

Background: 4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) is a highly potent nucleoside analog reverse transcriptase (RT) inhibitor with a 3′-OH.
Results: EFdA inhibits RT as an immediate or delayed chain terminator depending on the DNA substrate sequence. RT efficiently misincorporates EFdA, producing non-extendable mismatched primers protected from excision.
Conclusion: EFdA blocks RT by multiple mechanisms.
Significance: Understanding the EFdA inhibition mechanism will help develop better antivirals.
Keywords: AIDS, Enzyme Inhibitor, Human Immunodeficiency Virus (HIV), Nucleoside/Nucleotide Analogue, Reverse Transcription, EFdA, NRTIs, Antivirals, Reverse Transcriptase
Abstract
4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) is a nucleoside analog that, unlike approved anti-human immunodeficiency virus type 1 (HIV-1) nucleoside reverse transcriptase inhibitors, has a 3′-OH and exhibits remarkable potency against wild-type and drug-resistant HIVs. EFdA triphosphate (EFdA-TP) is unique among nucleoside reverse transcriptase inhibitors because it inhibits HIV-1 reverse transcriptase (RT) with multiple mechanisms. (a) EFdA-TP can block RT as a translocation-defective RT inhibitor that dramatically slows DNA synthesis, acting as a de facto immediate chain terminator. Although non-translocated EFdA-MP-terminated primers can be unblocked, they can be efficiently converted back to the EFdA-MP-terminated form. (b) EFdA-TP can function as a delayed chain terminator, allowing incorporation of an additional dNTP before blocking DNA synthesis. In such cases, EFdA-MP-terminated primers are protected from excision. (c) EFdA-MP can be efficiently misincorporated by RT, leading to mismatched primers that are extremely hard to extend and are also protected from excision. The context of template sequence defines the relative contribution of each mechanism and affects the affinity of EFdA-MP for potential incorporation sites, explaining in part the lack of antagonism between EFdA and tenofovir. Changes in the type of nucleotide before EFdA-MP incorporation can alter its mechanism of inhibition from delayed chain terminator to immediate chain terminator. The versatility of EFdA in inhibiting HIV replication by multiple mechanisms may explain why resistance to EFdA is more difficult to emerge.
Introduction
The enzyme reverse transcriptase (RT) of human immunodeficiency virus type 1 (HIV-1)4 has been the major target of antiretrovirals that belong to two general classes referred to as nucleoside RT inhibitors (NRTIs) and non-nucleoside RT inhibitors. Currently there are eight approved NRTIs for the treatment of HIV infection, and these are almost always components of first line therapies (1–8). Once phosphorylated, NRTI triphosphates compete with the natural dNTPs for incorporation into the viral nascent DNA chain, and because they lack a 3′-OH, they inhibit further DNA polymerization by acting as chain terminators. Despite the availability of potent antivirals, HIV can escape treatment by developing drug resistance mutations. Thus, there is a need for potent drugs that block wild-type and drug-resistant HIVs.
We have shown previously that nucleoside analogs that retain the 3′-OH group and have a modification at the 4′-position of the sugar ring can inhibit HIV-1 RT very efficiently (9). We also reported that 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) is a deoxyadenosine analog with anti-HIV efficacy at subnanomolar concentrations in primary cells, making it the most potent NRTI described to date (10–13). Unlike all approved anti-HIV NRTIs, EFdA possesses a 3′-OH, which mimics the structure of normal dNTPs (see Fig. 1A). It has been demonstrated that this property provides enhanced phosphorylation of the NRTIs by cellular kinases increasing their antiviral efficacy (14). In addition, EFdA has 4′-ethynyl and 2-fluoro substitutions, which also contribute to its extraordinary selectivity index of about 200,000 as determined after initial antiviral characterization of a series of 4′-substituted analogs (9, 11). We have shown that EFdA is resistant to degradation by adenosine deaminase, providing an enhanced half-life to the compound compared with other NRTIs (11, 15). EFdA is very effective not only against wild-type HIV but also against drug-resistant strains and has displayed synergistic antiviral effects when combined with other inhibitors such as the recently approved second generation non-NRTI rilpivirine (16). We have also shown that EFdA is significantly better than tenofovir (TFV), zidovudine, and emtricitabine in blocking simian immunodeficiency virus replication in monkey peripheral blood mononuclear cells and that it was highly effective at treating simian immunodeficiency virus infection and AIDS symptoms in simian immunodeficiency virus-infected macaques with advanced AIDS with no apparent signs of toxicity (17).
FIGURE 1.

EFdA-TP inhibits RT-catalyzed DNA synthesis as an ICT or DCT. A, structure of EFdA. B, DNA/DNA Td31/Pd18 (20 nm) was incubated with 20 nm RT for 15 min in the presence of 10 μm dNTPs, 6 mm MgCl2, and increasing concentrations of ddATP (lanes 1–4) or EFdA-TP (lanes 5–22). The sequence of the template and the mechanism that EFdA-TP uses to inhibit RT are shown next to different bands of the gel. C, the translocation state of RT after ddAMP (lanes 1–6 in both gels) or EFdA-MP (lanes 7–20 and 7–21) incorporation was determined using site-specific Fe2+ footprinting. Td43/Pd30 or Td43/Pd30+5 (100 nm) with 5′-Cy3-label on the DNA templates was incubated with 600 nm RT and various concentrations of the next incoming nucleotide (dTTP). The complexes were treated for 5 min with 1 mm ammonium iron sulfate. An excision at position −18 indicates a pre-translocation complex, whereas the one at position −17 represents a post-translocation complex. The sequences of the T/Ps that were used in the experiments are shown under the gels with underlined matched sequences in primer extension and footprinting assays. D, representative reactions that highlight the sites on the template sequences where EFdA-TP acts as an ICT or DCT. The numbers indicate the points (marked in red) of the dATP or dATP analog incorporation (P1, P6, and P10). In green color, we indicate the bases after dATP or dATP analog incorporation (P7 and P11). E, similar to A, the effect of ddATP and EFdA-TP on primer extension was observed using the DNA/DNA Td31/Pd18+5.
Here we used a series of biochemical experiments and demonstrated that EFdA triphosphate (EFdA-TP) blocks RT with multiple mechanisms. Specifically, we found that, unlike approved anti-HIV NRTIs, EFdA-TP can act as a de facto immediate or delayed chain terminator in a manner that depends on the sequence of the nucleic acid template. Moreover, EFdA monophosphate (EFdA-MP) is misincorporated by RT, resulting in mismatched primers that are very difficult to extend. Finally, the detailed analysis of the versatile mechanisms of RT inhibition by EFdA provides novel insights into a reduced resistance profile for EFdA because delayed termination seems to protect EFdA-MP-terminated primers from excision.
EXPERIMENTAL PROCEDURES
Enzymes, Nucleic Acids, Nucleotides, and Nucleotide Analogs
HIV-1 RT was expressed and purified as described previously (10, 18–23). RT was expressed in JM-109 cells (Invitrogen) and purified by nickel affinity chromatography and Mono Q anion exchange chromatography (24). Oligonucleotides used in this study were chemically synthesized and purchased from Integrated DNA Technologies (Coralville, IA). Sequences of the DNA substrates are shown in Table 1. Deoxynucleotide triphosphates and dideoxynucleotide triphosphates were purchased from Fermentas (Glen Burnie, MD). Concentrations of nucleotides were calculated spectrophotometrically on the basis of absorption at 260 nm and their extinction coefficients. All nucleotides were treated with inorganic pyrophosphatase (Roche Diagnostics) as described previously (25) to remove traces of PPi contamination that might interfere with the rescue assay.
TABLE 1.
DNA oligonucleotide sequences used in this study
An example of the oligonucleotide abbreviation is as follows: Td31(2T)/Pd18, template DNA of 31 nucleotides in length with a T in the second unpaired position after annealing to the corresponding 18-mer DNA primer. The bases that vary in similar sequences are marked in red. Pd18+5 is a primer that is annealed on a template five nucleotides further than Pd18. In each group of templates, the varying base is indicated inside parentheses: Td31(6G). For the SPR experiments, a thiol-modified DNA primer containing a disulfide on the N2 of a guanosine residue is indicated by X in the sequence. Cy3 or biotin labels are indicated next to the sequences.
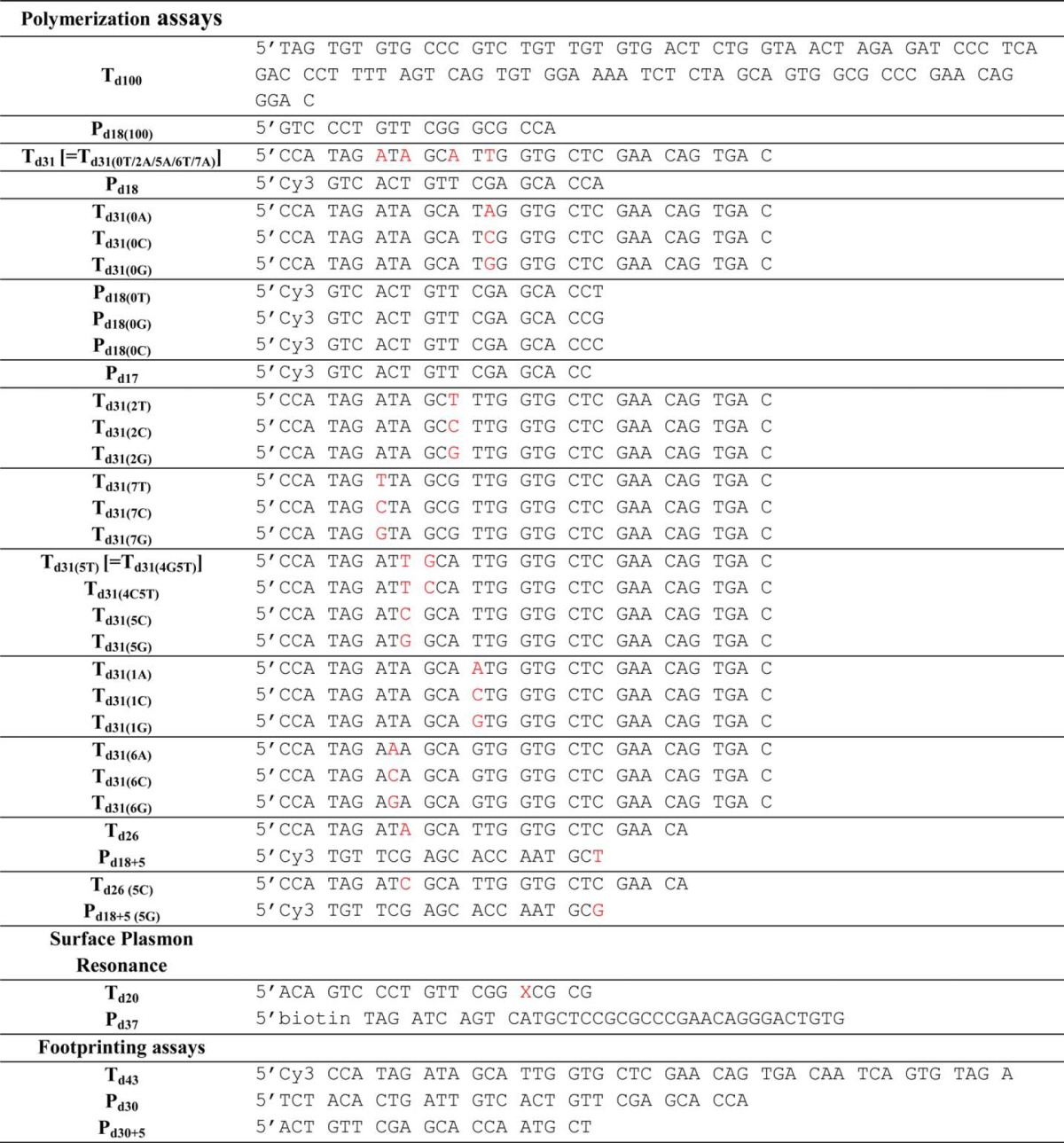
Active Site Titration of HIV-1 RT
To determine polymerization-competent RT populations used in this study, we first carried out active site titration assays using pre-steady state experiments (26, 27). A fixed concentration of RT (50 nm; determined by absorbance measurements) in RT buffer (50 mm NaCl and 50 mm Tris-HCl, pH 7.8) was incubated with increasing concentrations of template/primer (T/P; Td31/Pd18 where Td31 is the DNA template of 31 nucleotides in length annealed to Pd18, which is a DNA primer of 18 nucleotides in length) followed by rapidly mixing with a solution containing 5 mm MgCl2 and 50 μm dATP in the same RT buffer using a rapid quench-flow instrument (RQF-3, KinTek Corp., Austin, TX) at 37 °C for 0.005–1 s. The reactions were quenched by the addition of 150 mm EDTA. The products were resolved on 15% polyacrylamide and 7 m urea gels. In this and in subsequent assays, the gels were scanned with a Typhoon FLA 9000 phosphorimaging system (GE Healthcare), and densitometry analysis was performed using ImageQuant TL software (GE Healthcare). The amounts of extended primer (P) were plotted against time, and the data were fit to a biphasic equation (Equation 1) using GraphPad Prism 4.0 (GraphPad Software Inc., La Jolla, CA) (26),
where A is the amplitude of the burst phase that represents the E-DNA complex at the start of the reaction, kobs is the observed burst rate constant for dNTP incorporation, kss corresponds to the steady state rate constant, and t is the reaction time. Next, the active site concentration and T/P dissociation constant (Kd(DNA)) were determined by plotting the amplitude (A) against T/P concentration. The data were fit by non-linear regression to a quadratic equation,
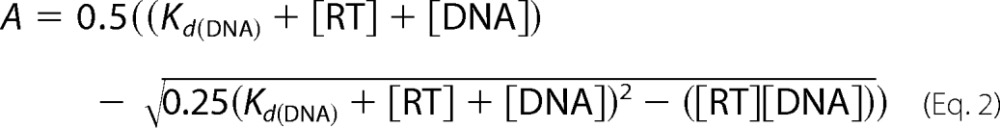 |
where [RT] is the concentration of actively binding polymerase molecules. Subsequent pre-steady state experiments were performed using corrected active site concentrations.
Gel-based Drug Susceptibility Assays: Inhibition of HIV-1 RT-catalyzed DNA Synthesis by EFdA-TP and ddATP
DNA template was annealed to 5′-Cy3-labeled DNA primer at a 3:1 molar ratio. For these experiments, we used Td31/Pd18 and Td26/Pd18+5 where Pd18+5 is an 18-mer DNA primer shifted by five nucleotides relative to the Pd18 primer. Throughout most of our work, we carried out our analyses using two types of primers (in this case, Pd18 and Pd18+5) that allowed us to interrogate two different template sites where RT incorporates dAMP or EFdA-MP at different affinities and different translocation efficiencies. To monitor primer extension, the DNA/DNA hybrid (20 nm) was incubated at 37 °C with RT (20 nm) in RT buffer. Varying amounts of EFdA-TP or ddATP were added, and the reactions were initiated by the addition of 6 mm MgCl2 in a final volume of 20 μl. All dNTPs were present at a final concentration of 10 μm. The reactions were terminated after 15 min by adding an equal volume of 100% formamide containing traces of bromophenol blue. The products were resolved on a 15% polyacrylamide and 7 m urea gel and analyzed as described before (10, 22, 23).
Effect of Template Sequence on RT Inhibition by EFdA-TP
Various T/Ps (oligonucleotide sequences are shown in Table 1) were used to determine the effect of the DNA sequence on the mechanism of inhibition by EFdA-TP. The experiments were carried out as described above.
Surface Plasmon Resonance Assays
Surface plasmon resonance (SPR) was used to determine the kinetic constants of nucleotide triphosphate or EFdA-TP binding to HIV-1 RT. To allow nucleotide binding to the reverse transcriptase, the enzyme was covalently cross-linked to a double-stranded DNA/DNA T/P as described before (24). Briefly, a thiol-modified DNA primer at the N2 position of a guanosine residue (indicated by X in 5′-ACAGTCCCTGTTCGGXCGCG-3′) was heat-annealed to a 5′-biotinylated DNA template (5′-biotin-TAGATCAGTCATGCTCCGCGCCCGAACAGGGACTGTG) at a 3:1 T/P ratio (Table 1). The double-stranded DNA was cross-linked to RT containing mutation Q258C by incubating overnight at 30 °C in a mixture containing 25 mm Tris, pH 7.8, 75 mm NaCl, 10 mm MgCl2, and 100 μm ddGTP. The incorporation of the dideoxynucleotide into the T/P (DNAddGMP) aligns the thiol-modified G with the cysteine 258 of RT and allows disulfide bond formation as we have previously described (24). The covalent RT-DNAddGMP complex was purified by tandem nickel affinity/ion exchange chromatography to remove unreacted DNA and enzyme (28). The purified complex was immobilized on a streptavidin sensor chip on a Biacore T100 system (GE Healthcare). RT-DNAddGMP was flowed over the chip surface of channel 2 until ∼8,000 response units were immobilized through the very strong interactions of the biotinylated RT-DNAddGMP with the streptavidin sensor chip. Channel 1 was left untouched and was later used as a control surface. Increasing concentrations of dATP (1–1,000 nm) or EFdA-TP (5–1,000 nm) were flowed in channels 1 and 2 in RT buffer and 10 mm MgCl2 for 2 min to allow nucleotide association and dissociation. The signal observed in channel 1 (control channel) was subtracted from the signal obtained from the RT-DNAddGMP channel 2. The ddGTP at the 3′-end of the DNA primer prevents incorporation of the dATP or EFdA-TP and allows estimation of their dissociation and association rates, the ratio of which provides the Kd equilibrium binding constant of the binding of the incoming nucleotide triphosphate to the RT-DNAddGMP complex. Optimal analysis of the binding signal was performed using a two-state reaction protocol that assumes a conformational change associated with substrate binding in the Biacore T100 software to obtain measured kinetic constants. Specifically, the overall equilibrium dissociation constant Kd for this type of two-state reaction protocol is defined by (kd1/ka1)·(kd2/(kd2 + ka2)) where ka1 is the association rate constant for substrate binding, kd1 is the dissociation rate constant for substrate from the complex, ka2 is the forward rate constant for the conformational change, and kd2 is the reverse rate constant for the conformational change.
Pre-steady State Kinetics of dATP and EFdA-TP Incorporation
Pre-steady state kinetics analysis was performed using dATP or EFdA-TP single nucleotide incorporation experiments catalyzed by RT. The optimal rate of polymerization (kpol) was determined using the rapid quench-flow method as described before (19, 26, 27, 29). Specifically, a solution containing 50 nm RT and 50 nm Td31/Pd18 or Td26/Pd18+5 in RT buffer was rapidly mixed with a solution of 5 mm MgCl2 and 1–50 μm dATP or EFdA-TP for reaction times ranging between 0.005 and 2 s followed by quenching with 150 mm EDTA. The reaction products were resolved and quantified as described above. The observed burst rates (kobs) at each dATP or EFdA-TP concentration were determined by fitting the data to Equation 1 and solving the equation by non-linear regression using GraphPad Prism 4.0.
To determine the optimal rates of dATP and EFdA-TP incorporation (kpol) and their binding to the enzyme-DNA complex (Kd(dATP) or Kd(EFdA-TP)), observed burst rates (kobs) were fit to the following hyperbolic equation.
All experiments were carried out independently at least two times, and the standard deviations of the kinetic parameters are reported in the respective tables.
Pre-steady State Kinetics of dATP and EFdA-TP Misincorporation
We used pre-steady state kinetics to monitor the misincorporation of dAMP and EFdA-MP opposite an A template base using the Td31(1A)/Pd18 or Td31(6A)/Pd18+5 template/primer, which has an A at the first template overhang position when Pd18 or Pd18+5 was used as the primer, respectively (DNA sequences are shown in Table 1). We used both Pd18 and Pd18+5 primers to interrogate potential differences in misincorporation efficiency at two template sites where substrates are incorporated at different efficiencies. Similarly, we monitored misincorporation of dATP and EFdA-TP opposite G using Td31(1G)/Pd18 or Td31(6G)/Pd18+5 and misincorporation opposite a C using Td31(1C)/Pd18 or Td31(6C)/Pd18+5. In these experiments, a solution containing 30 nm RT and a 20 nm concentration of the corresponding T/P in RT buffer was mixed with a solution of 6 mm MgCl2 and various concentrations of dATP or EFdA-TP for the indicated reaction times followed by quenching with an equal volume of formamide. The reaction products were resolved and quantified as described above. Further analysis was carried out as described for the match pre-steady state kinetics experiments.
Site-specific Fe2+ Footprinting Assays
Site-specific Fe2+ footprints were monitored on 5′-Cy3-labeled DNA templates. 100 nm 5′-Cy3-Td43/Pd30 or 5′-Cy3-Td43/Pd30+5 was incubated with 600 nm RT in a buffer containing 120 mm sodium cacodylate, pH 7.0, 20 mm NaCl, 6 mm MgCl2, and 1 μm EFdA-TP or 5 μm ddATP to allow quantitative chain termination. Prior to treatment with Fe2+, complexes were preincubated for 7 min with increasing concentrations of the next incoming nucleotide (dTTP). The complexes were treated with ammonium iron sulfate (1 mm) as described previously (10, 30). This reaction relies on autoxidation of Fe2+ (31) to create a local concentration of hydroxyl radicals that cleave the DNA at the nucleotide closest to the Fe2+ specifically bound to the RNase H active site. The two types of T/P systems carrying Pd18 and Pd18+5 primers allowed us to interrogate the translocation efficiencies in different nucleic acid sequence contexts.
Extension of EFdA-MP-terminated Primers
Td31/Pd18-EFdA-MP (chemically prepared T/P possessing EFdA-MP at the 3′-end of the primer) and Td26/Pd18+5-EFdA-MP were prepared by incubating 1 μm EFdA-TP, a 1 μm concentration of the corresponding T/P, 1 μm RT, and 6 mm MgCl2 in RT buffer. The reactions were carried out at 37 °C for 1 h. Similarly, for the extension of the mismatched primers, Td31(6C)/Pd18+5-EFdA-MP, Td31(6G)/Pd18+5-EFdA-MP, and Td31(6A)/Pd18+5-EFdA-MP were prepared by incubating 20 μm EFdA-TP, a 1 μm concentration of the corresponding T/P, 2 μm RT, and 6 mm MgCl2 in RT buffer. The reactions were carried out at 37 °C for 4 h. After incorporation of EFdA-MP, the various T/PEFdA-MP substrates were purified using the QIAquick nucleotide removal kit (Qiagen, Valencia, CA). Under these conditions, the extension of T/P to T/PEFdA-MP was complete. Purified Td31/Pd18-EFdA-MP, Td26/Pd18+5-EFdA-MP, Td31(6C)/Pd18+5-EFdA-MP, Td31(6G)/Pd18+5-EFdA-MP, or Td31(6A)/Pd18+5-EFdA-MP (5 nm) was incubated with 20 nm RT in RT buffer and 6 mm MgCl2. The first incoming nucleotide was added at different concentrations (0–50 μm) in the presence of the other dNTPs (1 μm). The reactions were incubated at 37 °C for 5 and 30 min, and products were analyzed as described above.
Pre-steady State Kinetics of dTTP Incorporation to T/PEFdA-MP
Pre-steady state kinetics analysis of dTTP incorporation to T/PEFdA-MP was performed using Td31/Pd18-EFdA-MP, Td26/Pd18+5-EFdA-MP, or Td26(5C)/Pd18+5(5G)-EFdA-MP. In these experiments, a solution containing 30 nm RT and a 20 nm concentration of the corresponding T/P in RT buffer was mixed with a solution of 6 mm MgCl2 and various concentrations of dTTP for 30 s to 10 min followed by quenching with an equal volume of formamide. The reaction products were resolved and quantified as described above. Further analysis was carried out as described for the match pre-steady state kinetics experiments.
PPi- and ATP-dependent Excision and Rescue of T/PEFdA-MP
PPi-dependent Excision of T/PEFdA-MP
Td26/Pd18+5-EFdA-MP-dTMP was prepared similarly to Td26/Pd18+5-EFdA-MP. After EFdA-MP incorporation, 100 μm dTTP was added to the reaction and incubated at 37 °C for an additional 1 h. The purification of the terminated primer was performed as described before. 20 nm purified Td31/Pd18-EFdA-MP or Td26/Pd18+5-EFdA-MP-dTMP was incubated at 37 °C with 60 nm RT in the presence of 150 μm PPi in RT buffer and 6 mm MgCl2. Aliquots of the reaction were stopped at different times (0–30 min) and analyzed as described above.
ATP-dependent Rescue of T/PEFdA-MP
20 nm purified Td31/Pd18-EFdA-MP was incubated with 60 nm RT in the presence of 3.5 mm ATP, 100 μm dATP, 0.5 μm dTTP, and 10 μm ddGTP in RT buffer and 10 mm MgCl2. Similarly, 20 nm purified Td26/Pd18+5-EFdA-MP+dTMP was incubated with 60 nm RT in the presence of 3.5 mm ATP, 100 μm dTTP, and 10 μm ddCTP in RT buffer and 10 mm MgCl2. Reactions were arrested at various time points (0–90 min) and analyzed as described above. Control reactions without dNTPs and ATP (no excision; no incorporation) or without ATP (no excision; incorporation) were incubated for the maximum time (90 min).
PPi-dependent Excision of T/PEFdA-MP in Which EFdA-MP Is Incorporated as a Match or Mismatch
20 nm purified Td31(6X)/Pd18+5-EFdA-MP (where X = T, A, C, or G) was incubated at 37 °C with 60 nm RT in the presence of 150 μm PPi in RT buffer and 6 mm MgCl2. Aliquots of the reaction were stopped at different times (0–20 min) and analyzed as described above.
EFdA and Tenofovir Combination Studies: Inhibition of HIV-1 RT-catalyzed DNA Synthesis and Stopping Patterns by EFdA-TP and TFV-DP
We monitored primer extension using two T/P systems. (a) A shorter T/P, Td31(7C)/Pd18 (20 nm), was incubated at 37 °C with RT (20 nm) in RT buffer. Varying amounts of EFdA-TP (0–8 μm) and tenofovir diphosphate (TFV-DP; 0–2 μm) were added, and the reactions were initiated by the addition of 6 mm MgCl2 in a final volume of 20 μl. All dNTPs were present at a final concentration of 1 μm. The reactions were terminated after 15 min by adding an equal volume of 100% formamide containing traces of bromophenol blue. The products were analyzed as described above. (b) Using a longer template, we compared the stopping patterns of EFdA-TP and TFV-DP. Specifically, Td100/Pd18(100) (50 nm) was incubated at 37 °C with RT (20 nm) in RT buffer, 6 mm MgCl2, 1 μm dNTPs, and 240 nm EFdA-TP or 2 μm TFV-DP. The reactions were terminated after 15 min by adding an equal volume of 100% formamide containing traces of bromophenol blue, and the products were analyzed as described above.
RESULTS
EFdA-TP Inhibits RT with Multiple Mechanisms: EFdA-TP Inhibits RT Primarily as an Effective Immediate or Delayed Chain Terminator (ICT or DCT)
All NRTIs approved for the treatment of HIV infection mimic the natural dNTPs, but because they lack a 3′-OH group, once incorporated into the nascent DNA they act as immediate (or obligate) chain terminators. Primer extension assays show that despite having a 3′-OH EFdA-TP inhibits RT-catalyzed DNA synthesis mainly at the point of incorporation as a de facto ICT, similar to other ICTs such as ddATP (Fig. 1B; strong stops at positions of incorporation P1 and to a lesser extent at P10). For the sake of simplicity, we use the term “immediate chain terminator” in lieu of the more accurate “de facto immediate chain terminator” as EFdA-TP causes a dramatic but not 100% complete suppression of primer extension (eventually some primer extension can be observed at high dNTP concentrations).
However, our data show that EFdA-TP can also inhibit RT as a DCT. Specifically, our primer extension assays revealed additional bands that do not correspond to positions where EFdA-TP or other dATP analogs are expected to be incorporated. Hence, there are strong bands at positions P7 and to a lesser extent at P11, suggesting that EFdA-TP can act as a DCT primarily when incorporated at P6 and to a lesser extent at P10. In these cases after RT incorporates EFdA-MP at the P6 and P10 positions, it continues incorporating the next dNTP before DNA synthesis is stalled. We confirmed these findings using an oligonucleotide substrate with a shorter template but longer primer sequence that allowed examination of EFdA-MP incorporation only at the P6 and P10 sites (Td26/Pd18+5; Fig. 1E). Hence, EFdA-TP acts exclusively as an ICT at P1 and mostly as an ICT at P10, whereas it acts primarily as a DCT at P6 and to a lesser extent at P10 (Fig. 1, B and E). Using hydroxyl radical footprinting assays of RT bound to an EFdA-MP-terminated T/P (T/PEFdA-MP) we demonstrated a correlation of ICT inhibition (such as at P1 site) and a decreased translocation of RT from the nucleotide-binding or pre-translocation site (N-site; cleavage at −18) to the primer-binding or post-translocation site (P-site; cleavage at −17) (Fig. 1C). Therefore, when EFdA-TP inhibits RT as an ICT it acts as a translocation-defective RT inhibitor. In turn, footprinting assays at sites where EFdA acts as a DCT (such as P6) demonstrated that T/PEFdA-MP is translocated to the P-site (Fig. 1C). Collectively, these experiments show that EFdA-TP can act both as an ICT and as a DCT depending on the template sequence and how it allows translocation of RT on T/PEFdA-MP (Figs. 1, 8, and 9).
FIGURE 8.
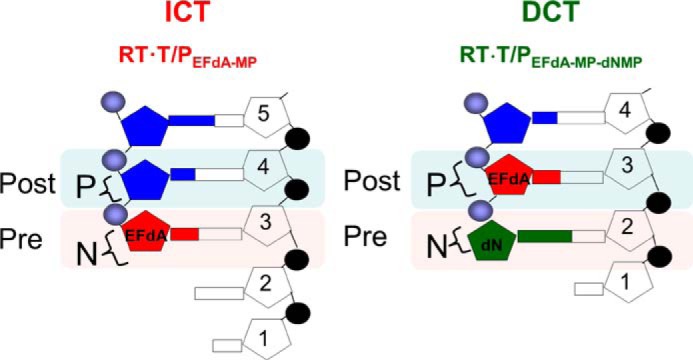
Schematic representation of EFdA-MP-containing complexes. EFdA acts as an ICT by occupying the N-site of RT, whereas it occupies the P-site of RT when it acts as a DCT. In this case, the N-site is free for the next incoming dNMP to be incorporated.
FIGURE 9.
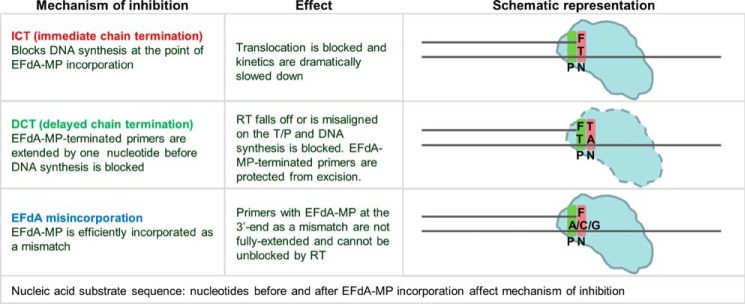
Overview of the multiple mechanisms of RT inhibition by EFdA.
The Mechanism of RT Inhibition by EFdA-TP Is Sequence-dependent
To further explore the effect of the T/P sequence on the EFdA-TP inhibition mechanism, we used a series of oligonucleotides that vary at positions upstream or downstream from the sites of EFdA-MP incorporation (Table 1). As a control we used ddATP, which always acts as a canonical ICT (data not shown).
With regard to the effect of changes before the site of EFdA-MP incorporation, remarkably we found that a single change in the template sequence can change the inhibition mechanism: the presence of C at a template position prior to the P6 site leads to exclusive ICT (Fig. 2A, Td31(5C)/Pd18). In contrast, the presence of A, G, or T at the same template position leads primarily to DCT (Fig. 2A and Table 2; Td31(5A)/Pd18, Td31(5G)/Pd18, and Td31(5T)/Pd18), although the presence of bands at the sites of EFdA incorporation (P6, P6, and P5, respectively) suggested an additional but less pronounced ICT mode of inhibition as well (none of these stopping patterns were observed in the absence of EFdA-TP).
FIGURE 2.

Effect of template sequence on mechanism of inhibition by EFdA-TP. Primer extension assays were performed by incubating various DNA/DNA T/Ps with 20 nm RT for 15 min in the presence of 10 μm dNTPs, 6 mm MgCl2, and increasing concentrations of EFdA-TP. The sequence of the template is shown next to the gels. Twenty templates with A, T, C, or G at the P0, P2, P5, and P7 positions were used. The nucleotides that vary are shown in blue color and indicated with an asterisk next to the gel bands. In red color, we highlight the T positions where EFdA-TP is expected to be incorporated. Immediate and delayed chain terminations are shown in red and green boxes, respectively. Blue color and asterisks highlight the positions of the nucleotides that vary among different gels.
TABLE 2.
Effect of template sequence on mechanism of action of EFdA-TP
The mechanism of inhibition by EFdA-TP was determined by the results in Fig. 2. Changes in the template sequences are indicated in bold.
| T/P | Mechanism of action |
|---|---|
| Effect of template sequence before EFdA-MP incorporation | |
| Td31(0A)/Pd17 | P2: ICT |
| Td31(0T)/Pd17 | P1: ICT; P2: DCT/ICT |
| Td31(0C)/Pd17 | P2: ICT |
| Td31(0G)/Pd17 | P2: ICT |
| Td31(0A)/Pd18 | P1: ICT |
| Td31(0T)/Pd18 | P1: ICT |
| Td31(0C)/Pd18 | P1: ICT |
| Td31(0G)/Pd18 | P1: ICT |
| Td31(5A)/Pd18 | P6: ICT; P7: DCT |
| Td31(5T)/Pd18 | P5: ICT; P6: DCT/ICT |
| Td31(4C5T)/Pd18 | P5: ICT; P6: DCT/ICT |
| Td31(5C)/Pd18 | P6: ICT |
| Td31(5G)/Pd18 | P6: ICT; P7: DCT |
| Effect of template sequence after EFdA-MP incorporation | |
| Td31(2A)/Pd18 | P1: ICT |
| Td31(2T)/Pd18 | P1: ICT; P2: DCT/ICT |
| Td31(2C)/Pd18 | P1: ICT |
| Td31(2G)/Pd18 | P1: ICT |
| Td31(7A)/Pd18 | P6: ICT; P7: DCT |
| Td31(7T)/Pd18 | P6: ICT; P7: DCT/ICT |
| Td31(7C)/Pd18 | P6: ICT; P7: DCT |
| Td31(7G)/Pd18 | P6: ICT; P7: DCT |
Introducing a C before the incorporation site of EFdA-MP does not appear to always result in strong ICT. When we changed the P4 site of Td31(4G5T)/Pd18 to a C (Td31(4C5T)/Pd18), we still observed a primarily DCT mode of inhibition (Fig. 2B), suggesting that sequences before and after the EFdA-MP incorporation site contribute to the preferred mechanism of inhibition.
EFdA-TP acted as an ICT at the P1 site regardless of the type of preceding nucleotide (Fig. 2A, gels in second column). To determine whether this strong preference is due to the upstream (P0) position being provided to the enzyme already in double-stranded DNA form (last primer nucleotide), we repeated the experiments using a shorter primer that stopped one position before the EFdA-MP incorporation site. The data shown in Fig. 2A, column 1, show that there was still ICT inhibition in all cases, whereas only in the case of Td31(0T)/Pd17 was there an apparent mixture of ICT and DCT.
We also tested the effect of template sequence at one position after the P6 or P1 site of EFdA-MP incorporation (P7 and P2 sites in Fig. 2A, columns 4 and 3, respectively). We generally found no significant changes in the type of inhibition mechanism with the exception of Td31(2T)/Pd18, which was now inhibited by both ICT and DCT instead of only ICT. Collectively, these results indicate that the mechanism of inhibition by EFdA can be influenced by the template sequence before and after the site of incorporation.
EFdA-TP Binds RT Tighter than the Natural Substrate dATP
Because EFdA-TP inhibits RT very efficiently, we wanted to examine and compare the binding affinity of the inhibitor and dATP with the enzyme. Therefore, we used SPR to determine the dissociation constant Kd for EFdA-TP and dATP. A two-state reaction protocol that assumes a 1:1 binding of substrate (EFdA-TP or dATP) to an immobilized ligand (RT) followed by a conformational change (“closing” of fingers subdomain) to form a stable complex (32–36) was used to analyze the SPR data. It is also assumed that a substrate in a complex that has undergone a conformational change can only dissociate if the conformational change is first reversed. This analysis generated the following kinetic values: ka1, the association rate constant for substrate binding; kd1, the dissociation rate constant for substrate from the complex; ka2, the forward rate constant for the conformational change; kd2, the reverse rate constant for the conformational change; and Kd, the overall equilibrium dissociation constant, which for this type of two-state reaction protocol is defined by (kd1/ka1)·(kd2/(kd2 + ka2)). Fig. 3 shows that, using the Td20/Pd37, the Kd for dATP is about 10 times higher than that of EFdA-TP. Hence, RT can bind EFdA-TP considerably more tightly than the natural substrate. This difference in the Kd of dATP and EFdA-TP is mostly due to an increased association rate for substrate binding (ka1) and increased forward rate constant for the conformational change (ka2) in the case of EFdA-TP (Fig. 3).
FIGURE 3.

SPR to determine the kinetic constants of EFdA-TP and dATP binding to HIV-1 RT. Nucleotide binding was performed by using RT covalently cross-linked to Td37/Pd20, which has a 5′-biotinylated DNA template and a ddGMP incorporated at the primer. The RT-DNAddGMP complex was immobilized on a streptavidin sensor chip, and increasing concentrations of dATP or EFdA-TP were flowed to allow nucleotide association and dissociation. A two-state reaction protocol was used to analyze the SPR data, which assume a 1:1 binding of substrate (EFdA-TP or dATP) to an immobilized ligand (RT) followed by a conformational change (closing of fingers subdomain) to form a stable complex. The graph shows the association and dissociation of EFdA-TP over time. This analysis generated the following kinetic values: ka1, the association rate constant for substrate binding; kd1, the dissociation rate constant for substrate from the complex; ka2, the forward rate constant for the conformational change; kd2, the reverse rate constant for the conformational change; and Kd, the overall equilibrium dissociation constant, which for this type of two-state reaction protocol is defined by Kd = (kd1/ka1)·(kd2/(kd2 + ka2)). The -fold change of the various constants is shown in parentheses.
RT Incorporates EFdA-TP Very Efficiently by Stalling Full Extension of the Terminated Primer
To further understand the biochemical basis of the strong RT inhibition by EFdA-TP, we compared the incorporation efficiency of EFdA-TP with that of the natural substrate dATP under pre-steady state conditions. Our results show that RT uses EFdA-TP very efficiently (Fig. 4). Interestingly, we demonstrated that depending on the sequence of the template the selectivity for EFdA-TP over the natural dATP substrate ranges from 0.8- to 2.3-fold. Another independent study using a different T/P showed that the incorporation efficiency of EFdA-TP is 2-fold higher than that of dATP (37). The high incorporation efficiency of EFdA-TP is mostly the result of the higher binding affinity of EFdA-TP (lower dissociation constant Kd). These results are consistent with the SPR data that also showed an increased RT binding affinity for EFdA-TP compared with dATP using another T/P sequence.
FIGURE 4.

Pre-steady state kinetics of correct single nucleotide incorporation of EFdA-TP and dATP by HIV-1 RT. Preincubated HIV-1 RT (50 nm) and Td31/Pd18 (A) or Td26/Pd18+5 (50 nm) (B) were mixed by a quench-flow instrument with various concentrations of EFdA-TP or dATP for reaction times varying between 5 ms and 2 s. The amount of product at different reaction times was fit to the burst equation (Equation 1) to obtain burst phase rates of nucleotide incorporation. Observed rate constants were then plotted using a hyperbolic equation (Equation 3) to estimate kpol and Kd(dNTP) (A and B). The sequences of the T/Ps are shown below the plots, and the calculated constants are shown in C. Error bars represent S.D. of at least two independent experiments.
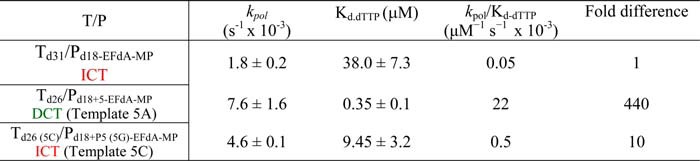
Because EFdA-TP differs from the conventional chain terminators and possesses a 3′-OH group, we examined the possibility that there can be additional DNA polymerization events at the EFdA-MP end of T/PEFdA-MP. Primer extension experiments showed that when EFdA-TP acts as an ICT there is eventually slow full extension of the EFdA-MP-terminated primers (Fig. 5A). When EFdA-TP acts as a DCT, there is extension of the EFdA-MP primers by only one nucleotide under these conditions (Fig. 5B) and very little full extension over the course of hours (not shown). The extension of EFdA-MP-terminated primers has also been reported by Muftuoglu et al. (37) in the context of a different sequence and in the presence of higher dNTP and RT concentrations and is consistent with our findings reported here.
FIGURE 5.

Incorporation of dNTPs into T/PEFdA-MP by HIV-1 RT. A, Td31/Pd18-EFdA-MP (5 nm) was incubated with 20 nm HIV-1 RT, 6 mm MgCl2, 1 μm dATP, dCTP, dGTP, and increasing concentrations of the next correct dNTP (dTTP, 0–50 μm). The reactions were terminated after 15 min. B, Td31/Pd18+5-EFdA-MP (5 nm) was incubated with 20 nm HIV-1 RT, 6 mm MgCl2, 1 μm dATP, dCTP, dGTP, and increasing concentrations of the next correct dNTP (dTTP, 0–50 μm). The reactions were terminated after 5 and 30 min. C–E, various Td31/Pd18+5-EFdA-MP (5 nm) where EFdA-MP is incorporated as a mismatch were incubated with 20 nm HIV-1 RT, 6 mm MgCl2, 1 μm dATP, dCTP, dGTP, and increasing concentrations of the next correct dNTP (dTTP, 0–50 μm). The reactions were terminated after 5 and 30 min. The sequences of the T/Ps are shown below the gels, and the lanes are numbered for clarity.
We used pre-steady state kinetics to quantify and compare the efficiencies of dNTP incorporation in EFdA-MP-terminated primers at ICT (Td31/Pd18-EFdA-MP) versus DCT (Td26/Pd18+5-EFdA-MP) sites. Consistent with the gel-based results of Fig. 5, A and B, there was indeed a >400-fold difference in the extension efficiency of the EFdA-MP-terminated primers when different T/Ps were used (Table 3). Interestingly, when we simply changed a single template base from A to C at one position before the EFdA-MP incorporation site (Table 3), we observed that the kinetics of dNTP incorporation on EFdA-MP-terminated primers were dramatically changed, showing a shift from a DCT to ICT mechanism. Using a single different T/P sequence, Muftuoglu et al. (37) recently showed that RT can incorporate dCTP after EFdA-MP 400 times more efficiently than it does after AMP (∼1.5 versus 0.0034 μm−1·s−1) (38).
TABLE 3.
Pre-steady state kinetic parameters of dTTP incorporation on T/PEFdA-MP by HIV-1 RT
Values were determined from three independent experiments. Positions where EFdA-TP acts as an ICT or DCT are shown in red and green, respectively.
EFdA-TP Is Efficiently Incorporated as a Mismatch but with No Further Extension
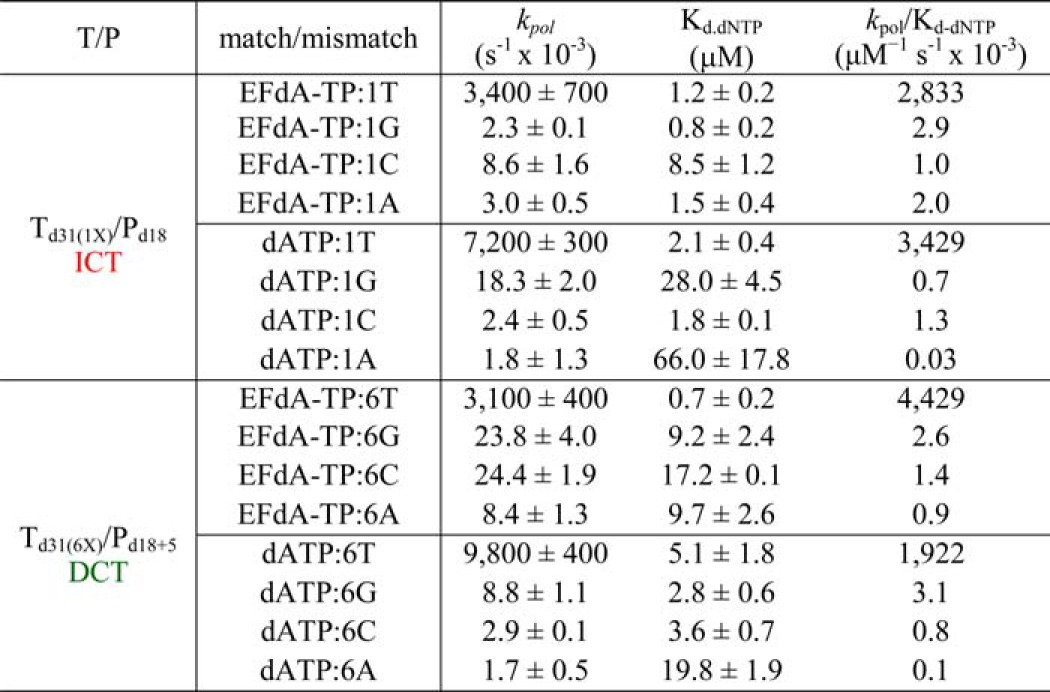
The inhibition and incorporation experiments suggest that EFdA-TP can be used by RT as a substrate for DNA polymerization at levels even better than the natural dATP substrate. Using pre-steady state kinetics, we extended this finding by showing that EFdA-TP can be incorporated very efficiently not only as a cognate substrate but also as a mismatched substrate (Table 4). Specifically, EFdA-TP is incorporated as a mismatch more efficiently against a G than an A or a C. We also found that the template sequence affects the misincorporation efficiency in different ways for EFdA-TP and dATP. Hence, EFdA-MP is misincorporated more efficiently than dAMP at the P1 sites (ICT mechanism) when the mispairing base is opposite G or A, but not C (Table 4). In contrast, the misincorporation efficiencies of EFdA-MP and dAMP are similar at the P6 site (DCT mechanism) opposite G and C, whereas EFdA-MP is misincorporated more efficiently than dAMP opposite A. In addition, we tested the extension of primers terminated with misincorporated EFdA-MP (EFdA-MP opposite a C, A, or G) and found that there is no dNTP addition even at higher dNTP concentrations or incubation times (Fig. 5, C, D, and E) in contrast to the one-nucleotide extension of T/PEFdA-MP with EFdA-MP opposite the cognate base T (Fig. 5B).
TABLE 4.
Pre-steady state kinetic parameters of EFdA-TP and dATP match and mismatch incorporation by HIV-1 RT
Values were determined from three independent experiments. Positions where EFdA-TP acts as an ICT or DCT are shown in red and green, respectively. The different templates vary in position X: G, C, or A.
Delayed Chain Termination Protects EFdA-MP from Excision
One of the major mechanisms of resistance to NRTIs is the ATP-based excision of the chain terminator by RT (39, 40). The product of this excision reaction is a tetraphosphate (NRTI-MP-ATP) and an unblocked primer that RT can use to resume DNA synthesis (41). For excision to occur, the NRTI-MP end of the chain-terminated primer should be positioned at the N-site (42). Our footprinting data in Fig. 1C establish that the translocation state, which reflects the position of RT on DNA, can be affected by the nucleic acid sequence. Specifically, in the absence of dNTP, Td43/Pd30-EFdA is bound by RT in a pre-translocation mode (Fig. 1C, lane 7, left gel), whereas Td43/Pd30+5-EFdA is bound in a post-translocation mode (Fig. 1C, lane 7, right gel). Importantly, these differences in binding lead to differences in the mechanism of EFdA inhibition with immediate chain termination at the site where translocation is not favored (P1 site of Td31/Pd18) or delayed chain termination at the site where translocation is indeed favored (P6 site of Td31/Pd18; Fig. 1, B and C). Because binding in a pre-translocation mode allows excision to occur (24, 28, 30), we hypothesized that the post-translocation bound Td26/Pd18+5-EFdA-MP-dTMP would be protected from excision. To test this hypothesis, we performed ATP- or PPi-based phosphorolysis experiments to unblock the following EFdA-MP-terminated template primers: (a) Td31/Pd18-EFdA-MP, which binds RT in a pre-translocation mode and is the product of ICT, and (b) Td26/Pd18+5-EFdA-MP-dTMP, which binds RT in a post-translocation mode and is the product of DCT with a dTMP incorporated into the EFdA-MP-terminated primer.
We performed the experiments in the presence of either PPi or ATP. In the latter case, the excision reaction was coupled with the reverse DNA polymerization because dNTPs were also added in the reaction (rescue conditions). When EFdA-TP is inserted opposite P1 it acts as an ICT (as in Td31/Pd18-EFdA-MP or Td43/Pd18-EFdA-MP), and the nucleic acid is bound in a pre-translocation binding mode (Fig. 1C). In that case, there is some PPi-based excision of EFdA-MP from the 3′-end of the primer (Fig. 6A, left panel, lanes 2–8), although even in this case it does not appear to be a significant problem because there is facile reincorporation of EFdA-TP as we have shown previously (10). In addition, ATP-based rescue experiments showed that when a high concentration of competing dATP is included in the reaction we can detect rescue of the EFdA-MP-terminated primers (Fig. 6B, lanes 4–11) that are the ICT inhibition product, suggesting that when EFdA-MP is incorporated at an ICT site it is excised even though this phenomenon does not account for high level excision-based resistance as shown by our published virological data (11). In contrast, when EFdA-TP is inserted opposite P6, it acts as a DCT and allows a single incorporation event, which in the case of Td26/Pd18+5-EFdA-MP-dTMP is a dTMP incorporation opposite P7. Under these conditions of DCT, there is a compelling suppression of PPi-based and ATP-based excision as shown in Fig. 6, right panels in A (lanes 10–16) and B (lanes 15–23). Hence, at sites of DCT, EFdA-MP-terminated primers are protected from excision, providing a strong advantage for EFdA-TP against excision-based resistance. Furthermore, mismatched primers that have EFdA-MP at the 3′-end are protected from excision (Fig. 6C, lanes 8–12, 14–18, and 20–24).
FIGURE 6.

PPi- and ATP-dependent unblocking of EFdA-MP-terminated primers. A, purified Td31/Pd18-EFdA-MP or Td26/Pd18+5-EFdA-MP-dTMP was incubated with HIV-1 RT in the presence of 6 mm MgCl2 and 150 μm PPi at 37 °C. Aliquots were removed, and reactions were stopped at the indicated time points (0–30 min). The excision products are shown in braces. B, purified Td31/Pd18-EFdA-MP or Td26/Pd18+5-EFdA-MP-dTMP was incubated at 37 °C with HIV-1 RT in the presence of 10 mm MgCl2, 3.5 mm ATP, and d(d)NTPs (100 μm dATP, 0.5 μm dTTP, and 10 μm ddGTP for Td31/Pd18-EFdA-MP or 100 μm dTTP and 10 μm ddCTP for Td26/Pd18+5-EFdA-MP-dTMP). Aliquots of the reactions were stopped at the indicated time points (0–90 min). C, purified Td31(6X)/Pd18+5-EFdA-MP (where X = T, A, C, or G) was incubated with HIV-1 RT in the presence of 6 mm MgCl2 and 150 μm PPi at 37 °C. Aliquots were removed, and reactions were stopped at the indicated time points (0–20 min). The excision products are shown in braces. The lane numbers are shown below each gel.
EFdA-TP and TFV-DP Do Not Always Compete for the Same Incorporation Sites
Because both EFdA and tenofovir are adenosine analogs, we wanted to see whether they would compete for the same incorporation sites. Surprisingly, when we used the active forms of the two analogs (EFdA-TP and TFV-DP) in parallel reactions at concentrations that resulted in approximately the same amount of primer extension (Fig. 7A, 2 versus lane 3), there were some differences in inhibitor incorporation as judged by stopping patterns. Specifically, although both inhibitors appeared to be incorporated opposite Ts (+6, +8, +10, +17, and +19) (Fig. 7A, lanes 2 and 3), there was a site that was strongly preferred by EFdA for DCT inhibition (+6 and +7 strong stops with no equivalent stops in the uninhibited first lane), whereas there was only moderate inhibition by TFV (weaker stop in the TFV-inhibited third lane). Furthermore, the +6 stop was weaker than the +10 stop for TFV but not for EFdA, providing initial evidence that the two adenosine analogs can have different incorporation efficiencies at different sites on the template. To further explore this possibility, we included both TFV-DP and EFdA-TP in the same reaction and in varying ratios using a shorter Td31(7C)/Pd18 that allowed us to better resolve and monitor the EFdA-MP-terminated versus the TFV-terminated products. In each of the eight panels of Fig. 7B, we used increasing amounts of EFdA-TP in the presence of constant amounts of TFV-DP. Conveniently, we can follow incorporation of EFdA-MP and TFV in the same gels: hence, at the P1 site, the EFdA-MP-terminated primers (Pd18-EFdA-MP) migrate slightly slower than the corresponding TFV-terminated primers (Pd18-TFV) (Fig. 7B, red boxes). At the P6 site, we can compare TFV-based inhibition (Pd18+5-TFV) with the EFdA-based inhibition (Pd18+5-EFdA-MP-dTMP at P7). We found that EFdA can compete with an excess of TFV more efficiently at the P6 versus at the P1 site: for example, in the presence of an excess of TFV (Fig. 7B, TFV = 1,000 nm), ∼50% of chain termination at P6 is caused by ∼31 nm EFdA (Fig. 7B, blue box), but the same ∼50% chain termination at P1 is caused by ∼500 nm EFdA (Fig. 7B, red box). Similar results can be seen at other TFV concentrations in Fig. 7B. Collectively, these data suggest that EFdA-TP and TFV-DP have different inhibition site specificities.
FIGURE 7.

EFdA-TP and TFV-DP have a different stopping pattern and do not always compete for the same incorporation sites. A, using a long template, Td100, we investigated the stopping pattern of EFdA-TP and TFV-DP. At some positions, both analogs appear to have similar incorporation efficiency (same intensity of bands). However, at position +6, EFdA-MP is incorporated more efficiently than TFV, or TFV is not incorporated at all (+7; DCT site). The sequence of template is shown next to the gels. B, Td31(7C)/Pd18 (20 nm) was incubated with 20 nm RT for 15 min in the presence of 1 μm dNTPs, 6 mm MgCl2, and various concentrations of EFdA-TP and TFV-DP. The sites where the inhibitors act as ICT or DCT are shown in red and green, respectively. In red boxes, we highlight some examples of ICT where both analogs are equally incorporated. In the blue box, we highlight the ICT and DCT sites at P6 and P7, respectively, where TFV and EFdA-MP, respectively, are equally incorporated.
DISCUSSION
NRTIs were the first drugs used for the treatment of HIV infection and have always been part of first line antiretroviral therapies (1–8). All approved anti-HIV nucleoside analogs block RT-catalyzed DNA synthesis due to the lack of a 3′-OH group, which is necessary for DNA polymerization. Previously, we have shown that a series of 4′-substituted analogs with a 3′-OH possess anti-HIV activity (9–11). EFdA is the most potent of this series and, to our knowledge, the most potent NRTI with subnanomolar EC50 both in T-cells and peripheral blood mononuclear cells (10, 11, 16). The strong potency of EFdA is the result of its unique mechanism of action as well as its resistance to degradation by adenosine deaminase (15). Previously, we have reported that the inability of EFdA-MP-terminated primers to translocate from the N- to the P-site of HIV-1 RT causes inhibition of DNA polymerization (10). Here we demonstrate that EFdA inhibits RT with multiple mechanisms, which depend on the sequence of the nucleic acid substrate (Figs. 8 and 9).
There is a small number of reports on the effect of T/P sequence on RT inhibition by NRTIs. Scott and co-workers (43) reported that the primer terminus and adjacent upstream base pairs interact with HIV-1 RT in a sequence-dependent manner that affects the unblocking of NRTI-terminated primers. In addition, Hughes and co-workers (44) published that there is a difference in the extent of delayed chain termination by 4′-methyl dNTP analogs depending on the nucleic acid sequence. We report here that, depending on the T/P sequence, EFdA-TP can block RT either as a de facto ICT or as a DCT (Figs. 1, 2, 7, 8, and 9). Although EFdA-TP can inhibit RT both as an ICT and a DCT, it appears that it predominantly inhibits at the point of incorporation (four of five sites in Fig. 7A), causing immediate chain termination and preventing enzyme translocation (Figs. 1, 8, and 9), thus acting as a translocation-defective RT inhibitor (10). Nonetheless, we have identified sites at various template sequences where EFdA-TP can also act as a DCT; it does not block enzyme translocation, allowing the incorporation of one additional dNTP before DNA synthesis is terminated.
What factors may lead to the differences in the mechanism of EFdA inhibition at the P1 (ICT) and P6-P7 (ICT and DCT) sites? We initially examined the effect of inhibitor incorporation efficiency as we anticipated that at sites of efficient EFdA-MP incorporation the EFdA-MP-terminated primer products (T/PEFdA-MP) would be less likely to translocate, leading to immediate chain termination. Surprisingly, although EFdA-MP is better incorporated at the P6 site (Fig. 4), the footprinting experiments showed increased translocation at P6 (Fig. 1C), allowing more access to the dNTP-binding site and incorporation of the next complementary nucleotide (T/PEFdA-MP-dNMP). Further polymerization may be inhibited by unfavorable interactions between the 4′-ethynyl of T/PEFdA-MP-dNMP and RT residues of the DNA-binding cleft.
We subsequently examined the effect of T/P sequence on the inhibition mechanism of EFdA. Our data definitively showed that a change of a single nucleotide can be sufficient to alter the inhibition mechanism from ICT to DCT (Fig. 2A, Td31(5C)/Pd18 versus Td31(5A)/Pd18), although in some cases, it does not have an effect (Fig. 2B, Td31(4G5T)/Pd18 versus Td31(4C5T)/Pd18). Hence, we conclude that the T/P sequence is a major factor that can determine the inhibition mechanism of EFdA. Ongoing structural studies are designed to determine the molecular basis for this specificity.
The ability of EFdA to inhibit RT with multiple mechanisms may have implications for current anti-HIV therapies. We recently showed that the combination of EFdA and TFV had an effect on the inhibition of HIV replication in cell culture (16). This was an unexpected finding as both NRTIs are analogs of dA. A partial explanation is provided in the present study where we show examples of template sites where TFV is not incorporated efficiently (Fig. 7A, lane 3, faint band at template position +6), whereas EFdA-MP very efficiently blocked RT with both ICT and DCT mechanisms (Fig. 7A, lane 2, strong bands at template positions +6 and +7). Differences in the mechanism of activation to their respective active forms may partially be responsible for the lack of competition between EFdA and TFV (16).
Although T/PEFdA-MP is generally ideally located to undergo phosphorolytic excision, with EFdA-MP positioned at the N-site (Fig. 8) (24, 30, 39), excision-based resistance is not a major pathway for resistance to EFdA (11).5 This is likely because of the apparently high reincorporation efficiency of the newly excised EFdA-TP. Furthermore, as shown in Fig. 6, when EFdA-MP is incorporated as a DCT, T/PEFdA-MP-dTMP is protected from excision, resulting in decreased drug resistance by the excision mechanism. Moreover, we did not observe excision of EFdA-MP-terminated primers in positions where EFdA-MP is incorporated as a mismatch (Fig. 6C). These data are consistent with our earlier cell-based data demonstrating that EFdA maintains its potency against HIV strains that carry excision-based zidovudine resistance mutations (11).
There are two nucleos(t)ide analog drugs that retain a 3′-OH and have been approved for the treatment of viral infections: entecavir, which is the most potent anti-hepatitis B virus nucleoside analog drug (45, 46), and sofosbuvir, which is a recently approved anti-hepatitis C virus nucleotide analog drug (47, 48). So far, there are no NRTIs with a 3′-OH group that are approved for the treatment of HIV infections. EFdA could be paired with approved anti-HIV drugs, leading to synergistic and additive effects as shown in cell-based assays, and it also suppressed viral loads in macaques to undetectable levels within a week of treatment and without any signs of toxicity (9, 11, 16, 17, 44). Its ability to inhibit RT by multiple mechanisms imparts excellent antiviral activity (11) and may contribute to its favorable resistance profile (49), thus rendering it a potentially promising new generation NRTI.
This work was supported, in whole or in part, by National Institutes of Health Grants R01AI076119, R01AI076119-S1, R01AI076119-02S1, R01AI099284, R01AI100890, R21AI112417, and P01GM103368 (to S. G. S.) and AI079801 (to M. A. P.). This work was also supported by Mizzou Advantage and the Ministry of Knowledge and Economy, Bilateral International Collaborative Research and Development Program, Republic of Korea. Co-authors Drs. Hiroaki Mitsuya and Eiichi N. Kodama are inventors of EFdA, and thus there is a potential conflict of interest.
B. Marchand, X. Tu, K. Kirby, E. Michailidis, O. Ihenacho, E. Kodama, H. Mitsuya, M. Parniak, and S. Sarafianos, unpublished data.
- HIV-1
- human immunodeficiency virus type 1
- NRTI
- nucleoside RT inhibitor
- EFdA
- 4′-ethynyl-2-fluoro-2′-deoxyadenosine
- TFV
- tenofovir
- MP
- monophosphate
- DP
- diphosphate
- TP
- triphosphate
- T/P
- template/primer
- ICT
- immediate chain terminator
- DCT
- delayed chain terminator
- SPR
- surface plasmon resonance
- N-site
- nucleotide-binding or pre-translocation site
- P-site
- primer-binding or post-translocation site.
REFERENCES
- 1. Hammer S. M., Saag M. S., Schechter M., Montaner J. S., Schooley R. T., Jacobsen D. M., Thompson M. A., Carpenter C. C., Fischl M. A., Gazzard B. G., Gatell J. M., Hirsch M. S., Katzenstein D. A., Richman D. D., Vella S., Yeni P. G., Volberding P. A. (2006) Treatment for adult HIV infection: 2006 recommendations of the International AIDS Society–U.S.A. panel. Top. HIV Med. 14, 827–843 [PubMed] [Google Scholar]
- 2. Schinazi R. F., Hernandez-Santiago B. I., Hurwitz S. J. (2006) Pharmacology of current and promising nucleosides for the treatment of human immunodeficiency viruses. Antiviral Res. 71, 322–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parniak M. A., Sluis-Cremer N. (2000) Inhibitors of HIV-1 reverse transcriptase. Adv. Pharmacol. 49, 67–109 [DOI] [PubMed] [Google Scholar]
- 4. De Clercq E. (2007) Anti-HIV drugs. Verh. K. Acad. Geneeskd. Belg. 69, 81–104 [PubMed] [Google Scholar]
- 5. Sluis-Cremer N., Tachedjian G. (2008) Mechanisms of inhibition of HIV replication by non-nucleoside reverse transcriptase inhibitors. Virus Res. 134, 147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deval J. (2009) Antimicrobial strategies: inhibition of viral polymerases by 3′-hydroxyl nucleosides. Drugs 69, 151–166 [DOI] [PubMed] [Google Scholar]
- 7. Sharma P. L., Nurpeisov V., Hernandez-Santiago B., Beltran T., Schinazi R. F. (2004) Nucleoside inhibitors of human immunodeficiency virus type 1 reverse transcriptase. Curr. Top. Med. Chem. 4, 895–919 [DOI] [PubMed] [Google Scholar]
- 8. Sarafianos S. G., Marchand B., Das K., Himmel D. M., Parniak M. A., Hughes S. H., Arnold E. (2009) Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 385, 693–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kodama E. I., Kohgo S., Kitano K., Machida H., Gatanaga H., Shigeta S., Matsuoka M., Ohrui H., Mitsuya H. (2001) 4′-Ethynyl nucleoside analogs: potent inhibitors of multidrug-resistant human immunodeficiency virus variants in vitro. Antimicrob. Agents Chemother. 45, 1539–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Michailidis E., Marchand B., Kodama E. N., Singh K., Matsuoka M., Kirby K. A., Ryan E. M., Sawani A. M., Nagy E., Ashida N., Mitsuya H., Parniak M. A., Sarafianos S. G. (2009) Mechanism of inhibition of HIV-1 reverse transcriptase by 4′-ethynyl-2-fluoro-2′-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J. Biol. Chem. 284, 35681–35691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kawamoto A., Kodama E., Sarafianos S. G., Sakagami Y., Kohgo S., Kitano K., Ashida N., Iwai Y., Hayakawa H., Nakata H., Mitsuya H., Arnold E., Matsuoka M. (2008) 2′-Deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int. J. Biochem. Cell Biol. 40, 2410–2420 [DOI] [PubMed] [Google Scholar]
- 12. Ohrui H. (2006) 2′-Deoxy-4′-C-ethynyl-2-fluoroadenosine, a nucleoside reverse transcriptase inhibitor, is highly potent against all human immunodeficiency viruses type 1 and has low toxicity. Chem. Rec. 6, 133–143 [DOI] [PubMed] [Google Scholar]
- 13. Kageyama M., Nagasawa T., Yoshida M., Ohrui H., Kuwahara S. (2011) Enantioselective total synthesis of the potent anti-HIV nucleoside EFdA. Org. Lett. 13, 5264–5266 [DOI] [PubMed] [Google Scholar]
- 14. Gallois-Montbrun S., Schneider B., Chen Y., Giacomoni-Fernandes V., Mulard L., Morera S., Janin J., Deville-Bonne D., Veron M. (2002) Improving nucleoside diphosphate kinase for antiviral nucleotide analogs activation. J. Biol. Chem. 277, 39953–39959 [DOI] [PubMed] [Google Scholar]
- 15. Kirby K. A., Michailidis E., Fetterly T. L., Steinbach M. A., Singh K., Marchand B., Leslie M. D., Hagedorn A. N., Kodama E. N., Marquez V. E., Hughes S. H., Mitsuya H., Parniak M. A., Sarafianos S. G. (2013) Effects of 4′- and 2-substitutions on the bioactivity of 4′-ethynyl-2-fluoro-2′-deoxyadenosine. Antimicrob. Agents Chemother. 57, 6254–6264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hachiya A., Reeve A. B., Marchand B., Michailidis E., Ong Y. T., Kirby K. A., Leslie M. D., Oka S., Kodama E. N., Rohan L. C., Mitsuya H., Parniak M. A., Sarafianos S. G. (2013) Evaluation of combinations of 4′-ethynyl-2-fluoro-2′-deoxyadenosine with clinically used antiretroviral drugs. Antimicrob. Agents Chemother. 57, 4554–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murphey-Corb M., Rajakumar P., Michael H., Nyaundi J., Didier P. J., Reeve A. B., Mitsuya H., Sarafianos S. G., Parniak M. A. (2012) Response of simian immunodeficiency virus to the novel nucleoside reverse transcriptase inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine in vitro and in vivo. Antimicrob. Agents Chemother. 56, 4707–4712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bauman J. D., Das K., Ho W. C., Baweja M., Himmel D. M., Clark A. D., Jr., Oren D. A., Boyer P. L., Hughes S. H., Shatkin A. J., Arnold E. (2008) Crystal engineering of HIV-1 reverse transcriptase for structure-based drug design. Nucleic Acids Res. 36, 5083–5092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schuckmann M. M., Marchand B., Hachiya A., Kodama E. N., Kirby K. A., Singh K., Sarafianos S. G. (2010) The N348I mutation at the connection subdomain of HIV-1 reverse transcriptase decreases binding to nevirapine. J. Biol. Chem. 285, 38700–38709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ndongwe T. P., Adedeji A. O., Michailidis E., Ong Y. T., Hachiya A., Marchand B., Ryan E. M., Rai D. K., Kirby K. A., Whatley A. S., Burke D. H., Johnson M., Ding S., Zheng Y. M., Liu S. L., Kodama E., Delviks-Frankenberry K. A., Pathak V. K., Mitsuya H., Parniak M. A., Singh K., Sarafianos S. G. (2012) Biochemical, inhibition and inhibitor resistance studies of xenotropic murine leukemia virus-related virus reverse transcriptase. Nucleic Acids Res. 40, 345–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kirby K. A., Marchand B., Ong Y. T., Ndongwe T. P., Hachiya A., Michailidis E., Leslie M. D., Sietsema D. V., Fetterly T. L., Dorst C. A., Singh K., Wang Z., Parniak M. A., Sarafianos S. G. (2012) Structural and inhibition studies of the RNase H function of xenotropic murine leukemia virus-related virus reverse transcriptase. Antimicrob. Agents Chemother. 56, 2048–2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Michailidis E., Ryan E. M., Hachiya A., Kirby K. A., Marchand B., Leslie M. D., Huber A. D., Ong Y. T., Jackson J. C., Singh K., Kodama E. N., Mitsuya H., Parniak M. A., Sarafianos S. G. (2013) Hypersusceptibility mechanism of tenofovir-resistant HIV to EFdA. Retrovirology 10, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Michailidis E., Singh K., Ryan E. M., Hachiya A., Ong Y. T., Kirby K. A., Marchand B., Kodama E. N., Mitsuya H., Parniak M. A., Sarafianos S. G. (2012) Effect of translocation defective reverse transcriptase inhibitors on the activity of N348I, a connection subdomain drug resistant HIV-1 reverse transcriptase mutant. Cell. Mol. Biol. 58, 187–195 [PMC free article] [PubMed] [Google Scholar]
- 24. Sarafianos S. G., Clark A. D., Jr., Tuske S., Squire C. J., Das K., Sheng D., Ilankumaran P., Ramesha A. R., Kroth H., Sayer J. M., Jerina D. M., Boyer P. L., Hughes S. H., Arnold E. (2003) Trapping HIV-1 reverse transcriptase before and after translocation on DNA. J. Biol. Chem. 278, 16280–16288 [DOI] [PubMed] [Google Scholar]
- 25. Meyer P. R., Matsuura S. E., So A. G., Scott W. A. (1998) Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 95, 13471–13476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh K., Marchand B., Rai D. K., Sharma B., Michailidis E., Ryan E. M., Matzek K. B., Leslie M. D., Hagedorn A. N., Li Z., Norden P. R., Hachiya A., Parniak M. A., Xu H. T., Wainberg M. A., Sarafianos S. G. (2012) Biochemical mechanism of HIV-1 resistance to rilpivirine. J. Biol. Chem. 45, 38110–38123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson K. A. (1993) Conformational coupling in DNA polymerase fidelity. Annu. Rev. Biochem. 62, 685–713 [DOI] [PubMed] [Google Scholar]
- 28. Sarafianos S. G., Clark A. D., Jr., Das K., Tuske S., Birktoft J. J., Ilankumaran P., Ramesha A. R., Sayer J. M., Jerina D. M., Boyer P. L., Hughes S. H., Arnold E. (2002) Structures of HIV-1 reverse transcriptase with pre- and post-translocation AZTMP-terminated DNA. EMBO J. 21, 6614–6624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hachiya A., Kodama E. N., Schuckmann M. M., Kirby K. A., Michailidis E., Sakagami Y., Oka S., Singh K., Sarafianos S. G. (2011) K70Q adds high-level tenofovir resistance to “Q151M complex” HIV reverse transcriptase through the enhanced discrimination mechanism. PLoS One 6, e16242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marchand B., Gotte M. (2003) Site-specific footprinting reveals differences in the translocation status of HIV-1 reverse transcriptase. Implications for polymerase translocation and drug resistance. J. Biol. Chem. 278, 35362–35372 [DOI] [PubMed] [Google Scholar]
- 31. Biaglow J. E., Kachur A. V. (1997) The generation of hydroxyl radicals in the reaction of molecular oxygen with polyphosphate complexes of ferrous ion. Radiat. Res. 148, 181–187 [PubMed] [Google Scholar]
- 32. Hu W. S., Hughes S. H. (2012) HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2, a006882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang H., Chopra R., Verdine G. L., Harrison S. C. (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282, 1669–1675 [DOI] [PubMed] [Google Scholar]
- 34. Tuske S., Sarafianos S. G., Clark A. D., Jr., Ding J., Naeger L. K., White K. L., Miller M. D., Gibbs C. S., Boyer P. L., Clark P., Wang G., Gaffney B. L., Jones R. A., Jerina D. M., Hughes S. H., Arnold E. (2004) Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat. Struct. Mol. Biol. 11, 469–474 [DOI] [PubMed] [Google Scholar]
- 35. Das K., Bandwar R. P., White K. L., Feng J. Y., Sarafianos S. G., Tuske S., Tu X., Clark A. D., Jr., Boyer P. L., Hou X., Gaffney B. L., Jones R. A., Miller M. D., Hughes S. H., Arnold E. (2009) Structural basis for the role of the K65R mutation in HIV-1 reverse transcriptase polymerization, excision antagonism, and tenofovir resistance. J. Biol. Chem. 284, 35092–35100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lansdon E. B., Samuel D., Lagpacan L., Brendza K. M., White K. L., Hung M., Liu X., Boojamra C. G., Mackman R. L., Cihlar T., Ray A. S., McGrath M. E., Swaminathan S. (2010) Visualizing the molecular interactions of a nucleotide analog, GS-9148, with HIV-1 reverse transcriptase-DNA complex. J. Mol. Biol. 397, 967–978 [DOI] [PubMed] [Google Scholar]
- 37. Muftuoglu Y., Sohl C. D., Mislak A. C., Mitsuya H., Sarafianos S. G., Anderson K. S. (2014) Probing the molecular mechanism of action of the HIV-1 reverse transcriptase inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) using pre-steady-state kinetics. Antiviral Res. 106, 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim J., Roberts A., Yuan H., Xiong Y., Anderson K. S. (2012) Nucleocapsid protein annealing of a primer-template enhances (+)-strand DNA synthesis and fidelity by HIV-1 reverse transcriptase. J. Mol. Biol. 415, 866–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arion D., Kaushik N., McCormick S., Borkow G., Parniak M. A. (1998) Phenotypic mechanism of HIV-1 resistance to 3′-azido-3′-deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry 37, 15908–15917 [DOI] [PubMed] [Google Scholar]
- 40. Meyer P. R., Matsuura S. E., Mian A. M., So A. G., Scott W. A. (1999) A mechanism of AZT resistance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell 4, 35–43 [DOI] [PubMed] [Google Scholar]
- 41. Sarafianos S. G., Hughes S. H., Arnold E. (2004) Designing anti-AIDS drugs targeting the major mechanism of HIV-1 RT resistance to nucleoside analog drugs. Int. J. Biochem. Cell Biol. 36, 1706–1715 [DOI] [PubMed] [Google Scholar]
- 42. Boyer P. L., Sarafianos S. G., Arnold E., Hughes S. H. (2001) Selective excision of AZTMP by drug-resistant human immunodeficiency virus reverse transcriptase. J. Virol. 75, 4832–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meyer P. R., Smith A. J., Matsuura S. E., Scott W. A. (2004) Effects of primer-template sequence on ATP-dependent removal of chain-terminating nucleotide analogues by HIV-1 reverse transcriptase. J. Biol. Chem. 279, 45389–45398 [DOI] [PubMed] [Google Scholar]
- 44. Vu B. C., Boyer P. L., Siddiqui M. A., Marquez V. E., Hughes S. H. (2011) 4′-C-Methyl-2′-deoxyadenosine and 4′-C-ethyl-2′-deoxyadenosine inhibit HIV-1 replication. Antimicrob. Agents Chemother. 55, 2379–2389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Woo G., Tomlinson G., Nishikawa Y., Kowgier M., Sherman M., Wong D. K., Pham B., Ungar W. J., Einarson T. R., Heathcote E. J., Krahn M. (2010) Tenofovir and entecavir are the most effective antiviral agents for chronic hepatitis B: a systematic review and Bayesian meta-analyses. Gastroenterology 139, 1218–1229 [DOI] [PubMed] [Google Scholar]
- 46. Michailidis E., Kirby K. A., Hachiya A., Yoo W., Hong S. P., Kim S. O., Folk W. R., Sarafianos S. G. (2012) Antiviral therapies: focus on hepatitis B reverse transcriptase. Int. J. Biochem. Cell Biol. 44, 1060–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lawitz E., Mangia A., Wyles D., Rodriguez-Torres M., Hassanein T., Gordon S. C., Schultz M., Davis M. N., Kayali Z., Reddy K. R., Jacobson I. M., Kowdley K. V., Nyberg L., Subramanian G. M., Hyland R. H., Arterburn S., Jiang D., McNally J., Brainard D., Symonds W. T., McHutchison J. G., Sheikh A. M., Younossi Z., Gane E. J. (2013) Sofosbuvir for previously untreated chronic hepatitis C infection. N. Engl. J. Med. 368, 1878–1887 [DOI] [PubMed] [Google Scholar]
- 48. Gentile I., Borgia F., Buonomo A. R., Castaldo G., Borgia G. (2013) A novel promising therapeutic option against hepatitis C virus: an oral nucleotide NS5B polymerase inhibitor sofosbuvir. Curr. Med. Chem. 20, 3733–3742 [DOI] [PubMed] [Google Scholar]
- 49. Maeda K., Desai D. V., Aoki M., Nakata H., Kodama E. N., Mitsuya H. (2014) Delayed emergence of HIV-1 variants resistant to 4′-ethynyl-2-fluoro-2′-deoxyadenosine: comparative sequential passage study with lamivudine, tenofovir, emtricitabine and BMS-986001. Antiviral Ther. 19, 179–189 [DOI] [PubMed] [Google Scholar]