

Background: Thioredoxin is a physiological inhibitor of ASK1.
Results: The catalytic motif of thioredoxin is essential for its binding to ASK1 and the interaction does not involve intermolecular disulfide bonds.
Conclusion: Thioredoxin-binding domain of ASK1 is a rigid domain that interacts with reduced thioredoxin through a large binding interface.
Significance: Structural basis of the interaction between ASK1 and reduced thioredoxin.
Keywords: Analytical Ultracentrifugation, Circular Dichroism (CD), Fluorescence, Small-angle X-ray Scattering (SAXS), Thioredoxin, Apoptosis Signal-regulating Kinase 1 (ASK1)
Abstract
Apoptosis signal-regulating kinase 1 (ASK1), a mitogen-activated protein kinase kinase kinase, plays a key role in the pathogenesis of multiple diseases. Its activity is regulated by thioredoxin (TRX1) but the precise mechanism of this regulation is unclear due to the lack of structural data. Here, we performed biophysical and structural characterization of the TRX1-binding domain of ASK1 (ASK1-TBD) and its complex with reduced TRX1. ASK1-TBD is a monomeric and rigid domain that forms a stable complex with reduced TRX1 with 1:1 molar stoichiometry. The binding interaction does not involve the formation of intermolecular disulfide bonds. Residues from the catalytic WCGPC motif of TRX1 are essential for complex stability with Trp31 being directly involved in the binding interaction as suggested by time-resolved fluorescence. Small-angle x-ray scattering data reveal a compact and slightly asymmetric shape of ASK1-TBD and suggest reduced TRX1 interacts with this domain through the large binding interface without inducing any dramatic conformational change.
Introduction
Apoptosis signal-regulating kinase 1 (ASK1)3 (MAP3K5), a member of the mitogen-activated protein kinase kinase kinase family, activates c-Jun N-terminal kinase and p38 MAP kinase signaling pathways in response to various stress stimuli, including oxidative stress, endoplasmic reticulum stress, and calcium ion influx (1–3). ASK1 plays a key role in the pathogenesis of multiple diseases including cancer, neurodegeneration, and cardiovascular diseases and is considered a promising therapeutic target (reviewed by Ref. 4). Human ASK1 consists of 1,374 amino acids with the serine/threonine kinase domain located in the middle of the molecule flanked by two coiled-coil (CC) motifs, which are important for homo-oligomerization of ASK1 (5, 6). ASK1 under non-stress conditions forms a homo-oligomer by direct interaction through the C-terminal CC domain and interacts with several other proteins including thioredoxin-1 (TRX1) and the 14-3-3 protein, thus forming a high molecular mass complex called ASK1 signalosome (1, 7, 8). Both TRX1 and the 14-3-3 protein are physiological inhibitors of ASK1. In response to oxidative stress they dissociate from ASK1, this allows the homo-oligomerization and recruitment of tumor necrosis factor receptor-associated factors 2 and 6 to the N-terminal region of ASK1 and accelerates the autophosphorylation of Thr838 within the activation loop resulting in ASK1 activation (1, 9, 10).
Thioredoxins (TRXs) are small dithiol oxidoreductases ubiquitously present in species ranging from archaea to mammals. TRXs perform various biological functions including the reduction of protein disulfide bonds in the reducing cellular compartments, the supply of reducing equivalents to redox enzymes, and the regulation of several transcription factors and proteins through either a direct reduction of their cysteine groups or different mechanisms (reviewed by Ref. 11). TRXs are involved in the regulation of NF-κB (12), the Escherichia coli T7 polymerase complex (13), or Jab-1 (14). The TRX molecule consists of a five-stranded β-pleated sheet that forms a hydrophobic core surrounded by four α-helices. The highly conserved redox catalytic motif 31WCGPC35 links the second β-strand to the second α-helix and the two cysteine residues within this sequence (Cys32 and Cys35 in human TRX1) are responsible for TRX-dependent redox activity (15).
TRX1 interacts with the ASK1 region located between residues 46 and 277 and this interaction is thought to inhibit the activation of ASK1 through blocking its homo-oligomerization via an adjacent N-terminal CC motif (1, 9). The Cys250 residue, located within this region of ASK1, is crucial for the oxidative stress-dependent signaling downstream of ASK1 and likely involved in TRX1 binding (16, 17). However, the precise mechanism of TRX1 binding to ASK1, as well as its dissociation, is still unclear due to the lack of structural data on the TRX-binding domain of ASK1 (ASK1-TBD). Several studies suggested that under oxidative stress conditions TRX1 is oxidized on Cys32 and Cys35 within the redox catalytic motif and the formation of an intramolecular disulfide bond between these two cysteine residues causes the dissociation of TRX1 from ASK1 through an unknown mechanism, thus allowing the activation of ASK1 (1, 9, 18). It is, however, unclear whether the interaction between TRX1 and ASK1 is based on the non-covalent interactions only or if it also involves the formation of intermolecular disulfide bond(s). The latter possibility was suggested by Nadeau et al. (17, 19) who proposed another mechanism for ASK1 activation. The authors suggest the oxidative stress induces the formation of intermolecular disulfide bonds between ASK1 molecules and this oxidation is required for the activation of ASK1 kinase function. In their model, the interaction between TRX1 and ASK1 involves a formation of intermolecular disulfide bond(s) and the TRX1-dependent inhibition of ASK1 is mediated by its thiol reductase activity because only the oxidized and disulfide bond-containing oligomeric form of ASK1 enables activation.
To better understand the interaction between TRX1 and ASK1, we prepared and performed detailed biophysical and structural characterization of the isolated ASK1-TBD (sequence 88–302) and its complex with reduced TRX1. The results show that ASK1-TBD is a monomeric and rigid domain that forms a stable complex with reduced TRX1 with 1:1 molar stoichiometry. The binding interaction does not involve the formation of intermolecular disulfide bonds. Residues Cys32 and Cys35 as well as Trp31 from the catalytic WCGPC motif of TRX1 are essential for complex stability with Trp31 being directly involved in binding interaction as suggested by time-resolved tryptophan fluorescence. SAXS data revealed a compact and slightly asymmetric shape of ASK1-TBD and suggest reduced TRX1 interacts with this domain through the large binding interface without inducing any dramatic conformational change. Molecular modeling indicated the TRX1 binding site is located close to the N-terminal end of a ∼50 residue long α-helix, which forms the C terminus of ASK1-TBD. Our results also show the ASK1 residue Cys250 is likely located either at or in close vicinity of TRX1-binding surface.
EXPERIMENTAL PROCEDURES
Expression and Purification of TRX-binding Domain of ASK1
DNA encoding four different N-terminal fragments of human ASK1 (residues 46–302, 88–302, 46–322, and 88–322) were ligated into pST39 (20) using the XbaI and BamHI sites and pRSFDuet-1 (Novagen) using BamHI and PstI sites. Modified pRSFDuet-1 containing the sequence of the His6-tagged GB1 domain of protein G inserted into the first multiple cloning site was a gift of Evzen Boura (Institute of Organic Chemistry and Biochemistry AS CR, Prague, Czech Republic). ASK1-(88–302) (in pST39) was expressed as a C-terminal His6 tag fusion protein by leakage expression at 30 °C for 20 h and purified from E. coli BL21 (DE3) cells using chelating Sepharose Fast Flow (GE Healthcare Life Sciences) according to standard protocols. Eluted ASK1-TBD was dialyzed against buffer containing 20 mm Tris-HCl (pH 7.5), 200 mm NaCl, 1 mm EDTA, 5 mm DTT, 10% (w/v) glycerol and purified using size-exclusion chromatography on a HiLoad 26/60 Superdex 75 column (GE Healthcare Life Sciences). All mutants were generated by using the QuikChange site-directed mutagenesis kit (Stratagene) and mutations were confirmed by sequencing.
Expression and Purification of TRX1
The expression construct for human TRX1 (C73S mutant) was a gift of Katja Becker (Justus-Liebig-Universität, Giessen, Germany). TRX1 was expressed as an N-terminal His6 tag fusion protein by isopropyl 1-thio-β-d-galactopyranoside induction for 20 h at 30 °C and purified from E. coli BL21 (DE3) cells using chelating Sepharose Fast Flow (GE Healthcare Life Sciences) according to standard protocols. Eluted TRX1 was dialyzed against buffer containing 20 mm Tris-HCl (pH 7.5), 200 mm NaCl, 1 mm EDTA, 5 mm DTT, 10% (w/v) glycerol and purified using size-exclusion chromatography on Superdex 75 10/300 GL column (GE Healthcare Life Sciences). All mutants were generated by using the QuikChange site-directed mutagenesis kit (Stratagene), and mutations were confirmed by sequencing.
Preparation of Oxidized TRX1
TRX1 (140 μm in buffer containing 20 mm Tris-HCl (pH 7.5) and 200 mm NaCl) was incubated with 100-fold molar excess of H2O2 in a total volume of 500 μl for 15 min at 37 °C (21). Oxidation reaction was stopped by adding 2 units of catalase (Sigma).
Time-resolved Fluorescence Measurements
Fluorescence intensity and anisotropy decays were measured on a time-correlated single photon counting apparatus, as described previously (22). Tryptophan emission was excited at 298 nm by a tripled output of the Ti:Sapphire laser. Tryptophan fluorescence was isolated at 355 nm by a combination of monochromator and a stack of UG1 and BG40 glass filters (Thorlabs) placed in front of the input slit. Fluorescence decays were typically accumulated in 1024 channels with a time resolution of 50 ps/channel until 105 counts in the decay maximum were reached. Samples were placed in a thermostatic holder, and all experiments were performed at 23 °C in a buffer containing 20 mm Tris-HCl (pH 7.5), 200 mm NaCl, and 5 mm DTT. The TRX1 concentration was 10 μm; the ASK1-TBD concentration was 30 μm (or 10 μm in experiments with Trp-containing mutants of ASK1-TBD). Fluorescence decays were assumed to be multiexponential according to the formula,
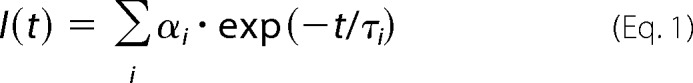 |
where τi and αi are the fluorescence lifetime components and the corresponding amplitudes, respectively. Emission decays I(t) were analyzed by a maximum entropy method (23). The program yields a set of amplitudes, αi, representing the lifetime distribution. Typically, we have chosen 100 lifetimes equidistantly spaced in a logarithmic scale, ranging from 20 ps to 20 ns. The mean emission lifetime was calculated as,
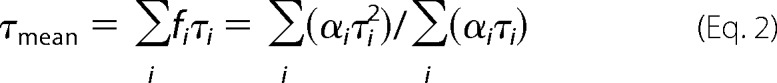 |
where fi are the fractional intensities of corresponding lifetime components. Fluorescence anisotropy r(t) was obtained by a simultaneous reconvolution of parallel I‖(t) and perpendicular I⊥(t) polarized components. The anisotropies r(t) were analyzed for a series of exponentials by a model-independent maximum entropy method without setting assumptions about the shape of the correlation time distributions (23),
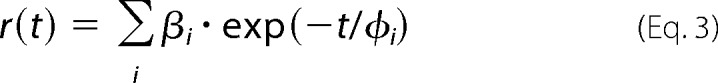 |
where amplitudes βi represent the distribution of the correlation times φi. They are related to the initial anisotropy r0 by the following formula.
 |
Typically we used 100 correlation times equidistantly spaced in the logarithmic scale from 100 ps to 200 ns.
Analytical Ultracentrifugation (AUC)
Sedimentation velocity (SV) experiments were performed using a ProteomLabTM XL-I analytical ultracentrifuge (Beckman Coulter). Samples were dialyzed against buffer containing 20 mm Tris-HCl (pH 7.5), 200 mm NaCl, and 2 mm 2-mercaptoethanol before analysis. Experiments with oxidized TRX1 were performed in buffer containing 20 mm Tris-HCl (pH 7.5), and 200 mm NaCl. The buffer density, viscosity, and partial specific volume of all proteins were estimated using the program SEDNTERP. SV experiments of ASK1-TBD and TRX1 were conducted at various loading concentrations and molar ratios in charcoal-filled Epon centerpieces with 12-mm optical path length, 20 °C, and 48,000 rpm rotor speed (An-50 Ti rotor, Beckman Coulter). All sedimentation profiles were recorded with interference optics. The diffusion-deconvoluted sedimentation coefficient distributions c(s) were calculated from raw interference data using the software package SEDFIT. For experiments with mixtures of TRX1 and ASK1-TBD at various molar ratios, this procedure was followed by the integration of calculated distributions to determine the overall weight-averaged s-values (sw). Constructed sw isotherms were fitted with A + B ⇌ AB model as implemented in the software package SEDPHAT with known sw values of individual components as prior knowledge. Resulting parameters were verified and loading concentrations were corrected using global Lamm equation modeling also implemented in SEDPHAT software (24, 25).
Small Angle X-ray Scattering
SAXS data were collected on the European Molecular Biology Laboratory (EMBL) P12 beamline on the storage ring PETRA III (Deutsches Elektronen Synchrotron (DESY), Hamburg, Germany). The ASK1-TBD, TRX1, and ASK1-TBD·TRX1 complex were measured in concentration ranges of 1.2–4.6, 1.4–12, and 1.5–11.9 mg ml−1, respectively. The data were averaged after normalization to the intensity of the transmitted beam, and the scattering of buffer was subtracted using PRIMUS (26). The forward scattering I(0) and the radius of gyration Rg were evaluated using the Guinier approximation (27). These parameters were also computed from the entire scattering pattern using the program GNOM (28), which provides the distance distribution functions P(r) and the maximum particle dimensions Dmax. The solute apparent molecular mass (MMexp) was estimated by comparison of the forward scattering with that from reference solutions of bovine serum albumin (molecular mass 66 kDa). Ab initio molecular envelopes were computed by the program DAMMIN (29), which represents the protein by an assembly of densely packed beads. Multiple iterations of DAMMIN were averaged using the program DAMAVER (30). The averaged envelopes were then used as final SAXS structures.
Circular Dichroism Measurements
The far-UV CD spectra were measured in a quartz cuvette with an optical path length of 1 mm (Starna, USA) using a J-810 spectropolarimeter (Jasco, Japan). The conditions of the measurements were as follows: a spectral region of 200–260 nm, a scanning speed of 10 nm min−1, a response time of 8 s, a resolution of 1 nm, a bandwidth of 1 nm, and a sensitivity of 100 mdeg. The final spectrum was obtained as an average of 5 accumulations. The spectra were corrected for a baseline by subtracting the spectra of the corresponding polypeptide-free solution. The CD measurements were conducted at 22 °C in buffer containing 20 mm Tris-HCl (pH 7.5), 200 mm NaCl, and 2 mm 2-mercaptoethanol. The ASK1-TBD and TRX1 concentrations were 8 μm. After baseline correction, the final spectra were expressed as a mean residue ellipticities QMRW (deg cm2 dmol−1 res−1) and calculated using the equation,
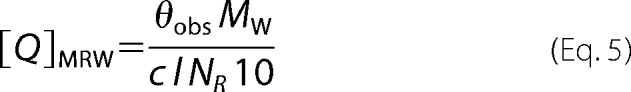 |
where θobs is the observed ellipticity in mdeg, c is the protein concentration in mg ml−1, l is the path length in cm, Mw is the protein molecular weight, and NR is the number of amino acids in the protein (31). The near-UV CD spectra were measured in a quartz cuvette with an optical path length of 1 cm (Starna, USA) in a spectral region of 250–320 nm. The final spectrum was obtained as an average of 15 accumulations. The ASK1-TBD and TRX1 concentrations were 26 μm.
Protein Structure Modeling
Because ASK1 sequence 88–302 does not show any homology to known structures, its structural models were created by ab initio modeling using I-TASSER (32), Phyre2 (33), and Robetta (34) servers. The agreement between the calculated scattering curves of theoretical models and the experimental SAXS data were evaluated using CRYSOL (35).
RESULTS
Preparation of ASK1-TBD and Its Complex with TRX1
It has previously been shown ASK1-TBD is located between residues 46 and 277 within the N-terminal part of ASK1 (1, 9). We expressed several constructs of human ASK1 consisting of residues 46–302, 88–302, 46–322, and 88–322 with either the C-terminal His6 tag or the N-terminal His6-GB1 tag and tested their solubility and stability. Only the construct consisting of the C terminally His-tagged ASK1 sequence 88–302 exhibited sufficient expression yield, solubility, and stability, and thus was used for further studies. To avoid TRX1 homo-dimerization due to the intermolecular disulfide bond formation by the non-active site Cys73 residue under high protein concentrations, we decided to use the mutant C73S of human TRX1 (denoted in this work as TRX1), rather than the wild-type protein, throughout this work. This mutation has no effect on the activity or the structure of human TRX1 (36, 37).
Biophysical Characterization of the Interaction between ASK1-TBD and Reduced TRX1
The AUC was used for the biophysical characterization of prepared ASK1-TBD and its interaction with TRX1 under reducing conditions. The normalized continuous sedimentation coefficient distributions c(s) from the SV AUC experiments revealed the reduced TRX1 and ASK1-TBD form a complex with a weight-averaged sedimentation coefficient (corrected to 20.0 °C and the density of water), sw(20,w), of 3.0 S, whereas TRX1 and ASK1-TBD alone show single peaks with sw(20,w) values of 1.6 and 2.4 S, respectively (Fig. 1). The observed values of sw(20,w) for TRX1 and ASK1-TBD correspond to molecular masses of ∼12 and ∼25 kDa, respectively, suggesting both proteins are monomers in solution (theoretical molecular mass of TRX1 and ASK1-TBD are 12.99 and 25.57 kDa, respectively). In addition, the observed value of sw(20,w) of 3.0 S for the ASK1-TBD·TRX1 complex corresponds to molecular mass of ∼33 kDa, suggesting the 1:1 molar stoichiometry of the complex (theoretical molecular mass = 38.6 kDa). To obtain the apparent equilibrium dissociation constant (KD) of the ASK1-TBD·TRX1 complex, a range of concentrations and different molar ratios of ASK1-TBD and reduced TRX1 were examined using SV AUC (Fig. 2A). Analysis of the isotherm of weight-averaged s values (sw isotherm) as a function of TRX1 concentration revealed the best-fit KD of 0.3 ± 0.1 μm using a 1:1 Langmuir binding model. This confirms the ASK1-TBD and TRX1 form a complex with a 1:1 molar stoichiometry. Because all SV AUC experiments were performed in the presence of the reducing agent 2-mercaptoethanol and obtained SV AUC data can be adequately fitted using the reversible Langmuir-type kinetic model (A + B ⇌ AB), it is reasonable to assume the interaction between ASK1-TBD and TRX1 under reducing conditions does not involve the formation of intermolecular disulfide bonds. No significant amount of the mixed disulfide ASK1-TBD·TRX1 complex was observed even upon the 36-h long incubation of an equimolar mixture of ASK1-TBD and TRX1 in the absence of the reducing agents (Fig. 3), further corroborating that intermolecular disulfide bonds are not involved in complex formation.
FIGURE 1.
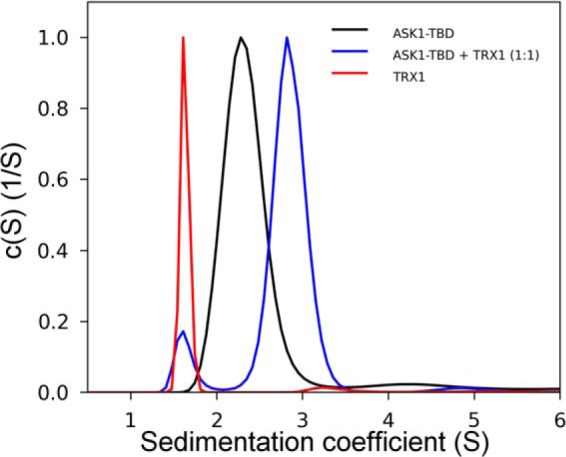
Sedimentation velocity ultracentrifugation. The normalized continuous sedimentation coefficient distributions, c(s), for ASK1-TBD alone (black), TRX1 alone (red), and ASK1-TBD and TRX1 mixed in the molar ratio 1:1 (blue) are shown. All experiments were performed under reducing conditions.
FIGURE 2.

Sedimentation velocity ultracentrifugation. A, isotherm of weight-averaged sedimentation coefficients sw (sw isotherm) obtained from SV experiments of mixtures of ASK1-TBD WT (5 μm) and reduced TRX1 (3–50 μm). B, the sw isotherm obtained from SV experiments of mixtures of ASK1-TBD WT (5 μm) and oxidized TRX1 (3–50 μm). TRX1 was oxidized by incubation with a 100-fold molar excess of H2O2 for 15 min at 37 °C (21). C, the sw isotherm obtained from SV experiments of mixtures of ASK1-TBD WT (20 μm) and reduced TRX1 CS mutant (5–100 μm). D, the sw isotherm obtained from SV experiments of mixtures of ASK1-TBD W31F mutant (20 μm) and reduced TRX1 (15–290 μm). The insets show the sedimentation coefficient distributions c(s) of mixtures of ASK1-TBD and TRX1 at various concentrations and molar ratios underlying the sw data points.
FIGURE 3.
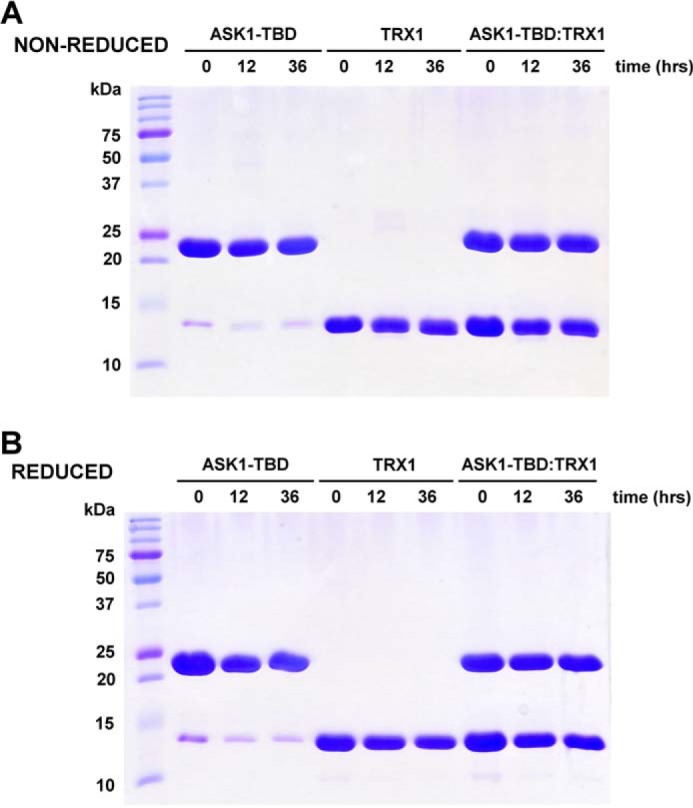
Non-reduced (A) and reduced (B) 15% SDS-PAGE of purified ASK1-TBD, TRX1, and their mixture (with 1:1 molar ratio) after the incubation in a buffer containing 20 mm Tris-HCl (pH 7.5), 200 mm NaCl, and 1 mm EDTA (and no reducing agents) for 0, 12, and 36 h at 4 °C.
Oxidized TRX1 Binds ASK1-TBD with Significantly Lower Binding Affinity Compared with Reduced TRX1
Several studies have shown the oxidation of TRX1 disrupts its binding to ASK1 (1, 9, 18). Therefore, we next examined the interaction between oxidized TRX1 and ASK1-TBD using SV AUC. Oxidized TRX1 was prepared by incubation with 100-fold molar excess of H2O2 for 15 min at 37 °C. Such oxidation was previously shown to produce well defined TRX1 containing two-disulfide bridges (Cys32-Cys35, Cys62-Cys69) (21). The sw isotherm was determined over a range of loading concentrations of oxidized TRX1 and ASK1-TBD (Fig. 2B). The analysis of obtained data revealed oxidized TRX1 exhibits a significantly lower binding affinity for ASK1-TBD compared with reduced TRX1 with the best-fit KD of 6 ± 2 μm using a 1:1 Langmuir binding model, confirming the oxidation of TRX1 disrupts its interaction with ASK1.
Structural Integrity of the Catalytic WCGPC Motif of TRX1 Is Essential for the Binding to ASK1-TBD
The redox-inactive mutant TRX1-CS where both Cys residues (Cys32 and Cys35) from the catalytic 31WCGPC35 motif were replaced by Ser does not bind to ASK1 (1, 9, 18). To test whether this is also true for the interaction with the isolated ASK1-TBD, SV AUC experiments were conducted and the sw isotherm was determined over a range of loading concentrations of TRX1-CS and ASK1-TBD (Fig. 2C). These data revealed negligible binding affinity (KD was estimated to be of ∼900 ± 200 μm) for the TRX1-CS mutant, in agreement with previous reports. In addition, the catalytic 31WCGPC35 motif of human TRX1 contains a conserved Trp31, which is located in close proximity to the catalytic Cys residues and undergoes a conformational change upon oxidation of Cys32 and Cys35 or their mutation to Ser (37, 38). Both oxidized TRX1 and the TRX1-CS mutant exhibit significantly reduced binding to ASK1 (1, 9, 18), suggesting possible involvement of Trp31 in this interaction. To check the importance of this residue, TRX1 mutant W31F was prepared and its binding to ASK1-TBD was investigated using SV AUC (Fig. 2D). Indeed, the obtained SV data revealed significantly lower binding affinity of the W31F mutant to ASK1-TBD (KD of 30 ± 5 μm), confirming the importance of this residue for ASK1-TBD·TRX1 complex stability.
Trp31 of TRX1 Is Directly Involved in the Interaction with ASK1-TBD
Because Trp31 is the only tryptophan residue in human TRX1 and ASK1-TBD does not contain any tryptophan residue, the time-resolved tryptophan fluorescence intensity and anisotropy decay measurements were used to further study the involvement of Trp31 in TRX1 binding to ASK1-TBD. Both time-resolved fluorescence intensity and anisotropy decays were analyzed using a singular-value decomposition maximum entropy method as previously described (23). Complex formation significantly increased the mean excited state lifetime (τmean) of Trp31 from 1.62 to 3.34 ns (Table 1). This could reflect the ASK1-induced conformational change in TRX1, which affects interaction of Trp31 with its surroundings. The observed increase in τmean could also reflect a direct interaction of ASK1-TBD with this residue, reducing its contacts with the polar environment or altering quenching interactions in its vicinity.
TABLE 1.
Summary of time-resolved fluorescence measurements of Trp31 of TRX1
| Sample | τmeana,b | β1c,d | φ1c,e | β2c,d | φ2c,e | β3c,d | φ3c,e | βlongc,d | φlongc,e |
|---|---|---|---|---|---|---|---|---|---|
| ns | ns | ns | ns | ns | |||||
| TRX1 alone | 1.62 | 0.018 | <0.1 | 0.069 | 1.7 | 0.133 | 10 | ||
| TRX1+ASK1-TBD WT | 3.34 | 0.079 | 3.3 | 0.141 | 30 | ||||
| TRX1+ASK1-TBD C250S | 4.52 | 0.026 | <0.1 | 0.024 | 1.3 | 0.034 | 3.8 | 0.136 | 38 |
a The mean fluorescence lifetime (τmean) was calculated using Equation 2.
b S.D. = 0.05 ns.
c The fluorescence anisotropies r(t) were analyzed for series of exponentials (Equation 3), where the amplitudes βi represent the distribution of the correlation times φi. Initial anisotropy of Trp31 for all samples is r0 = 0.22 ± 0.01. Amplitudes β1 were calculated as β1 = r0 − (β2+ β3 + βlong) for each sample.
d S.D. = 0.005.
e S.D. = 15%.
Measurements of the emission anisotropies revealed significantly different mobility of TRX1 Trp31 in the presence and absence of ASK1-TBD as documented by the raw data presented in Fig. 4A. Visual inspection of early depolarization phases in Fig. 4A clearly reveals fluorescence of Trp31 depolarizes significantly faster in the absence than in the presence of ASK1-TBD. The depolarization rate can be directly related to the rotational freedom of the Trp31 residue. The slower depolarization means slower and/or more restricted local and segmental motion of the fluorophore (39). From this point of view, the binding of ASK1-TBD reduces segmental flexibility of the catalytic motif where Trp31 is located. This observation correlates with the decrease in τmean (Table 1), which could indicate the motional restriction of the catalytic motif results in lower accessibility of Trp31 to polar environment and/or suppressed quenching interactions in its vicinity. Rigorous data analysis is in full agreement with the visual observation. TRX1 alone revealed two classes of short correlation times, one very short unresolved (φ1 < 100 ps) and the second, φ2, close to 1.7 ns (Table 1, Fig. 4B). In addition, the third correlation time φlong = 10 ns was also present in the data. The recovered φlong can be assigned to the overall rotational motion of TRX1, and its value is close to what would be expected for a globular protein with a molecular mass about 13 kDa (39). Complex formation resulted in a disappearance of the fastest decay component with the correlation time φ1 belonging to the fastest Trp31 motion. At the same time, the correlation time corresponding to the segmental motion increased from 1.7 to 3.3 ns (φ2 → φ3, Table 1 and Fig. 4B). Complex formation also slightly decreased the sum of amplitudes of the fast anisotropy decay components βshort (βshort = β1 + β2 + β3) indicating angular restriction of the motion. Altogether, these changes can be interpreted as a significantly reduced segmental flexibility of the catalytic motif, where Trp31 is located, upon the TRX1 binding to ASK1-TBD. In addition, the observed increase in the longest correlation time φlong from 10 to 30 ns likely reflects the higher molecular mass of the complex compared with TRX1 alone and its value corresponds with the expected molecular mass of the complex (38.6 kDa). These results suggest Trp31 from the catalytic motif of TRX1 could be directly involved in its interaction with ASK1-TBD.
FIGURE 4.

Time-resolved TRX1 Trp31 fluorescence anisotropy decay measurements. A, TRX1 Trp31 fluorescence anisotropy decays constructed from the raw polarized decay data for TRX1 in the absence (○) and presence (●) of ASK1-TBD. The weighted residuals of both fits (gray, TRX1 alone; black, TRX1 + ASK1-TBD) are shown in the lower panels. The fit quality is also demonstrated by the autocorrelation functions shown in the inset (gray, TRX1 alone; black, TRX1 + ASK1-TBD). B, rotational correlation time distributions of Trp31 (TRX1) in the absence and presence of ASK1-TBD. The unresolved component with very short correlation time (φ1 < 100 ps) observed in the fluorescence anisotropy decay of TRX1 alone is not shown. C, solution structure of reduced human TRX1 (38). Residues Trp31, Cys32, and Cys35 are shown as sticks.
ASK1-TBD Is a Rigid Domain That Does Not Change Its Structure Upon the Binding of TRX1
To investigate the structural flexibility of ASK1-TBD, the time-resolved tryptophan fluorescence intensity and anisotropy decay measurements of single tryptophan residues inserted at four different positions within the ASK1-TBD (Trp132, Trp175, Trp242, and Trp272) were performed. The sequence of ASK1-TBD does not contain any tryptophan residue; phenylalanines located at these positions were replaced by tryptophans. Results of these measurements are listed in Table 2. It can be noticed that all four mutants showed relatively long τmean ranging from 4.22 to 5.35 ns, indicating that all four Trp residues are likely buried and inaccessible to the polar environment (40). The analysis of fluorescence anisotropy decays revealed bimodal correlation time distributions (Table 2) with small amplitudes of the fast rotational and segmental motion of the fluorophore (β1), suggesting regions around all four Trp residues, especially Trp inserted at position 242, are rigid. These data suggest that ASK1-TBD is a compact and rigid domain.
TABLE 2.
Summary of time-resolved fluorescence measurements of single tryptophan (W) mutants of ASK1-TBD
| ASK1-TBD Trp mutant | τmeana,b | β1c,d | φ1c,e | β2c,d | φ2c,e |
|---|---|---|---|---|---|
| ns | ns | ns | |||
| W132 | 4.62 | 0.033 | 2.7 | 0.190 | 18 |
| W175 | 5.35 | 0.038 | 2.0 | 0.180 | 18 |
| W242 | 4.94 | 0.020 | 5.0 | 0.192 | 18 |
| W272 | 4.22 | 0.032 | 2.5 | 0.162 | 16 |
| 0.013 | Aggr. (>50) |
a The mean fluorescence lifetime (τmean) was calculated using Equation 2.
b S.D. = 0.05 ns.
c The fluorescence anisotropies r(t) were analyzed for series of exponentials (Equation 3), where the amplitudes βi represent the distribution of the correlation times φi.
d S.D. = 0.005.
e S.D. = 15%.
Next, CD spectroscopy was used to check whether the binding of TRX1 affects the overall structure of ASK1-TBD. The deconvolution of CD spectra using the K2D method (41) indicated that ASK1-TBD contains ∼35% of α, ∼20% of β, and ∼45% of random structure. This estimation is in agreement with the theoretical prediction using PSIPRED (35% α, 15% β, and 50% random structure) (42). The far-UV CD spectrum of the ASK1-TBD·TRX1 complex (with 1:1 molar stoichiometry) showed no significant difference when compared with the sum of the individual CD spectra of ASK1-TBD and TRX1 (Fig. 5A), suggesting no significant changes in overall secondary structure upon complex formation. The comparison of near-UV CD spectra that give us the information about the tertiary structure reveal significant differences only in the region from 275 to 295 nm (Fig. 5B). Because the CD signal in this region arises from the environments of Tyr and Trp residues (43), it is likely that the observed differences mainly reflect the structural change in the vicinity of TRX1 Trp31 upon complex formation as has also been suggested by time-resolved tryptophan fluorescence experiments (Table 1 and Fig. 4).
FIGURE 5.

Circular dichroism measurements. A, the comparison of the far-UV CD spectrum of the ASK41-TBD·TRX1 complex (solid line) with the sum of the individual far-UV CD spectra of ASK1-TBD and TRX1 (dotted line). B, the comparison of the near-UV CD spectrum of the ASK1-TBD·TRX1 complex (solid line) with the sum of the individual near-UV CD spectra of ASK1-TBD and TRX1 (dotted line). Proteins were mixed with the 1:1 molar stoichiometry. The mean residue ellipticity (MRE) is plotted as a function of the wavelength.
Low-resolution Structure of ASK1-TBD and Its Complex with TRX1 Obtained from SAXS Measurements
SAXS was used to obtain the visual insight into the structure of ASK1-TBD and its complex with TRX1. The experimental SAXS curves from the ASK1-TBD·TRX1 complex and ASK1-TBD alone are shown in Fig. 6A. The absence of aggregation in both samples was confirmed by the inspection of the SAXS data and the linearity of the Guinier region (inset in Fig. 6A). The apparent molecular masses of ASK1-TBD and the ASK1-TBD·TRX1 complex were estimated by comparison of the forward scattering intensity I(0) with that from reference solutions of bovine serum albumin (Table 3). The estimated mass of 37 kDa for the ASK1-TBD·TRX1 complex corresponds well to a 1:1 stoichiometry (theoretical molecular mass = 38.6 kDa) in agreement with the results from SV AUC. Values of the Rg calculated both from the slope of the Guinier plot and from the distance distribution (P(r)) function suggest the complex is more asymmetric compared with ASK1-TBD alone (Table 3). This was further confirmed by the P(r) function, which revealed maximum dimensions (Dmax) of ASK1-TBD alone and the ASK1-TBD·TRX1 complex to be 82 and 99 Å, respectively (Fig. 6B). These Dmax values corroborate a more extended and asymmetric shape of the complex compared with ASK1-TBD alone.
FIGURE 6.

Structural characterization of ASK1-TBD and its complex with reduced TRX1 by SAXS. A, scattering intensity as a function of the scattering vector s (s = 4π sin(θ)/λ, where 2θ is the scattering angle and λ is the wavelength). Inset shows Gunier plots of ASK1-TBD and the ASK1-TBD·TRX1 complex at concentrations 2.3 and 6 mg/ml, respectively. B, distance distribution function P(r). C, averaged and filtered DAMMIN shape envelope (spheres around the dummy residues) of ASK1-TBD. D, averaged and filtered DAMMIN shape envelope of the ASK1-TBD·TRX1 complex. The main difference between the envelope of the complex and those of ASK1-TBD alone is shown in red.
TABLE 3.
Structural parameters determined from SAXS data
| Sample | Rga | Rgb | Mw,I(0)c | Dmaxd |
|---|---|---|---|---|
| Å | kDa | Å | ||
| ASK1-TBD | 23.7 ± 0.3 | 24.2 ± 0.2 | ∼25 | 82 |
| ASK1-TBD·TRX1 | 28.9 ± 0.2 | 29.3 ± 0.1 | ∼37 | 99 |
a Determined by Guinier approximation.
b Determined from P(r) function.
c Estimated by comparison of the forward scattering intensity I(0) with that from reference solutions of bovine serum albumin.
d Determined by indirect Fourier transformation from SAXS data.
To obtain the information about the shape of these molecules, the ab initio envelopes were calculated from the scattering data using the program DAMMIN (Fig. 6, C and D). The reconstructed envelopes consist of an average of at least 10 individual reconstructions and the individual envelopes agreed well with each other, as determined using normalized spatial discrepancy. Normalized spatial discrepancy is a measure of the degree each of the selected envelopes differs from one another. Values <1 are considered to indicate no systematic differences. Normalized spatial discrepancy values of 0.58 and 0.49 were obtained for envelopes of ASK1-TBD and the ASK1-TBD·TRX1 complex, respectively. The envelope for ASK1-TBD alone (Fig. 6C) shows this domain adopts a compact and slightly asymmetric conformation with one side being narrower than the other. The envelope of the complex (Fig. 6D) is similar and shows a more extended thicker part of the molecule, suggesting that TRX1 interacts with this thicker end of the ASK1-TBD molecule. The size (∼20 × ∼35 × ∼30 Å) and the shape of this additional area (shown in red in Fig. 6D) correspond well with the size and the shape of the TRX1 molecule. The comparison of both envelopes also suggests the interaction between TRX1 and ASK1-TBD is mediated through the large binding interface, rather than one or few contacts. In good agreement with results of CD measurements, the high similarity of obtained envelopes also indicates that TRX1 binding does not induce any dramatic structural change within ASK1-TBD, although we cannot rule out the possibility of a local conformational change that is beyond the resolution of this method.
Structural Modeling of the ASK1-TBD·TRX1 Complex
To further refine the structural details of ASK1-TBD and its complex with TRX1, a structural model of ASK1-TBD was generated. Because the sequence 88–302 of ASK1 lacks homology to any known structures, its models were created by ab initio modeling using I-TASSER, Phyre2, and Robetta servers (32–34). However, only one of the models calculated by the Robetta server showed reasonable agreement not only with the SAXS-based envelope (Figs. 7, A and B, the back-calculated scattering curve based on this model fits the SAXS data with χ2 values of 0.91) but also with the secondary structure prediction and the results of time-resolved fluorescence measurements that suggested all Trp residues that replaced Phe residues at four different positions within ASK1-TBD are likely buried and located in relatively rigid areas (Table 2). The superposition of this model with the SAXS-based envelope is shown in Fig. 7A. The model indicates that the C-terminal part of ASK1-TBD contains a ∼50 residue long α-helix that protrudes from the more spherical N-terminal part with the 3-layer α/β sandwich architecture. The comparison of SAXS-based envelopes suggests TRX1 interacts with the N-terminal bulkier part of ASK1-TBD. Fig. 7C shows a superposition of the SAXS envelope of the ASK1-TBD·TRX1 complex with its model, which was created by inserting a crystal structure of TRX1 (37) into the empty part of the complex envelope. The shape and size of the SAXS-based envelope allowed for the TRX1 molecule to be oriented by its catalytic WCGPC motif (Fig. 7C, shown in yellow) toward to ASK1-TBD consistent with the tryptophan fluorescence data, which suggested the involvement of Trp31 in binding to ASK1-TBD. The accuracy of this model was also assessed by calculating and comparing its theoretical SAXS scattering profile with the experimental scattering curve and the calculated scattering curve fitted the SAXS data well with χ2 values of 1.10 (Fig. 7D).
FIGURE 7.

Superposition of SAXS envelopes with the ab initio models of ASK1-TBD and the ASK1-TBD·TRX1 complex. A, superposition of the SAXS envelope with the theoretical model of ASK1-TBD (sequence 88–302) obtained by ab initio modeling using Robetta (34). The N-terminal CC motif of ASK1 is shown in dark red. Phe residues that were mutated to Trp are shown in green. Cys250 is shown in red. B, comparison of the calculated scattering curve of the theoretical model of ASK1-TBD (red line) with the experimental scattering data (black line). C, superposition of the SAXS envelope with the model of the ASK1-TBD·TRX1 complex that was created using the theoretical model of ASK1-TBD and the crystal structure of human TRX1 (37). The catalytic 31WCGPC35 motif of TRX1 is shown in yellow. D, comparison of the calculated scattering curve of the theoretical model of the ASK1-TBD·TRX1 complex (red line) with the experimental scattering data (black line).
ASK1 Residue Cys250 Is Located in the Vicinity of TRX1-binding Surface
The ab initio model of ASK1-TBD indicated that the TRX1-binding site is located in the vicinity of the Cys250 residue located at the N-terminal end of the long α-helix, which forms the C terminus of modeled ASK1-TBD (Fig. 7C). The mutation of this residue inhibits the interaction between ASK1 and TRX1 (16, 17). To check whether the same holds true for the isolated ASK1-TBD, mutant C250S was prepared and its interaction with TRX1 was characterized using both SV AUC and time-resolved fluorescence measurements. Analysis of the sw isotherm as a function of reduced TRX1 concentration revealed that ASK1-TBD C250S exhibits a significantly lower binding affinity compared with WT with the best-fit KD of 50 ± 10 μm using 1:1 Langmuir binding model (Fig. 8A).
FIGURE 8.

ASK1 residue Cys250 is important for the interaction between ASK1-TBD and TRX1. A, sedimentation velocity ultracentrifugation. The sw isotherm obtained from SV AUC experiments of mixtures of ASK1-TBD C250S (20 μm) and TRX1 (5–100 μm). The inset shows the sedimentation coefficient distributions c(s) of mixtures of ASK1-TBD C250S and TRX1 at various concentrations and molar ratios underlying the sw data points. B, rotational correlation time distribution of Trp31 (TRX1) in the absence and presence of ASK1-TBD C250S. The unresolved component with very short correlation time (φ1 < 100 ps) observed in the fluorescence anisotropy decay of TRX1 alone is not shown.
To further investigate the effect of the C250S mutation on the interaction between ASK1 and TRX1, time-resolved fluorescence measurements of Trp31 of TRX1 were performed. Measurements of the emission anisotropies revealed different hydrodynamic properties of TRX1 Trp31 when interacting with the ASK1-TBD C250S mutant compared with WT (Table 1 and Fig. 8B). Data analysis revealed four classes of correlation times, one unresolved and very short (φ1 < 100 ps), two longer corresponding to segmental motions (φ2 and φ3 close to 1.3 and 3.8 ns, respectively), and the fourth correlation time φlong = 38 ns reflects the overall rotational motion of the complex (Table 1, Fig. 8B). The simultaneous presence of correlation times observed either for TRX1 alone (φ1 and φ2) or TRX1 bound to ASK1-TBD (φ3) suggests an incomplete complex formation when only a portion of TRX1 is bound to ASK1-TBD C250S. This conclusion is fully consistent with results of SV AUC measurements showing that TRX1 interacts with ASK1-TBD C250S with weaker affinity compared with ASK1-TBD WT. Moreover, a significantly longer mean excited state lifetime of Trp31 was observed in this case. In particular, τmean = 4.52 ns for the ASK1-TBD C250S·TRX1 complex compared with 3.34 ns for the ASK1-TBD WT·TRX1 complex. This suggests that Trp31 of TRX1 interacts with ASK1-TBD C250S by the altered way, likely as a result of either different conformations of ASK1-TBD or different interactions at the binding interface. Taken together, both SV AUC and time-resolved fluorescence measurements revealed the ASK1 Cys250 residue is likely located either at or in close vicinity of TRX1-binding site and is crucial for the interaction between ASK1 and TRX1.
DISCUSSION
In the present study, our main aim was to provide a structural insight into the interaction between ASK1 and reduced TRX1. TRX1, a ubiquitous oxidoreductase, was identified as a physiological inhibitor of ASK1, which interacts with the N-terminal region of ASK1 preventing homophilic oligomerization through the N-terminal CC domain of ASK1. Only the reduced form of TRX, but not the oxidized form where both Cys residues from the redox catalytic 31WCGPC35 motif form an intramolecular disulfide bond, interacts with ASK1 (1, 9, 10, 18). However, the precise mechanisms of TRX1 binding to ASK1 as well as its dissociation are still unclear as no structural data are available on ASK1-TBD and its interaction with TRX1.
Screening of several constructs containing ASK1 sequences between residues 46 and 302 showed that only the C terminally His-tagged sequence 88–302 enables the preparation of a soluble and stable protein that binds reduced TRX1 with 1:1 molar stoichiometry and KD of ∼300 nm (Fig. 2A). On the other hand, oxidized TRX1 showed significantly lower binding affinity with KD of 6 ± 2 μm (Fig. 2B), confirming the oxidation of TRX1 hinders its interaction with ASK1. However, the mechanism behind the lower binding affinity of oxidized TRX1 is still unclear. It has been suggested the oxidation of TRX1 generates an intramolecular disulfide bond between Cys32 and Cys35 within the redox catalytic motif and this, in turn, causes the dissociation of TRX1 from ASK1 (1, 9, 18). This hypothesis is supported by the fact the redox inactive TRX1-CS mutant, in which both Cys32 and Cys35 from the catalytic motif are replaced by Ser, does not bind to ASK1 (1, 9, 18). SV AUC experiments revealed that the TRX1-CS mutant exhibits negligible binding affinity for ASK1-TBD (with KD of ∼1 mm, Fig. 2C), confirming previous observations and suggesting these active site Cys residues play an important role in TRX1 binding to ASK1. In addition, our data also suggest that the interaction between ASK1-TBD and reduced TRX1 does not involve the formation of intermolecular disulfide bridges as SV AUC experiments were performed under reducing conditions and all obtained SV AUC data can be adequately fitted using the reversible Langmuir-type kinetic model.
Under strong oxidative conditions or at high protein concentrations human TRX1 forms homodimers covalently linked through the non-active site cysteine 73 (37). Because the TRX1 C73S mutant was used throughout this work to avoid formation of these homodimers, it is necessary to keep in mind data presented in this work cannot assess the potential role of this residue in the interaction between TRX1 and ASK1.
The catalytic motif of human TRX1 also contains a conserved tryptophan residue Trp31, which undergoes a subtle conformational change upon both TRX1 oxidation, when Cys32 and Cys35 are disulfide linked, and the replacement of Cys32 and Cys35 by Ser (the TRX1-CS mutant) (37, 38). Crystallographic analysis revealed that Trp31 is partially disordered in reduced form, but ordered in TRX1-CS and oxidized TRX1. This resemblance of TRX1-CS and oxidized TRX1 also likely contributes to the ability of the TRX1-CS mutant to act as a competitive inhibitor of thioredoxin reductase (44). Because both TRX1-CS and oxidized TRX1 were shown to be unable to bind to ASK1 (1, 9, 10, 18), it is entirely possible the similarity in the conformational behavior of Trp31 might contribute to their inability to bind to ASK1. Consistent with this hypothesis, results of our experiments strongly suggest that Trp31 is directly involved in TRX1 binding to ASK1-TBD. First, the W31F mutation significantly reduced TRX1 binding affinity for ASK1-TBD (Fig. 2D). Next, the time-resolved tryptophan fluorescence measurements showed the TRX1 binding to ASK1-TBD both increases τmean and suppresses the segmental dynamics of Trp31. In the free TRX1, Trp31 exhibits relatively short τmean and a fast emission depolarization, suggesting this residue is exposed to the solvent and highly mobile, in a good agreement with its surface-exposed location (Fig. 4C) as well as partially disordered nature observed in structural studies (37). The longer τmean and a slower and/or more restricted local and segmental motion of Trp31 in the presence of ASK1-TBD suggest the motional restriction of the catalytic motif and a lower accessibility of Trp31 to polar environment and/or suppressed quenching interactions in its vicinity upon complex formation (39, 40). Because Trp31 is located on the surface of the TRX1 molecule (Fig. 4C), it is reasonable to interpret observed changes as a direct involvement of this residue in TRX1 binding to ASK1-TBD.
The SAXS experiments revealed ASK1-TBD is monomeric in solution and adopts a compact and slightly asymmetric shape with one side being narrower than the other (Fig. 6C). The shape of the ASK1-TBD·TRX1 complex is similar but more extended within its thicker part (Fig. 6D). The comparison of obtained envelopes suggests that TRX1 interacts with the thicker end of the ASK1-TBD molecule through the large binding interface without inducing any dramatic structural change, although a local conformational change, which is beyond the resolution of this method, cannot be ruled out. Ab initio molecular modeling, although speculative as the 88–302 sequence of ASK1 lacks homology to any known structures, provided a structural model that shows reasonable agreement with the SAXS-based envelope, the secondary structure prediction, and the results of time-resolved tryptophan fluorescence measurements (Fig. 7, Table 2). This structural model suggests TRX1 interacts with the bulkier part of ASK1-TBD close to the N terminus of the long α-helix that protrudes from the more spherical part of ASK1-TBD and forms its C-terminal end. This long α-helix contains approximately one-half of the N-terminal CC motif of ASK1, based on the prediction using the COILS program (45), located between residues ∼285 and 320 (shown in red in Fig. 7). It has been suggested the TRX1 binding to the N-terminal part of ASK1 blocks its homophilic interaction through this N-terminal CC motif that is required for ASK1 autophosphorylation and activation (9). Our structural model is consistent with this hypothesis as TRX1 binding close to the N-terminal end of this long α-helix might affect its conformation and its coiled-coil interactions. In addition, both SV AUC and the time-resolved fluorescence measurements suggested that ASK1 residue Cys250, which has been shown to be important for both TRX1 binding and the oxidative stress-dependent signaling downstream of ASK1 (16, 17), is located either at or in close vicinity of the TRX1-binding site (Fig. 8, Table 1) consistent with our structural model (Fig. 7).
Taken together, biophysical and structural characterization of the isolated TRX1-binding region of ASK1 revealed that this part of ASK1 is a monomeric and rigid domain that forms a stable equimolar complex with reduced TRX1. Residues from the catalytic 31WCGPC35 motif of TRX1 are essential for TRX1 binding to ASK1-TBD and the interaction does not involve the formation of intermolecular disulfide bonds. Time-resolved tryptophan fluorescence suggested a direct involvement of Trp31 in the TRX1 binding to ASK1. SAXS data revealed a compact and slightly asymmetric shape of ASK1-TBD and suggested TRX1 interacts with this domain through the large binding interface without inducing any dramatic conformational change. Molecular modeling indicated the TRX1-binding site is located close to the N-terminal end of a ∼50-residue long α-helix that forms the C terminus of ASK1-TBD. In addition, our results also show that ASK1 residue Cys250 is likely located either at or in close vicinity to the TRX1-binding surface.
Acknowledgment
We thank Dr. Karen Iorio for critical reading of the manuscript.
This work was supported by Czech Science Foundation Project 14-10061S, Grant Agency of Charles University Project 568912, and Academy of Sciences of the Czech Republic Research Project RVO:67985823 of the Institute of Physiology.
- ASK1
- apoptosis signal-regulating kinase 1
- ASK1-TBD
- TRX1-binding domain of ASK1
- AUC
- analytical ultracentrifugation
- CC
- coiled-coil
- SAXS
- small angle x-ray scattering
- SV
- sedimentation velocity
- TRX1
- thioredoxin 1.
REFERENCES
- 1. Saitoh M., Nishitoh H., Fujii M., Takeda K., Tobiume K., Sawada Y., Kawabata M., Miyazono K., Ichijo H. (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 17, 2596–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nishitoh H., Matsuzawa A., Tobiume K., Saegusa K., Takeda K., Inoue K., Hori S., Kakizuka A., Ichijo H. (2002) ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16, 1345–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Takeda K., Matsuzawa A., Nishitoh H., Tobiume K., Kishida S., Ninomiya-Tsuji J., Matsumoto K., Ichijo H. (2004) Involvement of ASK1 in Ca2+-induced p38 MAP kinase activation. EMBO Rep. 5, 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kawarazaki Y., Ichijo H., Naguro I. (2014) Apoptosis signal-regulating kinase 1 as a therapeutic target. Expert Opin. Ther. Targets 18, 651–664 [DOI] [PubMed] [Google Scholar]
- 5. Tobiume K., Saitoh M., Ichijo H. (2002) Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J. Cell Physiol. 191, 95–104 [DOI] [PubMed] [Google Scholar]
- 6. Bunkoczi G., Salah E., Filippakopoulos P., Fedorov O., Müller S., Sobott F., Parker S. A., Zhang H., Min W., Turk B. E., Knapp S. (2007) Structural and functional characterization of the human protein kinase ASK1. Structure 15, 1215–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Noguchi T., Takeda K., Matsuzawa A., Saegusa K., Nakano H., Gohda J., Inoue J., Ichijo H. (2005) Recruitment of tumor necrosis factor receptor-associated factor family proteins to apoptosis signal-regulating kinase 1 signalosome is essential for oxidative stress-induced cell death. J. Biol. Chem. 280, 37033–37040 [DOI] [PubMed] [Google Scholar]
- 8. Zhang L., Chen J., Fu H. (1999) Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14–3-3 proteins. Proc. Natl. Acad. Sci. U.S.A. 96, 8511–8515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fujino G., Noguchi T., Matsuzawa A., Yamauchi S., Saitoh M., Takeda K., Ichijo H. (2007) Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Mol. Cell Biol. 27, 8152–8163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu H., Nishitoh H., Ichijo H., Kyriakis J. M. (2000) Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol. Cell. Biol. 20, 2198–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Powis G., Montfort W. R. (2001) Properties and biological activities of thioredoxins. Annu. Rev. Biophys. Biomol. Struct. 30, 421–455 [DOI] [PubMed] [Google Scholar]
- 12. Qin J., Clore G. M., Kennedy W. M., Huth J. R., Gronenborn A. M. (1995) Solution structure of human thioredoxin in a mixed disulfide intermediate complex with its target peptide from the transcription factor NFκB. Structure 3, 289–297 [DOI] [PubMed] [Google Scholar]
- 13. Huber H. E., Russel M., Model P., Richardson C. C. (1986) Interaction of mutant thioredoxins of Escherichia coli with the gene 5 protein of phage T7: the redox capacity of thioredoxin is not required for stimulation of DNA polymerase activity. J. Biol. Chem. 261, 15006–15012 [PubMed] [Google Scholar]
- 14. Hwang C. Y., Ryu Y. S., Chung M. S., Kim K. D., Park S. S., Chae S. K., Chae H. Z., Kwon K. S. (2004) Thioredoxin modulates activator protein 1 (AP-1) activity and p27 Kip1 degradation through direct interaction with Jab1. Oncogene 23, 8868–8875 [DOI] [PubMed] [Google Scholar]
- 15. Holmgren A., Söderberg B. O., Eklund H., Brändén C. I. (1975) Three-dimensional structure of Escherichia coli thioredoxin-S2 to 2.8-Å resolution. Proc. Natl. Acad. Sci. U.S.A. 72, 2305–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang R., Al-Lamki R., Bai L., Streb J. W., Miano J. M., Bradley J., Min W. (2004) Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ. Res. 94, 1483–1491 [DOI] [PubMed] [Google Scholar]
- 17. Nadeau P. J., Charette S. J., Landry J. (2009) REDOX reaction at ASK1-Cys250 is essential for activation of JNK and induction of apoptosis. Mol. Biol. Cell 20, 3628–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu Y., Min W. (2002) Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ. Res. 90, 1259–1266 [DOI] [PubMed] [Google Scholar]
- 19. Nadeau P. J., Charette S. J., Toledano M. B., Landry J. (2007) Disulfide bond-mediated multimerization of Ask1 and its reduction by thioredoxin-1 regulate H2O2-induced c-Jun NH2-terminal kinase activation and apoptosis. Mol. Biol. Cell 18, 3903–3913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan S. (2001) A modular polycistronic expression system for overexpressing protein complexes in Escherichia coli. Protein Expr. Purif. 21, 224–234 [DOI] [PubMed] [Google Scholar]
- 21. Hashemy S. I., Holmgren A. (2008) Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J. Biol. Chem. 283, 21890–21898 [DOI] [PubMed] [Google Scholar]
- 22. Rezabkova L., Kacirova M., Sulc M., Herman P., Vecer J., Stepanek M., Obsilova V., Obsil T. (2012) Structural modulation of phosducin by phosphorylation and 14-3-3 protein binding. Biophys. J. 103, 1960–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vecer J., Herman P. (2011) Maximum entropy analysis of analytically simulated complex fluorescence decays. J. Fluoresc. 21, 873–881 [DOI] [PubMed] [Google Scholar]
- 24. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dam J., Velikovsky C. A., Mariuzza R. A., Urbanke C., Schuck P. (2005) Sedimentation velocity analysis of heterogeneous protein-protein interactions: Lamm equation modeling and sedimentation coefficient distributions c(s). Biophys. J. 89, 619–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roessle M. W., Klaering R., Ristau U., Robrahn B., Jahn D., Gehrmann T., Konarev P., Round A., Fiedler S., Hermes C., Svergun D. (2007) Upgrade of the small-angle x-ray scattering beamline X33 at the European Molecular Biology Laboratory, Hamburg. J. Appl. Crystallogr. 40, S190–S194 [Google Scholar]
- 27. Guinier A. (1939) La diffraction des rayons X aux très faibles angles: applications à l'etude des phénomènes ultra-microscopies. Ann. Phys. Paris 12, 161–237 [Google Scholar]
- 28. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 29. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Whitmore L., Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 34. Song Y., DiMaio F., Wang R. Y., Kim D., Miles C., Brunette T., Thompson J., Baker D. (2013) High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Svergun D., Barberato C., Koch M. H. J. (1995) CRYSOL: a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 36. Forman-Kay J. D., Clore G. M., Stahl S. J., Gronenborn A. M. (1992) 1H and 15N resonance assignments and secondary structure of the human thioredoxin C62A, C69A, C73A mutant. J. Biomol. NMR 2, 431–445 [DOI] [PubMed] [Google Scholar]
- 37. Weichsel A., Gasdaska J. R., Powis G., Montfort W. R. (1996) Crystal structures of reduced, oxidized, and mutated human thioredoxins: evidence for a regulatory homodimer. Structure 4, 735–751 [DOI] [PubMed] [Google Scholar]
- 38. Qin J., Clore G. M., Gronenborn A. M. (1994) The high-resolution three-dimensional solution structures of the oxidized and reduced states of human thioredoxin. Structure 2, 503–522 [DOI] [PubMed] [Google Scholar]
- 39. Lakowicz J. R. (1999) Principles of Fluorescence Spectroscopy, Second Ed., Kluwer Academic/Plenum Publishers, New York [Google Scholar]
- 40. Schauerte J. A., Gafni A. (1989) Long-lived tryptophan fluorescence in phosphoglycerate mutase. Biochemistry 28, 3948–3954 [DOI] [PubMed] [Google Scholar]
- 41. Andrade M. A., Chacón P., Merelo J. J., Morán F. (1993) Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 6, 383–390 [DOI] [PubMed] [Google Scholar]
- 42. Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 43. Kelly S. M., Price N. C. (2000) The use of circular dichroism in the investigation of protein structure and function. Curr. Protein Peptide Sci. 1, 349–384 [DOI] [PubMed] [Google Scholar]
- 44. Oblong J. E., Berggren M., Gasdaska P. Y., Powis G. (1994) Site-directed mutagenesis of active site cysteines in human thioredoxin produces competitive inhibitors of human thioredoxin reductase and elimination of mitogenic properties of thioredoxin. J. Biol. Chem. 269, 11714–11720 [PubMed] [Google Scholar]
- 45. Lupas A., Van Dyke M., Stock J. (1991) Predicting coiled coils from protein sequences. Science 252, 1162–1164 [DOI] [PubMed] [Google Scholar]