

Background: Much evidence indicates that giant viruses, including Megavirus chilensis, possibly encode autonomous glycosylation systems.
Results: The Megavirus genome encodes proteins involved in the synthesis of 2-acetamido-2,6-dideoxy hexoses.
Conclusion: N-Acetyl-rhamnosamine that is found in the virion glycans is produced by a novel viral biosynthetic pathway.
Significance: These results will help in understanding the origin and function of the virally encoded glycosylation machineries.
Keywords: Glycosylation, Virus Structure, Nuclear Magnetic Resonance (NMR), Mass Spectrometry (MS), Gas Chromatography-Mass Spectrometry (GC-MS), Carbohydrate Metabolism, N-Acetyl-L-Rhamnosamine, Megavirus Chilensis, UDP-N-acetylglucosamine
Abstract
Giant viruses mimicking microbes, by the sizes of their particles and the heavily glycosylated fibrils surrounding their capsids, infect Acanthamoeba sp., which are ubiquitous unicellular eukaryotes. The glycans on fibrils are produced by virally encoded enzymes, organized in gene clusters. Like Mimivirus, Megavirus glycans are mainly composed of virally synthesized N-acetylglucosamine (GlcNAc). They also contain N-acetylrhamnosamine (RhaNAc), a rare sugar; the enzymes involved in its synthesis are encoded by a gene cluster specific to Megavirus close relatives. We combined activity assays on two enzymes of the pathway with mass spectrometry and NMR studies to characterize their specificities. Mg534 is a 4,6-dehydratase 5-epimerase; its three-dimensional structure suggests that it belongs to a third subfamily of inverting dehydratases. Mg535, next in the pathway, is a bifunctional 3-epimerase 4-reductase. The sequential activity of the two enzymes leads to the formation of UDP-l-RhaNAc. This study is another example of giant viruses performing their glycan synthesis using enzymes different from their cellular counterparts, raising again the question of the origin of these pathways.
Introduction
Nucleocytoplasmic large DNA viruses are a monophyletic group of viruses infecting eukaryotes (1). They are characterized by large particles and genome sizes, the largest, coined giant viruses, overlapping the cellular world. Large DNA viruses encode many genes that are not generally found in other viruses, such as elements of the translation machinery and enzymes of metabolic pathways. Recent evidence has indicated that some giant viruses also encode autonomous glycosylation systems (2, 3). These machineries include the glycosyltransferases, which are needed for the production of complex carbohydrates, and the enzymes necessary for the biosynthesis of the nucleotide sugar substrates. The presence of metabolic pathways for the production of the glycosyltransferase substrates makes the viruses independent from their host supply and could possibly extend the range of hosts that can be infected.
We have recently demonstrated that Mimivirus, belonging to the Mimiviridae family, encodes a functional pathway for UDP-N-acetyl-d-glucosamine (UDP-d-GlcNAc) production (4). Orthologs have been found also in other members of this family (i.e. in Megavirus chilensis, Phaeocystis globosa virus (PgV), and Cafeteria roenbergensis virus (CroV)), suggesting that the GlcNAc is a key component of glycans in several Mimiviridae species (4). A UDP-l-rhamnose pathway was identified in Mimivirus and in some members of the chlorovirus family (5); similarly, the presence of GDP-l-fucose biosynthetic enzymes is a feature of most chloroviruses identified so far (6). However, analyses of nucleocytoplasmic glycans have demonstrated that, beside sugars widely used by most organisms, they often contain unusual monosaccharides, which are very rare in nature. For instance, Mimivirus glycoconjugates contain viosamine and its O-methylated derivatives, and the corresponding enzymes required for its biosynthesis are virally encoded (7). Finally, the glycoconjugates associated with the major capsid protein of chlorella virus PBCV-1 are characterized by the presence of d-rhamnose, a stereoisomer that is very rarely used compared with its l-form (3); again, PBCV-1 encodes a bifunctional GDP-d-mannose dehydratase, which, besides its typical activity, is also able to catalyze GDP-d-rhamnose formation with high efficiency (8).
Megavirus chilensis was isolated from marine coastal water off of Chile; it is the largest member of the Mimiviridae family and is endowed with a genome of 1.26 kb encoding 1,120 proteins (9). The preliminary GC-MS analyses of Megavirus glycoconjugates revealed the presence of large amounts of GlcNAc but also of other sugars that did not correspond to any standard available. Inspection of the M. chilensis genome revealed the presence of a gene cluster, containing several genes potentially involved in glycosylation, including enzymes for nucleotide sugar production. Specifically, sequence analysis of two of them has revealed some homology with enzymes responsible for the biosynthesis of 2-acetamido-2,6-dideoxy-β-l-hexoses in bacteria. The l-enantiomers of 6-deoxy-hexosamines are rare sugars in nature and are found in LPS and other surface polysaccharides in some Gram-positive and Gram-negative bacteria (10–12).
The first reaction that leads to the formation of the UDP-2-acetamido-2,6-dideoxy-β-l-hexoses is the dehydration of UDP-d-GlcNAc. Two different types of enzymes have been identified. In one case, this step is a simple dehydration with formation of UDP-2-acetamido-2,6-dideoxy-α-d-xylo-4-hexulose, which is typical of the UDP-bacillosamine or UDP-N-acetyl-d-fucosamine (UDP-d-FucNAc)5 and UDP-N-acetyl-d-quinovosamine (UDP-d-QuiNAc) pathways (13, 14). In other cases, the enzymes are “inverting” 4,6-dehydratases, catalyzing also the epimerization of C-5, with the consequent inversion of the d-configuration to l-, and the formation of UDP-2-acetamido-2,6-dideoxy-β-l-arabino-4-hexulose (Fig. 1). This reaction is catalyzed by PseB/FlaA1 in the case of the pseudaminic acid (Pse) pathway, where the transfer of an amino group to the C-4 carbonyl follows this first step (13). On the other hand, several enzymes are involved in the formation of 2-acetamido-2,6-dideoxy-l-hexoses (Fig. 1). Three enzymes from Pseudomonas aeruginosa O11 (WbjB, WbjC, and WbjD) and Staphylococcus aureus type 5 (CapE, CapF, and CapG) are required for the conversion of UDP-d-GlcNAc to UDP-N-acetyl-l-fucosamine (UDP-l-FucNAc) (11, 12). For UDP-N-acetyl-l-quinovosamine (UDP-l-QuiNAc), a parallel pathway is present in Vibrio cholera O37, including WbvB, WbvR, and WbvD (10). UDP-l-FucNAc and UDP-l-QuiNAc only differ by the orientation of the newly generated 4-hydroxyl group, after the stereospecific reduction of the 4-keto group (Fig. 1). Conflicting data have been reported in the literature about the identification of enzymes that catalyze the different steps of these pathways. Specifically, 3-epimerase activity was initially assigned to WbjB/CapE and WbvB, together with the 4,6-dehydratase and 5-epimerase activities (10, 11). Hence, the following enzymes in the pathways, WbjC/CapF and WbvR, were proposed to only have a 4-reductase activity (10). Further studies have clarified this issue, demonstrating that WbjB/CapE behave as 4,6-dehdyratase-5-epimerase, whereas WbjC/CapF catalyze both 3-epimerization and 4-reduction, converting UDP-2-acetamido-2,6-dideoxy-β-l-arabino-4-hexulose to UDP-N-acetyl-l-pneumosamine (UDP-l-PneNAc) (12). This finding is also consistent with the structure of CapF proteins, which comprise two different domains: an N-terminal SDR-like reductase domain and a C-terminal cupin-like epimerase domain, which catalyze the two reactions independently (15). Conversely, there were no further studies on WbvR, responsible for the UDP-N-acetyl-l-rhamnosamine (UDP-RhaNAc) production, which is devoid of a cupin-like epimerase domain and displays only a single SDR-like domain. Finally, WbjD/CapG and WbvD are responsible for the epimerization of the 2-acetamido group, leading to UDP-l-FucNAc and UDP-l-QuiNAc, respectively (10).
FIGURE 1.

Pathway for the biosynthesis of UDP-2-acetamido-2,6-dideoxy-hexoses. 1, UDP-α-d-GlcNAc; 2, UDP-2-acetamido-2,6-dideoxy-β-l-arabino-4-hexulose; 3, UDP-2-acetamido-2,6-dideoxy-β-l-lyxo-4-hexulose; 4, UDP-β-l-RhaNAc; 5, UDP-β-l-QuiNAc; 6, UDP-β-l-PneNAc; 7, UDP-β-l-FucNAc. The bacterial enzymes involved in the different steps are indicated in italic type. From compound 2, a transamination reaction leads to the pseudamic acid pathway. For further details, see the Introduction.
The aim of this study was to identify and characterize the two first enzymes of the M. chilensis pathway for the production of 6-deoxy-hexosamines. We elucidated the reactions catalyzed by each protein, using combined HPLC, mass spectrometry, and NMR spectroscopy analyses and demonstrated that the viral enzymes produce UDP-RhaNAc. RhaNAc is present in the M. chilensis viral particles, suggesting a possible role of this sugar in the virus biology. Finally, we report the crystal structure of the first enzyme involved in this pathway and discuss the differences of the viral structure compared with its previously reported cellular counterparts.
EXPERIMENTAL PROCEDURES
Cloning and Expression of the Recombinant Proteins
M. chilensis genomic DNA was purified as described previously (9). mg534 (YP_004894585) and mg535 (YP_004894586) coding regions were amplified by PCR using Pfu polymerase and they were then cloned in pGEX-6P-1 vector (GE Healthcare) using BamHI and XhoI restriction enzymes following the standard procedures. The sequenced constructs were then used for transformation in Escherichia coli BL-21 GOLD cells (Stratagene).
Expression of the glutathione S-transferase (GST) fusion proteins, which were used to test the enzymatic activity, was performed as described previously (7). Cells were grown at 22 °C in Terrific broth medium; isopropyl β-d-1-thiogalactopyranoside was added at a final concentration of 0.1 mm when the culture A600 reached 0.6. After induction, cells were further incubated overnight at 22 °C. Purification and proteolytic removal of the GST fusion protein were done as reported (7). All purification steps, including proteolytic cleavage, were performed at 4 °C, with the exception of the binding to GSH-Sepharose, which was done at 20 °C. The purified proteins were concentrated using centrifugal filters Amicon® Ultra (Millipore). The absorbance spectrum of the purified protein was obtained using a Beckman Coulter DU800 spectrophotometer, and concentration was estimated by absorbance at 280 nm using a computed extinction coefficient of 32,320 m−1 cm−1 and 35,870 m−1 cm−1 for Mg534 and Mg535, respectively (16). Protein purity, assessed by SDS-PAGE, exceeded 95% in all preparations.
For crystallization, the mg534 gene was cloned into an in-house modified pETDuet vector (Novagen) to yield an N-terminally His6-tagged protein. The resulting vector was transformed into the E. coli Rosetta(DE3) strain. Cells were grown in 2YT medium (Difco) containing 100 μg.ml−1 ampicillin and 34 μg·ml−1 chloramphenicol at 37 °C to an A600 of 0.6, and then the temperature was shifted to 17 °C. After 30 min, protein expression was induced by the addition of 0.2 mm isopropyl β-thiogalactopyranoside. Cells were grown 14–16 h postinduction. Bacteria were harvested by centrifugation and resuspended in buffer A (50 mm Tris-HCl, 300 mm NaCl, pH 8.0) containing 0.01% (w/v) DNase, 0.01% (w/v) lysozyme, and antiprotease tablets (Roche Applied Science), and proteins were extracted by sonication. The crude lysate was clarified by centrifugation at 13,000 × g for 45 min before being applied to a 1-ml HisPure nickel-nitrilotriacetic acid column (Pierce) charged with Ni2+ and equilibrated with 50 mm Tris-HCl, 300 mm NaCl buffer, pH 8.5 (buffer A) on an AKTA Explorer 10S FPLC system (GE Healthcare). The column was washed with 10 column volumes of buffer A containing 25 mm imidazole and 20 column volumes of buffer A containing 50 mm imidazole. Elution was performed with a linear gradient over 20 column volumes from 50 to 500 mm imidazole.
The N-terminal His tag was removed by proteolytic cleavage using an in-house produced His-tagged human rhinovirus 3C protease, and a second run of purification was performed to separate the cleaved protein from the tag. The recombinant protein corresponds to the native enzyme in which the initial methionine is replaced by GPGS sequence resulting from the cleavage site. The protein was then desalted on a desalting column (HiPrep 26/10 desalting, GE Healthcare) in 10 mm Tris, pH 8.0, and concentrated to 6.5 mg·ml−1.
Mg534 Crystallization, Data Collection, and Refinement
The protein crystallization assays were performed at 293 K against 288 different conditions corresponding to commercially available solution sets (CrystalScreen-Hampton Research, Wizards-Emerald BioStructures) and “in-house” designed conditions using the SAmBA software (17, 18). The screening for crystallization conditions was performed on three 96-well crystallization plates (Greiner) loaded by an eight-needle dispensing robot (Tecan, WS 100/8 work station modified for our needs), mixing 0.5 μl of protein with 0.5 μl of reservoir in a sitting drop (19). Crystals appeared in malic acid buffer (0.1 m), PEG 3350 (20%) (w/v), pH 7.0. We refined these conditions at 293 K by hanging drop vapor diffusion using 24-well culture plates (Greiner) using the hanging drop vapor diffusion method by mixing 1 μl of protein with 0.5 μl of reservoir containing malic acid buffer (0.1 m), PEG 3350 (22–25%) (w/v), pH 7.0. Crystals were flash-frozen in liquid nitrogen. No cryopreservatives were used before freezing. A native data set was collected at 100 K at beamline ID23-eh2 at the European Synchrotron Radiation Facility (Grenoble, France). The crystal diffracted to a resolution of 2.2 Å. Data collection was performed with an oscillation angle of 0.2° and a crystal-detector distance of 236.2 mm, and 300 images were collected on a PILATUS detector. The crystal belongs to the hexagonal space group P6322 with cell parameters of a = b = 153.5 Å, c = 60.7 Å. MOSFLM (20) and SCALA (21, 22) from the CCP4 package (Collaborative Computational Project, Number 4) were used for the processing, scaling, and reduction of the data set. Statistics of the processed data are presented in Table 1.
TABLE 1.
Data collection and refinement statistics
| Mg534 | |
|---|---|
| Data collection | |
| Space group | P6322 |
| Cell dimensions | |
| a, b, c (Å) | 153.5, 153.5, 60.7 |
| α, β, γ (degrees) | 90.0, 90.0, 90.0 |
| Wavelength (Å) | 0.8729 |
| Resolution range (Å) | 38.7–2.2 |
| Total observations | 150,564 (19,931) |
| Unique observations | 21,901 (3,132) |
| Rmerge (%) | 8.7 (29.9) |
| I/σI | 15.5 (6.0) |
| Completeness (%) | 100 (100) |
| Multiplicity | 6.9 (6.4) |
| Refinement | |
| Rwork/Rfree | 19.71/23.4 |
| No. of atoms | |
| Protein | 2,381 |
| Ligand/ion | 48 |
| Water | 89 |
| B-factors | |
| Protein | 20.9 |
| Ligand/Ion | 28.2 |
| Water | 18.6 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (degrees) | 1.181 |
a Rsym = Σ|Ii − 〈Ii〉|/ΣIi, where Ii is the intensity of the ith observation and 〈Ii〉 is the mean intensity of the reflection.
b Rwork = Σ(‖Fo| − |Fc‖/Σ|Fo|).
c Rfree = R value for a randomly selected subset (5%) of the data that were not used for minimization of the crystallographic residual.
The structure was solved by molecular replacement using PHASER (23) implemented in the PHENIX suite (24) using the structure of Helicobacter pylori FlaA1 (PDB entry 2GN4 (25)). One unique solution with one molecule per asymmetric unit was found with a translation function Z-score of 11.6. The model was refined using PHENIX.REFINE, including simulated annealing, coordinates, and B-factor refinement. After several rounds of manual building, the final round of refinement resulted in a final Rwork of 19.7% and Rfree of 23.4%. The refined structure of Mg534 consists of residues 1–319. Residues 173–195, being disordered in the crystal structure, are absent of the final model. The quality of the model was validated using Molprobity. 96.6% of the residues are in the most favored regions of the Ramachandran plot, and 3.4% are in the additionally allowed regions. Refinement statistics are listed in Table 1. The statistics are provided on the model after a last round of refinement, including all reflections.
Enzymatic Activities
Enzymatic activity of the expressed proteins was determined using an HPLC assay. In the standard assay, the purified Mg534 protein was incubated in 50 mm Tris-HCl. 150 mm NaCl, pH 7.5, 25 °C, in the presence of 200 μm UDP-GlcNAc and 100 μm NADP+. All chemicals were obtained from Sigma-Aldrich. At different time points, aliquots were withdrawn, and protein was removed by heating at 99 °C for 3 min, followed by centrifugation. HPLC analysis was performed using a Wesca/R anion exchange column, as described (4, 7).
Mg535 activity was determined using Mg534 product as substrate. Mg534 was incubated with UDP-GlcNAc (0.5–1 mm) and NADP+ (0.1 mm) for 30 min to ensure complete conversion of UDP-GlcNAc to the intermediate product. Mg534 was then removed by ultrafiltration (Amicon, YM-10). To prevent degradation of this compound, Mg534 product was prepared immediately before use. Mg535 activity was analyzed in the same conditions reported above in the presence of a molar excess of NADPH with respect to the nucleotide sugar concentration or with the addition of an NADPH-regenerating system (1 mm glucose-6-P, 5 units·ml−1 glucose-6-phosphate dehydrogenase). Product formation was monitored by HPLC analysis. ESI-MS and MS/MS analysis was performed in direct infusion analysis on an LCQ FLEET (Thermo Scientific) in negative ion mode.
For NMR analysis of products obtained by Mg534 + Mg535 activity, the produced compounds were further purified by HPLC, using a Bio-Sil C18 HL90–10 column (Bio-Rad), using 100 mm NH4HCO3, pH 7.1, and a linear gradient to 30% CH3OH from 10 to 35 min. Peaks corresponding to the nucleotide sugars were collected and subjected to solid phase extraction using Superclean ENVI-Carb tubes (Supelco). The cartridge was sequentially treated with 6 ml of 60% CH3CN containing 0.3% NH4HCO3, pH 9.0, 6 ml of H2O, and 6 ml of 100 mm NH4HCO3, pH 7.1. The collected eluate was applied to the cartridge using a peristaltic pump, followed by a wash with 6 ml of H2O. Sequential elution of the nucleotide sugar were accomplished using 1 ml of 60% CH3CN containing 0.3% NH4HCO3, pH 9.0. The fractions showing UV absorbance at 260 nm were pooled and dried under vacuum.
NMR Analysis
All NMR experiments were carried out on a Bruker DRX-600 spectrometer equipped with a cryoprobe. Chemical shifts of spectra recorded in D2O are expressed in ppm relative to internal acetone (2.225 and 31.45 ppm). The spectral width was set to 10 ppm, and the frequency carrier was placed at the residual HOD peak, suppressed by presaturation. Two-dimensional spectra (double quantum-correlated spectroscopy, total correlation spectroscopy, gradient heteronuclear single quantum coherence, and gradient heteronuclear multiple bond coherence) were measured using standard Bruker software. For all of the experiments, 512 FIDs of 2,048 complex data points were collected, 32 scans/FID were acquired for homonuclear spectra, and a mixing time of 100 ms was used for total correlation spectroscopy spectrum acquisition. The gradient heteronuclear single quantum coherence spectrum was acquired with 50 scans/FID, and the GARP sequence was used for 13C decoupling during acquisition; gradient heteronuclear multiple bond coherence scans doubled those of the gradient heteronuclear single quantum coherence spectrum. Data processing and analysis were performed with Bruker Topspin 3 program.
GC-MS Analyses
To verify the presence of the product obtained by the combined activity of Mg534 and Mg535 in M. chilensis, both the product and the viral particles were analyzed by GC-MS. M. chilensis was purified from an Acantamoeba castellani culture as described (9). Viral proteins were isolated by incubating the viral particles (1 × 1011 in 100 μl) at 99 °C for 10 min in the presence of 100 mm DTT and 2% SDS. Following acetone precipitation, the pellet was resuspended in 200 μl of PBS and briefly sonicated; 10 units of Benzonase (Sigma) were added and incubated for 60 min at 37 °C. After a second acetone precipitation, the pellet was subjected to acid hydrolysis with 2 m TFA at 100 °C for 4 h. Standard UDP-RhaNAc was directly subjected to acid hydrolysis at the end of the incubation without any further purification. Preparation of alditol acetate derivatives and GC-MS analysis were done as described (7). Samples resuspended in dichloromethane were injected into an HP5890 series II gas chromatograph coupled to a HP5970 mass spectrometer equipped with an electron impact ionization source (Hewlett Packard). Energy of the electron impact ionization source was 70 eV. Separation was performed on a DB5ms capillary column (Phenomenex, 0.25 mm × 30 m); the helium gas flow was 1 ml·min−1. The oven temperature gradient was as follows: initial temperature of 80 °C, 80–120 °C (rate, 3 °C/min), isothermal at 120 °C for 1 min, 120–230 °C (rate 3 °C/min), and isothermal at 230 °C for 4 min. The MS analysis was performed in full-scan mode, from m/z 40 to m/z 550.
RESULTS
Sequence Analysis
Analysis of the Megavirus genome revealed the presence of a gene cluster containing several ORFs potentially involved in glycosylation (Fig. 2). We used a BLAST search to identify possible functional homologues of the corresponding gene products. mg534, the first gene of the cluster, encodes a 323-amino acid protein that showed significant similarities with inverting UDP-GlcNAc 4,6-dehydratases; specifically, best hits with cellular organisms (39% identity) were with bacterial sequences that are suggestive of an involvement in pseudaminic acid synthesis. Homology was slightly lower with the well characterized enzymes PseB from Helicobacter pylori (37% identity) and FlaA1 from P. aeruginosa (35% identity), both responsible for the first step of the pseudaminic pathway (25, 26). Moreover, it shared 37% identity with CapE from Staphylococcus aureus and WbjB from P. aeruginosa, both involved in UDP-FucNAc production (11), and with WbvB from V. cholerae, belonging to the UDP-l-QuiNAc pathway (10). Interestingly, whereas CapE and WbjB are characterized by motif MXXXK in the catalytic triad (11, 27), the mg534 gene product contains the canonical sequon, YXXXK, typical of SDR enzymes, also found in PseB/FlaA1 (26, 27). Mg534 lacks the so-called “latch” that has been identified in CapE (27), which is also absent in PseB/FlaA1 and related inverting 4,6-dehydratases.
FIGURE 2.

The M. chilensis six-gene cluster. The arrows indicate the coding strand.
The second ORF of the cluster, mg535, encodes a 270-amino acid long protein, which displayed 34% sequence identity with WbvR of V. cholerae. This enzyme is characterized by a Rossmann NADB domain and is responsible for the formation of UDP-l-RhaNAc, an intermediate of the UDP-l-QuiNAc pathway (10). No homology was observed with the P. aeruginosa WbjC and S. aureus CapF, both catalyzing the formation of UDP-PneNAc, the second step of the UDP-l-FucNac pathway (10, 15). The third gene of the cluster, mg536, has been annotated as a UDP-GlcNAc 2-epimerase; however, the similarity with V. cholerae O37 WbvD (50% identity) and P. aeruginosa WbjD (47% identity) suggested that it catalyzes the epimerization of C-2 in the 2-acetamido-2,6-dideoxy-sugars (10).
Altogether, these results indicated that the first three genes of the cluster (mg534, mg535, and mg536) could be required for the formation of UDP-2-acetamido-2,6-dideoxy-l-hexoses. It is worth noting that the cluster contains other genes potentially involved in glycan formation and modification. mg537 encodes a protein of unknown function, also found in bacterial clusters related to polysaccharide production. Mg538 is similar to proteins characterized by an EXOV-like domain, which is typical of polysaccharide pyruvyltransferases (28). Interestingly, a homologue of Mg538, L143, is also present in the UDP-d-viosamine gene cluster of Mimivirus (7). mg539 encodes a 1097-amino acid long protein, in which three GT-A domains are recognizable; the C-terminal domain displays 52% identity (128/248 residues) with Mimivirus R363, a putative glycosyltransferase. The M. chilensis gene cluster is shared with the closely related viruses Courdo7, Courdo11, LBA111, and Terra1 (29). On the other hand, no homologues for mg534, mg535, and mg536 were found in the genome of other giant viruses identified so far, suggesting that the presence of 6-deoxy-hexosamines is specific to the Megavirus group.
Crystal Structure of Mg534
As expected from their sequence identity (Fig. 3), the overall structure of Mg534 is very similar and superimposable to 4,6-dehydratase-5-epimerases, such as WbjB from Acinetobacter baumannii (PDB code 4J2O), the PseB protein FlaA1 (PDB 2GN4, (25)), and CapE structures (PDB code 4G5H (27)) (root mean square deviation 1.7, 1.36, and 1.65 Å, respectively). The structure is made of two subdomains, a large Rossmann-like NADP binding fold and a smaller subdomain responsible for the substrate binding. We tried to crystallize Mg534 alone and in complex with its substrate, but although we also obtained crystals in the presence of UDP-GlcNAc, it was never visible in the structure. By contrast, the NADP cofactor was always present, even when not added to the crystallization mixture.
FIGURE 3.

Structure-based alignment of Mg534 with WbjB from A. baumannii strain D1279779 (PDB code 4J2O), FlaA1 from Helicobacter Pylori (PDB code 2GN4), WbvB from Vibrio cholerae O37 (AAM22595), and CapE from S. aureus (PBD code 4G5H). Strictly conserved residues are boxed in red. The structural alignment was computed using the Expresso server, and the figure was produced using the ESPript server.
The Mg534 protein crystallizes in the hexagonal space group with one molecule per asymmetric unit, and the crystallographic hexamer is made of a trimer of dimers. The resulting buried surface area is of 25,850 A2. Residues 173–195 are disordered in the Mg534 structure and are absent in the final model. In the homologous structures, this segment corresponds to a helix covering the substrate binding pocket, and it is stabilized by interactions with the ligand. Given the structural alignment, it is likely that the missing part of Mg534 adopts the same conformation as its bacterial counterparts. The superposition of the NADPH binding domain of Mg534 with CapE highlights a large conformational change of the substrate binding domain that could reflect the dynamic of the structure during the catalysis. In absence of the substrate, the domain is rotated and closes the substrate binding pocket.
The Mg534 protein exhibits the canonical catalytic triad containing the 140YXXXK144 motif present in other SDR enzymes. UDP-GlcNAc “inverting” 4,6-dehydratases can be divided into two subfamilies, based on the conservation of the canonical YXXXK motif at the active site, like FlaA1 (25), or the presence of an altered motif, MXXXK, where the catalytic tyrosine is replaced by a methionine, represented by CapE; this subfamily contains an additional structural element, the so-called latch (27, 30) responsible of the dimer stabilization. It forms a long loop that folds on the top of the substrate binding site of the second molecule of the dimer. We suggest that the latch closes the cavity when the substrate is bound, and it could play a role in the regulation of the reaction. The comparison of the various structures highlights the absence of a latch in Mg534, as for FlaA1 (Fig. 4). However, there are differences between FlaA1 and Mg534 suggesting that Mg534 could correspond to a third subfamily of SDR. Although there is no bona fide latch in FlaA1, there is a long loop, which instead of folding on the second monomer of the dimer is folding back on the same monomer active site. In Mg534, the loop is much shorter (Fig. 4) and appears as a rigid linker between the two subunits. Another difference with the other inverting 4,6-dehydratases corresponds to two insertions in the Mg534 sequence (residues 72–73 and 115–119) (Fig. 3). Both lead to longer loops at the central channel of the hexamer, but surprisingly, the channel does not appear smaller in Mg534 than in the homologous structures.
FIGURE 4.
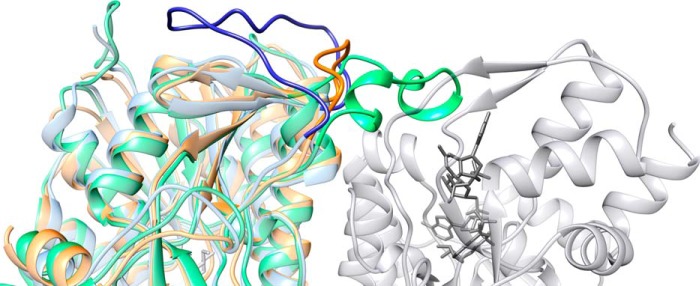
Ribbon representation of the Mg534 structure. Mg534 (orange) superimposed with CapE (green) and FlaA1 (blue) produced using Chimera (43) (University of California, San Francisco). The view is centered on the variable region corresponding to the CapE latch. The molecule in light gray corresponds to the second monomer of the dimer of CapE. The substrate and the coenzyme are colored in dark gray. Whereas the latch of CapE (dark green) associates with the substrate binding domain of the second monomer, the equivalent region of FlaA1 folds back on the first monomer (dark blue). This long loop is absent from the Mg534 structure.
Enzymatic Activity of Mg534 and Mg535
For characterization of the catalytic activity, mg534 and mg535 gene products were expressed in E. coli as GST fusion proteins. After removal of the fusion partner by proteolytic cleavage, both Mg534 and Mg535 resulted in single bands of the expected molecular weight, as judged by SDS-PAGE analysis (not shown).
Mg534 was incubated with UDP-GlcNAc and NADP+, and the product formation was monitored by HPLC analysis (Fig. 5). During the incubation time, a progressive reduction of UDP-GlcNAc was observed, with the concomitant formation of a new peak (Fig. 5B). ESI-MS analysis was performed on this product after the complete disappearance of the UDP-GlcNAc substrate. Two species were identified; one had m/z 588, 18 mass units less than the substrate, suggestive of a dehydration product of UDP-GlcNAc, and the second had the same molecular mass as UDP-GlcNAc (m/z 606) (not shown). This latter mass can be related to hydration of the 4-keto group with the formation of UDP-2-acetamido-2,6-dideoxy-hexose-4,4-diol, a process that has been well documented (25). Upon incubation with Mg535 and a system for NADPH regeneration, the Mg534 product was converted to a new compound with a retention time similar to UDP-GlcNAc (Fig. 5C). Indeed, ESI-MS analysis of the Mg535 product revealed an m/z of 590, which is compatible with the NADPH-dependent reduction of the C-4 carbonyl group of the intermediate compound (not shown). Altogether, the results obtained from HPLC and ESI-MS analyses were consistent with the formation of a UDP-2-acetamido-2,6-dideoxy-hexose, catalyzed by the sequential activity of Mg534 and Mg535.
FIGURE 5.

Anion exchange HPLC analysis of UDP-2-acetamido sugars. A, standard UDP-GlcNAc (peak 1). B, product (peak 2) obtained from the dehydration of UDP-GlcNAc catalyzed by Mg534. C, product (peak 3) derived from peak 2 in the presence of Mg535 and an NADPH-generating system, corresponding to UDP-l-RhaNAc.
To have a conclusive identification of the final compound produced by the combined activity of Mg534 and Mg535, we performed NMR analysis. The proton spectrum (Fig. 6A) showed two anomeric signals; the one at 5.98 (not shown) contained some of the nucleotide signals, whereas the second (5.35 ppm) belonged to the reaction product of the former glucosamine unit. COSY spectrum (not shown) analysis identified all sugar ring proton chemical shifts, and 3J coupling constant values disclosed the stereochemistry of the unit. H-1 at 5.35 was a doublet because coupled to a phosphorous atom only (3JH1,P = 8.9 Hz), whereas coupling with H-2 was undetectable. H-4 appeared as a triplet because the coupling constant value with H-3 and H-5 was the same (3JH3H4 = 3JH4H5 = 9.8 Hz), and the large 3J values found indicated that these three protons were in the axial position; having determined the stereochemistry at C-3, the shape of H-3 as a double doublet (3JH2,H3 = 4.2 Hz, 3JH3,H4 = 9.8 Hz) indicated that H-2 (4.54 ppm) occupied the equatorial position of the nearby C-2 atom. The high field chemical shift of H-5 (3.49 ppm) indicated the β-configuration of this unit. The correlation of H-5 with a methyl group at 1.33 identified this residue as a 6-deoxysugar. Proton and carbon chemical shift (Table 2) classified this residue as a β-rhamnosamine; differences in chemical shifts from those reported previously (10) reflect the occurrence of the UDP moiety; and the l-configuration was surmised by the biosynthetic pathway foreseen for these enzymes.
FIGURE 6.

(600 MHz, 298 K) expansion of proton spectra of the reaction products among UDP-GlcNAc. A, Mg534 and Mg535 together with NADPH. B, Mg534 only; C, Mg534 and NaBH4. Signals in the expansion at high field are traced at 4-fold reduction. Signals not labeled are impurities or related to the UDP moiety. A, 2-acetamido-2,6-dideoxy-α-d-arabino-4-hexulose with the not hydrated keto function at C-4; hA, 2-acetamido-2,6-dideoxy-α-d-arabino-4-hexulose with the hydrated keto function at C-4; G, α-d-glucosamine; I, β-l-6-deoxy-idosamine; Q, α-d-quinovosamine; R, β-l-rhamnosamine; X, 2-acetamido-2,6-dideoxy-α-d-xylo-4-hexulose. Signals not labeled are impurities or related to the UDP moiety.
TABLE 2.
Proton and carbon chemical shift (600 MHz, 298 K)
Chemical shifts are with respect to internal acetone (1H 2.225 ppm, 13C 31.45 ppm) measured for the reaction products between UDP-GlcNAc and Mg534 in the presence of NaBH4 (UDP-β-6-deoxy-IdoNAc and UDP-α-QuiNAc) or in the presence of Mg535 and NADPH (UDP-β-RhaNAc).
| 1 | 2 | 3 | 4 | 5 | 6 | NAc | |
|---|---|---|---|---|---|---|---|
| UDP-β-RhaNAc | |||||||
| 1H | 5.35 | 4.54 | 3.80 | 3.49 | 3.26 | 1.33 | 2.08 |
| 13C | 95.4 | 55.1 | 72.9 | 73.3 | 74.3 | 18.0 | 23.3 |
| UDP-α-QuiNAc | |||||||
| 1H | 5.44 | 4.00 | 7.74 | 3.25 | 4.00 | 1.29 | 2.06 |
| 13C | 97.5 | 55.2 | 72.3 | 76.7 | 70.5 | 18.3 | 23.3 |
| UDP-β-6-deoxy-IdoNAc | |||||||
| 1H | 5.52 | 4.05 | 3.98 | 3.72 | 4.27 | 1.36 | 2.06 |
| 13C | 97.7 | 54.4 | 69.1 | 72.2 | 74.1 | 17.6 | 23.3 |
Characterization of Mg534 Reaction Product
Previous studies performed on WbjB and CapE enzymes, which are involved in the UDP-FucNAc pathway, led to the final conclusion that these enzymes are inverting UDP-GlcNAc dehydratases, catalyzing 4,6-dehydration and 5-epimerization (12). Similar results have been obtained also for FlaA1 and PseB enzymes, which catalyze the first step of the Pse pathway (25–27). However, for FlaA1, PseB, and CapE, a slow 5-epimerase activity that converts UDP-2-acetamido-2,6-dideoxy-β-l-arabino-4-hexulose back to UDP-2-acetamido-2,6-dideoxy-α-d-xylo-4-hexulose was also reported, with the consequent accumulation of this latter compound at longer incubation times (25, 26, 30). To confirm that Mg534 also behaves as an inverting UDP-GlcNAc 4,6-dehydratase, we performed a direct NMR analysis of its product, as reported (26). For this set of experiments, Mg534 was maintained bound to the GSH-Sephadex beads as GST fusion protein. This allowed the rapid substitution of the normal buffer with the D2O buffer for the reaction, thus avoiding purification and lyophilization of the unstable Mg534 product.
After 60 min of reaction, more than 75% of UDP-GlcNAc substrate was converted into three new products (Fig. 6B) Comparison of proton chemical shifts of these products with those reported (31, 32) identified the main product (H-1 at 5.57 ppm) as UDP-2-acetamido-2,6-dideoxy-4-keto-β-arabino-hexose with the carbonyl function at C-4 in the hydrated form, whereas that at 5.70 had the carbonyl not hydrated; the H-1 signal at 5.47 ppm belonged to the expected UDP-derivative with the xylo stereochemistry.
To confirm these results, the C-4 keto group of Mg534 products was chemically reduced using NaBH4. The compounds obtained were then purified and analyzed by NMR (Fig. 6C and Table 2). Aside from signals deriving from UDP moieties, the proton spectrum contained two main anomeric protons at 5.55 and 5.44 ppm; COSY spectrum interpretation identified all protons connected to these anomeric signals, and the heteronuclear single quantum coherence spectrum assigned the corresponding carbon resonances (Table 2). H-1 at 5.55 ppm was a double doublet because coupled with the phosphorous atom (3JH1,P = 8.4 Hz) and with H-2 at 4.05 ppm (3JH1,H2 = 2.9 Hz); H-3 at 3.98 ppm appeared as a triplet because the coupling with both H-2 and H-4 was large and had the same value (3JH2H3 = 3JH3H4 = 7.2 Hz); H-4 coupling with H-5 was close to null, and H-6 was found at 1.36 ppm. The null coupling among H-4 and H-5 together with the coupling constants among H-2, H-3, and H-4 indicated that this residue had the ido stereochemistry; interestingly, the coupling among H-3 and H-4 (or H-2 and H-3) measured 7.2 Hz, a value that diverges from the typical value reported for trans diaxial protons (∼10 Hz) and indicates that the ring conformation is a distorted 4C1 chair or that an equilibrium among the 4C1 and a twisted form occurs, in agreement with what is reported for other ido configured residues, such as iduronic acid (33). Configuration at the anomeric center was β because of the small coupling constant value observed, and the l- absolute configuration was given on the basis of the reaction mechanism foreseen for this enzyme. By analog considerations, the product with the anomeric signal at 5.44 ppm was identified as an α-d-quinovosamine (Fig. 6C and Table 2). Indeed, NMR analysis of the two main products of the reaction identified UDP-6-deoxy-β-l-IdoNAc from UDP-2-acetamido-2,6-dideoxy-β-l-arabino-4-hexulose and UDP-α-d-QuiNAc from UDP-2-acetamido-2,6-dideoxy-α-d-xylo-4-hexulose; this result was expected by the stereochemistry of the NaBH4 reduction, which preferentially transforms the keto group into an equatorially oriented hydroxyl function.
These results proved that Mg534 is a 4,6-dehydratase-5-epimerase, and the identification of UDP-RhaNAc as the final product of Mg534 + Mg535 combined activity led us to conclude that, by analogy with WbjC enzyme, Mg535 should operate on UDP-2-acetamido-2,6-dideoxy-β-l-arabino-4-hexulose by a two-step mechanism; the first step inverts C-3 stereochemistry, yielding UDP-2-acetamido-2,6-dideoxy-β-l-lyxo-4-hexulose, whereas the second reduces the keto function at C-4 using NADPH.
GC-MS Analysis of M. chilensis
The final product obtained by the combined activity of Mg534 and Mg535 proteins, identified as UDP-RhaNAc, was then subjected to acid hydrolysis and GC-MS analysis. In parallel, we assessed the M. chilensis virion sugar composition to confirm the presence of this sugar in the viral particles. Analysis of RhaNAc standard is reported in Fig. 7A, whereas Fig. 7B reports the analysis of M. chilensis viral particles. Viral glycans resulted, mainly composed of GlcNAc, but three other prominent peaks were also observed; one showed the same retention time (41.6 min) and fragmentation spectrum of RhaNAc standard (Fig. 7C), confirming the presence of this sugar in the virion. A more abundant peak at 41.3 min (identified as Y) showed a spectrum identical that reported in Fig. 7C, suggesting the presence of a RhaNAc epimer, which could correspond to the QuiNAc possibly formed by the Mg536 activity. The presence of PneNAc was ruled out because the standard monosaccharide produced by P. aeruginosa WbjB + WbjC enzymes exhibited a different elution time (not shown). Finally, the fragmentation spectrum of the peak at 39.7 min (identified with X) indicated that this compound could represent a methylated form of a 2-acetamido-2,6-dideoxy-hexose (Fig. 7D).
FIGURE 7.

GC-MS analysis of standard RhaNAc and M. chilensis glycans. Alditol acetates were analyzed by electron impact GC-MS in full scan mode. Chromatograms show total ion currents. A, standard RhaNAc produced by Mg534 and Mg535; myoinositol (Myo) was used as internal reference. Ribose (Rib) derived from NADP that was present in the reaction for UDP-RhaNAc production. B, analysis of monosaccharides obtained after acid hydrolysis of M. chilensis glycans. One peak was identified as RhaNAc on the basis of the retention time and the fragmentation spectrum, which were identical to those reported in C. C, fragmentation spectrum of standard RhaNAc. The fragmentation spectrum of the unknown peak Y was also identical, suggesting that compound Y can represent an epimer. D, fragmentation spectrum of peak X, suggestive of a 4-O-methyl-2-acetamido-2,6-dideoxy-hexosamine.
DISCUSSION
The results obtained in the present study indicate that the M. chilensis genome encodes enzymes for the production of RhaNAc, a 6-deoxy-l-hexosamine. The first enzyme of this pathway is encoded by the mg534 gene and catalyzes the conversion of UDP-GlcNAc to UDP-2-acetamido-2,6-dideoxy-β-l-arabino-4-hexulose. Most organisms, including A. castellani, the host used for laboratory propagation of M. chilensis, produce the substrate UDP-d-GlcNAc; however, Mimiviridae also encode the enzymes for the production of this nucleotide sugar, thus providing a further supply of the substrate independently from its host (4). Mg534 proved to be an inverting 4,6-dehydratase, able to catalyze the elimination of a water molecule from C-4 and C-6 and the following epimerization of C-5. This first reaction is common to both the 6-deoxy-l-hexosamine and the pseudaminic acid pathways. Interestingly, both the sequence and structural comparison suggest that Mg534 is more related to PseB/FlaA1 enzymes of the pseudaminic acid biosynthesis rather than to the bacterial enzymes of the 6-deoxy-l-hexosamine pathways. Specifically, Mg534, like PseB/FlaA1, displays the typical YXXXK sequence in the active site, which is common to most members of the extended SDR family (34). On the other hand, in CapE and in the related inverting 4,6-dehdyratases, the catalytic tyrosine is substituted with a methionine residue (27). The presence of a methionine residue instead of a tyrosine in the active site of inverting 4,6-dhydratases has been related to a different optimal pH for catalysis (35).
Another feature, which characterizes CapE and the other enzymes involved in 6-deoxy-hexosamine production, is the presence of the so-called “latch” (27, 30). This structure is believed to play a role in controlling the rate of the side reaction that converts the UDP-arabino sugar back to the UDP-xylo configuration (30). Indeed, the rate of formation of the UDP-xylo by-product has been reported to be higher for CapE compared with FlaA1 and PseB enzymes, and it was even increased in mutants of the “latch” region of CapE (27, 30). The Mg534 crystal structure revealed the absence of the “latch” region and pointed out some differences also with the FlaA1 structure, suggesting that it may represent a third subfamily of the inverting UDP-GlcNAc 4,6-dehydratases. Formation of the UDP-xylo by-product was observed in the reaction catalyzed by Mg534 at longer incubation times. However, the rate of the conversion catalyzed by Mg534 did not seem as high as that observed for CapE; further experiments are needed to compare the rate of by-product formation catalyzed by Mg534, FlaA1, and CapE enzymes, using all of these enzymes in the same reaction conditions. The significance of by-product formation is not clear because UDP-2-acetamido-2,6-dideoxy-β-d-xylo-4-hexulose is not recognized by the following enzyme in the pathway; however, the low rate of its formation suggests that it probably does not have a relevant physiological role in vivo.
The demonstration that Mg534 is an inverting 4,6-dehydratase and that the final product of Mg534 and Mg535 combined activity is UDP-RhaNAc led us to conclude that, in analogy with WbjC and CapF, which catalyze the formation of UDP-PneNAc (10, 15), Mg535 is a 3-epimerase, 4-reductase. However, whereas WbjC and CapF are characterized by the presence of an N-terminal 4-reductase RmlD-like domain and a C-terminal epimerase domain belonging to the cupin superfamily, Mg535 displays a single RmlD-like domain. Bifunctional enzymes displaying epimerase and reductase activities at the same catalytic site have been extensively studied. The prototype is GDP-l-fucose synthase, which catalyzes the epimerization of C-3 and C-5 on GDP-4-keto-6-deoxy-d-mannose, followed by the NADPH-dependent reduction of the carbonyl at C-4 (36, 37). Another example is observed in the UDP-l-rhamnose pathway in plants, viruses, and fungi, where enzymes that exhibit a single RmlD-like domain catalyze both 3,5-epimerization and reduction of the 4-keto group (5, 38). Conversely, in the typical dTDP-l-rhamnose pathway of bacteria, the 3,5-epimerization and reduction are carried on by different enzymes, displaying an RmlC cupin domain and an RmlD domain, respectively (39). However, Mg535 is unusual among the reported bifunctional epimerase-reductases, because it catalyzes the epimerization on C-3 only, the C-5 epimerization being already performed by Mg534. The conversion of UDP-2-acetamido-2,6-dideoxy-β-l-arabino-4-hexulose to UDP-2-acetamido-2,6-dideoxy-β-l-lyxo-4-hexulose has been proven to occur also spontaneously in solution by keto-enol tautomerization (32); however, the rate of reaction is very low, and it is not clear if it may be physiologically relevant. Site-directed mutagenesis studies performed on GDP-l-fucose synthase of the GDP-l-fucose pathway revealed an ordered sequence of epimerization, on C-3 first, followed by C-5; moreover, these studies revealed a key role of cysteine 109 as a base, to deprotonate the substrate at C-3 (37). Interestingly, despite the low sequence homology between Mg535 and GDP-l-fucose synthase, the cysteine residue is conserved in the active site of both enzymes. This residue is also present in WbvR, the enzyme of V. cholerae, which leads to UDP-l-RhaNAc formation and is similarly devoid of the cupin epimerase domain (10). Indeed, WbvR was initially described by Kneideiger et al. (10) to be a 4-reductase only; however, in the same report, the preceding enzyme in the pathway was proposed to be trifunctional, being also a 3-epimerase, along with 4,6-dehydratase, 5-epimerase. Additional studies, and in particular the Mg535 structural characterization and site-directed mutagenesis, will help to clarify all of these issues.
mg534 and mg535 are found in a M. chilensis gene cluster that is also found in the genomes of other closely related viruses, which have been comprised in group C of the Mimiviridae (29), suggesting that the presence of modified acetamido sugars is a common feature in several members of this lineage. Specifically, highly similar gene clusters were found in viruses isolated from very different environments, such as Courdo11 found in fresh water, LBA111 obtained from the bronchio-alveolar lavage of a pneumonia patient, and Terra1 isolated from soil (40, 41). The cluster was not found in the Mimiviridae belonging to group A and B, even if some of its genes are shared with these other giant viruses. The occurrence of gene clusters is a common feature of several members of the Mimiviridae and Phycodnaviridae families. For instance, Mimivirus presents a cluster encoding enzymes involved in UDP-d-viosamine production together with several glycosyltransferases and glycan-modifying enzymes (7). Similarly, in CroV, three enzymes possibly involved in the sialic acid or 3-deoxy-d-manno-oct-2-ulosonic acid production are found together with glycosyltransferases in a well defined 38-kbp region of the genome (42). Moreover, inspection of other nucleocytoplasmic genomes revealed the presence of several other gene clusters encoding enzymes for glycan production,6 suggesting that physical proximity of the genes involved in these pathways, which need to function in a coordinate manner, is an advantageous strategy in giant viruses. However, it is important to point out that, in most cases, the enzymes for glycan production that are found in the clusters do not share a common origin, thus excluding the possibility that they could have been acquired “en bloc” from a bacterial source. For instance, phylogenetic studies have revealed that the first and second enzymes in the pathway that leads to UDP-viosamine in Mimivirus are derived from a eukaryotic and a prokaryotic source, respectively (7). The data obtained in the present study indicate that Mg534 is more related to the enzymes of the pseudaminic acid than to those of the 6-deoxy-l-hexosamine pathways, suggesting that it may have been acquired independently from Mg535. On the other hand, the different clusters also contain highly homologous genes, as for M. chilensis Mg538 and Mimivirus L143. Altogether, these findings indicate an active role of viruses in cluster formation. However, the origin of gene clusters and their role in horizontal gene transfer will require further investigation.
The presence in giant viruses of a glycosylation system that is independent from the host point out the importance of glycans in their biology. Together with previous reports (5–7), the data obtained in this study also highlight the need to use rare sugars, which are not produced by the eukaryotic hosts, to obtain unusual oligosaccharide structures. These are probably essential for the interaction of the viral particles with the host, in particular to promote phagocytosis, but they can also provide protection against the harsh environment in which the viruses need to propagate.
Acknowledgments
We thank the BM30, ID14, ID23, ID29 (ESRF), and PROXIMA (SOLEIL) beamline teams for expert assistance. We are grateful to Prof. J. S. Lam (University of Guelph, Ontario, Canada) for kindly providing the WbjB and WbjC expression vectors.
This work was supported in part by CNRS, IBISA, and the Provence-Alpes-Côte d'Azur region (to C. A.). This work was also supported in part by Regione Liguria (to M. T.).
The atomic coordinates and structure factors (code 4TQG) have been deposited in the Protein Data Bank (http://wwpdb.org/).
F. Piacente, C. Abergel, and M. G. Tonetti, unpublished results.
- UDP-d-FucNac and UDP-l-FucNac
- uridine 5′-diphosphate-N-acetyl-d-fucosamine and -l-fucosamine, respectively
- RhaNAc
- N-acetylrhamnosamine
- SDR
- short-chain dehydrogenase/reductase
- UDP-d-QuiNAc and UDP-l-QuiNAc
- uridine 5′-diphosphate-N-acetyl-d-quinovosamine and -l-quinovosamine, respectively
- Pse
- pseudaminic acid
- UDP-l-PneNAc
- uridine 5′-diphosphate-N-acetyl-l-pneumosamine
- ESI
- electrospray ionization
- FID
- free induction decay
- IdoNAc
- N-acetyl-idosamine.
REFERENCES
- 1. Iyer L. M., Balaji S., Koonin E. V., Aravind L. (2006) Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 117, 156–184 [DOI] [PubMed] [Google Scholar]
- 2. Van Etten J. L., Gurnon J. R., Yanai-Balser G. M., Dunigan D. D., Graves M. V. (2010) Chlorella viruses encode most, if not all, of the machinery to glycosylate their glycoproteins independent of the endoplasmic reticulum and Golgi. Biochim. Biophys. Acta 1800, 152–159 [DOI] [PubMed] [Google Scholar]
- 3. De Castro C., Molinaro A., Piacente F., Gurnon J. R., Sturiale L., Palmigiano A., Lanzetta R., Parrilli M., Garozzo D., Tonetti M. G., Van Etten J. L. (2013) Structure of N-linked oligosaccharides attached to chlorovirus PBCV-1 major capsid protein reveals unusual class of complex N-glycans. Proc. Natl. Acad. Sci. U.S.A. 110, 13956–13960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Piacente F., Bernardi C., Marin M., Blanc G., Abergel C., Tonetti M. G. (2014) Characterization of a UDP-N-acetylglucosamine biosynthetic pathway encoded by the giant DNA virus Mimivirus. Glycobiology 24, 51–61 [DOI] [PubMed] [Google Scholar]
- 5. Parakkottil Chothi M., Duncan G. A., Armirotti A., Abergel C., Gurnon J. R., Van Etten J. L., Bernardi C., Damonte G., Tonetti M. (2010) Identification of an l-rhamnose synthetic pathway in two nucleocytoplasmic large DNA viruses. J. Virol. 84, 8829–8838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tonetti M., Zanardi D., Gurnon J. R., Fruscione F., Armirotti A., Damonte G., Sturla L., De Flora A., Van Etten J. L. (2003) Paramecium bursaria chlorella virus 1 encodes two enzymes involved in the biosynthesis of GDP-l-fucose and GDP-d-rhamnose. J. Biol. Chem. 278, 21559–21565 [DOI] [PubMed] [Google Scholar]
- 7. Piacente F., Marin M., Molinaro A., De Castro C., Seltzer V., Salis A., Damonte G., Bernardi C., Claverie J. M., Abergel C., Tonetti M. (2012) Giant DNA virus mimivirus encodes pathway for biosynthesis of unusual sugar 4-amino-4,6-dideoxy-d-glucose (Viosamine). J. Biol. Chem. 287, 3009–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fruscione F., Sturla L., Duncan G., Van Etten J. L., Valbuzzi P., De Flora A., Di Zanni E., Tonetti M. (2008) Differential role of NADP+ and NADPH in the activity and structure of GDP-d-mannose 4,6-dehydratase from two chlorella viruses. J. Biol. Chem. 283, 184–193 [DOI] [PubMed] [Google Scholar]
- 9. Arslan D., Legendre M., Seltzer V., Abergel C., Claverie J. M. (2011) Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. U.S.A. 108, 17486–17491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kneidinger B., Larocque S., Brisson J. R., Cadotte N., Lam J. S. (2003) Biosynthesis of 2-acetamido-2,6-dideoxy-l-hexoses in bacteria follows a pattern distinct from those of the pathways of 6-deoxy-l-hexoses. Biochem. J. 371, 989–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kneidinger B., O'Riordan K., Li J., Brisson J. R., Lee J. C., Lam J. S. (2003) Three highly conserved proteins catalyze the conversion of UDP-N-acetyl-d-glucosamine to precursors for the biosynthesis of O antigen in Pseudomonas aeruginosa O11 and capsule in Staphylococcus aureus type 5: implications for the UDP-N-acetyl-l-fucosamine biosynthetic pathway. J. Biol. Chem. 278, 3615–3627 [DOI] [PubMed] [Google Scholar]
- 12. Mulrooney E. F., Poon K. K., McNally D. J., Brisson J. R., Lam J. S. (2005) Biosynthesis of UDP-N-acetyl-l-fucosamine, a precursor to the biosynthesis of lipopolysaccharide in Pseudomonas aeruginosa serotype O11. J. Biol. Chem. 280, 19535–19542 [DOI] [PubMed] [Google Scholar]
- 13. Morrison M. J., Imperiali B. (2014) The renaissance of bacillosamine and its derivatives: pathway characterization and implications in pathogenicity. Biochemistry 53, 624–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. King J. D., Kocíncová D., Westman E. L., Lam J. S. (2009) Review: lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun. 15, 261–312 [DOI] [PubMed] [Google Scholar]
- 15. Miyafusa T., Caaveiro J. M., Tanaka Y., Tsumoto K. (2012) Crystal structure of the enzyme CapF of Staphylococcus aureus reveals a unique architecture composed of two functional domains. Biochem. J. 443, 671–680 [DOI] [PubMed] [Google Scholar]
- 16. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D, Bairoch A. (2005) in The Proteomics Protocols Handbook (Walker J. M., ed), pp. 571–607, Humana Press, New York [Google Scholar]
- 17. Abergel C., Moulard M., Moreau H., Loret E., Cambillau C., Fontecilla-Camps J. C. (1991) Systematic use of the incomplete factorial approach in the design of protein crystallization experiments. J. Biol. Chem. 266, 20131–20138 [PubMed] [Google Scholar]
- 18. Audic S., Lopez F., Claverie J. M., Poirot O., Abergel C. (1997) SAmBA: an interactive software for optimizing the design of biological macromolecules crystallization experiments. Proteins 29, 252–257 [DOI] [PubMed] [Google Scholar]
- 19. Abergel C., Coutard B., Byrne D., Chenivesse S., Claude J. B., Deregnaucourt C., Fricaux T., Gianesini-Boutreux C., Jeudy S., Lebrun R., Maza C., Notredame C., Poirot O., Suhre K., Varagnol M., Claverie J. M. (2003) Structural genomics of highly conserved microbial genes of unknown function in search of new antibacterial targets. J. Struct. Funct. Genomics 4, 141–157 [DOI] [PubMed] [Google Scholar]
- 20. Leslie A. G. W., Powell H. R. (2007) in Evolving Methods for Macromolecular Crystallography (Read R. J., Sussman J. L., eds) pp. 41–51, Springer, Amsterdam [Google Scholar]
- 21. Evans P. R. (1997) Proceedings of the CCP4 Study Weekend: Recent Advances in Phasing (Wilson K. S., Davies G., Ashton A. W., Bailey S., ed) pp. 97–102, Daresbury Laboratory, Warrington, UK [Google Scholar]
- 22. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 23. Bunkóczi G., Echols N., McCoy A. J., Oeffner R. D., Adams P. D., Read R. J. (2013) Phaser.MRage: automated molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 69, 2276–2286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ishiyama N., Creuzenet C., Miller W. L., Demendi M., Anderson E. M., Harauz G., Lam J. S., Berghuis A. M. (2006) Structural studies of FlaA1 from Helicobacter pylori reveal the mechanism for inverting 4,6-dehydratase activity. J. Biol. Chem. 281, 24489–24495 [DOI] [PubMed] [Google Scholar]
- 26. Morrison J. P., Schoenhofen I. C., Tanner M. E. (2008) Mechanistic studies on PseB of pseudaminic acid biosynthesis: a UDP-N-acetylglucosamine 5-inverting 4,6-dehydratase. Bioorg. Chem. 36, 312–320 [DOI] [PubMed] [Google Scholar]
- 27. Miyafusa T., Caaveiro J. M., Tanaka Y., Tanner M. E., Tsumoto K. (2013) Crystal structure of the capsular polysaccharide synthesizing protein CapE of Staphylococcus aureus. Biosci. Rep. 33, e00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Becker A., Kleickmann A., Arnold W., Pühler A. (1993) Analysis of the Rhizobium meliloti exoH/exoK/exoL fragment: ExoK shows homology to excreted endo-β-1,3–1,4-glucanases and ExoH resembles membrane proteins. Mol. Gen. Genet. 238, 145–154 [DOI] [PubMed] [Google Scholar]
- 29. Colson P., De Lamballerie X., Yutin N., Asgari S., Bigot Y., Bideshi D. K., Cheng X. W., Federici B. A., Van Etten J. L., Koonin E. V., La Scola B., Raoult D. (2013) “Megavirales,” a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch. Virol. 158, 2517–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miyafusa T., Caaveiro J. M., Tanaka Y., Tsumoto K. (2013) Dynamic elements govern the catalytic activity of CapE, a capsular polysaccharide-synthesizing enzyme from Staphylococcus aureus. FEBS Lett. 587, 3824–3830 [DOI] [PubMed] [Google Scholar]
- 31. Schoenhofen I. C., McNally D. J., Vinogradov E., Whitfield D., Young N. M., Dick S., Wakarchuk W. W., Brisson J. R., Logan S. M. (2006) Functional characterization of dehydratase/aminotransferase pairs from Helicobacter and Campylobacter: enzymes distinguishing the pseudaminic acid and bacillosamine biosynthetic pathways. J. Biol. Chem. 281, 723–732 [DOI] [PubMed] [Google Scholar]
- 32. McNally D. J., Schoenhofen I. C., Mulrooney E. F., Whitfield D. M., Vinogradov E., Lam J. S., Logan S. M., Brisson J. R. (2006) Identification of labile UDP-ketosugars in Helicobacter pylori, Campylobacter jejuni and Pseudomonas aeruginosa: key metabolites used to make glycan virulence factors. Chembiochem 7, 1865–1868 [DOI] [PubMed] [Google Scholar]
- 33. Remko M., von der Lieth C. W. (2006) Gas-phase and solution conformations of the α-l-iduronic acid structural unit of heparin. J. Chem. Inf. Model. 46, 1194–1200 [DOI] [PubMed] [Google Scholar]
- 34. Filling C., Berndt K. D., Benach J., Knapp S., Prozorovski T., Nordling E., Ladenstein R., Jörnvall H., Oppermann U. (2002) Critical residues for structure and catalysis in short-chain dehydrogenases/reductases. J. Biol. Chem. 277, 25677–25684 [DOI] [PubMed] [Google Scholar]
- 35. Creuzenet C., Urbanic R. V., Lam J. S. (2002) Structure-function studies of two novel UDP-GlcNAc C6 dehydratases/C4 reductases. Variation from the SYK dogma. J. Biol. Chem. 277, 26769–26778 [DOI] [PubMed] [Google Scholar]
- 36. Tonetti M., Sturla L., Bisso A., Benatti U., De Flora A. (1996) Synthesis of GDP-l-fucose by the human FX protein. J. Biol. Chem. 271, 27274–27279 [DOI] [PubMed] [Google Scholar]
- 37. Lau S. T., Tanner M. E. (2008) Mechanism and active site residues of GDP-fucose synthase. J. Am. Chem. Soc. 130, 17593–17602 [DOI] [PubMed] [Google Scholar]
- 38. Oka T., Nemoto T., Jigami Y. (2007) Functional analysis of Arabidopsis thaliana RHM2/MUM4, a multidomain protein involved in UDP-d-glucose to UDP-l-rhamnose conversion. J. Biol. Chem. 282, 5389–5403 [DOI] [PubMed] [Google Scholar]
- 39. Dong C., Beis K., Giraud M. F., Blankenfeldt W., Allard S., Major L. L., Kerr I. D., Whitfield C., Naismith J. H. (2003) A structural perspective on the enzymes that convert dTDP-d-glucose into dTDP-l-rhamnose. Biochem. Soc. Trans. 31, 532–536 [DOI] [PubMed] [Google Scholar]
- 40. Yoosuf N., Pagnier I., Fournous G., Robert C., La Scola B., Raoult D., Colson P. (2014) Complete genome sequence of Courdo11 virus, a member of the family Mimiviridae. Virus Genes 48, 218–223 [DOI] [PubMed] [Google Scholar]
- 41. Saadi H., Pagnier I., Colson P., Cherif J. K., Beji M., Boughalmi M., Azza S., Armstrong N., Robert C., Fournous G., La Scola B., Raoult D. (2013) First isolation of Mimivirus in a patient with pneumonia. Clin. Infect. Dis. 57, e127–e134 [DOI] [PubMed] [Google Scholar]
- 42. Fischer M. G., Allen M. J., Wilson W. H., Suttle C. A. (2010) Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. U.S.A. 107, 19508–19513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]