA was reduced by an order of magnitude (Fig. 10C). Interestingly, the steady-state level of HCV replicon RNA was also reduced in SI/T2332A and SI/T2332E cells (Fig. 10C), suggesting that targets of PKA phosphorylation other than NS5A contribute to HCV RNA replication. Consistent with the reduction in the steady-state level of RNA in SI/T2332A and SI/T2332E cells caused by the PKA inhibitor (Fig. 10C), KT-5720 treatment led to even further disruption of the replication organelles in these cells (panels SI/T2332A and SI/T2332E in Fig. 10D). These data suggest that other viral and/or cellular substrates of PKA are required for assembly, stability, and/or function of the replication organelle. All four inhibitors of PKA caused disruption of the replication organelle, although the fate of the NS5A and NS4B proteins differed for each inhibitor (Fig. 11C). We conclude that PKA activity is essential for HCV genome replication.
Relocalization of PKA in Cells Replicating HCV RNA
Having established that PKA does indeed phosphorylate NS5A on Thr-2332, we asked whether this phosphorylation occurred by recruiting PKA into the replication organelle. In naive Huh-7.5 cells, PKAc showed a preferential localization to the Golgi (PKAc in panel mock in Fig. 12A). In this experiment, the Golgi was stained with an antibody to the protein giantin (giantin in panel mock in Fig. 12A). In cells replicating HCV RNA, the Golgi-localized fraction of PKA was lost (PKAc in panel SI in Fig. 12A). Importantly, the Golgi remained intact in cells replicating HCV RNA. (giantin in panel SI in Figs. 12A and 13). The loss of Golgi-associated PKA in cells replicating HCV RNA did not require the ability of NS5A to be phosphorylated on Thr-2332 because the T2332A and T2332E mutants showed this phenotype (Fig. 12B). Although PKA did not accumulate in the replication organelle, the catalytic subunit was clearly present in the replication organelle, based on its colocalization with NS5A (Fig. 12B). Disruption of the replication organelle by precluding Thr-2332 phosphorylation was also sufficient to disrupt colocalization of PKA with NS5A (Fig. 12B). The ability of PKA and NS5A to colocalize in cells may facilitate NS5A phosphorylation on Thr-2332. However, it is also possible that the relocalization of PKA from the Golgi to the cytoplasm may be sufficient for NS5A phosphorylation prior to its association with the replication organelle.
FIGURE 12.

HCV replication leads to changes in localization of PKAc. A, PKAc enrichment at the Golgi apparatus in Huh-7.5 cells. The localization of PKAc (red) was determined by immunofluorescence. PKAc was present throughout the cell but did seem to be concentrated in a portion of the cytoplasm. Staining for giantin (green), a marker of the Golgi apparatus, was consistent with enrichment of PKAc at the Golgi apparatus. The nucleus was stained with DAPI (purple). The bottom right quadrant is a merge of the three other panels. B, Golgi fragmentation and loss of PKAc localization in Huh-7.5 cells replicating HCV RNA. The localization of PKAc (red) was determined in SI cells as described in A. The staining was much more diffuse than observed in A. Giantin (green) staining was fragmented in these cells. Whether the former phenotype is caused by the latter or vice versa or whether they are unrelated could not be determined unambiguously. C, PKAc does not localize to the replication organelle. In order to determine whether PKAc was present in the replication organelle, wild-type and Thr-2332 mutant replicons were stained for NS5A (green) and PKAc (red). The last panel in each row is the region within the white box of the merge panel. Giantin staining was also performed (Fig. 13).
FIGURE 13.
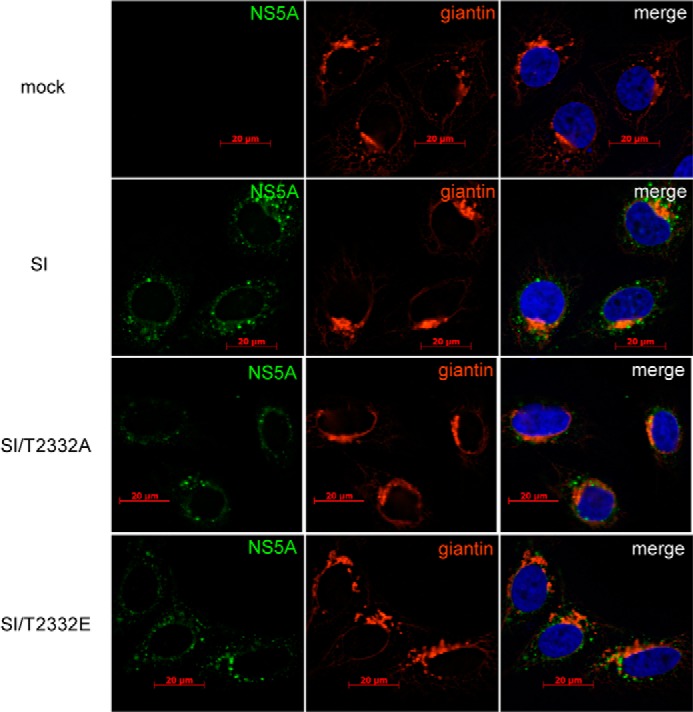
Replication of HCV RNA does not alter Golgi structure. Shown is immunofluorescence analysis of NS5A (green) and the Golgi marker giantin (red) in naive Huh-7.5 cells (mock) and SI, SI/T2332A, and SI/T2332E HCV replicon-containing cells. The stable replication of HCV RNA had no effect on Golgi structure relative to mock cells.
DISCUSSION
HCV NS5A contributes to genome replication, virion assembly, and myriad interactions with the host cell (7). How this protein interfaces with so many different viral and cellular pathways is unknown. It is well established that NS5A is a phosphoprotein (27). These early studies showed that NS5A migrated as 56- and 58-kDa species in SDS-polyacrylamide gels. The fact that the calculated molecular mass of NS5A is 49 kDa led to the conclusion that all NS5A produced in cells is phosphorylated, with basal (56-kDa) and hyperphosphorylated (58-kDa) forms (27). Later, it became clear that even unphosphorylated NS5A has a gel mobility of 56 kDa (18). The aberrant gel mobility is probably a reflection of the intrinsic disorder and/or proline-rich nature of the carboxyl-terminal two-thirds of NS5A (5, 6).
Phosphorylation and other post-translational modifications of an IDD of a protein can induce and/or stabilize structure, which, in turn, can confer unique function (8). Therefore, we hypothesized that single and multiple phosphorylation events of NS5A create forms of the viral protein with unique functions, thus explaining the promiscuous binding of NS5A. In order to test this possibility, a clear link between a phosphorylation site(s) on NS5A and a specific kinase is necessary. When a minimum cut-off of 1 eq of phosphate/eq of protein is required for that protein to be considered a substrate of a kinase, PKA is only capable of phosphorylating NS5A, although NetPhosK predicts numerous sites in NS3 and NS5B (Fig. 1). Mass spectrometry, reverse genetics, and a phospho-specific antibody showed that Thr-2332 (polyprotein numbering; residue 360 of processed NS5A) was the site of phosphorylation in vitro (Fig. 2). The latter two approaches permitted confirmation of this phosphorylation site in cells (Fig. 3).
Thr-2332 is located adjacent to two PPII motifs in NS5A (Fig. 6A). Our biophysical analysis (Fig. 6) is consistent with these motifs sampling multiple conformations on the millisecond time scale and Thr(P)-2332 restricting the sampling to conformations that are competent for binding to SH3 domains. It is also possible that the phosphorylation-dependent changes were caused by acceleration of the conformational dynamics into the picosecond to microsecond time scale. Phosphorylation-dependent changes in aggregation state were ruled out using dynamic light scattering (data not shown).
The ability of phosphorylation to regulate IDD conformation provides an explanation for cellular PPII motifs being located near phosphorylation sites (40). Phosphorylation need not always reduce conformational sampling; the context of the PPII motif would probably contribute to conformation. The relationship between conformational sampling and binding affinity remains to be determined. In addition, it is easy to imagine that other post-translational modifications affect function in a similar manner by modulating conformational sampling to promote or to interfere with a biomolecular interaction. Others have seen and/or proposed post-translational modification-dependent changes in motifs found in IDPs (50, 51). The novelty of this study is that we observe the prolines directly and provide an unambiguous link between a remote phosphorylation event and the conformation and dynamics of specific prolines. Therefore, these studies define the 15N,13C CON NMR strategy as a powerful approach for studying viral and cellular PPII motifs and interactions thereof.
The experimental paradigm used here to discover Thr(P)-2332 will be useful for identifying additional, specific sites of NS5A phosphorylation and the corresponding kinases. Two-dimensional gel analysis of NS5A present in Huh-7 cells persistently replicating HCV suggests many different phosphorylated species of the 56-kDa form, with pI values ranging from 4.0 to 5.5 (3). However, attempts to identify NS5A phosphorylation sites have yielded only a few sites (28–32). None of these studies identified Thr-2332. The Thr(P)-2332-NS5A species represents only 20% of total NS5A in cells (Fig. 3D). Therefore, this experimental paradigm clearly uncovers sites that have eluded direct approaches using NS5A isolated from HCV-replicating cells.
We conclude that Thr(P)-2332-NS5A, other PKA targets, and even other phosphorylated forms of NS5A contribute to form and function of the replication organelle. The NS5A IDD is among the most variable protein in the HCV proteome. It is possible that the NS5A phosphoproteome is equally variable. This variability may lead to differences in host factors used by different genotypes and the ability to antagonize host defenses required to establish persistence. Clearly, the NS5A phosphoproteome encoded by Con1b/S2204I and Jc1–2a genotypes of HCV used in this study is different, because Jc1–2a produces the p58 form of NS5A, and Con1b/S2204I does not. The magnitude of the response of these genotypes to loss of Thr(P)-2332 is substantially different (Fig. 4).
Dozens of host factors have been implicated in HCV genome replication and many (re)localize to the replication organelle (52). Of these, several are thought to be recruited by interacting with NS5A. Interestingly, two NS5A-interacting and replication organelle-associated host factors are regulated by PKA; these are annexin A2 (53, 54) and Raf-1 kinase (55). Annexin A2 interacts with phosphoinositides to form membrane microdomains of distinct protein composition, and these microdomains exhibit altered membrane dynamics (56). Phosphorylation of annexin A2 by PKA has been shown to promote its association with membranes. Raf-1 kinase interacts with Ras, and this interaction is disrupted by PKA phosphorylation of Raf-1 (57). Disruption of the Raf-1-Ras interaction may permit Raf-1 to localize to the replication organelle, thereby contributing to formation and/or stabilization of the replication organelle. Therefore, annexin A2 and Raf-1 are candidate host factors that may contribute to regulation of replication organelle structure by PKA.
It is possible that the ability to regulate assembly and disassembly of the replication organelle contributes to the ability of HCV to persist. This possibility is suggested by the observation that the difference in the steady-state level of RNA observed for the Thr-2332 mutants relative to wild type only manifests after multiple cell divisions (Fig. 4C). When the replication organelle is assembled, HCV replicase components are perinuclear and in an asymmetric organization (Fig. 3E). Therefore, it is possible that in this state partitioning between daughter cells may be asymmetric. When disassembled, components of the replication organelle are more homogeneous (Fig. 10D) and may be more equally distributed between daughter cells. Consistent with this possibility is the fact that cAMP concentration and PKA activity are highest at G1 and lowest at mitosis (58).
The NS5A phosphoproteome may be a highly efficacious target for development of anti-HCV agents. This idea is inspired by the recent discovery of daclatasvir, a drug thought to inhibit HCV genome replication by binding to NS5A, preventing hyperphosphorylation (14, 15) and disrupting the replication organelle (59). Evidence for direct binding of daclatasvir and related compounds to NS5A has been difficult to obtain (59). That NS5A is the target is supported primarily by drug resistance mutations (15). The effective concentration of daclatasvir is in the picomolar range; however, the concentration of NS5A in cells replicating HCV RNA is on the order of micromolar (14). Is it possible that the target is one of the many phosphorylated forms of NS5A? If this were the case, the picomolar efficacy could be related to binding of the drug to a NS5A species that is present at sub-picomolar levels. Analysis of the NS5A phosphoproteome after daclatasvir treatment may permit this possibility to be tested.
This study makes a very compelling case for the use of phosphorylation in the intrinsically disordered domain of NS5A to expand the functional proteome of HCV. Interestingly, evaluation of phosphorylated proteins of other RNA viruses reveals that these proteins have intrinsically disordered domains, and known phosphorylation sites map to these domains (Table 3). We speculate that phosphorylation in IDDs of viral proteins may be a general mechanism employed by RNA viruses to expand their functional proteomes.
TABLE 3.
Intrinsic disorder and phosphorylation of (+)-strand RNA viral proteins
| Virus | Protein | Disordera | Longest IDRb | PO4 in IDRc |
|---|---|---|---|---|
| Bovine viral diarrhea virus | NS5A | 31 | 82 | TBD |
| Cucumber mosaic virus | 2a | 23 | 61 | Yes |
| Cucumber necrosis virus | p33 | 27 | 70 | Yes |
| Dengue virus type 2 | NS5 | 37 | 45 | TBD |
| Hepatitis C virus | NS5A | 55 | 68 | Yes |
| Potato virus A | VPg | 32 | 20 | Yes |
| Semliki Forest virus | nsP3 | 32 | 81 | Yes |
| Sindbis virus | nsP3 | 49 | 58 | Yes |
| Tick-borne encephalitis virus | NS5 | 29 | 30 | TBD |
| Turnip crinkle virus | p28 | 38 | 40 | Yes |
| Turnip yellow mosaic virus | 66K | 53 | 115 | Yes |
| Yellow fever virus | NS5 | 25 | 47 | TBD |
a Percentage disorder was determined by PONDR (Predictor of Natrually Disordered Regions).
b Longest intrinsically disordered region in length of amino acids.
c If specific sites have been mapped as phosphorylated residues within the viral protein, this column documents if the residue(s) are located within a stretch of disordered amino acids. TBD, to be determined.
This work was supported, in whole or in part, by National Institutes of Health, NIGMS, Grant R01GM0089001 (to Kevin D. Raney and C. E. C.) and National Institutes of Health, NIAID, Grant R21 AI100590 (to C. E. C.).
- HCV
- hepatitis C virus
- SI
- S2204I mutation
- NS3, NS5A, and NS5B
- hepatitis C virus non-structural protein 3, 5A, and 5B, respectively
- IDD
- intrinsically disordered domain
- PPII
- poly-proline-II
- RT-qPCR
- real-time quantitative PCR
- CHX
- cycloheximide
- K25
- 25 μg/ml kanamycin
- C20
- 20 μg/ml chloramphenicol
- Ni-NTA
- nickel-nitrilotriacetic acid
- SH3
- Src homology 3
- HSQC
- heteronuclear single quantum correlation
- PKAc
- catalytic subunit of PKA.
REFERENCES
- 1. Love R. A., Brodsky O., Hickey M. J., Wells P. A., Cronin C. N. (2009) Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J. Virol. 83, 4395–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tellinghuisen T. L., Marcotrigiano J., Rice C. M. (2005) Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435, 374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang L., Hwang J., Sharma S. D., Hargittai M. R., Chen Y., Arnold J. J., Raney K. D., Cameron C. E. (2005) Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J. Biol. Chem. 280, 36417–36428 [DOI] [PubMed] [Google Scholar]
- 4. Hwang J., Huang L., Cordek D. G., Vaughan R., Reynolds S. L., Kihara G., Raney K. D., Kao C. C., Cameron C. E. (2010) Hepatitis C virus nonstructural protein 5A: biochemical characterization of a novel structural class of RNA-binding proteins. J. Virol. 84, 12480–12491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hanoulle X., Verdegem D., Badillo A., Wieruszeski J. M., Penin F., Lippens G. (2009) Domain 3 of non-structural protein 5A from hepatitis C virus is natively unfolded. Biochem. Biophys. Res. Commun. 381, 634–638 [DOI] [PubMed] [Google Scholar]
- 6. Liang Y., Kang C. B., Yoon H. S. (2006) Molecular and structural characterization of the domain 2 of hepatitis C virus non-structural protein 5A. Mol. Cells 22, 13–20 [PubMed] [Google Scholar]
- 7. Cordek D. G., Bechtel J. T., Maynard A. T., Kazmierski W. M., Cameron C. E. (2011) Targeting the NS5A protein of HCV: an emerging option. Drugs Future 36, 691–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Uversky V. N., Oldfield C. J., Dunker A. K. (2008) Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys. 37, 215–246 [DOI] [PubMed] [Google Scholar]
- 9. Blight K. J., Kolykhalov A. A., Rice C. M. (2000) Efficient initiation of HCV RNA replication in cell culture. Science 290, 1972–1974 [DOI] [PubMed] [Google Scholar]
- 10. Bukh J., Pietschmann T., Lohmann V., Krieger N., Faulk K., Engle R. E., Govindarajan S., Shapiro M., St Claire M., Bartenschlager R. (2002) Mutations that permit efficient replication of hepatitis C virus RNA in Huh-7 cells prevent productive replication in chimpanzees. Proc. Natl. Acad. Sci. U.S.A. 99, 14416–14421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yi M., Hu F., Joyce M., Saxena V., Welsch C., Chavez D., Guerra B., Yamane D., Veselenak R., Pyles R., Walker C. M., Tyrrell L., Bourne N., Lanford R. E., Lemon S. M. (2014) Evolution of a cell culture-derived genotype 1a hepatitis C virus (H77S.2) during persistent infection with chronic hepatitis in a chimpanzee. J. Virol. 88, 3678–3694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quintavalle M., Sambucini S., Di Pietro C., De Francesco R., Neddermann P. (2006) The α isoform of protein kinase CKI is responsible for hepatitis C virus NS5A hyperphosphorylation. J. Virol. 80, 11305–11312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neddermann P., Quintavalle M., Di Pietro C., Clementi A., Cerretani M., Altamura S., Bartholomew L., De Francesco R. (2004) Reduction of hepatitis C virus NS5A hyperphosphorylation by selective inhibition of cellular kinases activates viral RNA replication in cell culture. J. Virol. 78, 13306–13314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao M., Nettles R. E., Belema M., Snyder L. B., Nguyen V. N., Fridell R. A., Serrano-Wu M. H., Langley D. R., Sun J. H., O'Boyle D. R., 2nd, Lemm J. A., Wang C., Knipe J. O., Chien C., Colonno R. J., Grasela D. M., Meanwell N. A., Hamann L. G. (2010) Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465, 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lemm J. A., O'Boyle D., 2nd, Liu M., Nower P. T., Colonno R., Deshpande M. S., Snyder L. B., Martin S. W., St Laurent D. R., Serrano-Wu M. H., Romine J. L., Meanwell N. A., Gao M. (2010) Identification of hepatitis C virus NS5A inhibitors. J. Virol. 84, 482–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Appel N., Pietschmann T., Bartenschlager R. (2005) Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J. Virol. 79, 3187–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tellinghuisen T. L., Foss K. L., Treadaway J. (2008) Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 4, e1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang L., Sineva E. V., Hargittai M. R., Sharma S. D., Suthar M., Raney K. D., Cameron C. E. (2004) Purification and characterization of hepatitis C virus non-structural protein 5A expressed in Escherichia coli. Protein Expr. Purif. 37, 144–153 [DOI] [PubMed] [Google Scholar]
- 19. Phan T., Beran R. K., Peters C., Lorenz I. C., Lindenbach B. D. (2009) Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 83, 8379–8395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Q., Arnold J. J., Uchida A., Raney K. D., Cameron C. E. (2010) Phosphate release contributes to the rate-limiting step for unwinding by an RNA helicase. Nucleic Acids Res. 38, 1312–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhong W., Ferrari E., Lesburg C. A., Maag D., Ghosh S. K., Cameron C. E., Lau J. Y., Hong Z. (2000) Template/primer requirements and single nucleotide incorporation by hepatitis C virus nonstructural protein 5B polymerase. J. Virol. 74, 9134–9143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuiken C., Yusim K., Boykin L., Richardson R. (2005) The Los Alamos hepatitis C sequence database. Bioinformatics 21, 379–384 [DOI] [PubMed] [Google Scholar]
- 23. Gohara D. W., Ha C. S., Kumar S., Ghosh B., Arnold J. J., Wisniewski T. J., Cameron C. E. (1999) Production of “authentic” poliovirus RNA-dependent RNA polymerase (3D(pol)) by ubiquitin-protease-mediated cleavage in Escherichia coli. Protein Expr. Purif. 17, 128–138 [DOI] [PubMed] [Google Scholar]
- 24. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 25. Sahu D., Bastidas M., Showalter S. (2014) Generating NMR chemical shift assignments of intrinsically disordered proteins using carbon-detected NMR methods. Anal. Biochem. 449, 17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goddard T. D., Kneller D. G. (2006) SPARKY, version 3.113, University of California, San Francisco [Google Scholar]
- 27. Kaneko T., Tanji Y., Satoh S., Hijikata M., Asabe S., Kimura K., Shimotohno K. (1994) Production of two phosphoproteins from the NS5A region of the hepatitis C viral genome. Biochem. Biophys. Res. Commun. 205, 320–326 [DOI] [PubMed] [Google Scholar]
- 28. Katze M. G., Kwieciszewski B., Goodlett D. R., Blakely C. M., Neddermann P., Tan S. L., Aebersold R. (2000) Ser2194 is a highly conserved major phosphorylation site of the hepatitis C virus nonstructural protein NS5A. Virology 278, 501–513 [DOI] [PubMed] [Google Scholar]
- 29. Nordle Gilliver A., Griffin S., Harris M. (2010) Identification of a novel phosphorylation site in hepatitis C virus NS5A. J. Gen. Virol. 91, 2428–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reed K. E., Rice C. M. (1999) Identification of the major phosphorylation site of the hepatitis C virus H strain NS5A protein as serine 2321. J. Biol. Chem. 274, 28011–28018 [DOI] [PubMed] [Google Scholar]
- 31. Lemay K. L., Treadaway J., Angulo I., Tellinghuisen T. L. (2013) A hepatitis C virus NS5A phosphorylation site that regulates RNA replication. J. Virol. 87, 1255–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ross-Thriepland D., Harris M. (2014) Insights into the complexity and functionality of hepatitis C virus NS5A phosphorylation. J. Virol. 88, 1421–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ide Y., Tanimoto A., Sasaguri Y., Padmanabhan R. (1997) Hepatitis C virus NS5A protein is phosphorylated in vitro by a stably bound protein kinase from HeLa cells and by cAMP-dependent protein kinase A-α catalytic subunit. Gene 201, 151–158 [DOI] [PubMed] [Google Scholar]
- 34. Ide Y., Zhang L., Chen M., Inchauspe G., Bahl C., Sasaguri Y., Padmanabhan R. (1996) Characterization of the nuclear localization signal and subcellular distribution of hepatitis C virus nonstructural protein NS5A. Gene 182, 203–211 [DOI] [PubMed] [Google Scholar]
- 35. Macdonald A., Crowder K., Street A., McCormick C., Harris M. (2004) The hepatitis C virus NS5A protein binds to members of the Src family of tyrosine kinases and regulates kinase activity. J. Gen. Virol. 85, 721–729 [DOI] [PubMed] [Google Scholar]
- 36. Tan S. L., Nakao H., He Y., Vijaysri S., Neddermann P., Jacobs B. L., Mayer B. J., Katze M. G. (1999) NS5A, a nonstructural protein of hepatitis C virus, binds growth factor receptor-bound protein 2 adaptor protein in a Src homology 3 domain/ligand-dependent manner and perturbs mitogenic signaling. Proc. Natl. Acad. Sci. U.S.A. 96, 5533–5538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lohmann V., Körner F., Koch J., Herian U., Theilmann L., Bartenschlager R. (1999) Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285, 110–113 [DOI] [PubMed] [Google Scholar]
- 38. Macdonald A., Mazaleyrat S., McCormick C., Street A., Burgoyne N. J., Jackson R. M., Cazeaux V., Shelton H., Saksela K., Harris M. (2005) Further studies on hepatitis C virus NS5A-SH3 domain interactions: identification of residues critical for binding and implications for viral RNA replication and modulation of cell signalling. J. Gen. Virol. 86, 1035–1044 [DOI] [PubMed] [Google Scholar]
- 39. Feuerstein S., Solyom Z., Aladag A., Favier A., Schwarten M., Hoffmann S., Willbold D., Brutscher B. (2012) Transient structure and SH3 interaction sites in an intrinsically disordered fragment of the hepatitis C virus protein NS5A. J. Mol. Biol. 420, 310–323 [DOI] [PubMed] [Google Scholar]
- 40. Elam W. A., Schrank T. P., Campagnolo A. J., Hilser V. J. (2013) Evolutionary conservation of the polyproline II conformation surrounding intrinsically disordered phosphorylation sites. Protein Sci. 22, 405–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hughes M., Gretton S., Shelton H., Brown D. D., McCormick C. J., Angus A. G., Patel A. H., Griffin S., Harris M. (2009) A conserved proline between domains II and III of hepatitis C virus NS5A influences both RNA replication and virus assembly. J. Virol. 83, 10788–10796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pfannkuche A., Büther K., Karthe J., Poenisch M., Bartenschlager R., Trilling M., Hengel H., Willbold D., Häussinger D., Bode J. G. (2011) c-Src is required for complex formation between the hepatitis C virus-encoded proteins NS5A and NS5B: a prerequisite for replication. Hepatology 53, 1127–1136 [DOI] [PubMed] [Google Scholar]
- 43. Martin-Garcia J. M., Luque I., Ruiz-Sanz J., Camara-Artigas A. (2012) The promiscuous binding of the Fyn SH3 domain to a peptide from the NS5A protein. Acta Crystallogr. D Biol. Crystallogr. 68, 1030–1040 [DOI] [PubMed] [Google Scholar]
- 44. Egger D., Wölk B., Gosert R., Bianchi L., Blum H. E., Moradpour D., Bienz K. (2002) Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76, 5974–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Adler J., Parmryd I. (2010) Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander's overlap coefficient. Cytometry A 77, 733–742 [DOI] [PubMed] [Google Scholar]
- 46. Kase H., Iwahashi K., Nakanishi S., Matsuda Y., Yamada K., Takahashi M., Murakata C., Sato A., Kaneko M. (1987) K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem. Biophys. Res. Commun. 142, 436–440 [DOI] [PubMed] [Google Scholar]
- 47. Hidaka H., Inagaki M., Kawamoto S., Sasaki Y. (1984) Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry 23, 5036–5041 [DOI] [PubMed] [Google Scholar]
- 48. Chijiwa T., Mishima A., Hagiwara M., Sano M., Hayashi K., Inoue T., Naito K., Toshioka T., Hidaka H. (1990) Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J. Biol. Chem. 265, 5267–5272 [PubMed] [Google Scholar]
- 49. Glass D. B., Cheng H. C., Mende-Mueller L., Reed J., Walsh D. A. (1989) Primary structural determinants essential for potent inhibition of cAMP-dependent protein kinase by inhibitory peptides corresponding to the active portion of the heat-stable inhibitor protein. J. Biol. Chem. 264, 8802–8810 [PubMed] [Google Scholar]
- 50. Dyson H. J. (2011) Expanding the proteome: disordered and alternatively folded proteins. Q. Rev. Biophys. 44, 467–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Joerger A. C., Fersht A. R. (2010) The tumor suppressor p53: from structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2, a000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Moriishi K., Matsuura Y. (2007) Host factors involved in the replication of hepatitis C virus. Rev. Med. Virol. 17, 343–354 [DOI] [PubMed] [Google Scholar]
- 53. Backes P., Quinkert D., Reiss S., Binder M., Zayas M., Rescher U., Gerke V., Bartenschlager R., Lohmann V. (2010) Role of annexin A2 in the production of infectious hepatitis C virus particles. J. Virol. 84, 5775–5789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Saxena V., Lai C. K., Chao T. C., Jeng K. S., Lai M. M. (2012) Annexin A2 is involved in the formation of hepatitis C virus replication complex on the lipid raft. J. Virol. 86, 4139–4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Burckstümmer T., Kriegs M., Lupberger J., Pauli E. K., Schmittel S., Hildt E. (2006) Raf-1 kinase associates with hepatitis C virus NS5A and regulates viral replication. FEBS Lett. 580, 575–580 [DOI] [PubMed] [Google Scholar]
- 56. Tamma G., Procino G., Mola M. G., Svelto M., Valenti G. (2008) Functional involvement of Annexin-2 in cAMP induced AQP2 trafficking. Pflugers Arch. 456, 729–736 [DOI] [PubMed] [Google Scholar]
- 57. Dumaz N., Marais R. (2003) Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J. Biol. Chem. 278, 29819–29823 [DOI] [PubMed] [Google Scholar]
- 58. Grieco D., Porcellini A., Avvedimento E. V., Gottesman M. E. (1996) Requirement for cAMP-PKA pathway activation by M phase-promoting factor in the transition from mitosis to interphase. Science 271, 1718–1723 [DOI] [PubMed] [Google Scholar]
- 59. Targett-Adams P., Graham E. J., Middleton J., Palmer A., Shaw S. M., Lavender H., Brain P., Tran T. D., Jones L. H., Wakenhut F., Stammen B., Pryde D., Pickford C., Westby M. (2011) Small molecules targeting hepatitis C virus-encoded NS5A cause subcellular redistribution of their target: insights into compound modes of action. J. Virol. 85, 6353–6368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Blight K. J., McKeating J. A., Rice C. M. (2002) Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76, 13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu W., Harrison S. C., Eck M. J. (1997) Three-dimensional structure of the tyrosine kinase c-Src. Nature 385, 595–602 [DOI] [PubMed] [Google Scholar]