

Background: Exosome, a type of extracellular vesicles, can associate with Aβ in vitro.
Results: Intracerebrally injected exosomes trapped Aβ on surface glycosphingolipids and transported it into microglia in AD mouse brains, resulting in reductions in Aβ pathology.
Conclusion: Exogenous exosomes act as potent scavengers for Aβ in mouse brains.
Significance: The findings provide a novel therapeutic approach for AD.
Keywords: Alzheimer Disease, Amyloid-β (AB), Exosome, Glycolipid, Microglia
Abstract
Elevated levels of amyloid-β peptide (Aβ) in the human brain are linked to the pathogenesis of Alzheimer disease. Recent in vitro studies have demonstrated that extracellular Aβ can bind to exosomes, which are cell-secreted nanovesicles with lipid membranes that are known to transport their cargos intercellularly. Such findings suggest that the exosomes are involved in Aβ metabolism in brain. Here, we found that neuroblastoma-derived exosomes exogenously injected into mouse brains trapped Aβ and with the associated Aβ were internalized into brain-resident phagocyte microglia. Accordingly, continuous intracerebral administration of the exosomes into amyloid-β precursor protein transgenic mice resulted in marked reductions in Aβ levels, amyloid depositions, and Aβ-mediated synaptotoxicity in the hippocampus. In addition, we determined that glycosphingolipids (GSLs), a group of membrane glycolipids, are highly abundant in the exosomes, and the enriched glycans of the GSLs are essential for Aβ binding and assembly on the exosomes both in vitro and in vivo. Our data demonstrate that intracerebrally administered exosomes can act as potent scavengers for Aβ by carrying it on the exosome surface GSLs and suggest a role of exosomes in Aβ clearance in the central nervous system. Improving Aβ clearance by exosome administration would provide a novel therapeutic intervention for Alzheimer disease.
Introduction
Alzheimer disease (AD),2 a common dementia, is pathologically characterized by the presence of amyloid-β peptide (Aβ)-containing senile plaques within the brain. In familial AD, genetic mutations cause increased production of Aβ (1), whereas in far more common sporadic cases, Aβ generation is normal, but its clearance is impaired (2). Elevated levels of Aβ, caused by an imbalance in its metabolism, are linked to synaptic and nerve loss, which likely manifest as progressive cognitive deficits in AD (3).
Exosomes represent a subtype of secreted membrane vesicles (40–100 nm in diameter) of endosomal origin that are released from various types of cells including neurons (4). Exosomes serve to remove and discard unwanted proteins into a drainage system; they are also known to intercellularly shuttle their cargo: a specific set of proteins, RNAs, and lipids (5). Recently, exosomes were reported to associate with a portion of extracellular amyloid-β precursor protein (APP) and its metabolites, including C-terminal fragments (CTFs), amyloid intracellular domain (AICD), and Aβ, in cultures of human wild-type or mutant human APP-expressing neuroblastoma cells (6, 7). In addition, exosomal proteins such as Alix and flottilin-1 were identified around neuritic plaques in AD brains (6). Similarly, our previous study demonstrated that exosomes released from neuroblastoma or primary cortical neurons can bind to synthetic or endogenous Aβ and promote Aβ fibril formation on their surface in vitro (8). Furthermore, exosome-bound Aβ is incorporated into microglia for degradation, suggesting that exosomes may act as a mediator for Aβ elimination in brains (8). Here, we demonstrated that long term intracerebral administration of exosomes to the brain of APP transgenic mice resulted in a marked reduction in Aβ levels, amyloid depositions, and Aβ-mediated synaptotoxicity. We also clarified that glycosphingolipids (GSLs) abundant in the exosomes were essential for Aβ binding on the exosome surface.
EXPERIMENTAL PROCEDURES
Cell Cultures
Murine neuroblastoma Neuro2a (N2a) cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum. The murine microglial cell line BV-2 was purchased from National Cancer Institute (Istituto Nazionale per la Ricerca sul Cancro, Genova, Italy) and was cultured in RPMI1640 (Invitrogen) supplemented with 10% fetal bovine serum and 2 mm l-glutamine.
Animals
All animal experiments were conducted under a protocol approved by the animal care committees of Hokkaido University. Wild-type C57BL/6 mice were purchased from Japan SLC Inc. (Hamamatsu, Japan). Heterozygotic transgenic mice that express the human APP bearing the Swedish and Indiana (KM670/671NL, V717F) mutations (APPsweInd or J20 strain) were from The Jackson Laboratory (Bar Harbor, ME) and maintained in barrier facilities.
Exosome Isolation
Exosomes were prepared from culture supernatants of N2a cells as described previously (9). Briefly, 1 day before exosome isolation, culture medium was replaced with serum-free medium. The culture supernatants were collected and sequentially centrifuged at 3000 × g for 10 min, 4,000 × g for 10 min, and 10,000 × g for 30 min to remove cells, dead cells, and debris then spun again at 100,000 × g for 1 h to obtain exosomes as pellets.
For sucrose gradient analysis, each exosome pellet (100 μg of protein) was loaded onto 10 ml of a sucrose gradient (0.25–2.3 m sucrose in 20 mm HEPES) and centrifuged at 100,000 × g for 18 h. After centrifugation, 1-ml fractions were collected, diluted with 20 mm HEPES, and precipitated by centrifugation for 1 h at 100,000 × g. The resulting pellets were resuspended in PBS and subjected to Western blot analysis.
Electron Microscopy
Exosomes (100 μg of protein/ml) were resuspended in 50 mm Tris, 150 mm NaCl buffer (pH 7.6) (TBS), and applied to a grid covered with collodion. For Aβ binding experiments exosomes (100 μg of protein/ml) were incubated with Aβ1–42 (15 μm) in TBS at 37 °C for 5 h after pretreating with or without EGCase. Exosome mixtures were then applied to the grid. Exosomes were negatively stained with 2% phosphotungstic acid. Transmission images were acquired using an HD-2000 (Hitachi, Tokyo, Japan) or JEM-1400Plus (JEOL Ltd. Tokyo, Japan) transmission electron microscope.
Dynamic Light Scattering
Exosomes (untreated or treated with EGCase) were suspended in TBS at 100 μg of protein/ml. The particle size of the exosomes was measured by dynamic light scattering using a DelsaNano HC (Beckman Coulter).
Injection and Isolation of Biotinylated Exosomes
Exosomes were biotinylated with EZ-link sulfo-NHS Biotin (Pierce) according to the manufacturer's protocol with minor modifications. Briefly, the exosomes were suspended in PBS (150 μg of protein/ml) and incubated with biotin reagent (1 mg/ml) at room temperature for 30 min. The biotinylated exosomes were isolated by ultracentrifugation at 100,000 × g for 1 h at 4 °C and resuspended in PBS. For detecting co-precipitated Aβ, 2 μl of the 5 μg/μl biotin-exosome solution was injected into the right hippocampus of APP mouse (4 months old) using stereotaxic coordinates and kept for 3 h. The biotinylated exosomes in the homogenates of the hippocampus were precipitated using streptavidin microbeads according to the protocol in a μMACS streptavidin kit (Miltenyi Biotech, Bergisch Gladbach, German). Co-precipitated Aβ was analyzed with Western blotting or enzyme-linked immunosorbent assay (ELISA) following the solubilization of the exosomes in SDS sample buffer or guanidine buffer, respectively.
SDS-PAGE and Western Blotting
SDS-PAGE and Western blot analysis were performed according to the standard methods of Laemmli. To detect target proteins, we employed as a primary antibody monoclonal antibodies against Alix (BD Bioscience), APP CTFs (Sigma), actin (Sigma), Aβ (6E10, Signet, Dedham, MA), or neprilysin (Santa Cruz Biotechnology) or rabbit polyclonal antibodies against flottilin-1, endothelin converting enzyme-1 (ECE-1), (Santa Cruz Biotechnology), or insulin degrading enzyme (IDE, Abcam) and as a secondary antibody an anti-mouse IgG-HRP antibody(GE Healthcare), or anti-rabbit IgG-HRP antibody (GE Healthcare). To detect the ganglioside GM1, we used horseradish peroxidase-conjugated cholera toxin B subunit from Sigma. Bands were visualized using a combination of an ECL Plus kit (GE Healthcare) and an LAS4000 imaging system (Fuji Film, Tokyo).
Fluorescence Labeling for the Exosomes
Exosomes were stained with the red fluorescence dye PKH26 (Sigma) as described previously (8). Briefly, the exosomes were resuspended in diluent C (Sigma) and incubated with PKH26 at room temperature for 5 min. The reaction was stopped by the addition of 1% bovine serum albumin. The PKH26-labeled exosomes were precipitated again by ultracentrifugation at 100,000 × g at 4 °C for 1 h.
Exosome Isolation from Murine Cerebrospinal Fluid (CSF)
CSF was collected from the cisterna magna of 2-month-old C57BL/6 mice as previously described (10). Exosomes were isolated from the CSF using a method similar to that described above for isolation from culture medium.
Analysis of Exosomal Particle Number
A qNano System (Izon Science, Ltd) was employed to analyze the particle densities of N2a- and mouse CSF-derived exosomes resuspended in PBS.
Exosome Administration in Mouse Brains
Mice were continuously treated with exosome solution (2 mg protein/ml) or vehicle (PBS) by Alzet minipump (model 1002) at 0.25 μl/h for 14 days. Mice were placed in a stereotactic instrument (NARISHIGE, Tokyo, Japan), and stainless steel cannulas of Alzet Brain Infusion Kit3 were implanted into the right lateral ventricle (mediolateral, −0.8 mm; dorsoventral −3.0 mm) or hippocampus (anteroposterior, −2.0 mm; mediolateral, −1.3 mm; dorsoventral −2.2 mm). After a 14-day infusion, mice were then transcardially perfused with cold heparin/PBS. The right hemibrain was fixed with 4% paraformaldehyde, PBS at 4 °C for 48 h for use in immunohistochemistry, and the left hemibrain was rapidly frozen with liquid nitrogen and stored at −80 °C for later analysis.
For single injection studies, PKH-labeled exosomes or a conjugate of PKH-exosomes with fluorescent Aβ (4 μg of exosome protein in 2 μl PBS) were injected into the right hippocampus or the lateral ventricle of non-transgenic mice using stereotaxic coordinates as described above. To obtain the conjugates of the exosomes with Aβ, PKH-exosomes (100 μg/ml) were incubated with Aβ1–40 in TBS at 37 °C for 24 h then centrifuged at 100,000 × g for 1 h to remove free Aβ. At 3 or 24 h post injection, the mouse brains were prepared as described above for immunohistochemistry.
Immunohistochemistry
The tissue sections were cut with a cryostat (Leica CM3050S) and post-fixed with 4% paraformaldehyde, PBS. After blocking with 5% bovine serum albumin (BSA), 16-μm-thick sections were immunostained with monoclonal antibodies against Iba1 (Wako), βIII tublin (Promega), or glial fibrillary acidic protein (SHIMA laboratory) followed by visualization with AlexaFluor488-conjugated anti-IgG. Serial 30-μm thickness of brain sections were immunostained with monoclonal antibody against Aβ (4G8, Covance) after a brief formic acid treatment, and the signals were visualized using ABC elite kit (Vector Laboratories). Confocal images were obtained using an Olympus Fluoview FV10i microscope. The Aβ plaques were estimated as the percentage of the immunopositive area (positive pixel) to the examined area (total pixel) using ImageJ software.
Aβ ELISA
Aβ levels were determined using a sandwich ELISA. The kits for Aβ1–40 and Aβ1–42 were obtained from Wako (Osaka, Japan), and that for Aβ1–38 was from IBL (Gunma, Japan). Mouse hippocampus or exosomes were homogenized in 4 m guanidine-HCl buffer (pH 8.0) with an ultrasonic homogenizer (TAITEC, Saitama, Japan). After incubation at room temperature for 3 h, the homogenates were further diluted with 0.1% BSA, PBS and centrifuged at 16,000 × g for 20 min. The resulting supernatants were then applied to the ELISA. All samples were measured in duplicate.
Evaluation of Synaptic Densities
Synaptophysin-immunoreactive synaptic densities were quantified according to the methods of Mucke et al. (11) with minor modifications. The right hemisphere of each APP mouse brain was sagittally cut into 16-μm-thick sections using a freezing microtome. The serial sections were incubated with a monoclonal antibody against synaptophysin (D35E4, Cell Signaling) followed by incubation with AlexaFluor488-bound anti-IgG. Immunofluorescent signals were visualized using an Olympus Fluoview FV10i microscope. The linear range of the synaptophysin-positive fluorescence intensities in nontransgenic control sections was determined, and the same setting was used to analyze all of the following images. For each mouse 9 confocal images were captured in three sections per the right hemisphere of the brain, and each image covered an area 5500 μm2 in the molecular layer of the dentate gyrus. The synaptophysin-immunoreactive synaptic densities were estimated as a percentage of the immunostained area (positive pixel) to the selected image area (total pixel) using ImageJ software.
Thioflavin-S Staining
Brain sections (30 μm thick) were oxidized with 0.25% potassium permanganate for 20 min followed by 3 min of bleaching in 2% potassium metabisulfite and 1% oxalic acid. Sections were stained with 0.015% thioflavin-S in 50% ethanol in the dark for 10 min. After developing in two changes of 50% ethanol for 4 min each, images were captured with Olympus Fluoview FV10i microscope, and thioflavin-S-positive plaques were counted in three sections per mouse hippocampus.
Measurement of GSLs
The extraction of GSLs from the culture cells and the exosomes, the enzymatic digestion of GSL-glycans with EGCase I and II (Takara Bio, Shiga, Japan), and the purification of glycans using glycoblotting were performed as described previously (12). Purified GSL-glycans were analyzed by MALDI-TOF MS using an Ultraflex II TOF/TOF mass spectrometer equipped with a reflector, which was controlled by the FlexControl 3.0 software package (Bruker Daltonics, Bremen, Germany). All spectra were obtained as positive ions and were annotated using the FlexAnalysis 3.0 software package (Bruker Daltonics). The glycan structures were then identified by online database SphinGOMAP.
Quantification of Cholesterol, Sphingomyelin (SM), Ceramide (Cer), and Phosphatidylcholine
Total lipids were extracted from the exosomes or N2a cells by adding chloroform/methanol (1:2, v/v). Levels of sphingomyelin SM and Cer were determined by electron ionization-mass spectrometry (TripleTOFTM 5600) coupling with PeakView software (AB SCIEX, Framingham, MA). Cer (C16:0, d18:1) and SM (C16:0, d18:1) were purchased from Avanti Polar Lipids (Alabaster, AL) and were used as standards. The amounts of and cholesterol were measured by a phosphatidylcholine assay kit (BioVision, Milpitas, CA) and cholesterol E-test kit (Wako), respectively.
Proinflammatory Cytokine ELISA
The levels of proinflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-1β, and interferon (IFN-γ), were determined by ELISA (Multi-Analyte ELISArray, Qiagen) according to the manufacturer's instructions. Briefly, each APP mouse hippocampus was homogenized in 4 m guanidine-HCl buffer (pH 8.0) using an ultrasonic homogenizer (TAITEC). After an incubation at room temperature for 3 h, the homogenates were further diluted in 0.1% BSA, PBS and centrifuged at 16,000 × g at 4 °C for 20 min. The resulting supernatants were applied to the ELISA. All samples were handled in duplicate.
EGCase and Sialidase Treatment
Exosomes (1 mg of protein/ml) were incubated with 0.5 units/ml EGCase II (Takara Bio Inc., Shiga, Japan) at 37 °C for 15 h in PBS containing 20 mm HEPES (pH 7.4) or 1 unit/ml of sialidase from Clostridium perfringens (Sigma) at 37 °C for 16 h in 50 mm acetate buffer (pH 5.5). Each mixture was centrifuged at 100,000 × g for 1 h. The resultant precipitates were resuspended in TBS or Hepes-buffered saline and used for further examination.
Seed-free Aβ Preparation
Seed-free Aβ solutions were prepared essentially according to a published report (13).
Aβ Binding Assay
PKH26-labeled exosomes (untreated or treated with EGCase) were plated on chamber slideglass (Thermo Fisher Scientific, Waltham, MA) by staying in PBS for 1 h at room temperature. Fluorescence-labeled Aβ1–42 (1 μm) was then added into the chamber of cultured N2a cells or the labeled exosomes and co-incubated in serum-free medium at 37 °C for 5 h. After a wash with PBS to remove free Aβ, fluorescent images were captured using Olympus Fluoview FV10i microscope.
Binding Analysis by Surface Plasmon Resonance
The binding studies of N2a-derived exosomes (untreated or pretreated with EGCase) with immobilized Aβ peptide (Aβ1–40, Aβ1–42, Aβ1–38, or Aβ42–1) were performed using BIACORE T200 instrument (GE Healthcare). Briefly, seed-free Aβ was independently immobilized onto a carboxymethylated (CM) dextran-coated gold surface (CM5 sensor chip) by amine coupling. The amount (resonance units.) of immobilized Aβ1–40, Aβ1–42, Aβ1–38, or Aβ42–1 was 1143.0, 1266.7, 1316.3, or 948.0, respectively. Then the exosomes were suspended in Hepes-buffered saline (10 mm HEPES, 150 mm NaCl (pH 7.4)) and injected over the surface at 25 °C for 1 min at a flow rate of 30 μl/min. The resultant responses were subtracted from a blank that was immobilized with BSA or prepared by ethanolamine deactivation. Finally, the exosomes were regenerated from the Aβ-immobilized surface by injecting 5 m guanidine-HCl, 10 mm Tris-HCl (pH 8.0).
Thioflavin-T Assay
Seed-free Aβ1–42 solutions (15 μm) were incubated at 37 °C for various times with the exosomes (100 μg of protein of exosomes in 100 μl of TBS) that had been untreated or treated with EGCase. Fluorescence intensities of Thioflavin-T (Sigma) were determined as described before (8) using an Appliskan spectrofluorophotometer (Thermo Fisher Scientific).
Exosome Uptake Assay
Uptake of PKH26-labeled exosomes into microglial BV-2 cells were measured as described previously (8). Briefly, fluorescent exosomes were administered to BV-2 cells and incubated for various times in serum-free conditions. After a wash, the cells were then fixed, and confocal images were acquired using an Olympus Fluoview FV10i microscope. The fluorescence intensity of each sample was analyzed with ImageJ software.
RESULTS
Exogenously Injected Exosomes Trap Aβ and Are Incorporated into Microglia in Mouse Brains
To explore the hypothesis that providing exogenous exosomes in vivo would enhance Aβ clearance by facilitating engulfment of exosome-bound Aβ by microglia, we loaded isolated exosomes into mouse brains and evaluated the effect on the balance of Aβ metabolism. Exosomes were collected from culture supernatants of mouse neuroblastoma N2a cells using sequential ultracentrifugation; the exosomes typically consisted of membrane vesicles of 70–120 nm in diameter (Fig. 1, A and B) as previously described (14). The presence of exosomes was confirmed by detecting the exosomal markers Alix and flotillin-1 as well as the membrane glycolipid GM1 ganglioside in sucrose density gradient fractions corresponding to a density of 1.12 and 1.16 g/ml (Fig. 1C). Aβ was not identified in N2a cells and the exosomes used in this study (Fig. 1C). Exosomes isolated from N2a cells were biotinylated and injected into the hippocampus of APPSweInd transgenic (APP) mice. Hippocampal Aβ was detectable in streptavidin-precipitated exosomes together with the marker Alix 3 h after the injection (Fig. 1D). Accompanied by Aβ, the intrahippocampal-injected exosomes co-localized with the microglial marker Iba1 (Fig. 1E), agreeing with our previous in vitro study (8). These results demonstrate that in mouse brains exogenous exosomes bind Aβ and are then incorporated into microglia together with the bound Aβ for degradation.
FIGURE 1.
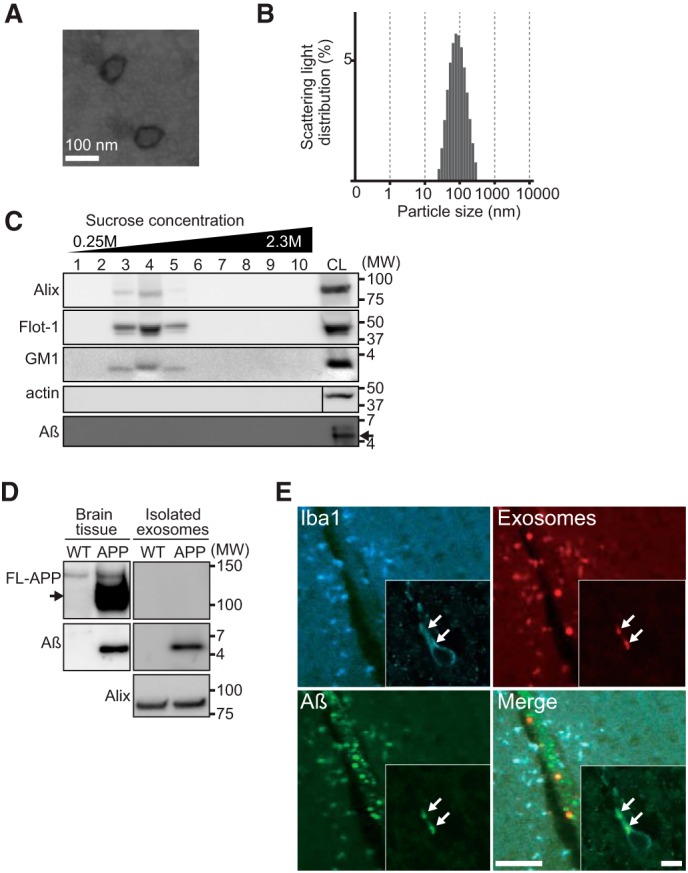
Exogenously injected exosomes trap Aβ and are internalized into microglia in mouse brains. Exosomes were isolated from culture supernatant of N2a cells by sequential centrifugation, eventually to 100,000 × g pellets. A, an electron microscopic image of phosphotungstic acid-stained exosomes. B, exosomes were measured by dynamic light scattering. C, exosomes were density-fractionated by sucrose gradient, and the fractions were analyzed by Western blotting to detect the exosome markers Alix, flotillin-1 (Flot-1), and ganglioside GM1 (GM1) as well as actin and Aβ. CL, cell lysates. D, biotinylated exosomes stereotaxically injected into the hippocampus of 4-month-old non-transgenic or APP mice were isolated using biotin binding affinity beads, then analyzed by Western blotting to detect co-precipitated full-length (FL)-APP, Aβ, and Alix. E, conjugates of fluorescence-labeled Aβ (green) and exosomes (red) were administered into the hippocampus of non-transgenic mice. The hippocampal images were captured 3 h after the injection, following antibody staining for the microglial marker Iba1 (blue). Scale bar, 50 μm and 10 μm (inset).
Continuous Exosome Administration Ameliorates Aβ Pathology and Synaptic Dysfunction in APP Mouse Brains
we next continuously administered exosomes into the lateral ventricles of 4-month-old APP mice for 14 days using osmotic minipumps. The exosome solution (2 mg of protein/ml) contained 2.67 × 1012 particles/ml, which is ∼35 times higher than the concentration of the exosomes in mouse CSF (7.51 × 1010 particles/ml). The intraventricularly injected exosomes have been reported to penetrate into brain parenchyma (15). After 2 weeks, there was an approximate 50% reduction in total Aβ1–40 and Aβ1–42 levels and 35% reduction in Aβ1–38 levels in the hippocampus of the exosome-treated APP mice compared with those infused with vehicle (Fig. 2A). A similar decline in Aβ levels was confirmed by direct administration of the exosomes into mouse hippocampus, with ∼50% less observed in the ipsilateral injection side than the contralateral (Fig. 2B). APP mice given vehicle treatment exhibited ∼50% decreased densities in synaptophysin immunoreactivities in the hippocampus as compared with wild-type mice (Fig. 2, C and D), agreeing with previous reports (11). After a 14-day infusion of the exosomes, the synaptophysin immunoreactivities were markedly increased in the APP mouse hippocampus (85 and 50% of those observed in WT mice after exosome and PBS treatment, respectively; Fig. 2D). Thus, the exosomes mediated significant recovery from synaptic impairment in the APP mice. Taken together, these findings validated that in vivo, exogenously added exosomes induce reductions in Aβ levels and Aβ-associated synaptotoxicity.
FIGURE 2.
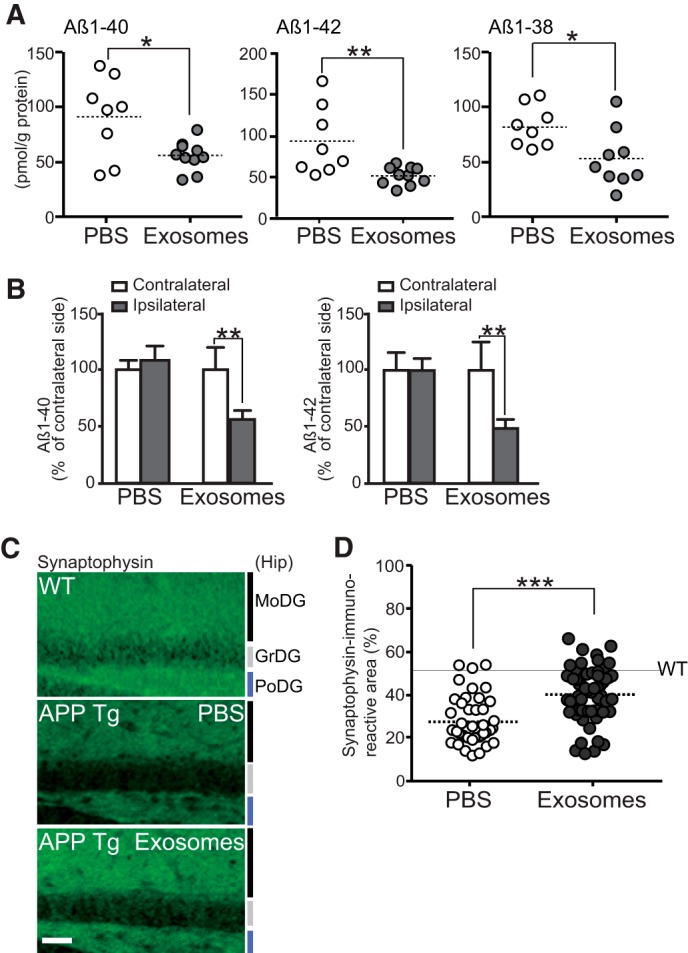
Intracerebral administration of N2a-exosomes induces Aβ clearance. Exosomes (12 μg of protein/PBS/day) or vehicles were continuously infused into lateral ventricle (A, C, and D) or right hippocampus (B) of APP mice (4 months) for 14 days. A, after the infusion, hippocampal levels of Aβ were measured by ELISA (n ≥ 5 animals per group; mean ± S.D.; *, p < 0.05; **, p < 0.01; Student's t test). B, hippocampal Aβ levels in ipsilateral and contralateral side were measured by ELISA. Values are represented as the percentages of the Aβ levels in the contralateral side (PBS, n = 3; exosome, n = 4; mean ± S.D.; **, p < 0.01; Student's t test). C, representative hippocampal sections of exosome- or vehicle-infused APP mice or age-matched nontransgenic controls stained with antibody against synaptophysin. MoDG, molecular dentate gyrus (DG); GrDG, granular DG; PoDG, polymorph DG. Scale bar, 100 μm. D, densities of synaptophysin-positive presynaptic terminals in the hippocampal sections in C were quantified (5 sections/mouse, 5 mice per group). Data presented are the mean ± S.D. ***, p < 0.001.
To assess the effect of exogenous exosomes on brain amyloid deposition, we continuously administered the exosomes into the hippocampus of 13-month-old APP mice for 14 days. We found that the exosomes markedly decreased the Aβ immunoreactive burden (65% reduction; Fig. 3, A and B) and number of thioflavin-S-positive plaques (38%; Fig. 3C) in the treated hippocampus compared with the untreated side. Supporting these histological results, tissue levels of Aβ1–40 and Aβ1–42 were also significantly decreased after exosome infusion, as determined by ELISA (Fig. 3D). We also performed exosome infusion into the lateral ventricles of 13-month-old APP mice for 14 days and found significant reductions in Aβ1–40 and Aβ1–42 levels in the mouse hippocampus (Fig. 3E). These findings demonstrate the evident efficacy of long term treatment with exogenous exosomes in Aβ deposition, even deposition of the fibrillar species of Aβ aggregates, in APP mice.
FIGURE 3.
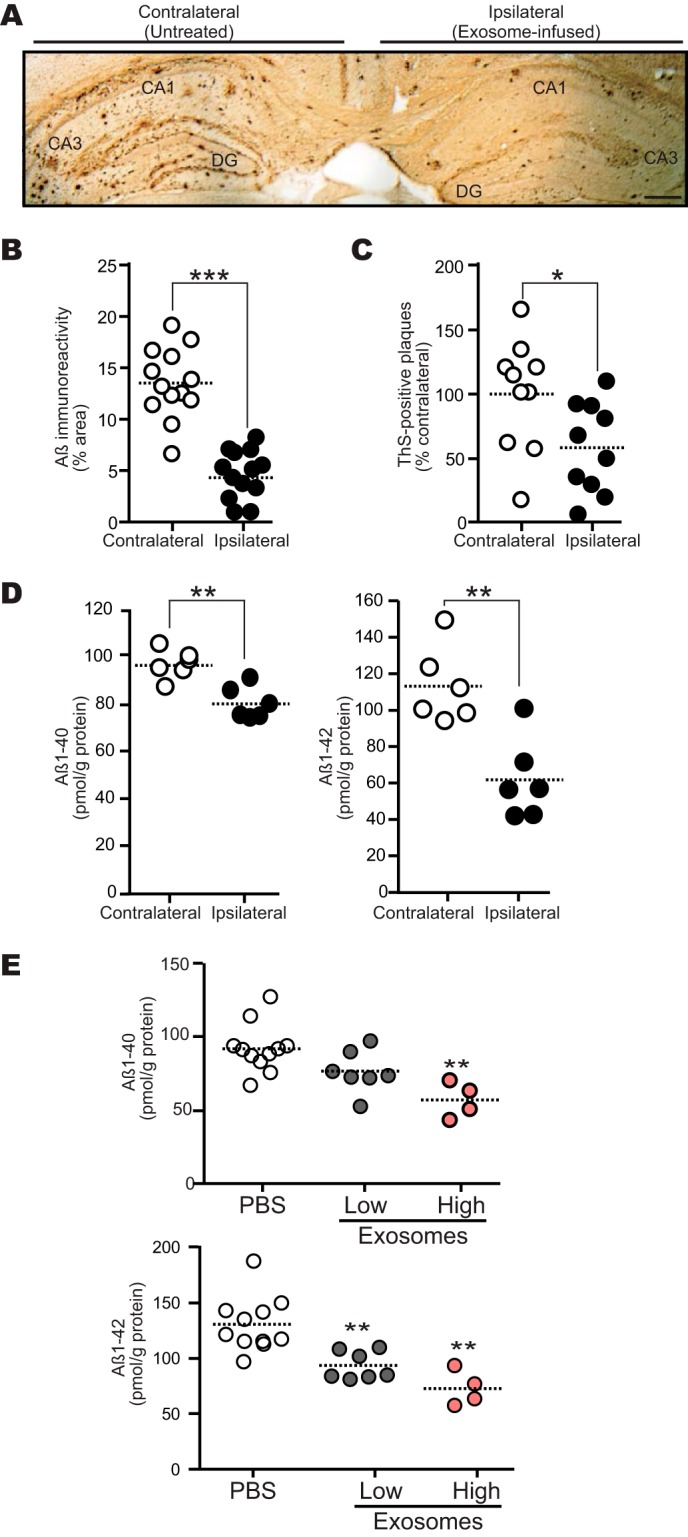
Intracerebral administration of N2a-exosomes reduces Aβ deposition. Exosomes (12 μg of protein/PBS/day) were continuously infused into the hippocampus (A–D) or lateral ventricle (E) of 13-month-old APP mouse for 14 days. A, representative image of APP mouse hippocampal section stained with antibody against Aβ (4G8). DG, dentate gyrus. Scale bar, 200 μm. B, Aβ-immunopositive areas in each hippocampal region were quantified (n = 4 animals, 3 or 4 sections per mouse brain; ***, p < 0.001). C, the number of thioflavin-S (ThS)-positive plaques in each hippocampus was determined (n = 4 animals, 2 or 3 sections per a brain; *, p < 0.05). D, the levels of hippocampal Aβ1–40 and Aβ1–42 were measured by ELISA (n = 3 animals, assayed in duplicate; **, p < 0.01). E, exosomes (low, 12 μg of protein/PBS/day; high, 24 μg of protein/PBS/day) were infused. Hippocampal Aβs were measured by ELISA (n ≥ 4 animals per group; **, p < 0.01 compared with PBS).
To exclude the possibility that the changes observed in vivo are due to effects of exosomes on Aβ generation and enzymatic degradation, we examined the expression levels of APP and its cleaved fragments, including CTF-α and CTF-β, in the hippocampus. No obvious differences in the expression levels were apparent between the exosome- and PBS-treated APP mice (Fig. 4A). In addition, the expression levels of well known proteases for Aβ peptides, including insulin-degrading enzyme, neprilysin, and angiotensin-converting enzyme, were investigated, but there were no variations even after exosome infusion (Fig. 4A). Although the exogenous exosomes were taken up by the microglia, there was no obvious activation in releasing the proinflammatory cytokines like TNFα, IL-6, IL-1β, or IFN-γ in the exosome-treated mouse hippocampus (Fig. 4B). The above findings indicate that the exosomes reduce Aβ levels by promoting an alternative pathway for Aβ clearance, i.e. exosome-associated Aβ uptake by microglia, without any stimulation in APP processing, Aβ degradation, or microglial inflammatory reactions.
FIGURE 4.
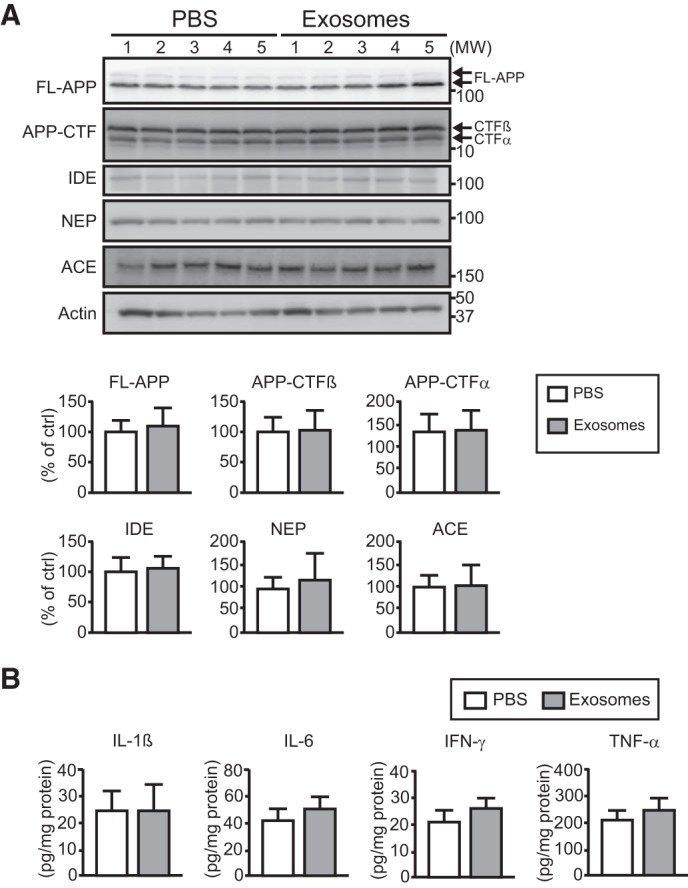
Exogenous exosomes do not stimulate APP processing, expressions of Aβ-degrading enzymes, or inflammatory response. Exosomes or PBS were continuously infused into the lateral ventricles of APP mice (4 months old) for 14 days (A and B). n ≥ 5 animals per group. A, full-length (FL)-APP, APP-CTFs, and the Aβ-degrading enzymes insulin-degrading enzyme (IDE), neprilysin (NEP), and angiotensin-converting enzyme (ACE) were detected in the hippocampus by Western blotting, and the intensity for each band was quantified. Data presented are the mean ± S.D. B, expression levels of proinflammatory cytokines (IL-1β, IL-6, IFN-γ, and TNF-α) in the hippocampus were measured by ELISA. Data are the mean ± S.D.
Exosomal GSL-glycans Are Critical for Their Association with Aβ in Vitro and in Vivo
The above experiments clearly indicate that the exogenous exosomes associated with the Aβ in the mouse brains, although how they interact with each other remained to be determined. Increasing evidence with synthetic liposomes or membranes has demonstrated that gangliosides (sialic acid-containing GSLs) clustering on the membrane surface bind to Aβ and that this Aβ-GSL complex then acts as a template for catalyzing the reaction of Aβ fibril formation (16, 17). In vivo, the monosialoganglioside GM1 was found to associate with Aβ in human brains that exhibit AD pathology (18, 19). GSLs are reported to exist in exosomes (6), but details regarding which species of GSLs have not yet been described. We summarized the profiles of GSL-derived glycans from the exosomes and the cells from which the exosomes derived by quantitative GSL-glycomics (Table 1). The total amount of GSLs was much higher in exosomes than in the parent cells (∼2300%, Fig. 5A). In either, the vast majority of GSLs were GM2 (>84%), with distinct minor compositions (Table 1). Except for GM2, the levels of sialylated GSLs were much higher in the exosomes (Fig. 5B). To investigate whether the GSLs abundant in the exosomes might affect Aβ binding and fibril formation, we deglycosylated exosomal GSLs using EGCase, which specifically cleaves the linkage between the oligosaccharide and glucosylceramide in GSLs (20). The particle size of the exosomes was stable up to 5 h after EGCase digestion, although larger aggregates commonly formed during the 24-h incubation (Fig. 5C). Within the 5-h period, the EGCase-treated exosomes associated very little with the Aβ, as compared with intact exosomes, which overtly colocalized with the Aβ (Fig. 5D).
TABLE 1.
Summary of GSL-glycans found in exosomes and their parental N2a cells
Gg, ganglio-series; Gb, globo-series; Lc, lacto-series.
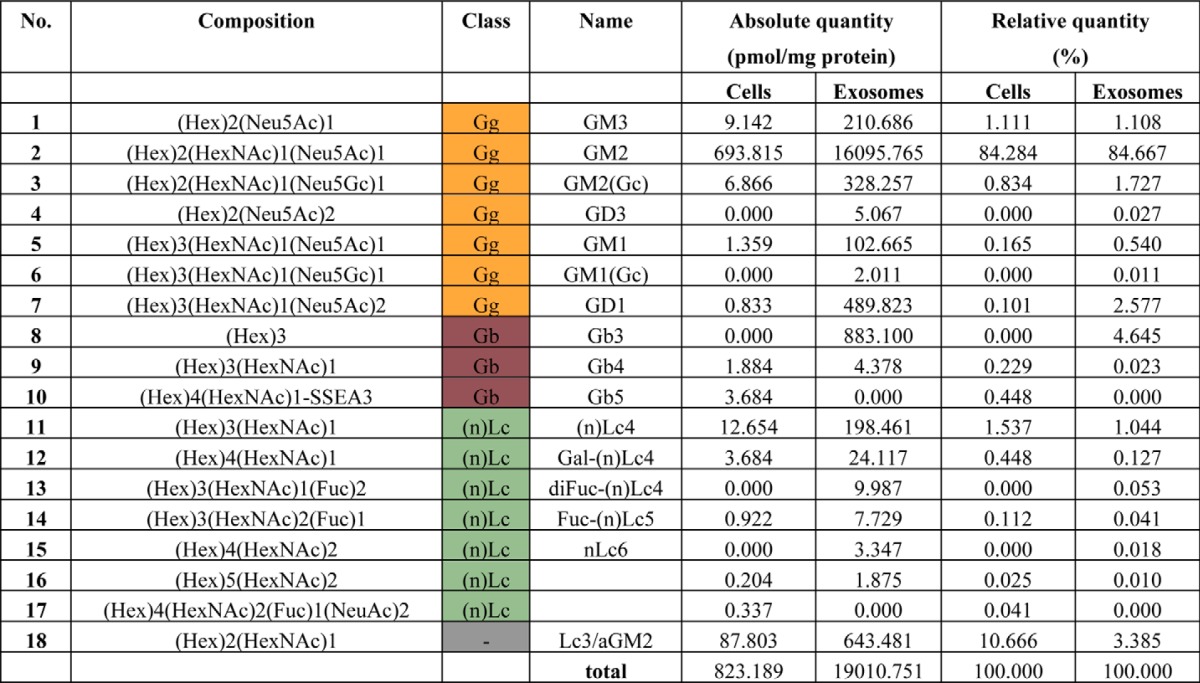
FIGURE 5.
Exosomal GSLs are responsible for Aβ binding on the vesicles. Exosomal and cellular glycomes of GSLs were surveyed by mass spectrometry. A, total amounts of GSL-glycans in exosomes and their originating cells were determined by standardization with protein or phosphatidylcholine (PC) content. B, GSLs other than GM2 detected in exosomes or cells were classified according to the number of sialic acid moieties. C, particle size of exosomes (untreated or treated with EGCase) was determined by dynamic light scattering analysis after incubating them at 37 °C for 0, 5, and 24 h. D, representative images of Aβ binding on N2a cells and exosomes (untreated as Ctrl or treated with EGCase, red) after 5 h incubation with fluorescent Aβ1–42 (1 μm, green). The cells were stained with DAPI. Arrows indicate Aβ fluorescence co-localized with exosomal signals. Scale bar, 25 μm (N2a cells) and 10 μm (exosomes) E, surface plasmon resonance sensorgrams showing the interactions of N2a-derived exosomes (1 μg of protein/μl) with immobilized Aβ1–42 or Aβ42–1. The responses were subtracted from a blank surface prepared by ethanolamine deactivation. RU, resonance units. F, sensorgrams showing the interactions of the exosomes (untreated (Ctrl) or pretreated with EGCase, 1 μg of protein/μl) with immobilized Aβ1–40, Aβ1–42, or Aβ1–38. The resultant responses were subtracted from a surface that was immobilized with BSA.
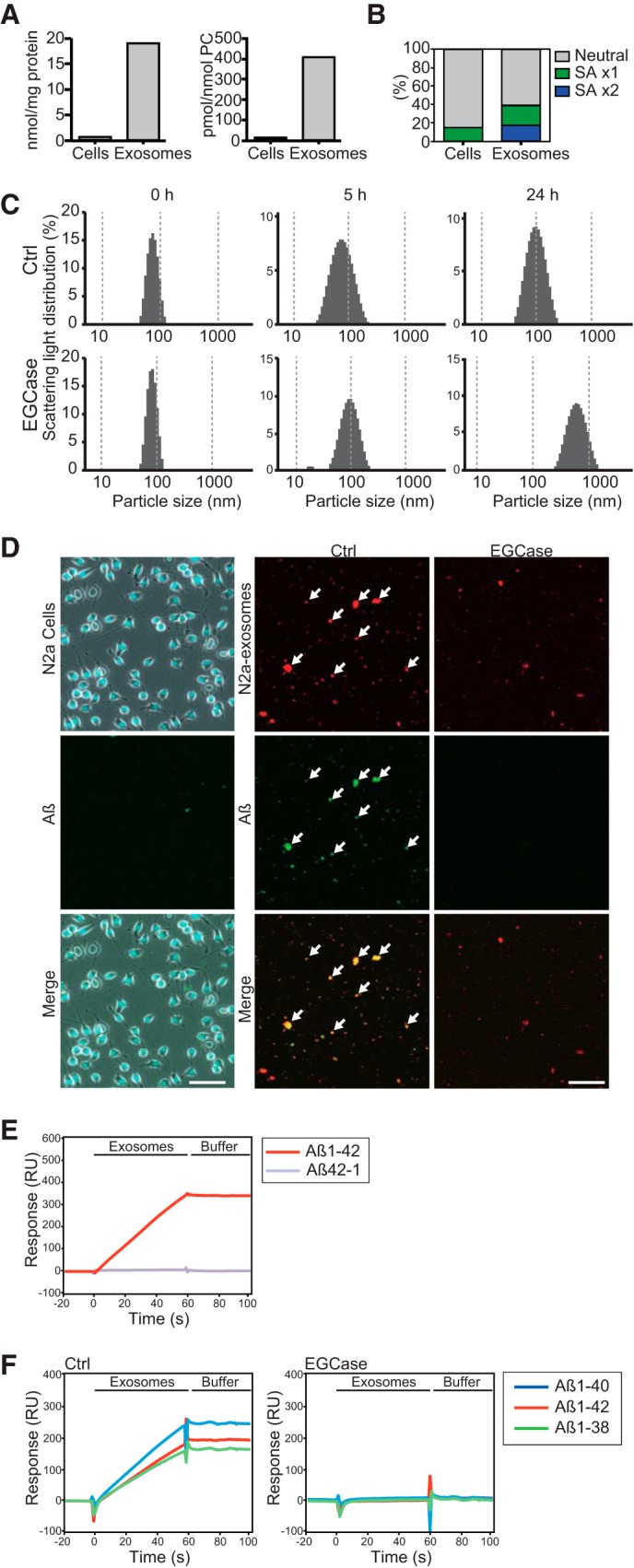
We performed surface plasmon resonance studies to evaluate the specificity of the interaction between N2a-derived exosomes and individual Aβs. As shown in Fig. 5E, when the exosomes were injected onto the immobilized Aβ1–42 and Aβ42–1, a peptide with reverse sequence of Aβ1–42, only the former gave a significant increase in resonance signal, demonstrating the specific nature of the interaction. Specific interactions of the exosomes were observed not only with immobilized Aβ1–42 but also with Aβ1–40 and Aβ1–38, and we found that the interactions were almost completely diminished when the exosomes were pretreated with EGCase (Fig. 5F). These results suggest that Aβs directly binds to the exosomes through the GSL glycans, particularly those sialic acid moieties on their surface. Pretreatment with EGCase also inhibited exosome-dependent amyloid fibril formation in incubation mixtures of exosomes and Aβ1–42, as assessed by thioflavin-T assay and electron microscopic observation (Fig. 6, A and B). Cleavage of sialic acids with sialidase resulted in similar reductions in fibril formation (Fig. 6C). Steric blocking of GM1 or GM2 ganglioside by cholera toxin subunit B or anti-GM2 antibody, respectively, also partially but significantly suppressed amyloid formation (Fig. 6C). In contrast to intact exosomes, the EGCase-treated exosomes nearly failed to coprecipitate with Aβ when injected into the hippocampus of APP mice (Fig. 6D). Our data verify that cleavage of GSL glycans from exosomes can sufficiently prevent the association of exosomes with Aβ. This suggests that there may be multiple species of GSLs, especially sialylated forms, on exosomal membranes that organize into unique sites of high potency able to induce Aβ binding and assembly.
FIGURE 6.
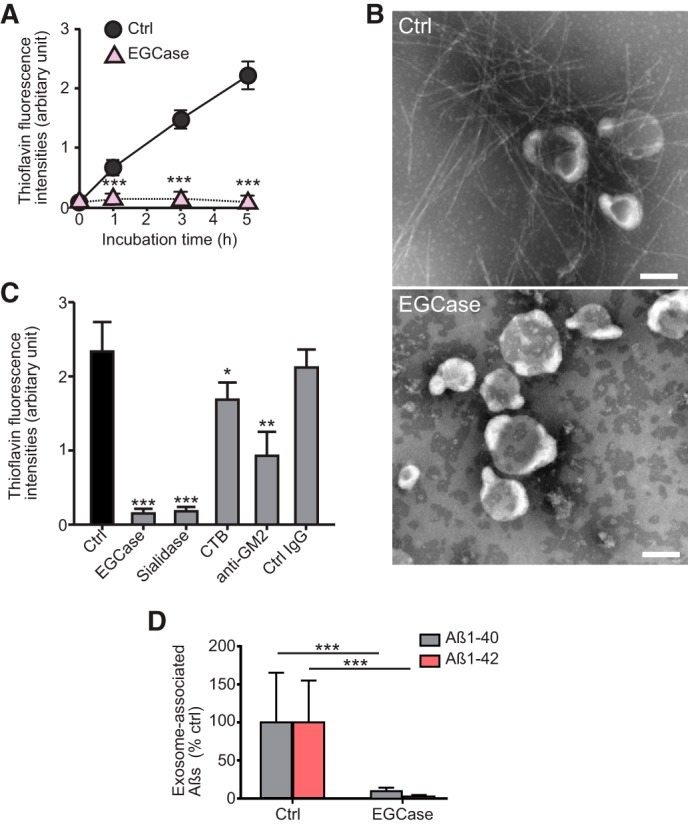
Exosomal GSLs are involved in Aβ assembly. A, thioflavin fluorescence intensities were measured in mixtures of exosomes (untreated as Ctrl or treated with EGCase) incubated with 15 μm Aβ1–42. Data are presented as the mean ± S.D.; ***, p < 0.001 (n = 4). B, representative electron microscopic images of exosomes incubated for 5 h with 15 μm Aβ1–42 are shown. Scale bar, 100 nm. C, the exosomes (untreated as Ctrl or treated with EGCase or sialidase) were incubated for 5 h with 15 μm Aβ1–42. The untreated exosomes were reacted with Aβ in the presence of cholera toxin B subunit (CTB) or anti-GM2 antibody. Fluorescence intensities of thioflavin-T were then measured. Values in each column are the mean ± S.D. of five values. *, p < 0.05; **, p < 0.01; ***, p < 0.001. D, biotinylated exosomes (untreated as Ctrl or treated with EGCase) stereotaxically injected into the hippocampus of APP mice (4 months) were isolated at 3 h after the injection, and the levels of exosome-associated Aβ were quantified by ELISA. Values are the mean ± S.D., ***, p < 0.001 (n = 4).
In addition to gangliosides, cholesterol and SM are also known to promote Aβ assembly via the lateral packing of gangliosides on membranes (21, 22). We found that both cholesterol and SM were highly abundant in exosomes compared with their parent cells (Fig. 7), suggesting that high densities of these two lipids would promote GSL binding to Aβ. Another lipid, Cer, which is the hydrophilic backbone of GSLs, is known to be involved in exosome generation (23). We found higher levels of ceramide in exosomes than in cells (Fig. 7), consistent with a previous report (23).
FIGURE 7.
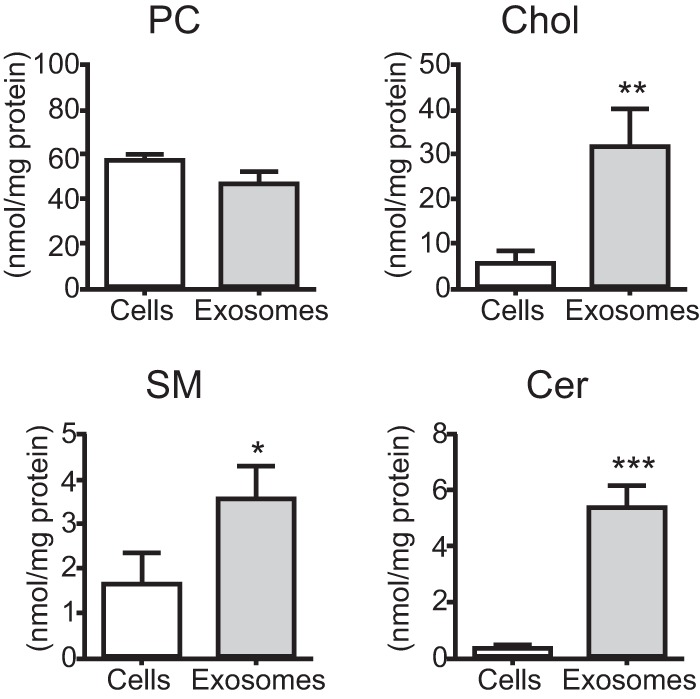
Exosomal and cellular lipid analysis. Levels of phosphatidylcholine (PC), cholesterol (Chol), sphingomyelin (SM), and ceramide (Cer) were measured in N2a cells and the isolated exosomes. The data presented are the mean ± S.D. from three independent experiments; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Exosomes Are Incorporated into Microglia in Vitro and in Vivo, in a GSL-glycan-independent Manner
Our previous in vitro experiments demonstrated that engulfment of exosomes by mouse primary microglia occurred in a partially phosphatidylserine-dependent manner (8). However, deglycosylated proteins, e.g. immunoglobulin FC receptor, reportedly exhibit low affinity toward microglia (24). To determine whether cleavage of GSL-glycans would affect microglial uptake of exosomes, we exposed fluorescent-labeled exosomes pretreated with EGCase or PBS to microglial BV-2 cells. We found no decrease in microglial uptake of EGCase-treated exosomes (Fig. 8A). Both of the labeled exosomes were co-localized with the microglial marker Iba1 when intracerebrally injected into mouse brains (Fig. 8B), and there were no obvious differences observed between untreated or EGCase-treated exosomes. Few fluorescent exosomes were apparent in merged images of cells stained for either a neuronal or astroglial marker (Fig. 8C). The above data indicate that exosomes can be incorporated into microglia undisturbed by the absence of GSL-glycans on the membrane.
FIGURE 8.
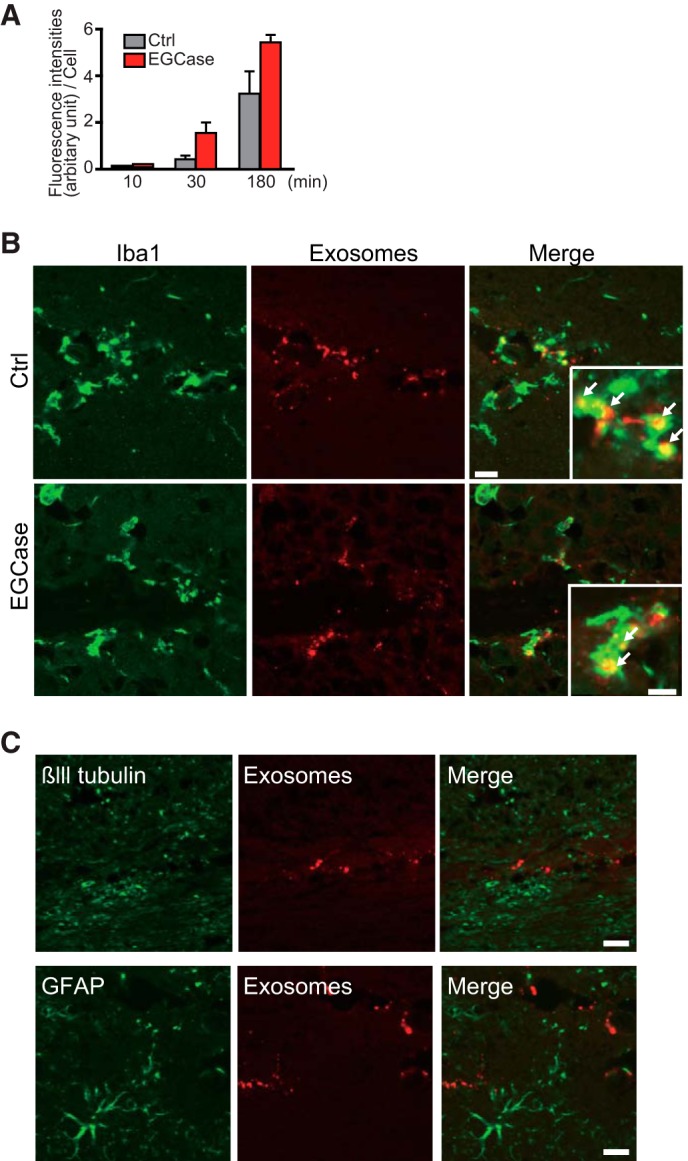
Cleavage of exosomal GSL-glycans does not affect their uptake by microglia. A, fluorescence-labeled exosomes (untreated as Ctrl or treated with EGCase) were exposed to microglial BV-2 cells for 3 h, and the fluorescence intensities of exosomes taken up into the cells were determined by confocal microscopy. B, representative hippocampal sections of non-transgenic mice (4 months) injected with fluorescence-labeled exosomes (untreated as Ctrl or treated with EGCase, red) and stained with anti-Iba1 antibody. Arrows indicate significant exosomal fluorescence in Iba1-positive microglia. Scale bars, 50 and 10 μm (insets). C, hippocampal sections from non-transgenic mice (4 months old) injected with fluorescence-labeled exosomes (untreated as Ctrl or treated with EGCase) stained with antibodies against the neuronal marker βIII tubulin or the astroglial marker glial fibrillary acidic protein (GFAP). Bar, 50 μm.
DISCUSSION
Our study presented here clearly demonstrated that intracerebral exosome infusion leads to a decrease in Aβ levels and ameliorates Aβ-related pathologies in APP mice. In the mouse brains, Aβ was trapped at the exosome surface by glycan moieties of GSLs and transported into microglia for degradation. Mass spectrometry-based analysis has revealed that GSLs are abundant in exosomes compared with parental cells. Just how GSLs are packed so much more into the exosomes than in parental cells remains an unanswered question. Exosomes are produced by intraluminal budding of the limited membrane of endosomes. Accumulation of Cer, which is generated by the hydrolysis of SM, has been reported to initiate the budding (23). Cer can induce a coalescence of small microdomains into larger microdomains to drive domain-induced budding of biological membranes (25), which results in highly loading Cer and its vicinal lipid molecules into the generated vesicle. Indeed, Cer and SM are concentrated in exosome membranes (Fig. 7) (23). In addition, SM forms a distinct membrane domain, namely a lipid raft, in the plasma membrane together with GSLs and cholesterol (26). Various raft-resident proteins have also been reported to be abundant in exosomes (27).
In the present study, providing GSLs-enriched exosomes to the APP mouse brains resulted in recovering synaptic impairment and decreasing Aβ plaques. However, the effect of GSLs on AD pathogenesis is a controversial issue. GSL storage disorders, which are subtypes of lysosomal storage diseases caused by genetic dysfunction in GSL catabolism, share pathological features with AD, such as Aβ burden (28, 29). Accumulated gangliosides are observed in human brains exhibiting AD, and they are proposed to contribute to AD development through promoting Aβ fibril formation (16). These discrepant effects of GSLs are likely to stem from their life span in brain tissues. Pathologically accumulated GSLs are pooled within cells to form complexes with Aβ and its polymer (19, 28), which might be retained to exert neuronal damages. On the other hand, exosomal GSLs capture Aβ in extracellular fluid and are rapidly taken up by phagocytes without persistent harm to the brain.
Our present study demonstrated that exosomes derived from N2a cells can promote Aβ fibril formation on their surface (Fig. 6, A and B). The exosome-bound Aβ was then incorporated into microglia for degradation (Figs. 1E and 8, A and B) (8). Therefore, continuous infusion of exosomes induced a reduction in amyloid depositions in aged APP mouse brains (Fig. 3C). These results provide a notion that the exosomes in the brains are rapidly cleared by microglia before the exosome-bound Aβs form amyloid fibrils for depositions. Amyloid plaques were reported to change their sizes over days in the brains of AD model mice (30). The exogenously added exosomes might prevent further Aβ depositions by blocking the supply of the soluble Aβ. Alternatively, exosomes might support the clearance of amyloid deposit, which already formed. The complex of GM1-Aβ has been reported to localize at the ends of extended Aβ fibrils in the incubation mixture of GM1 and Aβ (19). Alix, a marker for exosomes was enriched around the small Aβ plaques in brain sections from AD patients (6). Similarly, once the exogenous exosome-associated Aβs are attached to the Aβ fibrils in the amyloid plaques, they might provoke microglia gathering toward the plaques and accelerate their clearance.
Here, we used seed-free Aβ to perform the Aβ binding assay (Fig. 5D) and the surface plasmon resonance analysis (Fig. 5E and F) and demonstrated that Aβs directly bind to the exosomes through the GSL glycans on their surface. Seed-free Aβ was reported to contain soluble species of Aβ but not insoluble amyloid forms (13). The GM1-Aβ complex, which acts as a seed for Aβ amyloidogenesis, is known to consist of a clustered GM1 and a monomeric Aβ molecule (17, 31). Accordingly, our previous report has demonstrated that the exosomes derived from N2a cells almost prevented the oligomeric Aβ formation from seed-free Aβ, but not those preformed Aβ oligomers, which are recognized by A11, a specific antibody against oligomer (8). Thus, the above findings suggested that the exosomes released from N2a cells would be mostly associated with monomeric Aβ through their surface GSLs. However, a recent study demonstrated that soluble Aβ oligomer strongly binds to GM1-containing membranes in vitro and in vivo, and GM1-bound Aβ is detected in human cerebrospinal fluid (32). An additional investigation may be required in the future to clarify which form of Aβs can be associated with the exosomes.
It is worth noting that other aggregate-prone proteins, including α-synuclein and prion protein, which cause Parkinson and Creutzfeldt-Jakob diseases, respectively, are also associated with exosomes (33, 34). In addition, α-synuclein and prion protein have been reported to associate with GSLs on the surface of synthetic liposomes (35, 36). A challenging subject of future studies will be determining whether exosomes are involved in the clearance of these proteins.
The normal phagocytotic function of microglia is conceivably important for exosome-bound Aβ clearance in this study. Increasing evidence has indicated that a large portion of secreted exosomes is convincingly taken up by microglia (8, 37). In contrast, a small amount of exosomes can be incorporated into neurons (38). If the clearance function of microglia is decreased or absent, then the exosome-bearing aggregate-prone proteins would trigger pathological events (i.e. formation of senile plaque) or even perform minor interneuronal transfer to propagate their toxic assemblies. Indeed, exosome-associated prion proteins, in which their folded species are infectious, can spread between neuronal cells in a monoculture system (33). The transmissibility of amyloids, a characteristic feature of many neurodegenerative diseases including Alzheimer disease and spongiform encephalitis, might emerge under a lack of glial activity for removing exosomes.
Improvement of Aβ clearance by exosome administration or enhancement of exosome generation provides a novel therapeutic approach for AD therapy. It is noteworthy that the Aβ-degrading enzymes, insulin-degrading enzyme and neprilysin, have been reported to be found in exosomes secreted from microglia and adipose tissue-derived mesenchymal stem cells, respectively (39, 40). Exosomes have been used as a delivery platform, encapsulating reagents or siRNAs (41, 42). Peripheral injection of the exosomes holding siRNA (against an APP-processing enzyme, BACE1) succeeded in brain targeting and specific gene knockdown in mice (41). In the future, development of engineered nanovesicles that regulate multiple processes in AD pathogenesis might be a valuable tool for the therapy.
Acknowledgments
Part of this work was conducted at Hokkaido University, supported by “Nanotechnology Platform” Program of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan.
This work was supported by Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program, Ministry of Education, Culture, Sports, Science, and Technology, Japan.
- AD
- Alzheimer disease
- Aβ
- amyloid-β peptide
- APP
- amyloid-β precursor protein
- GSL
- glycosphingolipid
- CTF
- C-terminal fragment
- N2a
- Neuro2a
- EGCase
- endoglycoceramidase
- CSF
- cerebrospinal fluid
- SM
- sphingomyelin
- Cer
- ceramide
- GM1
- Neu5Acα2-3(Galβ1-3GalNAcβ1-4)Galβ1-4Glcβ1-Cer.
REFERENCES
- 1. Scheuner D., Eckman C., Jensen M., Song X., Citron M., Suzuki N., Bird T. D., Hardy J., Hutton M., Kukull W., Larson E., Levy-Lahad E., Viitanen M., Peskind E., Poorkaj P., Schellenberg G., Tanzi R., Wasco W., Lannfelt L., Selkoe D., Younkin S. (1996) Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat. Med. 2, 864–870 [DOI] [PubMed] [Google Scholar]
- 2. Mawuenyega K. G., Sigurdson W., Ovod V., Munsell L., Kasten T., Morris J. C., Yarasheski K. E., Bateman R. J. (2010) Decreased clearance of CNS β-amyloid in Alzheimer's disease. Science 330, 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 4. Simons M., Raposo G. (2009) Exosomes: vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 21, 575–581 [DOI] [PubMed] [Google Scholar]
- 5. Vlassov A. V., Magdaleno S., Setterquist R., Conrad R. (2012) Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 1820, 940–948 [DOI] [PubMed] [Google Scholar]
- 6. Rajendran L., Honsho M., Zahn T. R., Keller P., Geiger K. D., Verkade P., Simons K. (2006) Alzheimer's disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11172–11177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vingtdeux V., Hamdane M., Loyens A., Gelé P., Drobeck H., Bégard S., Galas M. C., Delacourte A., Beauvillain J. C., Buée L., Sergeant N. (2007) Alkalizing drugs induce accumulation of amyloid precursor protein byproducts in luminal vesicles of multivesicular bodies. J. Biol. Chem. 282, 18197–18205 [DOI] [PubMed] [Google Scholar]
- 8. Yuyama K., Sun H., Mitsutake S., Igarashi Y. (2012) Sphingolipid-modulated exosome secretion promotes clearance of amyloid-β by microglia. J. Biol. Chem. 287, 10977–10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Théry C., Amigorena S., Raposo G., Clayton A. (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 3, 22. [DOI] [PubMed] [Google Scholar]
- 10. DeMattos R. B., Bales K. R., Parsadanian M., O'Dell M. A., Foss E. M., Paul S. M., Holtzman D. M. (2002) Plaque-associated disruption of CSF and plasma amyloid-β (Aβ) equilibrium in a mouse model of Alzheimer's disease. J. Neurochem. 81, 229–236 [DOI] [PubMed] [Google Scholar]
- 11. Mucke L., Masliah E., Yu G. Q., Mallory M., Rockenstein E. M., Tatsuno G., Hu K., Kholodenko D., Johnson-Wood K., McConlogue L. (2000) High-level neuronal expression of Aβ 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fujitani N., Furukawa J., Araki K., Fujioka T., Takegawa Y., Piao J., Nishioka T., Tamura T., Nikaido T., Ito M., Nakamura Y., Shinohara Y. (2013) Total cellular glycomics allows characterizing cells and streamlining the discovery process for cellular biomarkers. Proc. Natl. Acad. Sci. U.S.A. 110, 2105–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Naiki H., Gejyo F. (1999) Kinetic analysis of amyloid fibril formation. Methods Enzymol. 309, 305–318 [DOI] [PubMed] [Google Scholar]
- 14. Théry C. (2011) Exosomes: secreted vesicles and intercellular communications. F1000 Biol. Rep. 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grapp M., Wrede A., Schweizer M., Hüwel S., Galla H. J., Snaidero N., Simons M., Bückers J., Low P. S., Urlaub H., Gärtner J., Steinfeld R. (2013) Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat. Commun. 4, 2123. [DOI] [PubMed] [Google Scholar]
- 16. Ariga T., McDonald M. P., Yu R. K. (2008) Role of ganglioside metabolism in the pathogenesis of Alzheimer's disease: a review. J. Lipid Res. 49, 1157–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Utsumi M., Yamaguchi Y., Sasakawa H., Yamamoto N., Yanagisawa K., Kato K. (2009) Up-and-down topological mode of amyloid β-peptide lying on hydrophilic/hydrophobic interface of ganglioside clusters. Glycoconj. J. 26, 999–1006 [DOI] [PubMed] [Google Scholar]
- 18. Yanagisawa K., Odaka A., Suzuki N., Ihara Y. (1995) GM1 ganglioside-bound amyloid β-protein (A β): a possible form of preamyloid in Alzheimer's disease. Nat. Med. 1, 1062–1066 [DOI] [PubMed] [Google Scholar]
- 19. Hayashi H., Kimura N., Yamaguchi H., Hasegawa K., Yokoseki T., Shibata M., Yamamoto N., Michikawa M., Yoshikawa Y., Terao K., Matsuzaki K., Lemere C. A., Selkoe D. J., Naiki H., Yanagisawa K. (2004) A seed for Alzheimer amyloid in the brain. J. Neurosci. 24, 4894–4902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fujitani N., Takegawa Y., Ishibashi Y., Araki K., Furukawa J., Mitsutake S., Igarashi Y., Ito M., Shinohara Y. (2011) Qualitative and quantitative cellular glycomics of glycosphingolipids based on rhodococcal endoglycosylceramidase-assisted glycan cleavage, glycoblotting-assisted sample preparation, and matrix-assisted laser desorption ionization tandem time-of-flight mass spectrometry analysis. J. Biol. Chem. 286, 41669–41679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yanagisawa K., Matsuzaki K. (2002) Cholesterol-dependent aggregation of amyloid β-protein. Ann. N.Y. Acad. Sci. 977, 384–386 [DOI] [PubMed] [Google Scholar]
- 22. Yuyama K., Yanagisawa K. (2010) Sphingomyelin accumulation provides a favorable milieu for GM1 ganglioside-induced assembly of amyloid β-protein. Neurosci. Lett. 481, 168–172 [DOI] [PubMed] [Google Scholar]
- 23. Trajkovic K., Hsu C., Chiantia S., Rajendran L., Wenzel D., Wieland F., Schwille P., Brügger B., Simons M. (2008) Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319, 1244–1247 [DOI] [PubMed] [Google Scholar]
- 24. Takata K., Hirata-Fukae C., Becker A. G., Chishiro S., Gray A. J., Nishitomi K., Franz A. H., Sakaguchi G., Kato A., Mattson M. P., Laferla F. M., Aisen P. S., Kitamura Y., Matsuoka Y. (2007) Deglycosylated anti-amyloid β antibodies reduce microglial phagocytosis and cytokine production while retaining the capacity to induce amyloid β sequestration. Eur. J. Neurosci. 26, 2458–2468 [DOI] [PubMed] [Google Scholar]
- 25. Gulbins E., Kolesnick R. (2003) Raft ceramide in molecular medicine. Oncogene 22, 7070–7077 [DOI] [PubMed] [Google Scholar]
- 26. Simons K., Ikonen E. (1997) Functional rafts in cell membranes. Nature 387, 569–572 [DOI] [PubMed] [Google Scholar]
- 27. de Gassart A., Geminard C., Fevrier B., Raposo G., Vidal M. (2003) Lipid raft-associated protein sorting in exosomes. Blood 102, 4336–4344 [DOI] [PubMed] [Google Scholar]
- 28. Keilani S., Lun Y., Stevens A. C., Williams H. N., Sjoberg E. R., Khanna R., Valenzano K. J., Checler F., Buxbaum J. D., Yanagisawa K., Lockhart D. J., Wustman B. A., Gandy S. (2012) Lysosomal dysfunction in a mouse model of Sandhoff disease leads to accumulation of ganglioside-bound amyloid-β peptide. J. Neurosci. 32, 5223–5236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu Y. H., Barnes S., Sun Y., Grabowski G. A. (2010) Multi-system disorders of glycosphingolipid and ganglioside metabolism. J. Lipid Res. 51, 1643–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bolmont T., Haiss F., Eicke D., Radde R., Mathis C. A., Klunk W. E., Kohsaka S., Jucker M., Calhoun M. E. (2008) Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 28, 4283–4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matsuzaki K., Kato K., Yanagisawa K. (2010) Aβ polymerization through interaction with membrane gangliosides. Biochim. Biophys. Acta 1801, 868–877 [DOI] [PubMed] [Google Scholar]
- 32. Hong S., Ostaszewski B. L., Yang T., O'Malley T. T., Jin M., Yanagisawa K., Li S., Bartels T., Selkoe D. J. (2014) Soluble Aβ oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 82, 308–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fevrier B., Vilette D., Archer F., Loew D., Faigle W., Vidal M., Laude H., Raposo G. (2004) Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 101, 9683–9688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emmanouilidou E., Melachroinou K., Roumeliotis T., Garbis S. D., Ntzouni M., Margaritis L. H., Stefanis L., Vekrellis K. (2010) Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martinez Z., Zhu M., Han S., Fink A. L. (2007) GM1 specifically interacts with α-synuclein and inhibits fibrillation. Biochemistry 46, 1868–1877 [DOI] [PubMed] [Google Scholar]
- 36. Sanghera N., Correia B. E., Correia J. R., Ludwig C., Agarwal S., Nakamura H. K., Kuwata K., Samain E., Gill A. C., Bonev B. B., Pinheiro T. J. (2011) Deciphering the molecular details for the binding of the prion protein to main ganglioside GM1 of neuronal membranes. Chem. Biol. 18, 1422–1431 [DOI] [PubMed] [Google Scholar]
- 37. Fitzner D., Schnaars M., van Rossum D., Krishnamoorthy G., Dibaj P., Bakhti M., Regen T., Hanisch U. K., Simons M. (2011) Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 124, 447–458 [DOI] [PubMed] [Google Scholar]
- 38. Frühbeis C., Fröhlich D., Kuo W. P., Amphornrat J., Thilemann S., Saab A. S., Kirchhoff F., Möbius W., Goebbels S., Nave K. A., Schneider A., Simons M., Klugmann M., Trotter J., Krämer-Albers E. M. (2013) Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 11, e1001604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tamboli I. Y., Barth E., Christian L., Siepmann M., Kumar S., Singh S., Tolksdorf K., Heneka M. T., Lütjohann D., Wunderlich P., Walter J. (2010) Statins promote the degradation of extracellular amyloid β-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J. Biol. Chem. 285, 37405–37414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Katsuda T., Tsuchiya R., Kosaka N., Yoshioka Y., Takagaki K., Oki K., Takeshita F., Sakai Y., Kuroda M., Ochiya T. (2013) Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci. Rep. 3, 1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M. J. (2011) Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 29, 341–345 [DOI] [PubMed] [Google Scholar]
- 42. Zhuang X., Xiang X., Grizzle W., Sun D., Zhang S., Axtell R. C., Ju S., Mu J., Zhang L., Steinman L., Miller D., Zhang H. G. (2011) Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 19, 1769–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]