

Background: Apelin receptor represents a therapeutic target for cardiovascular diseases.
Results: Apelin 17 activates ERK1/2 in a β-arrestin-dependent and G protein-dependent manner, whereas apelin 17 with deleted C-terminal phenylalanine only signals through the G protein.
Conclusion: Biased signaling promoted by an apelin fragment lacking the C-terminal phenylalanine is favoring Gi over β-arrestin.
Significance: Apelin receptor β-arrestin signaling may account for apelin hypotensive activity.
Keywords: Arrestin, Cell Signaling, Extracellular Signal-regulated Kinase (ERK), G Protein, G Protein-coupled Receptor (GPCR), Mitogen-activated Protein Kinase (MAPK), Pertussis Toxin, Apelin, Apelin Receptor
Abstract
Apelin plays a prominent role in body fluid and cardiovascular homeostasis. We previously showed that the C-terminal Phe of apelin 17 (K17F) is crucial for triggering apelin receptor internalization and decreasing blood pressure (BP) but is not required for apelin binding or Gi protein coupling. Based on these findings, we hypothesized that the important role of the C-terminal Phe in BP decrease may be as a Gi-independent but β-arrestin-dependent signaling pathway that could involve MAPKs. For this purpose, we have used apelin fragments K17F and K16P (K17F with the C-terminal Phe deleted), which exhibit opposite profiles on apelin receptor internalization and BP. Using BRET-based biosensors, we showed that whereas K17F activates Gi and promotes β-arrestin recruitment to the receptor, K16P had a much reduced ability to promote β-arrestin recruitment while maintaining its Gi activating property, revealing the biased agonist character of K16P. We further show that both β-arrestin recruitment and apelin receptor internalization contribute to the K17F-stimulated ERK1/2 activity, whereas the K16P-promoted ERK1/2 activity is entirely Gi-dependent. In addition to providing new insights on the structural basis underlying the functional selectivity of apelin peptides, our study indicates that the β-arrestin-dependent ERK1/2 activation and not the Gi-dependent signaling may participate in K17F-induced BP decrease.
Introduction
Apelin is a neuro-vasoactive peptide initially isolated from bovine stomach extracts (1) and was identified as the endogenous ligand of the human orphan receptor APJ (putative receptor protein related to the angiotensin II receptor type 1, AT1), which shares 31% sequence identity with the AT1 receptor (2). This receptor is 380 amino acids long and is a class A member of the seven transmembrane-domain G protein-coupled receptor (GPCR)3 family. It has also been cloned from mice (3) and rats (4, 5).
Apelin is a 36-amino acid peptide derived from a 77-amino acid precursor, preproapelin (1, 6, 7). The alignment of preproapelin amino acids from mammalian species demonstrates a fully conserved C-terminal 17-amino acid sequence, apelin 17 (Lys-Phe-Arg-Arg-Gln-Arg-Pro-Arg-Leu-Ser-His-Lys-Gly-Pro-Met-Pro-Phe, K17F), including the peptide encompassing the last C-terminal 13 amino acids, apelin 13. The N-terminal glutamine residue of apelin 13 is pyroglutamylated, producing the pyroglutamyl form of apelin 13 (pGlu-Arg-Pro-Arg-Leu-Ser-His-Lys-Gly-Pro-Met-Pro-Phe, pE13F) (1, 6, 7). K17F and pE13F are the predominant molecular forms of apelin found in rat brain and in rat and human plasma (8, 9). They exhibit high affinity (subnanomolar) for the human and the rat apelin receptors (ApelinR), although K17F has a 10-fold higher affinity than pE13F for both receptors (10–12). They similarly inhibit forskolin-induced cAMP production in eukaryotic cells stably expressing either the human (6, 11) or the rat ApelinR (4, 10). Both peptides promote phosphorylation of ERKs, Akt, and p70 S6 kinase (13) and are highly potent inducers of ApelinR internalization in a clathrin-dependent manner (14–16). It is important to underline that K17F induces internalization of rat apelin receptor more efficiently than pE13F by a factor of 30 (14).
Apelin and its receptor are both widely distributed in the rat brain (4, 17–19), where they colocalize with arginine vasopressin (AVP) in magnocellular neurons (9, 18, 19). Central injection of K17F in lactating rats inhibits the phasic electrical activity of AVP neurons, thereby decreasing AVP release into the bloodstream and thus increasing aqueous diuresis (9). Moreover, plasma AVP and apelin levels are conversely regulated by osmotic and volemic stimuli in humans and rodents to maintain body fluid homeostasis (8, 9, 19, 20).
The apelinergic system is also present in the cardiovascular system (21), and several studies suggest a role for apelin in the control of cardiovascular functions. Apelin increases the contractile force of the myocardium (22–24), and apelin gene-deficient mice subjected to chronic pressure overload developed severe and progressive heart failure (25). Moreover, intravenous injection of K17F or pE13F in rodents decreases arterial blood pressure (BP) and increases heart rate (7, 14, 16) via a NO production (26) with a greater effect for K17F than for pE13F (14). In contrast, the deletion of the C-terminal phenylalanine in K17F (K16P) or its substitution by an alanine in pE13F (pE13A) suppresses the ability of these peptides to decrease BP (14, 27), demonstrating the central role of the C-terminal Phe for the apelin-induced BP decrease.
We previously showed that the deletion or Ala substitution of the C-terminal Phe in K17F does not modify its ability to bind ApelinR or to inhibit forskolin-induced cAMP production (14, 28), indicating that the C-terminal Phe of apelin does not play a major role in apelin binding to the receptor or its capacity to activate Gi. In contrast, the ability of K17A and K16P to induce ApelinR internalization is drastically reduced when compared with K17F, suggesting an important role for the C-terminal Phe of apelin in agonist-promoted receptor internalization (14, 28).
To understand how the C-terminal Phe of K17F promotes ApelinR internalization, we built a three-dimensional model of the human ApelinR by homology using the validated cholecystokinin-8 receptor type 1 three-dimensional model as a template, and we subsequently docked pE13F or K17F into this model. We identified an hydrophobic cavity at the bottom of the binding pocket in which the C-terminal Phe of pE13F or K17F is embedded by Trp-152 in transmembrane IV and Trp-259 and Phe-255 in transmembrane VI. Site-directed mutagenesis revealed that Phe-255 and Trp-259 are required to trigger ApelinR internalization without playing a role in apelin binding nor in Gi protein coupling (28).
The functional dissociation between Gi protein coupling and receptor internalization suggests that ApelinR exists in different active conformations depending on the bound ligand leading to the activation of different signaling pathways, which is in line with the concept of biased agonists (29, 30). Such agonists stabilize distinct receptor conformations that differ in their signaling partner preference, leading to different biological responses (29, 31–33).
In the last decade, it has become clear that, in addition to their canonical G protein- and second messenger-dependent pathways, GPCRs also use alternative signaling pathways independently of their coupling to G proteins. One of them involves β-arrestins 1 and 2 (also named arrestin 2 and 3), a small family of cytosolic proteins known for their role in GPCR desensitization and internalization (34). Several studies have shown that, in addition to their classical role, β-arrestins can also act as signaling scaffolds for many signaling pathways, and in particular those of MAPKs (34), which lead to a second wave of signal transduction (35). Based on these findings, we hypothesized that the important role of the C-terminal Phe of apelin in its hypotensive action may result from a Gi-independent but β-arrestin-dependent signaling pathway. To test this hypothesis, we took advantage of the apelin fragments K17F and K16P, which exhibit opposite profiles on ApelinR internalization and BP. Using bioluminescence resonance energy transfer (BRET)-based biosensors, we first compared the capacity of K17F or K16P to activate Gαi and promote β-arrestin recruitment. We then investigated the ability of K17F and K16P to activate p42/44 MAPK (ERK) in CHO cells stably expressing the rat ApelinR in the absence or presence of pertussis toxin (PTX), a Gi protein inhibitor, or in the absence or presence of a mutant of β-arrestin (β-arrestin-2-K296A) that prevents the scaffolding of ERK1/2. Finally, we confirmed the involvement of β-arrestin-dependent ERK signaling pathway by using a mutated ApelinR previously shown to be unable to internalize upon K17F stimulation (28).
EXPERIMENTAL PROCEDURES
Chemicals were obtained from Sigma-Aldrich unless specified otherwise. The K16P- and K17F-apelin was synthetized by GL Biochem (Shanghai, China). Pertussis toxin was purchased from Sigma. The anti-phospho-p44/42 MAPK (ERK1/2) and anti-MAPK (3A7) antibodies were obtained from Cell Signaling Technology (Beverly, MA).
Animals
All procedures involving animals were carried out in accordance with institutional guidelines for the care and use of laboratory animals. Male Sprague-Dawley rats of 130–180 g (Charles River Breeding Laboratories, L'Arbresle, France) were used. They were fed a normal standard diet and offered water ad libitum.
Microdissection and Measurement of the Glomerular Arterioles Diameter
Rats were anesthetized by intraperitoneal injection of pentobarbital (60 mg/kg). The left kidney was prepared for microdissection of arterioles as previously described (36). Afferent arterioles were microdissected attached to the glomeruli. Sequential photographies were recorded on the same arteriole with a digital camera (Coolpix 5400; Nikon) under three experimental conditions at 1.5-min intervals: basal condition, 10−9 m Ang II and 10−9 m Ang II + 10−7 m K17F or + 10−7 m K16P. Diameters were measured on a distance equal to 20, 60, and 100 μm upstream of the glomerulus with ImageJ 1.43u, and the average diameter was calculated. Calibration was made using a stage micrometer.
Constructs
The wild-type and mutated (F255A) rat ApelinRs tagged at their C-terminal part with YFP were constructed by subcloning the open reading frame of the wild-type or F255A ApelinR from previous constructs encoding the wild-type or F255A rat ApelinR tagged at its C-terminal part with EGFP (ApelinR-EGFP) (4, 28) to pYFP-N1 (Clontech) using HindIII-BamHI restriction sites. C terminus HA-tagged wild-type and F255A-ApelinR were made by PCR using as template wild-type and F255/A-ApelinR-EGFP constructs and 5′-CATTGACGCAAATGGGCGGTAGGCGTG-3′ and 5′-GGATCCTAAGCGTAATCGGGCACATCGTAAGGGTAAGCGTAATCGGGCACATCGTAAGGGTAGTCCACAAGGGTTTCTTGGCTATAG-3′ as forward primer and reverse primer, respectively. The PCR products were then digested by HindIII-BamHI restriction enzymes (New England Biolabs) and cloned into pcDNA3 (Invitrogen). Wild-type rat β-arrestin 2 fused at its C terminus with DsRed was constructed by amplifying rat β-arrestin2 cDNA by PCR using 5′-CTAGCTAGCACCATGGGTGAAAAACCCGGGAC-3′ and 5′-CCCAAGCTTGCAGAACTGGTCATCACAGTC-3′ as forward and reverse primers, respectively, and by subcloning the PCR product into pDsRed-monomer-N1 (Clontech) using NheI-HindIII restriction sites. K296/A rat β-arrestin 2-DsRed was made by performing a first PCR to generate a megaprimer using 5′-TGGGCAACTCGCGCACGAAGACA-3′ (underlined nucleotides encode the alanine residue replacing the natural lysine residue) and 5′-CCCAAGCTTGCAGAACTGGTCATCACAGTC-3′ as forward and reverse primers, respectively. A second PCR was then performed using the product of the first PCR as a megaprimer (reverse primer) and the 5′-CTAGCTAGCACCATGGGTGAAAAACCCGGGAC-3′ as a forward primer. The sequences of all constructs were checked by sequencing.
The Gαi-RlucII was previously described by Breton et al. (37). Gβ1 and Gγ5 coding pcDNA3.1± vectors were obtained from Missouri University of Science and Technology. A GFP10-Gγ5 coding vector was subsequently produced by inserting the GFP10 sequence using the restriction sites NheI and Acc65I. The EPAC-based cAMP biosensor human β-arrestin1-RlucII and β-arrestin 2-RlucII plasmids were described by Breton et al. (37) and Zimmerman et al. (38).
Cell Culture and Transfection
For BRET assay, HEK293T (American Type Culture Collection, Manassas, VA) were maintained in DMEM (Wisent Bioproducts, St-Bruno, Canada) supplemented with 10% fetal bovine serum (Wisent Bioproducts). For ERK1/2 phosphorylation assay, CHO cells (American Type Culture Collection, Manassas, VA) stably expressing the wild-type or F255A rat ApelinR-EGFP (28) were maintained in Ham's F-12 medium supplemented with 10% heat-inactivated fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (all from Invitrogen). All cells were cultivated in a 37 °C humidified incubator with a 5% CO2 atmosphere. Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations.
Gαi Activation Assay
To directly monitor Gαi activation by ApelinR, we monitored the BRET between the RlucII-tagged Gαi and the GFP10-tagged Gγ5 subunits, which are known to separate after receptor's activation (39). Briefly, HEK293T cells were transfected with plasmids coding for Gαi1-RlucII, Gβ1, and Gγ5-GFP10 along with the wild-type HA-tagged ApelinR constructs. 24 h post-transfection, the cells were detached in PBS and seeded into a white opaque 96-well plate (PerkinElmer Life Sciences) at a density of 8 to 10 × 104 cells/well. 48 h post-transfection, varying concentrations of K16P or K17F were added for the indicated times to the wells along with the luciferase substrate DeepBlue C (2.5 mm final concentration; Goldbiotechnology, Inc., St. Louis, MO). The plates were then read in a Mithras LB940 instrument (Berthold Technologies, Bad Wildbad, Germany), and data were collected using the MicroWin 2000 software (Berthold Technologies). BRET signals were determined by calculating the ratio of the light emitted at 495–535 nm (GFP10) to the light emitted at 330–470 nm (Luciferase). This BRET signal is defined as BRET2.
cAMP Production Measurements
cAMP production was monitored using the EPAC-BRET biosensor as previously described (37). Briefly, HEK293T cells were transfected with the EPAC-BRET construct along with the wild-type ApelinR coding plasmid. 24 h post-transfection, the cells were seeded at a density of 8–10 × 104 cells/well in white opaque 96-well plates in PBS. 48 h post-transfection, vehicle, forskolin (10−5 m) or forskolin (10−5 m) + K17F or K16P (10−6 m) were added to the cells, and the plates were incubated at 37 °C for 15 min prior to BRET reading. Luciferase substrate DeepBlue C (2.5 × 10−6 m final, Goldbiotechnology, Inc.) was added 5 min prior to reading. BRET2 signals were determined by calculating the ratio of the light emitted at 495–535 nm (GFP10) to the light emitted at 330–470 nm (Luciferase) using the Mithras LB940 apparatus. BRET2 values obtained from vehicle-treated wells were subtracted from values obtained in the presence of forskolin and forskolin + ligands to yield ligand-induced ΔBRET2 values that represent ligand promoted cAMP production. A decrease in BRET2 signal represents an increase in cAMP production.
β-Arrestin Recruitment Assays
The interactions between β-arrestins and ApelinR were monitored by BRET, as previously described (40). Briefly, the β-arrestin1-RlucII or the β-arrestin2-RlucII-encoding plasmids were transfected along with the YFP-tagged wild-type ApelinR construct in HEK293T cells. 24 h post-transfection, the cells were transferred into a white opaque 96-well plate at a density of 8–10 × 104 cells/well. At 48 h post-transfection, the medium was replaced with PBS, and the cells were exposed to varying concentration of either K17F or K16P for 15 min prior to BRET reading to yield dose-response curves. The RLuc substrate coelentherazine H (2.5 μm; Nanolight Technology, Pinetop, AZ) was added 5 min prior to reading. For kinetic studies, Coelentherazine H was first added. Five min following the substrate addition, wells were treated with either vehicle, K16P- or K17F-apelin (both at 10−6 m), and the plates were immediately read repetitively for 15 min in the Mithras LB940 instrument. The BRET signal is determined by calculating the ratio of the light emitted at 505–555 nm (YFP) to the light emitted at 465–505 nm (Luciferase). This signal is defined as BRET1.
ERK1/2 Phosphorylation
Assessed by Western Blotting
CHO-K1 cells stably expressing wild-type ApelinR were grown in 12-well plates and starved for 16 h in a serum-free medium without or with PTX (25 ng/ml) prior to different stimulations: 10−7 m K17F or K16P for time course experiments, different concentrations from 10−11 to 10−6 m of K17F or K16P for 10 min for dose-response experiments. After stimulation, the medium was removed, 100 μl of SDS sample buffer (pH 6.8) containing 10 mm of dithiothreitol was added to each well, and the cells were placed on ice. Whole cell lysates were sonicated, heated for 5 min at 95 °C, then resolved on 10% Tris/glycine polyacrylamide gel, and transferred onto polyvinylidene difluoride membranes for immunoblotting. The membranes were incubated overnight with a 1:1000 dilution of phospho-ERK1/2 antibodies in 3% (w/v) bovine serum albumin Tris-buffered saline Tween (TBST) at 4 °C. The membranes were subsequently washed three times in TBST for 10 min and then incubated with a 1:10000 dilution of HRP-conjugated antibodies for chemiluminescence detection (Amersham Biosciences) for 1 h at room temperature. For the total ERK1/2 assessment, the membranes were stripped and reprobed using anti-mouse ERK1/2 antibodies (dilution 1:1000). ERK phosphorylation was normalized according to the loading of proteins by expressing the data as a ratio of pERK1/2 over total ERK1/2. Chemiluminescence detection was performed using ECL (GE Healthcare) and was quantified by densitometry with the ImageJ gel analysis software (Image Processing and Analysis in Java).
ERK1/2 Assessed by Alphascreen
CHO-K1 cells stably expressing the wild-type or F255A mutated ApelinR-EGFP were plated in 96-well plates at a density of 3 × 104 cells/well and grown for 24 h. The cells were then starved for 16 h in a serum-free medium with or without PTX (25 ng/ml) and treated with K17F or K16P at 10−7 m for different times. ERK1/2 phosphorylation was measured using the ERK1/2 Alphascreen Surefire kit (PerkinElmer Life Sciences). After stimulation by K17F or K16P, cells were lysed on ice in the lysis buffer provided by the manufacturer. 4 μl of lysates were then transferred to a 384-well white Optiplate (PerkinElmer Life Sciences), and ERK1/2 activation was measured according to the manufacturer's instructions. ERK1/2 phosphorylation values obtained from wells treated with vehicle were subtracted from values obtained in presence of ligands to get specific ligand-promoted ERK phosphorylation values.
Bias Calculation
All of the data were analyzed using the nonlinear curve fitting equations in GraphPad Prism (v6.0) to estimate the pEC50 values of the curves for the different pathways. Ligand bias was quantified by analyzing the concentration-response curves using the operational model of agonism, as described previously (41–43).
The operational model was also used to determine the transduction ratios (τ/KA) of both K17F and K16P as previously described (42–45) using the following equation,
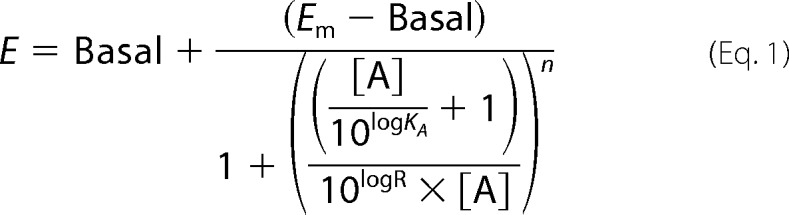 |
where E is the effect of the ligand, [A] is the concentration of agonist, Em is the maximal response of the system, Basal is the basal level of response in the absence of agonist, LogKA is the logarithm of the functional equilibrium dissociation constant of the agonist, n is the slope of the transducer function that links occupancy to response, and LogR is the logarithm of the “transduction coefficient” (or “transduction ratio”), τ/KA, where τ is an index of the coupling efficiency (or efficacy) of the agonist. To eliminate the influence of the system and the observation biases, the activity of K16P at a given signaling pathway was compared with that of K17F used as the reference compound using the following equation.
Ligand bias was calculated using the following,
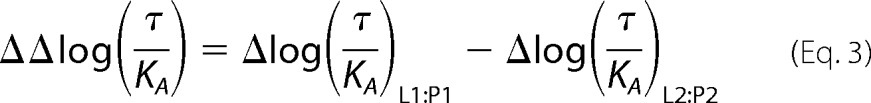 |
where L1 and L2 are ligands 1 and 2, respectively; P1 is pathway 1; and P2 is pathway 2 as recently described (45). The bias between two ligands was expressed as 10ΔΔlog(τ/KA).
Statistical Analysis
Statistical significance of the difference between conditions for BRET assays were assessed by performing two-way analysis of variance followed by Bonferroni post-tests using Prism 5.0 software (GraphPad Software, La Jolla, CA). Statistical comparisons for ERK1/2 phosphorylation experiment data were analyzed with GraphPad Prism and were performed using the paired Student's t test.
For the pairwise comparisons between K16P and K17F signaling bias, statistical analysis was performed using a two-way unpaired Student's t test on the ΔLog(τ/KA). Differences were considered significant when p was < 0.05.
RESULTS
Effects of K17F and K16P on Wild-type Apelin Receptor-mediated Gαi Activation
To characterize the effects of K17F and K16P on ApelinR-mediated Gαi signaling, HEK293 cells were transiently transfected with the HA-tagged wild-type rat ApelinR. BRET2-based assays were used to assess the activation of the Gi protein by monitoring the dissociation of the Gαi-Gγ5 complex and to investigate the inhibition of forskolin-induced cAMP production using the BRET2-based EPAC biosensor (46). Dose-response curves of K17F- and K16P-promoted Gi activation (Fig. 1A) showed very similar activities of both peptides to activate Gi as illustrated by the ability of the peptides to inhibit the Gαi1-Gγ5 BRET2 signal with comparable maximal responses and similar efficacies (IC50 values: 2.7 ± 0.52 10−10 and 8.1 ± 0.52 10−10 m, respectively). The similar efficacy of the two peptides for Gi activation was further confirmed by the similar ability of K17F and K16P to inhibit forskolin-stimulated cAMP production, as illustrated by the blockade of the forskolin-promoted decrease in EPAC BRET2 signal (Fig. 1B).
FIGURE 1.
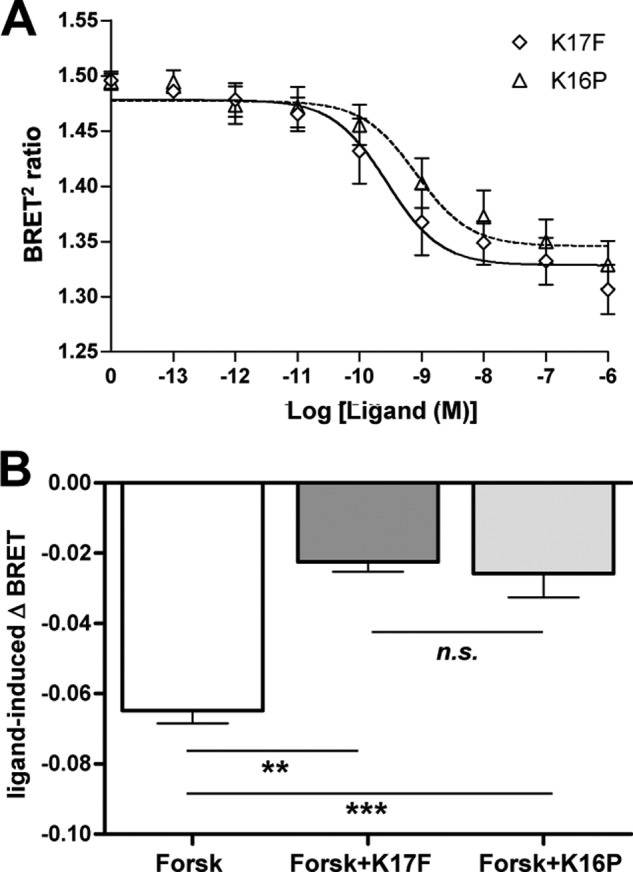
Gαi activation and adenylyl cyclase inhibition by ApelinR upon K17F and K16P stimulation. A, effects of increasing concentrations of K17F or K16P on Gαi activation monitored by BRET2 measurements of the dissociation between Gαi-RlucII and Gγ5-GFP10 upon a 5-min stimulation of HEK293T cells expressing the wild-type ApelinR. B, ligand-promoted changes in cAMP production in HEK293T cells expressing the wild-type ApelinR monitored by BRET2 using the EPAC biosensor (46) after forskolin (Forsk) treatment (10−5 m) alone or in combination with 10−6 m of either K17F or K16P. Forskolin and ligands (where applicable) were added 15 min prior to BRET2 measurements. The BRET2 values were subtracted from the BRET2 obtained with vehicle-treated cells to yield ligand-induced Δ BRET2 ratios. The data represent the means ± S.E. from three independent experiments. **, p < 0.01; ***, p < 0.001; n.s., nonsignificant.
Effects of K17F and K16P on the Recruitment of β-Arrestin 1 or β-Arrestin 2 by the Wild-type Apelin Receptor
Recruitment of β-arrestin 1 and 2 by ApelinR following K17F or K16P treatment was then investigated. To directly assess the physical interaction between the ligand-bound ApelinR and β-arrestin 1 or 2, the wild-type rat ApelinR fused to YFP at its C terminus and the human β-arrestin 1 or β-arrestin 2 fused to RLucII were transiently coexpressed in HEK293 cells. Fig. 2 (A and B, upper panels) shows that K17F and K16P increased the BRET1 signal for both β-arrestins 1 and 2 recruitment in a dose-dependent manner. Interestingly a greater recruitment was observed for β-arrestin 2 versus β-arrestin 1 This difference in BRET1 signal is consistent with the previously documented reduced ability of class A GPCRs to recruit β-arrestin 1 versus β-arrestin 2 (47). However, the lower signal could also represent different conformations for the β-arrestin/receptor complex for the two β-arrestin isoforms. For both β-arrestin 1 and β-arrestin 2, the maximal response was significantly higher for K17F than for that observed with K16P. Despite the significant difference in the efficacy of the two apelin peptides to promote β-arrestin recruitment, they showed similar potencies (EC50; K17F: 1.7 ± 0.74 × 10−8 m and 1.4 ± 0.69 × 10−8 m; K16P: 7.1 ± 0.5 × 10−8 m and 6.8 ± 0.57 × 10−8 m for β-arrestin 1 and β-arrestin 2, respectively) as would be predicted by their similar binding affinities (28). The apparent potencies of the peptides to promote the recruitment of β-arrestins is lower than that observed for the activation of Gi and cAMP production. This most likely reflects the different levels of amplification of the assays used but could also reflect a preference of the ligand-bound receptor for the Gi pathway. Kinetic experiments for both β-arrestins 1 and 2 recruitment reported in Fig. 2 (A and B, lower panels) confirmed the significantly greater propensity of K17F to promote β-arrestins recruitment compared with K16P. Indeed, K17F induced a rapid and strong increase in the BRET1 signal, whereas K16P induced a slower and much smaller increase for both β-arrestins. To compare the relative efficiency of K16P and K17F to engage β-arrestin 1 and 2 versus Gi, biased signaling was quantified using the operational model (see “Experimental Procedures” for details). As shown in Table 1, K16P was found to be 8.9- and 6.1-fold less efficient to engage β-arrestin 2 and 1 than K17F, whereas the two peptides had similar ability to activate Gi.
FIGURE 2.
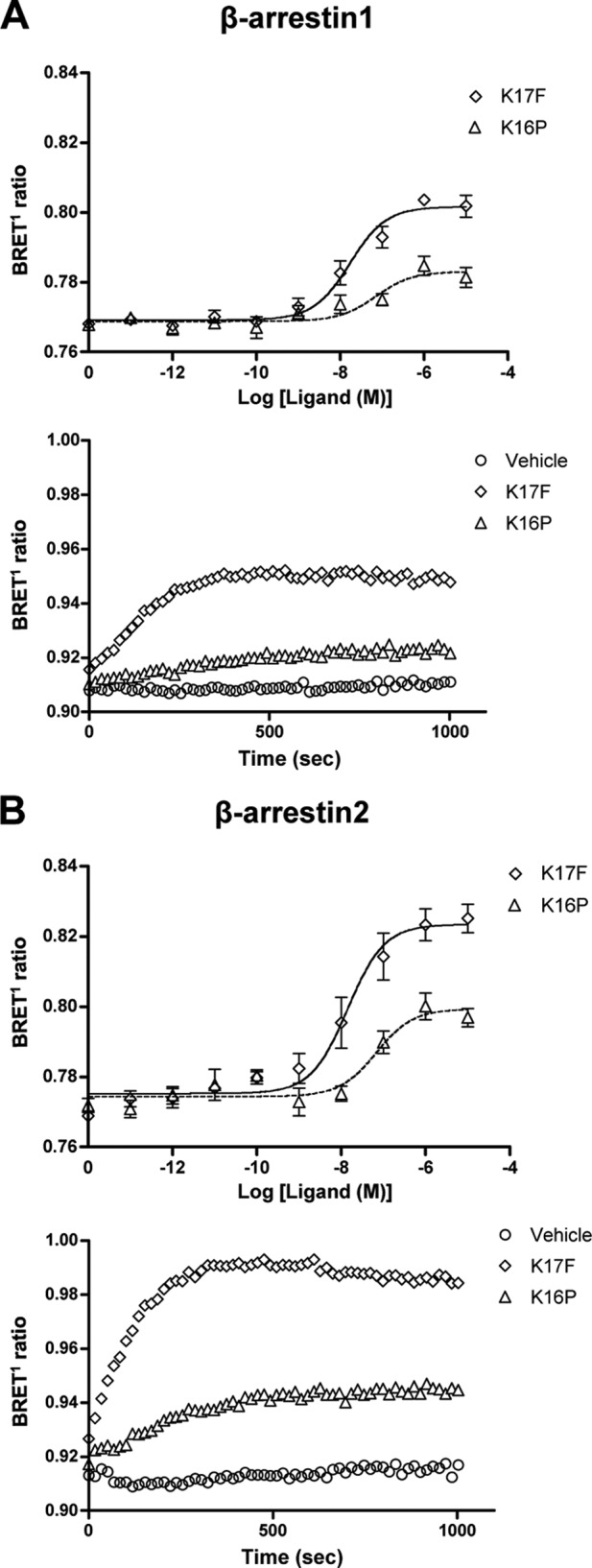
β-Arrestin recruitment to the wild-type ApelinR after stimulation with K17F or K16P. The effects of increasing concentrations of K17F or K16P on the recruitment of β-arrestin 1-Rluc (A, upper panel) or β-arrestin 2-Rluc (B, upper panel) to wild-type ApelinR-YFP were measured by BRET1. The effects of time on the recruitment of β-arrestin 1-Rluc (A, lower panel) or β-arrestin 2-Rluc (B, lower panel) to wild-type ApelinR-YFP were measured by BRET1 following stimulation with vehicle or 10−6 m of either K17F or K16P. The data in the upper panels represent the means ± S.E. of three independent experiments, whereas the lower panels are a representative illustration of three independent experiments.
TABLE 1.
ΔΔlog(τ/KA) ratios and bias factors for K17F and K16P
Statistical analysis was performed using a two-way unpaired Student's t test on the Δlog(τ/KA).
| Peptide | log τ/KA | Δlog τ/KA | ΔΔlog τ/KA | Bias | |
|---|---|---|---|---|---|
| Gi activation | K17F | 8.48 ± 0.19 | 0.00 ± 0.27 | ||
| K16P | 8.37 ± 0.18 | −0.11 ± 0.26 | |||
| βarr1 engagement | K17F | 7.88 ± 0.14 | 0.00 ± 0.20 | 0.00 ± 0.34 | 1.00 |
| K16P | 6.99 ± 0.24 | −0.89 ± 0.28 | 0.79 ± 0.38 | 6.10 | |
| βarr2 engagement | K17F | 7.64 ± 0.11 | 0.00 ± 0.15 | 0.00 ± 0.31 | 1.00 |
| K16P | 6.58 ± 0.20 | −1.06 ± 0.22 | 0.95 ± 0.34 | 8.93a |
a p < 0.05.
Effects of K17F and K16P on the Wild-type Apelin Receptor-mediated ERK1/2 Activation
To compare the ability of K17F and K16P to induce ERK1/2 phosphorylation, CHO-K1 cells stably expressing the wild-type ApelinR-EGFP were treated with either K17F or K16P (10−7 m) for different times (from 5 to 120 min) (Fig. 3, A and B) or treated with increasing concentrations of K17F and K16P (from 10−11 to 10−6 m) for 10 min (Fig. 3, C and D). Cells were then lysed, and ERK1/2 phosphorylation was measured by Western blot analysis using anti-phospho-ERK antibodies. As shown in Fig. 3A, K17F treatment led to a significant ERK1/2 phosphorylation. A similar ERK1/2 phosphorylation profile was observed following K16P treatment. K17F- or K16P-induced ERK1/2 phosphorylation reached a peak value at 5–10 min and remained greater than 2.5-fold over basal levels for at least 60 min. Dose-response curves of ERK1/2 phosphorylation in response to K17F or K16P treatment (Fig. 3, C and D) showed a similar maximal effect with an EC50 value for K17F of 5.6 ± 1.8 × 10−11 m, 13-fold lower than that of K16P (7.4 ± 2.6 × 10−10 m).
FIGURE 3.
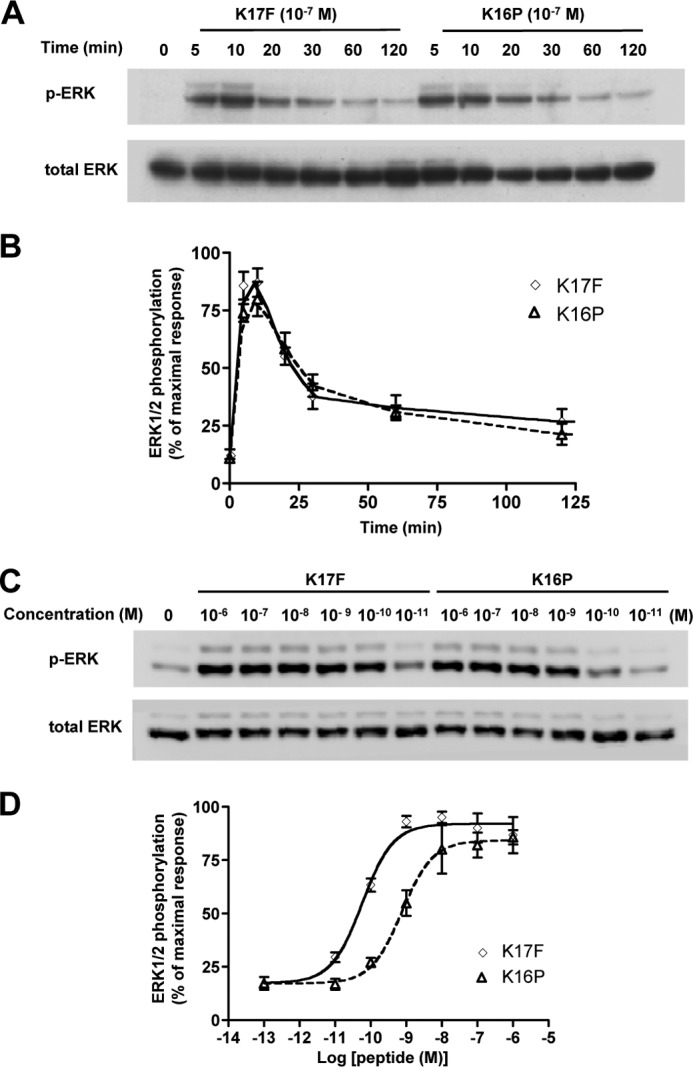
Apelin receptor-mediated ERK1/2 activation induced by K17F and K16P. A and B, time course of apelin receptor-mediated ERK1/2 phosphorylation induced by 10−7 m K17F or K16P. A, CHO cells stably expressing wild-type apelin receptor were treated with 10−7 m of K17F or K16P for 120 min. ERK1/2 phosphorylation was detected by Western blotting analysis using anti-phospho-ERK1/2 antibody (p-ERK), and total ERK1/2 was detected by total anti-ERK1/2 antibody (total ERK) in the same immunoblot for loading control. B, quantification of the bands corresponding to 44 and 42 kDa was performed by densitometry using ImageJ software. The data are expressed as percentages of pERK/total ERK. C and D, dose response of apelin receptor-mediated ERK1/2 phosphorylation induced by K17F and K16P stimulation. C, cells were treated with the indicated concentrations of K17F and K16P for 10 min, and ERK1/2 phosphorylation was measured. D, quantification of Western blotting bands was performed by densitometry analysis. The data correspond to the means ± S.E. from at least three independent experiments.
Involvement of a Gi Protein-dependent and -independent Mechanism in ERK1/2 Phosphorylation Induced by K17F and K16P
To determine the contribution of a Gαi-dependent mechanism to trigger K17F-induced ERK1/2 phosphorylation, we assessed the effect of a PTX treatment (25 ng/ml) for 16 h prior to stimulation of apelinR-expressing CHO-K1 cells with K17F (10−7 m). Time course analysis of ERK1/2 phosphorylation induced by K17F in absence of PTX showed a peak of maximal phosphorylation at 5–10 min and then a progressive decrease from 30 to 120 min. In contrast, in the presence of PTX, the ERK1/2 phosphorylation induced by K17F was strongly decreased but remained present up to 25 min post-stimulation (Fig. 4, A and B). The effect of the PTX treatment was more drastic on the K16P-induced ERK1/2 phosphorylation. Indeed, although some residual activity could be detected at 10 min, it was totally abolished after 20 min (Fig. 4C). The different effect of PTX treatment on K17F and K16P responses is particularly well illustrated in Fig. 4 (E and F), respectively, in which ERK1/2 phosphorylation induced by K17F (10−7 m) in the presence of PTX was monitored by the more quantitative Alphascreen assay. As shown in Fig. 4 (E and F), the level of ERK1/2 phosphorylation in PTX-treated cells measured over a period of 10 min was 3.4 times higher than vehicle upon stimulation with K17F (Fig. 4E), whereas only a marginal <2-fold increase, which did not reach statistical significance, was observed upon stimulation with K16P (Fig. 4F). Interestingly, the PTX-resistant response corresponds to the early phase of the K17F-promoted ERK1/2 response, suggesting that the latter phase is entirely Gi-dependent. This is in contrast with what was observed for other GPCRs such as the AT1 receptor, for which β-arrestin 2 has been shown to contribute to the late phase of the ERK1/2 response (34, 48, 49). However, a lack of a positive contribution of β-arrestin to late ERK1/2 response has been previously reported for the M3-muscarinic receptor (50).
FIGURE 4.
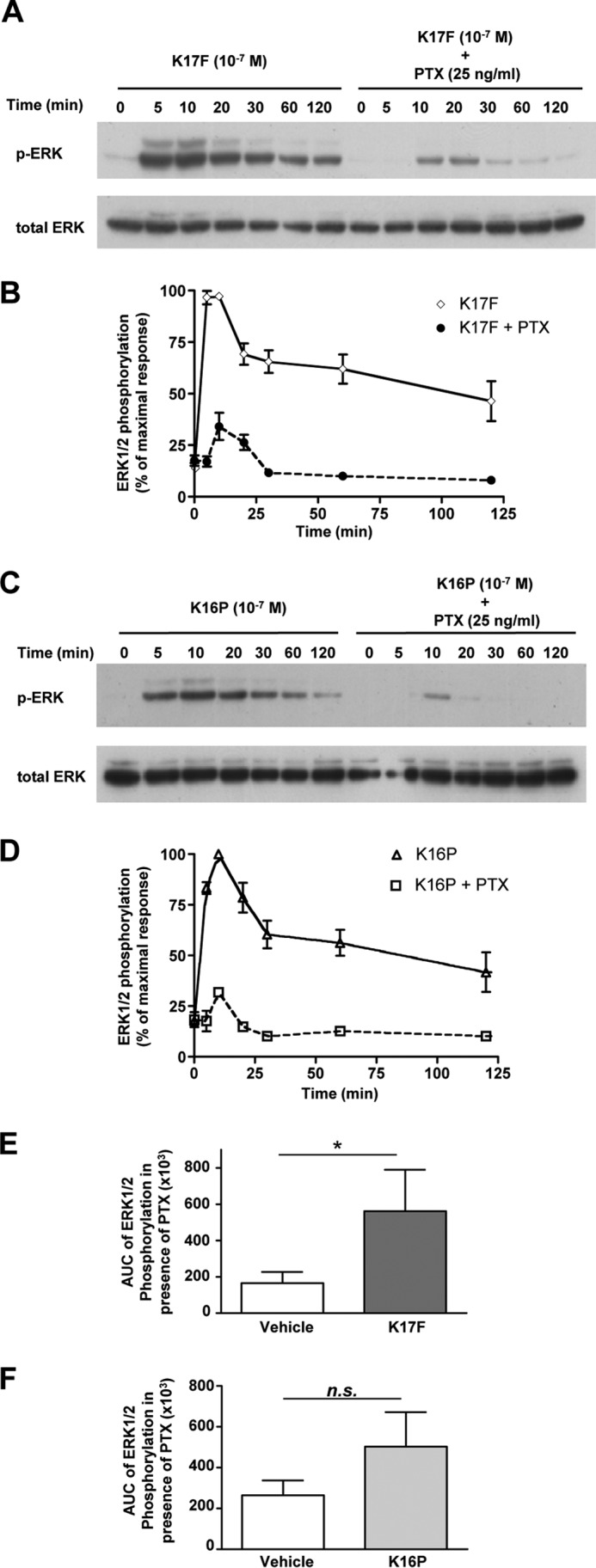
Apelin receptor-mediated ERK1/2 activation induced by K17F or K16P in the presence or absence of pertussis toxin. A and C, time course of apelin receptor-mediated ERK1/2 phosphorylation induced by K17F (A) or K16P (C) without or with PTX. CHO cells stably expressing the rat apelin receptor were pretreated with PTX (25 ng/ml) for 16 h and were treated with 10−7 m of K17F (A) or K16P (C) for 120 min. ERK1/2 phosphorylation was detected by Western blotting analysis using anti-phospho-ERK1/2 antibody (p-ERK) and anti-ERK1/2 antibody (total ERK) for loading control. B and D, quantification of Western blotting bands for K17F (B) or K16P (D) corresponding to 44 and 42 kDa was obtained by densitometry analysis. The data are expressed as percentages of pERK/total ERK and correspond to the means ± S.E. from at least three independent experiments. E and F, phosphorylation of ERK1/2 in CHO cells stably expressing the rat apelin receptor after stimulation by K17F (E) or K16P (F) in presence of PTX (25 ng/ml). Phosphorylation was quantified using the Alphascreen Surefire ERK1/2 assay kit. The values are expressed as the area under the curve (AUC) of ERK1/2 phosphorylation obtained from a time course of 20 min of stimulation. The data represent the means ± S.E. from at least four independent experiments. Statistical differences were assessed using Student's t test. *, p < 0.05; n.s., nonsignificant.
Involvement of β-Arrestin 2 Recruitment in K17F-induced Gαi Protein-independent ERK1/2 Phosphorylation
To investigate whether ERK1/2 phosphorylation could in part involve β-arrestin 2 recruitment to the receptor, CHO-K1 cells stably expressing the wild-type ApelinR-EGFP were transiently transfected with a control empty vector (DsRed mono), or a mutated form of β-arrestin 2 that inhibits β-arrestin-2 self-association, prevents the scaffolding of ERK1/2 and cannot support the β2-adrenergic receptor-stimulated ERK1/2 (DsRed mono-β-arrestin 2-K296A) (51). Cells were thus stimulated for 5–20 min with K17F or K16P (10−6 m) following pretreatment with PTX (25 ng/ml) or vehicle, and ERK1/2 phosphorylation was quantified using Alphascreen technology (Fig. 5, A–D). Under basal condition, ERK1/2 phosphorylation induced by K17F or K16P is not significantly affected by the transfection of either DsRed mono or β-arrestin 2 K296A. As expected, treatment with PTX reduced ERK1/2 phosphorylation induced by K17F (a factor of 4.2) or K16P (a factor of 9.6) (Fig. 5, A and C), yielding responses that remained significantly different from vehicle for K17F but not for K16P, which further illustrates the greater importance of the Gi component for the K16P-stimulated ERK pathway. Interestingly, in cells treated with PTX, expression of the β-arrestin 2 K296A completely abolished the K17F-induced ERK1/2 phosphorylation (Fig. 5, B and D), highlighting the significant β-arrestin-dependent component of the K17F-stimulated ERK1/2 response.
FIGURE 5.

Apelin receptor-mediated ERK1/2 activation induced by K17F or K16P in absence or presence of β-arrestin-2-K296A. A–D, phosphorylation of ERK1/2 in CHO cells stably expressing the rat apelin receptor transiently transfected with DsRed2 mono (empty vector) (A and C) or DsRed2 mono-β-arr2-K296A (B and D) stimulated with 10−6 m of K17F (A and B) or K16P (C and D) for 5–20 min without or with PTX (25 ng/ml). The values in graphics are expressed as area under the curve (AUC) values of ERK1/2 phosphorylation obtained for a time course of 20 min of stimulation. ERK1/2 phosphorylation was quantified using the Alphascreen Surefire ERK1/2 assay kit. The data correspond to the means ± S.E. from at least four independent experiments. Statistical differences were assessed using Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., nonsignificant.
Effects of K17F and K16P on ERK1/2 Phosphorylation in Cells Expressing the F255A Mutated Apelin receptor
We then investigated the capacity of the F255A mutated ApelinR (a mutant form of the receptor that does not undergo internalization upon K17F stimulation) to trigger ERK1/2 phosphorylation. We compared the ability of K17F and K16P to induce ERK1/2 phosphorylation following stimulation of the wild-type or the F255A mutated ApelinR in absence or in presence of PTX (Fig. 6, A–D). In the absence of PTX treatment, K17F and K16P induced a strong and similar ERK1/2 phosphorylation with a 26-fold increase over vehicle in CHO-K1 cells stably expressing the wild-type ApelinR (Fig. 6, A and C), whereas a significantly blunted ERK1/2 phosphorylation was observed in cells stably expressing the F255A mutated ApelinR, with a 6.9- and 3.6-fold increase for K17F and K16P, respectively, over vehicle (Fig. 6, B and D). As reported above, pretreatment with PTX significantly inhibited both K17F- and K16P-induced ERK1/2 phosphorylation in cells expressing the wild-type ApelinR (Fig. 6, A and C). Whereas the K16P-stimulated response was fully inhibited, a 4-fold increase over vehicle was still observed for K17F. This PTX-resistant response was fully abolished in cells expressing the F255A mutated ApelinR. Indeed, neither K17F nor K16P promoted ERK1/2 phosphorylation in PTX-treated cells expressing F255A-ApelinR (Fig. 6, B and D).
FIGURE 6.

Gαi protein-independent activation of MAPK cascade could be correlated to K17F-induced apelin receptor internalization. A–D, phosphorylation of endogenous ERK1/2 in CHO cells stably expressing wild-type apelin receptor-EGFP (A and C) or mutated F255A apelin receptor-EGFP (B and D) stimulated with 10−6 m of K17F (A and B) or K16P (C and D) for 5–20 min without (left two columns) or with (right two columns) PTX (25 ng/ml). The values in graphs are expressed as area under the curve (AUC) of ERK1/2 phosphorylation obtained for a time course of 20 min of stimulation with each of both peptides. ERK1/2 phosphorylation was quantified with Alphascreen technology. The data are the means ± S.E. from at least four independent experiments. Statistical differences were assessed using Student's t test. *, p < 0.05; **, p < 0.01; n.s., nonsignificant.
Vasoactive Role of K17F versus K16P in Rat Glomerular Afferent Arterioles
We finally investigated the ability of K17F and K16P to induce vasorelaxation of glomerular afferent arterioles preconstricted by Ang II (Fig. 7). Treatment of isolated glomerular afferent arterioles with 10−9 m of Ang II induced a vasoconstriction of the arterioles because we observed a significantly reduced arteriole diameter (12.77 ± 0.41 μm) compared with values measured under baseline conditions (14.78 ± 0.48 μm). Addition of 10−7 m K17F to preconstricted arterioles by 10−9 m of Ang II increased the arteriole diameter to 14.2 ± 0.54 μm, whereas 10−7 m K16P had no significant effect (13.13 ± 0.43 μm).
FIGURE 7.
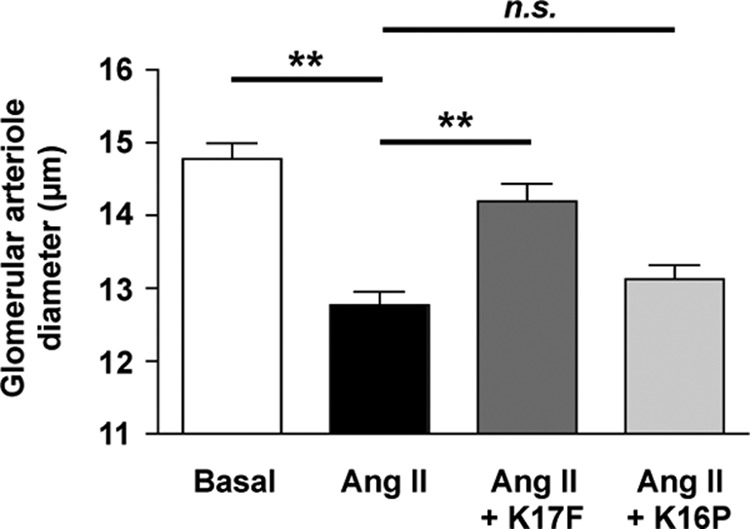
Effects of K17F and K16P on the afferent arteriole contractile response to Ang II. Arteriolar diameters were measured in the basal conditions, then 1.5 min after adding 10−9 M Ang II, and 1.5 min after addition of 10−7 m K17F or K16P on vasoconstricted arterioles (n = 5). **, p < 0.001; n.s., nonsignificant.
DISCUSSION
In the present work, we show that the C-terminal Phe of apelin plays a critical role in β-arrestin recruitment to the ApelinR but is not involved in Gαi protein activation nor adenylyl cyclase inhibition. We also demonstrate that apelin-induced ERK1/2 MAPK activation involves both Gαi- and β-arrestin-dependent pathways and that the β-arrestin-dependent ERK1/2 phosphorylation requires the presence of the C-terminal Phe of apelin. We also show that stimulation by K17F of an ApelinR in which the residue Phe-255 (predicted to interact with the C-terminal Phe of K17F (28)) is replaced by an alanine, did not internalize, and did not induce G protein-independent ERK1/2 phosphorylation. Together, these data demonstrate that stimulation of ApelinR by K17F but not by K16P activates a Gαi protein-independent but β-arrestin-dependent signaling pathway leading to MAPK activation.
Previous pharmacological studies showed that the C-terminal Phe in K17F does not play a major role in determining apelin binding affinity or the ability of the apelin peptides to inhibit forskolin-induced cAMP production (11, 14, 28) but is crucial to trigger ApelinR internalization. In addition, the deletion of the C-terminal Phe in K17F (K16P) or its substitution by an Ala in pE13F (pE13A) suppresses the ability of these peptides to decrease BP (14, 27), demonstrating the central role of the C-terminal Phe for the apelin-induced BP decrease. Altogether, this suggests that the decrease in BP requiring the C-terminal Phe of apelin may result from a Gi-independent but β-arrestin-dependent signaling pathway. To test this hypothesis, we first characterized the capacity of K17F and K16P to activate Gαi and to induce β-arrestin 1/2 recruitment. We showed that K17F and K16P are potent inducers of Gαi protein activation and adenylyl cyclase inhibition, in agreement with their effects on the inhibition of forskolin-induced cAMP production (28). In contrast, whereas K17F was found to be a potent inducer of β-arrestin 1/2 recruitment, K16P had a very low activity toward this pathway, even at a supramaximal concentration of 10−6 m. These data further support that K16P behaves as a biased ligand favoring G protein-coupling versus β-arrestin recruitment, consistent with the previous study demonstrating that K16P does not promote ApelinR internalization (28). This bias is clearly evident with the β-arrestin 2 recruitment-based assay as previously observed with the internalization-based assay. Indeed, the maximal BRET signal obtained with K16P at a supramaximal concentration of 10−6 m is decreased by 60% as compared with that obtained with K17F at the same concentration. Formal quantification of the bias revealed a 8.9-fold lower efficiency of K16P to promote β-arrestin 2 engagement than K17F, whereas the two peptides have equivalent abilities to activate Gαi. Taking into account the respective effects of these two peptides on BP, it is therefore tempting to propose that the apelin-induced BP decrease promoted by K17F results from the activation of one or several Gαi-independent but β-arrestin-dependent signaling pathway(s).
Because β-arrestin acts a scaffold protein that plays a role in the activation of MAPK, we hypothesized that the stimulation of ApelinR by K17F, inducing β-arrestin recruitment, should result in the activation of MAPK as previously shown for the angiotensin II receptor type 1a (AT1a) (49). We showed here that K17F is 13 times more potent than K16P to activate ERK1/2 phosphorylation, in agreement with the reduced ability of the peptide to promote β-arrestins engagement and ApelinR internalization (28). Altogether, these findings suggest that the difference in efficiency of both peptides to activate ERK1/2 phosphorylation could be linked to their differential ability to induce β-arrestin 1 or 2 recruitment and receptor internalization. This hypothesis is supported by our observation that although PTX-promoted inactivation of Gαi led to a strong decrease in both K17F- and K16P-stimulated ERK1/2 phosphorylation. However, there was a significant PTX-resistant response evoked by K17F, even if the total of ERK1/2 phosphorylation is mainly Gi-dependent. This PTX-resistant response occurs in a short window of time between 10 and 20 min after stimulation and disappears in the long-term sustained ERK1/2. This was in contrast with the complete abrogation by PTX treatment of the pE13F-induced ERK phosphorylation (13, 52), which is in line with the observed lower efficiency of pE13F as compared with K17F to induce ApelinR internalization and to decrease BP (14). In addition, the PTX-resistant ERK1/2 phosphorylation evoked by K17F was completely blocked by β-arrestin-2-K296A, which does not bind ERK1/2 efficiently (51). The observation that the F255A mutated ApelinR, which does not undergo endocytosis upon K17F stimulation while keeping its capacity to activate Gαi coupling (28), was unable to trigger ERK1/2 phosphorylation further confirms that the G protein-independent ERK1/2 phosphorylation is mediated by β-arrestin recruitment.
Altogether, we demonstrated that ApelinR MAPK signaling occurs through at least two distinct signaling pathways: one dependent on Gαi activation and the other dependent on β-arrestin recruitment. This dual signaling property of GPCRs has been reported for several receptors (35, 48, 53–58) and has given birth to the concept of biased agonists. This implies that a specific ligand might trigger distinct receptor conformations leading to different signaling pathways through interaction with different effector proteins. Such a mechanism has been shown for β2-adrenergic receptor (59) and more recently for V2 receptor (60). In this context, K16P could be considered as a G protein-biased agonist that conserves its ability to activate G protein signaling, whereas it lacks its capacity to trigger β-arrestin signaling. K17F and K16P will thus trigger different signalings by inducing different ApelinR conformations.
The physiological consequences of a G protein-independent and β-arrestin-dependent signaling pathway has been illustrated for AT1a receptor in cardiomyocytes. Stimulation by Ang II or [Sar1, Ile4, Ile8] Ang II of AT1a receptor led to positive inotropic and lusitropic responses in wild-type cardiomyocytes, whereas this response was abolished in β-arrestin 2 knock-out cardiomyocytes (61). The therapeutic potential of such compounds has been illustrated by the development of TR120027, a AT1a receptor β-arrestin-biased agonist, which improved cardiac performance and had cardioprotective activity by blocking the G protein-mediated vasoconstriction while increasing the cardiac contractility through β-arrestin signaling (62, 63). The activation of ApelinR β-arrestin-mediated signaling could have also a biological role because only apelin fragments that fully activate β-arrestin induced a decrease in arterial BP (14). So far, the lack of β-arrestin recruitment following K16P application has never been demonstrated in cells that endogenously express apelin receptors. Nevertheless, we recently observed in glomerular afferent arterioles from rat kidney, expressing apelin receptor mRNA (36) that 10−7 m K17F significantly (p < 0.001) induced a vasodilatation of afferent arterioles preconstricted by 10−9 m angiotensin II. In contrast, K16P, which is biased against β-arrestin-dependent signaling, is ineffective, suggesting a direct link between the biased signaling and the differential vasodilatory response promoted by distinct apelin fragments. In addition, Scimia et al. (64) have recently shown that a mechanical stretch of cardiomyocytes activates ERK1/2 phosphorylation in an ApelinR-dependent manner, independently of Gαi activation, and promotes β-arrestin recruitment. ERK1/2 activation by β-arrestin-dependent signaling pathway leads to a redistribution of ERK1/2 into endosomal vesicles that contain receptor-β-arrestin complexes as previously shown for AT1a receptor (65). Such redistribution of ERK1/2 might occur in endothelial cells or in cardiomyocytes upon ApelinR activation and might contribute to the actions of apelin on blood vessels and heart.
In conclusion, we have demonstrated that ApelinR triggers ERK1/2 phosphorylation in both a G protein- and β-arrestin-dependent manner. We further showed that K16P is a G protein-biased agonist displaying a strongly impaired β-arrestin signaling pathway that may account for its lack of activity in vivo on arterial blood pressure. This study brings new insights into ApelinR signaling mechanisms. Further studies are required to better understand the ApelinR molecular signaling features required for the development of ApelinR biased agonists that could be therapeutically useful. The development of such compounds will be interesting to further explore the physiopathological roles of apelin and to be able to target a specific apelin biological action.
Acknowledgment
We thank Dr Adrien Flahault for the statistical analysis of the data.
This work was supported by the Institut National de la Sante et de la Recherche Medicale, the College de France and the Agence Nationale pour la Recherche Physique et Chimie du Vivant. This work was also supported by funds from the Fondation pour la Recherche Medicale (to I. B.-F.) and the Cardiovasculaires-Obesite-Rein-Diabete from Region Ile-de-France (to E. C.).
- GPCR
- G protein-coupled receptor
- ApelinR
- apelin receptor
- pERK
- phospho-ERK
- BRET
- bioluminescence resonance energy transfer
- EPAC
- exchange protein activated by cAMP
- PTX
- pertussis toxin
- Ang II
- angiotensin II
- BP
- blood pressure
- AVP
- arginine vasopressin.
REFERENCES
- 1. Tatemoto K., Hosoya M., Habata Y., Fujii R., Kakegawa T., Zou M. X., Kawamata Y., Fukusumi S., Hinuma S., Kitada C., Kurokawa T., Onda H., Fujino M. (1998) Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 251, 471–476 [DOI] [PubMed] [Google Scholar]
- 2. O'Dowd B. F., Heiber M., Chan A., Heng H. H., Tsui L. C., Kennedy J. L., Shi X., Petronis A., George S. R., Nguyen T. (1993) A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 136, 355–360 [DOI] [PubMed] [Google Scholar]
- 3. Devic E., Rizzoti K., Bodin S., Knibiehler B., Audigier Y. (1999) Amino acid sequence and embryonic expression of msr/apj, the mouse homolog of Xenopus X-msr and human APJ. Mech. Dev. 84, 199–203 [DOI] [PubMed] [Google Scholar]
- 4. De Mota N., Lenkei Z., Llorens-Cortès C. (2000) Cloning, pharmacological characterization and brain distribution of the rat apelin receptor. Neuroendocrinology 72, 400–407 [DOI] [PubMed] [Google Scholar]
- 5. O'Carroll A. M., Selby T. L., Palkovits M., Lolait S. J. (2000) Distribution of mRNA encoding B78/apj, the rat homologue of the human APJ receptor, and its endogenous ligand apelin in brain and peripheral tissues. Biochim. Biophys. Acta 1492, 72–80 [DOI] [PubMed] [Google Scholar]
- 6. Habata Y., Fujii R., Hosoya M., Fukusumi S., Kawamata Y., Hinuma S., Kitada C., Nishizawa N., Murosaki S., Kurokawa T., Onda H., Tatemoto K., Fujino M. (1999) Apelin, the natural ligand of the orphan receptor APJ, is abundantly secreted in the colostrum. Biochim. Biophys. Acta 1452, 25–35 [DOI] [PubMed] [Google Scholar]
- 7. Lee D. K., Cheng R., Nguyen T., Fan T., Kariyawasam A. P., Liu Y., Osmond D. H., George S. R., O'Dowd B. F. (2000) Characterization of apelin, the ligand for the APJ receptor. J. Neurochem. 74, 34–41 [DOI] [PubMed] [Google Scholar]
- 8. Azizi M., Iturrioz X., Blanchard A., Peyrard S., De Mota N., Chartrel N., Vaudry H., Corvol P., Llorens-Cortes C. (2008) Reciprocal regulation of plasma apelin and vasopressin by osmotic stimuli. J. Am. Soc. Nephrol. 19, 1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Mota N., Reaux-Le Goazigo A., El Messari S., Chartrel N., Roesch D., Dujardin C., Kordon C., Vaudry H., Moos F., Llorens-Cortes C. (2004) Apelin, a potent diuretic neuropeptide counteracting vasopressin actions through inhibition of vasopressin neuron activity and vasopressin release. Proc. Natl. Acad. Sci. U.S.A. 101, 10464–10469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iturrioz X., Alvear-Perez R., De Mota N., Franchet C., Guillier F., Leroux V., Dabire H., Le Jouan M., Chabane H., Gerbier R., Bonnet D., Berdeaux A., Maigret B., Galzi J. L., Hibert M., Llorens-Cortes C. (2010) Identification and pharmacological properties of E339–3D6, the first nonpeptidic apelin receptor agonist. FASEB J. 24, 1506–1517 [DOI] [PubMed] [Google Scholar]
- 11. Medhurst A. D., Jennings C. A., Robbins M. J., Davis R. P., Ellis C., Winborn K. Y., Lawrie K. W., Hervieu G., Riley G., Bolaky J. E., Herrity N. C., Murdock P., Darker J. G. (2003) Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J. Neurochem. 84, 1162–1172 [DOI] [PubMed] [Google Scholar]
- 12. Zhou N., Fan X., Mukhtar M., Fang J., Patel C. A., DuBois G. C., Pomerantz R. J. (2003) Cell-cell fusion and internalization of the CNS-based, HIV-1 co-receptor, APJ. Virology 307, 22–36 [DOI] [PubMed] [Google Scholar]
- 13. Masri B., Morin N., Cornu M., Knibiehler B., Audigier Y. (2004) Apelin (65–77) activates p70 S6 kinase and is mitogenic for umbilical endothelial cells. FASEB J. 18, 1909–1911 [DOI] [PubMed] [Google Scholar]
- 14. El Messari S., Iturrioz X., Fassot C., De Mota N., Roesch D., Llorens-Cortes C. (2004) Functional dissociation of apelin receptor signaling and endocytosis: implications for the effects of apelin on arterial blood pressure. J. Neurochem. 90, 1290–1301 [DOI] [PubMed] [Google Scholar]
- 15. Evans N. A., Groarke D. A., Warrack J., Greenwood C. J., Dodgson K., Milligan G., Wilson S. (2001) Visualizing differences in ligand-induced β-arrestin-GFP interactions and trafficking between three recently characterized G protein-coupled receptors. J. Neurochem. 77, 476–485 [DOI] [PubMed] [Google Scholar]
- 16. Reaux A., De Mota N., Skultetyova I., Lenkei Z., El Messari S., Gallatz K., Corvol P., Palkovits M., Llorens-Cortès C. (2001) Physiological role of a novel neuropeptide, apelin, and its receptor in the rat brain. J. Neurochem. 77, 1085–1096 [DOI] [PubMed] [Google Scholar]
- 17. Brailoiu G. C., Dun S. L., Yang J., Ohsawa M., Chang J. K., Dun N. J. (2002) Apelin-immunoreactivity in the rat hypothalamus and pituitary. Neurosci. Lett. 327, 193–197 [DOI] [PubMed] [Google Scholar]
- 18. O'Carroll A. M., Don A. L., Lolait S. J. (2003) APJ receptor mRNA expression in the rat hypothalamic paraventricular nucleus: regulation by stress and glucocorticoids. J. Neuroendocrinol. 15, 1095–1101 [DOI] [PubMed] [Google Scholar]
- 19. Reaux-Le Goazigo A., Morinville A., Burlet A., Llorens-Cortes C., Beaudet A. (2004) Dehydration-induced cross-regulation of apelin and vasopressin immunoreactivity levels in magnocellular hypothalamic neurons. Endocrinology 145, 4392–4400 [DOI] [PubMed] [Google Scholar]
- 20. Blanchard A., Steichen O., De Mota N., Curis E., Gauci C., Frank M., Wuerzner G., Kamenicky P., Passeron A., Azizi M., Llorens-Cortes C. (2013) An abnormal apelin/vasopressin balance may contribute to water retention in patients with the syndrome of inappropriate antidiuretic hormone (SIADH) and heart failure. J. Clin. Endocrinol. Metab. 98, 2084–2089 [DOI] [PubMed] [Google Scholar]
- 21. Japp A. G., Cruden N. L., Amer D. A., Li V. K., Goudie E. B., Johnston N. R., Sharma S., Neilson I., Webb D. J., Megson I. L., Flapan A. D., Newby D. E. (2008) Vascular effects of apelin in vivo in man. J. Am. Coll. Cardiol. 52, 908–913 [DOI] [PubMed] [Google Scholar]
- 22. Ashley E. A., Powers J., Chen M., Kundu R., Finsterbach T., Caffarelli A., Deng A., Eichhorn J., Mahajan R., Agrawal R., Greve J., Robbins R., Patterson A. J., Bernstein D., Quertermous T. (2005) The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc. Res. 65, 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berry M. F., Pirolli T. J., Jayasankar V., Burdick J., Morine K. J., Gardner T. J., Woo Y. J. (2004) Apelin has in vivo inotropic effects on normal and failing hearts. Circulation 110, II187–II193 [DOI] [PubMed] [Google Scholar]
- 24. Szokodi I., Tavi P., Földes G., Voutilainen-Myllylä S., Ilves M., Tokola H., Pikkarainen S., Piuhola J., Rysä J., Tóth M., Ruskoaho H. (2002) Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ. Res. 91, 434–440 [DOI] [PubMed] [Google Scholar]
- 25. Kuba K., Zhang L., Imai Y., Arab S., Chen M., Maekawa Y., Leschnik M., Leibbrandt A., Makovic M., Schwaighofer J., Beetz N., Musialek R., Neely G. G., Komnenovic V., Kolm U., Metzler B., Ricci R., Hara H., Meixner A., Nghiem M., Chen X., Dawood F., Wong K. M., Sarao R., Cukerman E., Kimura A., Hein L., Thalhammer J., Liu P. P., Penninger J. M. (2007) Impaired heart contractility in Apelin gene-deficient mice associated with aging and pressure overload. Circ. Res. 101, e32–e42 [DOI] [PubMed] [Google Scholar]
- 26. Tatemoto K., Takayama K., Zou M. X., Kumaki I., Zhang W., Kumano K., Fujimiya M. (2001) The novel peptide apelin lowers blood pressure via a nitric oxide-dependent mechanism. Regul. Pept. 99, 87–92 [DOI] [PubMed] [Google Scholar]
- 27. Lee D. K., Saldivia V. R., Nguyen T., Cheng R., George S. R., O'Dowd B. F. (2005) Modification of the terminal residue of apelin-13 antagonizes its hypotensive action. Endocrinology 146, 231–236 [DOI] [PubMed] [Google Scholar]
- 28. Iturrioz X., Gerbier R., Leroux V., Alvear-Perez R., Maigret B., Llorens-Cortes C. (2010) By interacting with the C-terminal Phe of apelin, Phe255 and Trp259 in helix VI of the apelin receptor are critical for internalization. J. Biol. Chem. 285, 32627–32637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Galandrin S., Bouvier M. (2006) Distinct signaling profiles of β1 and β2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Mol. Pharmacol. 70, 1575–1584 [DOI] [PubMed] [Google Scholar]
- 30. Galandrin S., Oligny-Longpré G., Bonin H., Ogawa K., Galés C., Bouvier M. (2008) Conformational rearrangements and signaling cascades involved in ligand-biased mitogen-activated protein kinase signaling through the β1-adrenergic receptor. Mol. Pharmacol. 74, 162–172 [DOI] [PubMed] [Google Scholar]
- 31. Drake M. T., Violin J. D., Whalen E. J., Wisler J. W., Shenoy S. K., Lefkowitz R. J. (2008) β-Arrestin-biased agonism at the β2-adrenergic receptor. J. Biol. Chem. 283, 5669–5676 [DOI] [PubMed] [Google Scholar]
- 32. Shukla A. K., Violin J. D., Whalen E. J., Gesty-Palmer D., Shenoy S. K., Lefkowitz R. J. (2008) Distinct conformational changes in β-arrestin report biased agonism at seven-transmembrane receptors. Proc. Natl. Acad. Sci. U.S.A. 105, 9988–9993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Swaminath G., Xiang Y., Lee T. W., Steenhuis J., Parnot C., Kobilka B. K. (2004) Sequential binding of agonists to the β2 adrenoceptor. Kinetic evidence for intermediate conformational states. J. Biol. Chem. 279, 686–691 [DOI] [PubMed] [Google Scholar]
- 34. DeWire S. M., Ahn S., Lefkowitz R. J., Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 35. DeFea K. A., Zalevsky J., Thoma M. S., Déry O., Mullins R. D., Bunnett N. W. (2000) β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 148, 1267–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hus-Citharel A., Bouby N., Frugière A., Bodineau L., Gasc J. M., Llorens-Cortes C. (2008) Effect of apelin on glomerular hemodynamic function in the rat kidney. Kidney Int. 74, 486–494 [DOI] [PubMed] [Google Scholar]
- 37. Breton B., Sauvageau É., Zhou J., Bonin H., Le Gouill C., Bouvier M. (2010) Multiplexing of multicolor bioluminescence resonance energy transfer. Biophys. J. 99, 4037–4046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zimmerman B., Beautrait A., Aguila B., Charles R., Escher E., Claing A., Bouvier M., Laporte S. A. (2012) Differential β-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci. Signal. 5, ra33. [DOI] [PubMed] [Google Scholar]
- 39. Galés C., Rebois R. V., Hogue M., Trieu P., Breit A., Hébert T. E., Bouvier M. (2005) Real-time monitoring of receptor and G-protein interactions in living cells. Nat. Methods 2, 177–184 [DOI] [PubMed] [Google Scholar]
- 40. Rochdi M. D., Vargas G. A., Carpentier E., Oligny-Longpré G., Chen S., Kovoor A., Gitelman S. E., Rosenthal S. M., von Zastrow M., Bouvier M. (2010) Functional characterization of vasopressin type 2 receptor substitutions (R137H/C/L) leading to nephrogenic diabetes insipidus and nephrogenic syndrome of inappropriate antidiuresis: implications for treatments. Mol. Pharmacol. 77, 836–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Black J. W., Leff P. (1983) Operational models of pharmacological agonism. Proc. R. Soc. Lond. B Biol. Sci. 220, 141–162 [DOI] [PubMed] [Google Scholar]
- 42. Evans B. A., Broxton N., Merlin J., Sato M., Hutchinson D. S., Christopoulos A., Summers R. J. (2011) Quantification of functional selectivity at the human α1A-adrenoceptor. Mol. Pharmacol. 79, 298–307 [DOI] [PubMed] [Google Scholar]
- 43. Kenakin T., Watson C., Muniz-Medina V., Christopoulos A., Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 3, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kenakin T., Christopoulos A. (2013) Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 12, 205–216 [DOI] [PubMed] [Google Scholar]
- 45. van der Westhuizen E. T., Breton B., Christopoulos A., Bouvier M. (2014) Quantification of ligand bias for clinically relevant β2-adrenergic receptor ligands: implications for drug taxonomy. Mol. Pharmacol. 85, 492–509 [DOI] [PubMed] [Google Scholar]
- 46. Breton B., Lagacé M., Bouvier M. (2010) Combining resonance energy transfer methods reveals a complex between the α2A-adrenergic receptor, Gαi1β1γ2, and GRK2. FASEB J. 24, 4733–4743 [DOI] [PubMed] [Google Scholar]
- 47. Oakley R. H., Laporte S. A., Holt J. A., Caron M. G., Barak L. S. (2000) Differential affinities of visual arrestin, β arrestin1, and β arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 275, 17201–17210 [DOI] [PubMed] [Google Scholar]
- 48. Gesty-Palmer D., Chen M., Reiter E., Ahn S., Nelson C. D., Wang S., Eckhardt A. E., Cowan C. L., Spurney R. F., Luttrell L. M., Lefkowitz R. J. (2006) Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem. 281, 10856–10864 [DOI] [PubMed] [Google Scholar]
- 49. Tohgo A., Choy E. W., Gesty-Palmer D., Pierce K. L., Laporte S., Oakley R. H., Caron M. G., Lefkowitz R. J., Luttrell L. M. (2003) The stability of the G protein-coupled receptor-β-arrestin interaction determines the mechanism and functional consequence of ERK activation. J. Biol. Chem. 278, 6258–6267 [DOI] [PubMed] [Google Scholar]
- 50. Luo J., Busillo J. M., Benovic J. L. (2008) M3 muscarinic acetylcholine receptor-mediated signaling is regulated by distinct mechanisms. Mol. Pharmacol. 74, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu T. R., Baillie G. S., Bhari N., Houslay T. M., Pitt A. M., Adams D. R., Kolch W., Houslay M. D., Milligan G. (2008) Mutations of β-arrestin 2 that limit self-association also interfere with interactions with the β2-adrenoceptor and the ERK1/2 MAPKs: implications for β2-adrenoceptor signalling via the ERK1/2 MAPKs. Biochem. J. 413, 51–60 [DOI] [PubMed] [Google Scholar]
- 52. Masri B., Lahlou H., Mazarguil H., Knibiehler B., Audigier Y. (2002) Apelin (65–77) activates extracellular signal-regulated kinases via a PTX-sensitive G protein. Biochem. Biophys. Res. Commun. 290, 539–545 [DOI] [PubMed] [Google Scholar]
- 53. Charest P. G., Oligny-Longpré G., Bonin H., Azzi M., Bouvier M. (2007) The V2 vasopressin receptor stimulates ERK1/2 activity independently of heterotrimeric G protein signalling. Cell Signal. 19, 32–41 [DOI] [PubMed] [Google Scholar]
- 54. Shenoy S. K., Drake M. T., Nelson C. D., Houtz D. A., Xiao K., Madabushi S., Reiter E., Premont R. T., Lichtarge O., Lefkowitz R. J. (2006) β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J. Biol. Chem. 281, 1261–1273 [DOI] [PubMed] [Google Scholar]
- 55. Stalheim L., Ding Y., Gullapalli A., Paing M. M., Wolfe B. L., Morris D. R., Trejo J. (2005) Multiple independent functions of arrestins in the regulation of protease-activated receptor-2 signaling and trafficking. Mol. Pharmacol. 67, 78–87 [DOI] [PubMed] [Google Scholar]
- 56. Tohgo A., Pierce K. L., Choy E. W., Lefkowitz R. J., Luttrell L. M. (2002) β-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J. Biol. Chem. 277, 9429–9436 [DOI] [PubMed] [Google Scholar]
- 57. Wei H., Ahn S., Shenoy S. K., Karnik S. S., Hunyady L., Luttrell L. M., Lefkowitz R. J. (2003) Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 100, 10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ahn S., Shenoy S. K., Wei H., Lefkowitz R. J. (2004) Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J. Biol. Chem. 279, 35518–35525 [DOI] [PubMed] [Google Scholar]
- 59. Granier S., Kim S., Shafer A. M., Ratnala V. R., Fung J. J., Zare R. N., Kobilka B. (2007) Structure and conformational changes in the C-terminal domain of the β2-adrenoceptor: insights from fluorescence resonance energy transfer studies. J. Biol. Chem. 282, 13895–13905 [DOI] [PubMed] [Google Scholar]
- 60. Rahmeh R., Damian M., Cottet M., Orcel H., Mendre C., Durroux T., Sharma K. S., Durand G., Pucci B., Trinquet E., Zwier J. M., Deupi X., Bron P., Banères J. L., Mouillac B., Granier S. (2012) Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 109, 6733–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rajagopal K., Whalen E. J., Violin J. D., Stiber J. A., Rosenberg P. B., Premont R. T., Coffman T. M., Rockman H. A., Lefkowitz R. J. (2006) β-Arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc. Natl. Acad. Sci. U.S.A. 103, 16284–16289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Boerrigter G., Lark M. W., Whalen E. J., Soergel D. G., Violin J. D., Burnett J. C., Jr. (2011) Cardiorenal actions of TRV120027, a novel β-arrestin-biased ligand at the angiotensin II type I receptor, in healthy and heart failure canines: a novel therapeutic strategy for acute heart failure. Circ Heart Fail. 4, 770–778 [DOI] [PubMed] [Google Scholar]
- 63. Violin J. D., DeWire S. M., Yamashita D., Rominger D. H., Nguyen L., Schiller K., Whalen E. J., Gowen M., Lark M. W. (2010) Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 335, 572–579 [DOI] [PubMed] [Google Scholar]
- 64. Scimia M. C., Hurtado C., Ray S., Metzler S., Wei K., Wang J., Woods C. E., Purcell N. H., Catalucci D., Akasaka T., Bueno O. F., Vlasuk G. P., Kaliman P., Bodmer R., Smith L. H., Ashley E., Mercola M., Brown J. H., Ruiz-Lozano P. (2012) APJ acts as a dual receptor in cardiac hypertrophy. Nature 488, 394–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Luttrell L. M., Roudabush F. L., Choy E. W., Miller W. E., Field M. E., Pierce K. L., Lefkowitz R. J. (2001) Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. U.S.A. 98, 2449–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]