

Background: AMPAR trafficking plays an important role in synaptic plasticity, but how ubiquitinated AMPARs internalize remains largely unknown.
Results: The endocytic adaptor EPS15 interacts with ubiquitinated AMPARs and facilitates receptor internalization.
Conclusion: EPS15 is required for the internalization of ubiquitinated AMPARs.
Significance: Selective targeting of a distinct pool of surface AMPARs for internalization provides novel insights into the mechanisms of synaptic regulation.
Keywords: Adaptor Protein; α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid Receptor (AMPA receptor, AMPAR); Receptor Internalization; Trafficking; Ubiquitylation (Ubiquitination); EPS15; Nedd4
Abstract
AMPA-type glutamate receptors (AMPARs) play a critical role in mediating fast excitatory synaptic transmission in the brain. Alterations in receptor expression, distribution, and trafficking have been shown to underlie synaptic plasticity and higher brain functions, including learning and memory, as well as brain dysfunctions such as drug addiction and psychological disorders. Therefore, it is essential to elucidate the molecular mechanisms that regulate AMPAR dynamics. We have shown previously that mammalian AMPARs are subject to posttranslational modification by ubiquitin, with AMPAR ubiquitination enhancing receptor internalization and reducing AMPAR cell surface expression. Here we report a crucial role for epidermal growth factor receptor substrate 15 (Eps15), an endocytic adaptor, in ubiquitination-dependent AMPAR internalization. We find that suppression or overexpression of Eps15 results in changes in AMPAR surface expression. Eps15 interacts with AMPARs, which requires Nedd4-mediated GluA1 ubiquitination and the ubiquitin-interacting motif of Eps15. Importantly, we find that Eps15 plays an important role in AMPAR internalization. Knockdown of Eps15 suppresses the internalization of GluA1 but not the mutant GluA1 that lacks ubiquitination sites, indicating a role of Eps15 for the internalization of ubiquitinated AMPARs. These results reveal a novel molecular mechanism employed specifically for the trafficking of the ubiquitin-modified AMPARs.
Introduction
AMPA receptors (AMPARs)2 are glutamate-gated heterotetrameric ion channels responsible for mediating the majority of fast excitatory neurotransmission in the brain. Modifications in AMPAR synaptic expression have long been considered the critical molecular mechanism underlying both Hebbian-type (1, 2) and homeostatic (3–5) synaptic plasticity. AMPARs traffic rapidly between the plasma membrane and intracellular compartments, and although total AMPAR abundance is maintained through a balance between receptor synthesis and degradation, AMPAR surface accumulation is regulated by means of receptor insertion, internalization, and recycling. However, how specific surface AMPARs are selected and recognized by trafficking machinery remains unclear. One mechanism is posttranslational modification of surface proteins via ubiquitination.
Ubiquitin is a highly conserved, 8.5-kDa, 76-amino acid protein that can be conjugated covalently to a lysine residue in a target substrate. The specificity is determined by the E3 ligase. Ubiquitinated membrane proteins are recognized by the endocytotic machinery for internalization, with polyubiquitinated proteins often being sorted to the proteasome or lysosome for degradation (6). Ubiquitination has been implicated in the trafficking of glutamate receptors, including NMDA receptors (7) and AMPARs (8–10). Recent studies have shown that mammalian AMPARs are subject to direct ubiquitination (11–13).
AMPARs can be internalized via the clathrin-coated pits pathway via binding of the adaptor protein AP2 with the intracellular C termini of AMPAR subunits (14–17). However, because AP2 lacks ubiquitin-dependent regulation, a distinct adaptor may be required to recognize ubiquitin-modified receptors. To serve this function, EGF receptor protein tyrosine kinase substrate 15 (Eps15) emerged as an excellent candidate. Structurally, Eps15 is divided into four domains. Domain III contains several DPF motifs that interact with the adaptor proteins AP1 (18) and AP2 (19). Of particular interest is regulatory domain IV, which contains two ubiquitin-interacting motifs (UIMs). These UIM domains play a critical role in the association with and sorting of ubiquitinated receptors (20–22).
Here we report an important role for Eps15 in mediating ubiquitinated AMPAR trafficking. We find that Eps15 is localized synaptically and that alterations in Eps15 expression significantly affect surface levels of GluA1. Furthermore, we demonstrate that the interaction between Eps15 and GluA1 is dependent upon GluA1 ubiquitination and the Eps15 UIM regions. We also show that the E3 ligase Nedd4 is involved in this interaction and that the internalization pathway of ubiquitinated AMPARs is mediated by the clathrin-coated pits pathway. These results collectively reveal a novel ubiquitination-specific aspect of the molecular assembly utilized in AMPAR trafficking.
EXPERIMENTAL PROCEDURES
Immunofluorescence
Primary cultured hippocampal neurons from embryonic day 18 rat embryos were cultured onto coverslips as described previously (23, 24). Cells were fixed at 14–15 days in vitro with ice-cold 4% paraformaldehyde in artificial CSF (ACSF) containing 150 mm NaCl, 10 mm HEPES (pH 7.4), 3 mm KCl, 2 mm CaCl2, and 10 mm glucose for 10 min, washed twice with ACSF, and permeabilized with 0.3% Triton X-100/ACSF for 10 min before a 1-h incubation at room temperature in a blocking solution of 10% normal goat serum in ACSF. For double staining of endogenous Eps15 with either PSD-95 or GluA1N, cells were incubated overnight at 4 °C with either a primary anti-PSD-95 mouse antibody (1:500, NeuroMab) or a primary anti-GluA1N mouse antibody (1:300, Millipore) in blocking solution. Following incubation overnight, the cells were washed three times with ACSF and then incubated with primary anti-Eps15 rabbit antibody (1:100, Santa Cruz Biotechnology, Inc.) in blocking solution for 2 h at room temperature. Cells were again washed three times with ACSF and then incubated for 1 h at room temperature in the dark with 1:500 Alexa Fluor 555 goat anti-rabbit (Invitrogen) and 1:500 goat anti-mouse Alexa Fluor 488 (Invitrogen) secondary antibodies in ACSF. After another set of washes, coverslips were mounted onto slides with Prolong Gold Antifade (Invitrogen) and cured for at least 4 h. Images were collected with an inverted fluorescence microscope at a ×63 oil objective (Zeiss Axiovert 200 M). The exposure time for the fluorescence signal was first set automatically by the software and then adjusted manually so that the signals were within the full dynamic range. Either the glow scale lookup table or the histogram was used to monitor the saturation level.
Synaptosome Purification
For purification of synaptosomes from adult rat brains, dissected cortical tissue was minced and homogenized in either ice-cold radioimmune precipitation assay (RIPA) lysis buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% SDS) for the, and 0.1% SDS) for the control lysate or in ice-cold synaptosome solution (0.32 m sucrose, 1 mm NaHCO3, 1 mm MgCl2, and 0.5 mm CaCl2), both containing mini complete protease inhibitors (Roche Applied Sciences). Samples were transferred to fresh 15-ml conical tubes, solubilized further by 30 min of extraction at 4 °C, and then centrifuged at 1400 × g for 10 min. The supernatant (S1) was transferred to a new tube and centrifuged at 13,800 × g for 10 min. The remaining pellet (P2) containing the synaptosomes was resuspended in RIPA lysis buffer.
Primary cultured high-density cortical neurons from embryonic day 18 rat embryos were cultured on 60-mm dishes as described previously (23, 24). To purify synaptosomes from these cultures, the cells from two dishes were scraped into 500 μl of HEPES-buffered sucrose (0.32 m sucrose and 4 mm HEPES (pH 7.4)) containing mini complete protease and PhosSTOP phosphatase inhibitors (Roche). Cells were then homogenized with 30 strokes of a glass pestle, transferred to a sterile microcentrifuge tube, and solubilized by rotation at 4 °C for 2 h. A small volume of lysate was reserved as “total lysate.” To isolate cell nuclei (P1) from the lysate, solubilized lysates were centrifuged at 800–1000 × g at 4 °C for 1 min. The resulting supernatant (S1) was then centrifuged at about 10,000 × g at 4 °C for 15 min to yield a crude synaptosomal pellet (P2). Reserved total and P2 lysates were then lysed in modified RIPA lysis buffer containing protease and phosphatase inhibitors.
Protein amounts for both control and synaptosomal samples from adult rat brain and cultured cortical neurons were obtained using the BCA protein determination kit (Thermo Scientific), and samples were diluted to the same protein concentration with RIPA lysis buffer. 2× Laemmli buffer was then added, and samples were denatured on a 95 °C heat block for 10 min. Purity was further confirmed by Western analysis.
Eps15 Knockdown and Overexpression
For Eps15 knockdown experiments, control siRNA (20 nm scrambled siRNA, Ambion) or Eps15 siRNA (20 nm, Ambion, catalog no. s162462, GGCUUUUCACUUAAUCAAUtt) were cotransfected with an EGFP construct into hippocampal neurons cultured onto coverslips at 11 days in vitro with Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. For Eps15 overexpression experiments, pcDNA or Eps15 plasmids were cotransfected with a DsRed construct. Cells were fixed at 14–15 days in vitro and processed for immunostaining. Endogenous Eps15 was visualized with incubation in primary anti-Eps15 rabbit antibody (1:100, Santa Cruz Biotechnology, Inc.) overnight at 4 °C, followed by incubation in goat anti-rabbit secondary antibody conjugated to either Alexa Fluor 555 for knockdown experiments or Alexa Fluor 488 for overexpression experiments. For experiments examining the effect of Eps15 knockdown or overexpression on surface expression of GluA1N only, cells were fixed and stained under non-permeant conditions. Cells were then incubated overnight at 4 °C with a primary anti-GluA1N mouse antibody (1:300, Millipore), followed by incubation in goat anti-mouse secondary antibody conjugated to either Alexa Fluor 555 for knockdown experiments or Alexa Fluor 488 for overexpression experiments. Cells were imaged and collected as described above, and original images were analyzed directly using ImageJ software (available for download from the National Institutes of Health website) to assess total protein levels. All values were reported as mean ± S.E. Statistical analysis was performed using two-population Student's t test.
Immunoprecipitation
To examine the effect of ubiquitin on putative Eps15 protein interaction with GluA1, HEK 293T cells were cotransfected with GFP-tagged GluA1, as described previously (24), and either pcDNA or HA-tagged ubiquitin using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. Two days post-transfection, cells were rinsed with ice-cold PBS and resuspended in 100 μl of modified RIPA lysis buffer containing mini complete protease inhibitor (Roche). To examine the effect of ubiquitin on endogenous Eps15 and GluA1 in rat cortical neurons, cultures were incubated for 24 h with the proteasomal inhibitor MG-132 (5 μm) to increase ubiquitinated species and then washed in ice-cold ACSF and lysed as above. To examine how lysine residues available for ubiquitination on the GluA1 C terminus affect Eps15 and GluA1 interaction, GFP-tagged GluA1 and lysine mutants (F3R, K868R, and 4KR) created as previously described (13) were cotransfected with pcDNA or with HA-tagged ubiquitin in HEK 293T cells. Two days post-transfection, cells were washed with PBS and lysed as above.
All lysates were solubilized by sonication, incubated for 10 min on ice, and then centrifuged for 10 min at 13,000 × g to remove insolubilities. A small volume from each sample was reserved as total input. The remaining sample volumes were adjusted to 500 μl with Nonidet P-40 buffer (50 mm HEPES (pH 7.5), 150 mm NaCl, 5 mm EDTA, and 1% Nonidet P-40 plus mini complete) and incubated overnight for 8–12 h on rotation at 4 °C with 30 μl of a 50% slurry of protein A-Sepharose beads (Santa Cruz Biotechnology) in Nonidet P-40 buffer and 2 μl of Eps15 antibody (Santa Cruz Biotechnology). Immunocomplexes were washed three times with ice-cold Nonidet P-40 buffer, resuspended in 30 μl of 2× Laemmli buffer, and denatured on a 95 °C heat block for 10 min. Immunoprecipitates were resolved by Western blot analysis.
Western Blot Analysis
Lysates were resolved by SDS-PAGE, transferred to PVDF membranes (Bio-Rad), and blocked with 5% nonfat dry milk for 1 h at room temperature. Blots were incubated at 4 °C overnight with primary antibodies diluted in 5% nonfat dry milk. Antibodies used for immunoblots included 1:500 GluA1Nt α-Rb (Millipore), 1:1000 Eps15 α-Rb (Santa Cruz Biotechnology or BD Biosciences), 1:4000 β-tubulin α-Ms (Sigma), 1:4000 Nedd4 α-Rb (Abcam), and 1:500 α-Ms PSD-95 (NeuroMab). The following day, membranes were washed and then incubated in peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody (Sigma) for 1 h at room temperature. After further washing, immunoreactive bands were visualized by ECL (GE Healthcare) and measured by densitometry using ImageJ software. GluA1 protein immunointensity values were normalized to corresponding inputs where appropriate and then normalized to controls prior to statistical analysis. All values were reported as mean ± S.E. Statistical analysis was performed using two-population Student's t test.
Internalization Assays
GFP-tagged GluA1 and the GFP-tagged lysine mutants F3R, K868R, and 4KR were cotransfected with either control siRNA (20 nm scrambled, Ambion) or Eps15 siRNA (20 nm scrambled, Ambion) into 11 days in vitro hippocampal neurons cultured on coverslips using Lipofectamine 2000 according to the instructions of the manufacturer. Internalization assays were performed 2 days post-transfection. Live hippocampal neurons were incubated in medium containing 1:500 GFP α-Rb antibodies (Abcam) for 10 min on ice to label surface GFP-tagged constructs. Cells were washed in ACSF and then incubated in medium containing 25 μm glutamate for 10 min in a 37 °C incubator to promote receptor endocytosis. The medium was replaced, and cells were returned to the incubator for 20 min to allow further receptor internalization. Following the time chase, cells were fixed with ice-cold 4% paraformaldehyde for 10 min, washed with ACSF, and blocked for at least 30 min in 10% normal goat serum/ACSF. Then the remaining surface-associated antibodies were labeled with anti-Rb Alexa Fluor 405-conjugated secondary antibodies under non-permeant conditions (1:300, Invitrogen) for 1 h at room temperature in the dark, washed in ACSF, and permeabilized with 0.3% Triton X-100/ACSF for 10 min at room temperature. Cells were blocked again for at least 30 min in 10% normal goat serum/ACSF before the remaining internalized antibody-bound AMPARs were incubated with anti-Rb Alexa Fluor 555-conjugated secondary antibodies (1:700) for 1 h at room temperature in the dark.
Nedd4 RNA Interference
siRNA was designed and prepared by Santa Cruz Biotechnology, Inc. to specifically target NEDD4 mRNA. NEDD4 siRNA is a pool of three target-specific siRNAs (A, B, and C) against the Mus musculus NEDD4 sequence (catalog no. sc-41080). The NEDD4 siRNAs were as follows: A, CCAUGAAUCUAGAAGAACA; B, GAUCACCUCUCAUACUUCA; and C, CUGUUCACUUGUCCAGUUA. The pool was originally chosen to knock down Nedd4 in both rat and human cell lines. The human Nedd4 sequence shares target sites A (100% homology) and B (94% homology), whereas the rat Nedd4 sequence shares all three sites (A, 89%; B, 100%; and C, 89%). Nedd4 siRNA and control siRNA (scrambled siRNA, Ambion) were transfected into HEK 293T cells at 25 nm using Lipofectamine 2000 according to the instructions of the manufacturer. Two days post-transfection, cells were lysed and processed for Western blot analysis.
Statistical Analysis
Fluorescence intensities were quantified using ImageJ for the soma and the puncta. The “soma” area was defined as the major cellular body excluding dendritic projections, whereas “puncta” refers to individual spine intensities. Puncta were measured collectively along the length of a dendrite by masking the spines according to measurements set to exclude particles outside the normal intensity range of a typical spine. Both soma and puncta were measured to assess whether localized populations of AMPARs were affected by experimental conditions.
RESULTS
Synaptic Localization of Eps15
To examine the subcellular distribution pattern of endogenous Eps15, cultured rat hippocampal neurons were immunostained for Eps15 along with either the synaptic marker protein PSD-95 or AMPAR GluA1 subunits. Eps15 immunosignals were detected throughout the neuron but formed intense clusters along the dendrites, partially colocalizing with the postsynaptic density scaffolding molecule PSD-95 (Fig. 1A). Similarly, most of the Eps15 clusters appeared to codistribute with GluA1 along the dendrites (Fig. 1B), indicating a synaptic localization of Eps15. To further examine the relative protein distribution of Eps15 at synapses, we prepared synaptosomes from both adult rat brain lysate and cultured rat cortical cell lysates. Western blotting demonstrated a strong enrichment of Eps15 in synaptosomal preparations compared with cell lysates (Fig. 1, C and D). Synaptosomal purification was confirmed by an enrichment of the known synaptic proteins PSD-95 and GluA1.
FIGURE 1.
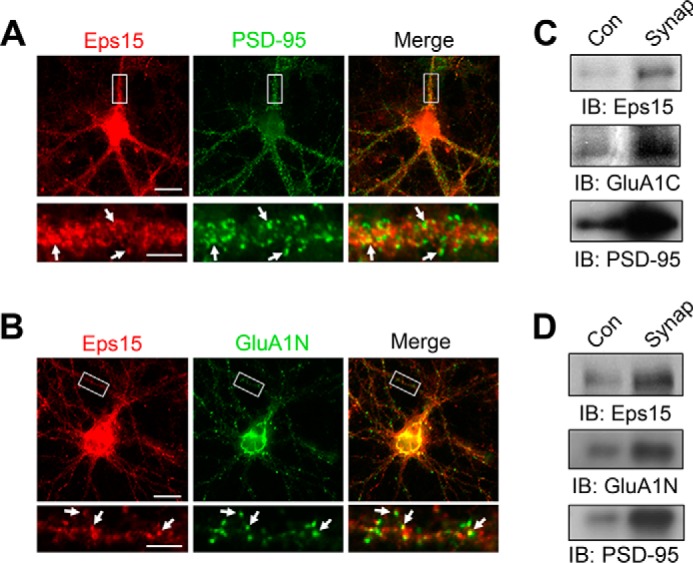
Synaptic localization of Eps15. A and B, double staining in cultured rat hippocampal neurons indicates partial colocalization of either Eps15 (red) and PSD-95 (green) (A) or Eps15 (red) and GluA1N (green) (B). The boxed area was enlarged (bottom panel) for clarity. Arrows indicate puncta of codistribution. Scale bars = 20 μm (images) and 5 μm (dendrite enlargement). C and D, synaptosomal fractions (Synap) of rat cortical lysate (C) and cultured rat cortical neurons (D) show synaptic enrichment of Eps15, GluA1, and the synaptic marker PSD-95. Con, control; IB, immunoblot.
Regulation of AMPAR Surface Expression by Eps15
As an endocytic adaptor, Eps15 is involved in the regulation of membrane protein dynamic distribution. Given the enrichment of Eps15 in the synapse, we next wanted to examine whether Eps15 is implicated in AMPAR surface expression. If Eps15 is involved in promoting AMPAR internalization, a depletion of Eps15 should cause an accumulation of surface AMPARs. To examine this possibility, a siRNA against Eps15 was utilized, and at least a 60% reduction of endogenous Eps15 was observed in siRNA-expressing neurons compared with cells expressing scrambled siRNA (Fig. 2, A and B). To determine the effect of Eps15 knockdown on AMPAR surface expression, transfected neurons were immunostained with anti-GluA1 N-terminal antibodies under non-permeant conditions. In cells transfected with Eps15 siRNA, we found a significant increase in surface GluA1 immunofluorescence intensity compared with cells transfected with scrambled siRNA (soma, 145.0% ± 3.5%; puncta 182.7% ± 5.8% of the control) (Fig. 3, A and B).
FIGURE 2.
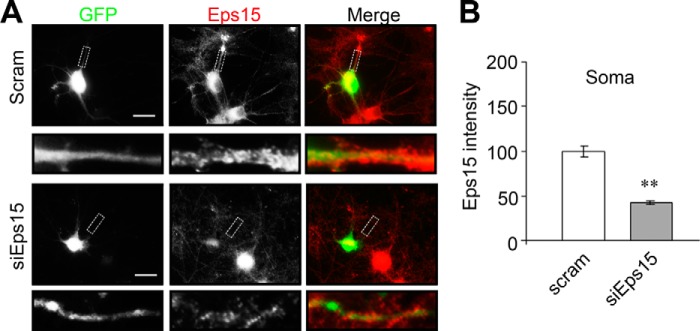
Basal Eps15 levels are affected by siRNA knockdown and overexpression. A, Eps15 siRNA (siEps15) or scrambled control siRNA (Scram) were cotransfected in cultured rat hippocampal neurons with GFP, followed by immunostaining of Eps15 (red) 2 days after transfection. B, pooled data on Eps15 expression. Eps15 level was reduced in neurons transfected with siRNA (Soma, n = 65–73 cells). Scale bars = 20 μm.
FIGURE 3.

Eps15 regulates GluA1 surface expression in neurons. A, Eps15 siRNA (siEps15) or scrambled control siRNA (Scram) were cotransfected with GFP, and cell surface GluA1 (Surf GluA1) was stained with anti-GluA1N antibodies (red) under non-permeant conditions. Scale bars = 20 μm. B, pooled data from A (soma, n = 57–63 cells; puncta, n = 250–300 puncta). Data are mean ± S.E. **, p < 0.01, Student's t test. C, either pcDNA (Con) or Eps15 was cotransfected with GFP, and cell surface GluA1 was stained with anti-GluA1N antibodies (red) under non-permeant conditions. Scale bars = 20 μm. D, pooled data from C (soma, n = 151–154 cells; puncta, n = 350–300 puncta). Con, control. Data are mean ± S.E. **, p < 0.01, Student's t test.
Next, we wanted to know whether overexpression of Eps15 would have the opposite effect. If Eps15 knockdown increases surface AMPARs by impeding AMPAR internalization, Eps15 overexpression should lead to decreased surface AMPARs by enhancing internalization. Toward this end, an Eps15 construct was transfected in hippocampal neurons. Immunostaining revealed an ∼50% increase in Eps15 expression in Eps15-transfected cells compared with pcDNA control cells. As expected, non-permeant staining of GluA1 showed a significant decrease in surface GluA1 in both soma (73.6% ± 2.3%) and puncta (45.5% ± 3.0%) (Fig. 3, C and D). It is interesting to note that Eps15 overexpression produced a lesser overall effect on GluA1 surface localization than Eps15 knockdown, suggesting a high level of endogenous Eps15 amount or activity in the regulation of AMPAR trafficking.
Eps15 Regulates AMPAR Internalization
We find that Eps15 overexpression or knockdown alters surface AMPAR expression, suggesting a role for Eps15 in AMPAR internalization. To examine this possibility, we knocked down Eps15 in cultured hippocampal neurons and then performed internalization assays. In brief, surface AMPARs were live-labeled using anti-GluA1N antibodies. After washing, cells were incubated at 37 °C for 20 min to allow receptor internalization. Any remaining surface-bound antibodies were then blocked with a secondary antibody, whereas internalized receptors were specifically labeled with fluorescent antibodies for visualization. We found that, under basal conditions, Eps15 knockdown caused a modest but significant decrease in receptor internalization both in soma (75.2% ± 2.2%) and puncta (61.4% ± 2.0%) (Fig. 4, A and B). Under glutamate-induced internalization, a similar but more pronounced reduction was observed (soma, 65.9% ± 3.4%; puncta, 51.0% ± 1.5%) (Fig. 5, A and B). Next, we performed similar internalization assays in neurons transfected with Eps15 to overexpress the adaptor protein. We found that overexpression of Eps15 resulted in an increase in the intensity of internalized AMPARs under basal conditions (soma 116.4% ± 3.3%; puncta 143.8% ± 5.7%) (Fig. 4, C and D). A more dramatic effect was observed in glutamate-induced internalization (Fig. 5, C and D) in both soma (127.0% ± 5.8%) and puncta (158.4% ± 8.36%).
FIGURE 4.

Eps15 regulates basal AMPAR endocytic trafficking. In hippocampal neurons, GFP was cotransfected with siEps15 or scrambled (Scram) siRNA as a control. Surface GluA1 subunits were labeled with anti-GluA1N antibodies, and internalization (Intern) assays of constitutive AMPAR internalization were performed. Knockdown of Eps15 (siEps15) inhibited basal AMPAR internalization, whereas overexpression of Eps15 (Eps15 O/E) promoted AMPAR internalization. A and B, soma, n = 145–150 cells; puncta, n = 250–300 puncta. Scale bars = 20 μm. C and D, soma, n = 120–140 cells; puncta, n = 200–250 puncta). Con, control. Data are mean ± S.E. **, p < 0.01, Student's t test. Scale bars = 20 μm.
FIGURE 5.

Eps15 regulates glutamate-induced AMPAR endocytic trafficking. In hippocampal neurons, GFP was cotransfected with Eps1 or pcDNA as a control. Surface GluA1 subunits were labeled with anti-GluA1N antibodies, and internalization (Intern) assays of glutamate-induced (50 μm, 10-min treatment and time-chased for 20 min) AMPAR internalization were performed. A and B, knockdown of Eps15 inhibited induced AMPAR internalization (soma, n = 40–50 cells; puncta, n = 500–550 puncta). Scram, scrambled. Scale bars = 20 μm. C and D, overexpression of Eps15 (Eps15 O/E) promoted AMPAR internalization (soma, n = 50–55 cells; puncta, n = 550–600 puncta). Con, control. Data are mean ± S.E. **, p < 0.01, Student's t test. Scale bars = 20 μm.
Eps15 and GluA1 Interaction Is Ubiquitin-dependent
Although it is clear that Eps15 overexpression and knockdown have an effect on both surface AMPAR localization and AMPAR trafficking, the mechanisms underlying its interaction with AMPARs remain unclear. A commonly recognized cellular signaling mechanism for the internalization of membrane proteins via the endocytotic machinery is ubiquitination. We and other laboratories have found that AMPARs are subject to ubiquitination (11–13). We show that surface GluA1 subunits are ubiquitinated preferentially via the E3 ligase Nedd4, leading to an increase in receptor internalization and degradation and a decrease in receptor surface expression (13). However, how ubiquitinated AMPARs are recognized selectively for internalization remains unknown. Because Eps15 can specifically interact with ubiquitin moieties, it may function as an adaptor to associate with ubiquitinated AMPARs. Therefore, we decided to examine whether ubiquitination is implicated in Eps15 and GluA1 interaction. In HEK 293T cells, GFP-tagged GluA1 (GFP-GluA1) was cotransfected with either pcDNA as a control or HA-tagged ubiquitin. Two days after transfection, Eps15 was immunoprecipitated with anti-Eps15 antibodies, and the immunocomplex was probed with anti-GluA1N antibodies to confirm protein interaction. In lysates cotransfected with pcDNA, only a minimal level of GluA1 was detected, indicating a weak interaction under basal conditions. However, in lysates cotransfected with ubiquitin, a significantly higher level of GluA1 was coimmunoprecipitated with Eps15 (374% ± 49.2% of the control, n = 6) (Fig. 6A), strongly indicating that ubiquitination plays a positive role in Eps15 interaction with GluA1. To confirm this effect in neurons, cultured cortical neurons were treated for 24 h with the proteasomal inhibitor MG-132 to increase the amount of ubiquitinated species. Immunoprecipitation assays showed that proteasome inhibition also significantly increased the association of GluA1 with Eps15 (315.3% ± 35.7%, n = 5) (Fig. 6B). Because AMPAR ubiquitination is regulated by glutamatergic activities (11, 25), we wanted to know whether glutamate affects Eps15 interaction. Toward this end, we treated cultured cortical neurons with 50 μm glutamate for 10 min. Compared with the control, glutamate treatment significantly increased GluA1 and Eps15 interaction (171.2% ± 20.2%, n = 5) (Fig. 6C), consistent with the requirement of AMPAR ubiquitination in receptor-Eps15 interaction.
FIGURE 6.

Eps15 and GluA1 interaction is ubiquitin-dependent. A, GFP-GluA1 was cotransfected in HEK cells with or without HA-tagged Ub. The presence of ubiquitin increased the amount of GluA1 coimmunoprecipitated (IP) with Eps15 (top left panel), although a similar amount of Eps15 was pulled down (bottom left panel). Similar amounts of proteins were shown in the lysates (input). IB, immunoblot. B, cultured rat cortical neurons were treated with 5 μm of the proteasome inhibitor MG-132 (MG). MG significantly increased the amount of GluA1 and Eps15 interaction in five individual experiments. Con, control. C, cortical neurons were treated with glutamate (50 μm, 10 min), and Eps15-GluA1 interaction was examined by coimmunoprecipitation. Glu treatment significantly increased GluA1 and Eps15 interaction in five individual experiments. D, illustration of wild-type Eps15 and Eps15 deletion mutants. EH, Eps15 homology domain; PRM, proline-rich motif. E, GFP-tagged Eps15 deletion mutants were cotransfected with non-tagged GluA1 in HEK 293 cells. Deletion of a region containing two ubiquitin interacting motifs (EΔC) abolished Eps15 and GluA1 interaction, whereas deletion of the AP2 binding region of Eps15 (EΔAP) had no effect. Similar results were observed in three individual experiments.
The UIM Domain Mediates Eps15 Interaction with AMPARs
Structurally, Eps15 has two domains of particular interest: two UIM regions at its C terminus and an AP2 binding region (Fig. 6D). We suspected, because of the increased interaction between Eps15 and GluA1 in the presence of ubiquitin, that the UIM regions were the critical sites of Eps15-GluA1 interaction. Also, the adaptor protein AP2 is known to interact with AMPAR to initiate receptor internalization. Given the AP2 binding domain in Eps15, it is possible that the EPS-GluA1 association is indirect, mediated via AP2. To address these possibilities, we used a set of GFP-tagged Eps15 domain deletion mutants that lack either the C terminus UIM regions (EΔC) or the AP2 binding region (EΔAP) (Fig. 6D). We then transfected HEK 293T cells with either GFP-tagged wild-type or mutant Eps15 together with non-tagged GluA1. Eps15 immunoprecipitates obtained with anti-GFP antibodies were probed for GluA1. In support of a role for ubiquitination and the requirement of the UIM motif in Eps15-GluA1 interaction, we found that deletion of the UIM regions of Eps15 completely abolished Eps15-GluA1 interaction (Fig. 6E). However, deletion of the AP2 binding region of Eps15 had no effect on Eps15-GluA1 interaction (Fig. 5E).
Ubiquitination of the GluA1 K868 Site Is Critical for Eps15 and GluA1 Interaction
Despite the observations that ubiquitin increases the interaction between Eps15 and GluA1, it remained uncertain whether this was a consequence of direct ubiquitination of GluA1 or an indirect result of ubiquitination on some intermediates. Therefore, we turned to closer examination of the role of GluA1 ubiquitination. During ubiquitination, a ubiquitin molecule is covalently conjugated to a lysine residue on its target substrate. Within the GluA1 intracellular domains, there are four lysine residues at the C terminus available for ubiquitin modification. In an earlier study, we found that although all lysine residues can be targeted, the last lysine on the GluA1 C terminus (Lys-868) was a primary site for GluA1 ubiquitination (13). Therefore, we reasoned that this site might also be the critical site in the interaction of GluA1 with Eps15. To examine this possibility, we replaced the first three lysine residues (K813R, K819R, and K822) at the GluA1 C terminus with arginine to create a triple mutant (F3R), leaving Lys-868 as the sole site for ubiquitination (Fig. 7A). The K868R and 4KR mutants described previously (13) were also used (Fig. 7A). Wild-type GluA1 and mutant constructs (F3R, K868R, and 4KR) were cotransfected with ubiquitin in HEK 293T cells, and GluA1-Eps15 interaction was analyzed by coimmunoprecipitation. As described above (Fig. 6A), ubiquitin overexpression enhanced Eps15 interaction with GluA1. Interestingly, a similar increase in protein interaction by ubiquitin was still observed in F3R, indicating that a single intact Lys-868 site is sufficient for ubiquitination-dependent GluA1 interaction with Eps15. In line with this, despite the three remaining lysine residues in the K868R mutant, ubiquitin expression showed only a minimal amount of interaction between Eps15 and GluA1, comparable with the 4KR mutant in which interaction of Eps15 with GluA1 is abolished even in the presence of ubiquitin.
FIGURE 7.

GluA1 ubiquitination is required for Eps15 association. A, illustration of lysine residues at the C terminus (C-term) of GFP-tagged GluA1 and various forms of KR mutants. B and C, GluA1 lysine mutants were cotransfected with or without HA-ubiquitin in HEK 293 cells, and coimmunoprecipitations (IP) were performed to examine GluA1-Eps15 interaction. Ub expression increased Eps15 association with GluA1 and F3K but not K868R or 4KR (n = 4–7). Values were normalized to paired controls. IB, immunoblot. D—F, siEps15 or scrambled control siRNAs (Scram) were cotransfected with GFP-tagged WT or KR mutants of GluA1 in cultured rat hippocampal neurons. Internalization assays were performed by labeling surface GluA1 with anti-GFP antibodies, followed by 10 min of glutamate treatment (50 μm). Compared with paired controls, Eps15 knockdown significantly decreased internalization of GluA1 and F3K but had less of an effect on K868R and no significant effect on 4KR. D, soma, n = 63–110 cells. Scale bars = 40 μm. E, puncta, n = 450–500 puncta. Data are mean ± S.E. *, p < 0.05; **, p < 0.01; Student's t test; n.s., not significant.
To directly examine the requirement of receptor ubiquitination in Eps15-dependent internalization, we transfected cultured hippocampal neurons with the varying lysine mutants and either an Eps15 siRNA or a control scrambled siRNA and performed GluA1 internalization assays 2 days after transfection using glutamate to promote internalization. In brief, surface GFP-GluA1 was labeled with anti-GFP antibodies, and internalization was triggered by glutamate treatment (50 μm, 10 min) at 37 °C. Cells were time-chased for an additional 20 min at 37 °C to allow for further internalization. Among cells expressing scrambled siRNA, F3R was internalized to a level comparable with that of wild-type GluA, whereas the internalization of both K868R and 4KR was reduced markedly (Fig. 7, D–F). siRNA knockdown of Eps15 caused at least a 40% reduction in wild-type GluA1 and F3R. In contrast, Eps15 siRNA resulted in smaller changes in the internalization rate of K868R and 4KR compared with their own respective controls (Fig. 7, D–F). These findings strongly indicate that GluA1 ubiquitination, primarily at the residue of Lys-868, is required for Eps15-mediated internalization.
Nedd4 Enhances Eps15 Interaction with GluA1
During ubiquitination, the final conjugation of a ubiquitin molecule to a lysine residue on the target substrate is mediated by an E3 ligase that confers target specificity. Recent studies have identified Nedd4 as the E3 ligase responsible for AMPAR GluA1 ubiquitination (11, 13). Therefore, we wanted to know whether Nedd4, via ubiquitinating GluA1, may facilitate Eps15 interaction with AMPARs. In HEK cells, GFP-tagged GluA1 was cotransfected with either pcDNA as a control or Nedd4, and anti-Eps15 antibodies were used to examine coimmunoprecipitation of GluA1. In lysates transfected with Nedd4, the amount of GluA1 in Eps15 immunoprecipitates was increased markedly compared with the control (210.1% ± 35.5%, n = 5) (Fig. 8A). To further confirm the effect of Nedd4 in neurons, viral Nedd4 and pHAGE control constructs were introduced to cortical neuronal cultures as described previously (13). Similar to the results from HEK cells, immunoprecipitation assays showed that Nedd4 overexpression resulted in a higher level of interaction between Eps15 and GluA1 (425.8% ± 60.2%, n = 4) (Fig. 8B). We next decided to examine the effect of Nedd4 knockdown on Eps15-GluA1 interaction. HEK cells were transfected with GFP-GluA1 and ubiquitin together with siRNAs targeting Nedd4 or scrambled siRNAs as a control. Eps15 was immunoprecipitated for GluA1 detection. Although the presence of ubiquitin was able to enhance GluA1 and Eps15 interaction in scrambled siRNA controls (170.0% ± 22.1%, n = 4), ubiquitin failed to affect GluA1-Eps15 interaction in neurons expressing Nedd4 siRNA (105.3% ± 15.6%, n = 4) (Fig. 8, C and D). Interestingly, we found that lysates overexpressing Nedd4 or ubiquitin showed higher total GluA1 expression, suggesting that GluA1 protein synthesis may be overregulated by these proteins to compensate for amounts degraded initially. These experiments collectively support a scenario in which Nedd4 targets GluA1 for ubiquitination, which then recruits Eps15 for interaction.
FIGURE 8.

Nedd4 enhances Eps15 association with GluA1. A, GFP-tagged GluA1 was cotransfected in HEK cells with either pcDNA (pc) or Nedd4 (N4). Coimmunoprecipitation (IP) of GluA1 by Eps15 was enhanced significantly by N4, observed consistently in five individual experiments. IB, immunoblot. B, cultured rat cortical neurons were transduced with either a control or N4 virus. The N4 virus significantly increased GluA1 and Eps15 interaction. Similar results were observed in four individual experiments. C and D, GFP-GluA1 was cotransfected in HEK cells with either Ub or pcDNA as control. Each set was additionally cotransfected with either scrambled siRNA (scram) or siRNA against Nedd4 (siN4). Knockdown of Nedd4 markedly reduced basal and ubiquitin-enhanced Eps15-GluA1 association. Data are mean ± S.E. (n = 4). *, p < 0.05,Student's t test.
Eps15-mediated AMPAR Internalization Is Clathrin-dependent
Having established that Eps15 interacts with ubiquitinated AMPARs to cause receptor internalization, we were curious about the internalization machinery involved. The clathrin-coated pits pathway is the canonical mechanism for the internalization of most of the membrane receptors, including AMPARs. AMPAR internalization begins with an association with the adaptor protein AP2, or Eps15 for the ubiquitinated receptors, followed by the recruitment of clathrin to form clathrin-coated pits. Clathrin interacts with amphiphysin, which then brings in dynamin to subsequently pinch off the coated pits to form endocytic vesicles. Because Eps15 is known to mediate receptor internalization through a clathrin-independent pathway (26, 27), we wanted to confirmed whether the Eps15-dependent internalization of ubiquitinated AMPARs utilized the clathrin pathway. We took advantage of a newly developed clathrin inhibitor, Pitstop (28), which binds to clathrin and competitively blocks the recruitment of amphiphysin, leading to potent suppression of clathrin-dependent internalization. We first tested the efficiency of Pitstop by treating cultured hippocampal neurons with Pitstop for 0, 10, or 30 min and examining receptor surface expression. We found that, at a concentration of 15 μm, Pitstop caused a marked increase in GluA1 surface accumulation (time 0, 100% ± 4.5%; time 30, 137.5% ± 5.6%; and time 60, 162.1% ± 8.0%), indicating its effectiveness in blocking receptor internalization (Fig. 9, A and B). To determine the dependence of the clathrin pathway for the internalization of ubiquitinated AMPARs, we examined constitutive and glutamate-induced receptor internalization in neurons transfected with GFP-GluA1 and ubiquitin. Under constitutive conditions, ubiquitin significantly increased the amount of GFP-GluA1 internalization (125.5% ± 6.6%). Application of Pitstop significantly suppressed AMPAR internalization (56.8% ± 2.8%) and abolished the ubiquitin-caused increase in AMPAR internalization (46.4% ± 2.1%) (Fig. 9, C and D). Glutamate-treated neurons showed a significantly increased amount of GluA1 internalization compared with the control (Fig. 9, E and F, Glu, 123.0% ± 4.9%) (). Interestingly, glutamate had less of an effect in neurons overexpressing ubiquitin (Fig. 9, E and F, Glu + Ub, 130.7% ± 5.3%), likely because of a ubiquitin-caused saturation in internalization prior to glutamate treatment. In neurons treated with Pitstop, glutamate-induced GluA1 internalization was decreased significantly even in the presence of Ub (Fig. 9, E and F, Pit + Glu, 73.4% ± 5.2%; Pit + Glu + Ubi, 68.7% ± 3.7%). These data support the utilization of the clathrin-dependent pathway for Eps15-mediated internalization of ubiquitinated AMPARs (Fig. 10).
FIGURE 9.

Eps15-mediated AMPAR internalization is clathrin-dependent. A and B, treatment of cultured hippocampal neurons with the clathrin inhibitor Pitstop (Pit, 15 μm) efficiently blocked AMPAR internalization (n = 18–19 cells). Scale bars = 40 μm. Surf, surface. C and D, neurons were transfected with GFP-GluA1 together with either ubiquitin or pcDNA as control (Con). Two days after transfection, surface GluA1 was labeled with anti-GFP antibodies, and internalization was allowed in the presence of Pitstop for 10 min. Pitstop effectively blocked basal and ubiquitin-enhanced GluA1 internalization (n = 45–95 cells). E and F, the experiment in C was repeated to examine glutamate-induced internalization. Cells were treated with glutamate (50 μm, 10 min) to induce internalization. Pitstop was applied prior to and during glutamate treatment. Pitstop effectively blocked both glutamate- and ubiquitin-stimulated GluA1 internalization (n = 43–174 cells). Data are mean ± S.E. *, p < 0.05; **, p < 0.01; Student's t test; n.s., not significant.
FIGURE 10.

An illustration of Eps15-mediated ubiquitination-dependent AMPAR endocytosis. Left, during basal activity, AMPAR endocytosis is AP2-mediated, and internalized receptors are mostly shuttled to recycling pathways. Right, in contrast, ubiquitination of GluA1, particularly at Lys-868, recruits the adaptor protein Eps15 via its UIM domain with or without AP2 participation. Following Eps15-mediated internalization, the ubiquitinated AMPARs are destined for degradation.
DISCUSSION
Upon conjugation with ubiquitin, AMPARs are recognized for internalization (11, 13), but the molecular steps involved in ubiquitination-triggered internalization are unknown. We show that the Eps15 adaptor protein plays an important role in this process by interacting specifically with ubiquitinated GluA1 subunits to initiate AMPAR internalization via the clathrin-coated pits pathway. In support of this process, we find that Eps15 interacts with GluA1 subunits in a ubiquitination-dependent manner. This interaction is enhanced when GluA1 ubiquitination is induced by overexpressing ubiquitin or the E3 ligase Nedd4 (13) or by glutamate treatment (11). In contrast, the interaction is abolished by mutation of GluA1 ubiquitination sites or deletion of the ubiquitin binding motifs in Eps15. Furthermore, Eps15 overexpression enhances AMPAR internalization and reduces receptor surface expression, which is abolished in cells expressing GluA1 without ubiquitination sites or by inhibition of the clathrin-dependent pathway.
Ubiquitin is conjugated to lysine residues during ubiquitination. Among the intracellular domains in GluA1, there are four lysines in total, all within the C terminus. Although all the lysine residues are targeted, the last lysine (Lys-868) appears to be the primary site for Nedd4-mediated ubiquitination (13). Consistently, we find that the expression of ubiquitin, to enhance GluA1 ubiquitination, strengthens GluA1-Eps15 association. This effect remains in GluA1-F3KR but is completely abolished in K868R and 4KR, indicating Lys-868 as the dominant site for ubiquitination.
In the AMPAR endocytic process, AP2 has been well studied as a clathrin adaptor interacting with GluA1, GluA2, and GluA3 (14–17). On GluA2, AP2 binds to a domain containing the core sequence KRMKV located at the C terminus proximal to the plasma membrane (16). A corresponding sequence, KRMKG, exists in GluA1 and is highly homologous to that in GluA2. Interestingly, this AP2 binding domain contains two lysine residues available for ubiquitin modification. It is possible that ubiquitination of these sites affects AP2 binding affinity. In line with this idea, mutation of the first lysine within this binding sequence abolishes AP2-GluA2 interaction (16). Therefore, ubiquitination may block AP2 binding and switch the endocytic adaptor to Eps15.
Because Eps15 is known to be constitutively associated with AP2 (19, 29), it is possible that Eps15 associates with AMPARs indirectly via AP2. Utilizing an Eps15 mutant lacking the AP2 binding region, we find that the mutant Eps15 remains able to interact with GluA1. We also find that the UIM domains in Eps15 as well as GluA1 ubiquitin modification are required for their interaction. These findings strongly indicate a more direct interaction between the Eps15 UIM motifs and ubiquitin modification at the GluA1 C terminus. Endogenous AMPARs are heterotetramers mostly composed of GluA1/A2 or GluA2/A3. Although both GluA1 and GluA2 are known to be regulated by ubiquitination, GluA2 does not appear to bind Eps15 (17). Also, although Nedd4 selectively targets GluA1 as well as Eps15, it does not appear to target GluA2 (Ref. 11 and Fig. 4E). Therefore, GluA1 C-terminal ubiquitination and Eps15-mediated internalization may specifically regulate GluA1-containing AMPAR trafficking. Eps15 appears to be self-sufficient in mediating AMPAR internalization. In a mutant GluA1-K868R that should, presumably, have only minimal levels of ubiquitin modification but retains an intact AP2 binding domain, we show that glutamate-induced internalization is suppressed significantly.
Several types of adaptor proteins are involved in membrane receptor internalization (21). Of these, epsins are members of the same family as Eps15. Because epsins and Eps15 show overall structural similarity and functional redundancy, it is reasonable to postulate that epsins may also be involved in ubiquitination-dependent AMPAR trafficking. This may explain, together with insufficient Eps15 knockdown, the incomplete abolishment of GluA1 internalization following Eps15 siRNA application.
The relative contribution of AP2 and Eps15 may depend on the cellular and synaptic activity status. Under basal conditions, there is only a low level of AMPAR ubiquitination (11, 13, 30) where Eps15 interaction is minimal and receptor internalization is mainly mediated by AP2. During neuronal activation, which is accompanied by elevated receptor ubiquitination, Eps15 plays a more important role in AMPAR internalization. In line with this notion, at basal conditions, siRNA knockdown of Eps15 decreased GluA1 internalization at the soma by 20% (Fig. 4, A and B). In contrast, under glutamate incubation, which causes AMPAR activation and ubiquitination (11), Eps15 knockdown reduced GluA1 internalization by 40% (Fig. 5, A and B). It is intriguing to postulate that AP2 is responsible for constitutive internalization, whereas Eps15 is used for activity-dependent facilitated trafficking (Fig. 8). Indeed, in GluA2 mutants that lack AP2 association, although NMDA-induced internalization is inhibited, AMPA treatment remains able to stimulate internalization (16). Given that GluA1 ubiquitination is induced by application of AMPA but not NMDA (11), it may indicate that AMPAR activation utilizes ubiquitination/Eps15-dependent internalization, whereas NMDAR-dependent internalization is ubiquitination-independent and is mediated mainly via AP2.
How ubiquitination-dependent AMPAR internalization is regulated remains unclear. Clearly, ubiquitination of AMPARs and their interaction with Eps15 can be regulated by the amount and activity of the E3 ligase Nedd4 and deubiquitinating enzymes (31). In addition to the extent of general ubiquitination, types of ubiquitination can also serve as a regulatory element. AMPARs are subject to mono- and polyubiquitination (11, 13, 30), but polyubiquitination seems to be the dominant form in mammalian neurons (13). Because monoubiquitin appears to have a low binding affinity to UIMs compared with polyubiquitin chains (32, 33), preferential polyubiquitination will strengthen AMPAR association with Eps15. Furthermore, different types of ubiquitin chains can be formed by the further conjugation of ubiquitin units to one of seven lysines on a ubiquitin molecule. It remains unknown which type of polyubiquitination is formed at AMPARs. However, if GluA1 conjugates with multiple forms of ubiquitin chains, it may offer a distinct affinity for Eps15 binding. It has been shown that Eps15 prefers to interact with Lys-63-linked polyubiquitin because of the conformational selectivity of the two UIM domains (34, 35). In line with this, Nedd4, the identified GluA1 E3 ligase, preferentially catalyzes Lys-63 ubiquitination (36). Furthermore, phosphorylation can regulate, and in some cases is a prerequisite for, ubiquitination (37). Given that GluA1 is under constant modification by phosphorylation, which plays an important role in receptor trafficking (38), it is interesting to postulate that GluA1 ubiquitination and, therefore, Eps15-mediated internalization is regulated upstream by protein kinases and receptor phosphorylation.
Ubiquitinated receptors are internalized via clathrin pathways. However, clathrin-independent processes have also been observed (26, 27, 39). For instance, following agonist binding and ubiquitin modification, the TGF-β receptors rapidly internalize via both clathrin-dependent and -independent lipid raft-mediated pathways, which direct internalized receptors for recycling and degradation, respectively (40). In the case of the EGF receptor, a low level of EGF stimulation that does not cause ubiquitination leads to internalization via the clathrin pathway, whereas a high concentration of EGF leads to EGF receptor ubiquitination and recruitment to lipid rafts for internalization (26). Using a clathrin-specific inhibitor, we found that blocking the clathrin-dependent pathway increases surface AMPAR expression. Importantly, both basal and glutamate or ubiquitin-enhanced internalization are equally suppressed by clathrin inhibition, indicating that the clathrin-dependent pathway is utilized in Eps15-mediated internalization of ubiquitinated AMPARS, consistent with other studies showing the involvement of the clathrin route in ubiquitination/Eps15-mediated receptor internalization (41, 42).
Acknowledgments
We thank Dr. Alexandre Benmerah (Institut Cochin, Université Paris Descartes) for the Eps15 constructs and the members of the Man laboratory members for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant MH 079407 (to H. Y.M.).
- AMPAR
- AMPA receptor
- UIM
- ubiquitin-interacting motif
- ACSF
- artificial CSF
- RIPA
- radioimmune precipitation assay
- Ub
- ubiquitin
- N4
- Nedd4.
REFERENCES
- 1. Malinow R., Malenka R. C. (2002) AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126 [DOI] [PubMed] [Google Scholar]
- 2. Collingridge G. L., Isaac J. T., Wang Y. T. (2004) Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 5, 952–962 [DOI] [PubMed] [Google Scholar]
- 3. Turrigiano G. G. (2008) The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang G., Gilbert J., Man H. Y. (2012) AMPA receptor trafficking in homeostatic synaptic plasticity: functional molecules and signaling cascades. Neural Plast. 2012, 825364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pozo K., Goda Y. (2010) Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 66, 337–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nandi D., Tahiliani P., Kumar A., Chandu D. (2006) The ubiquitin-proteasome system. J. Biosci. 31, 137–155 [DOI] [PubMed] [Google Scholar]
- 7. Kato A., Rouach N., Nicoll R. A., Bredt D. S. (2005) Activity-dependent NMDA receptor degradation mediated by retrotranslocation and ubiquitination. Proc. Natl. Acad. Sci. U.S.A. 102, 5600–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patrick G. N., Bingol B., Weld H. A., Schuman E. M. (2003) Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr. Biol. 13, 2073–2081 [DOI] [PubMed] [Google Scholar]
- 9. Bingol B., Schuman E. M. (2004) A proteasome-sensitive connection between PSD-95 and GluR1 endocytosis. Neuropharmacology 47, 755–763 [DOI] [PubMed] [Google Scholar]
- 10. Hou Q., Gilbert J., Man H. Y. (2011) Homeostatic regulation of AMPA receptor trafficking and degradation by light-controlled single-synaptic activation. Neuron 72, 806–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schwarz L. A., Hall B. J., Patrick G. N. (2010) Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J. Neurosci. 30, 16718–16729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lussier M. P., Nasu-Nishimura Y., Roche K. W. (2011) Activity-dependent ubiquitination of the AMPA receptor subunit GluA2. J. Neurosci. 31, 3077–3081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin A., Hou Q., Jarzylo L., Amato S., Gilbert J., Shang F., Man H. Y. (2011) Nedd4-mediated AMPA receptor ubiquitination regulates receptor turnover and trafficking. J. Neurochem. 119, 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carroll R. C., Beattie E. C., Xia H., Lüscher C., Altschuler Y., Nicoll R. A., Malenka R. C., von Zastrow M. (1999) Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 96, 14112–14117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Man H. Y., Lin J. W., Ju W. H., Ahmadian G., Liu L., Becker L. E., Sheng M., Wang Y. T. (2000) Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron 25, 649–662 [DOI] [PubMed] [Google Scholar]
- 16. Lee S. H., Liu L., Wang Y. T., Sheng M. (2002) Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron 36, 661–674 [DOI] [PubMed] [Google Scholar]
- 17. Kastning K., Kukhtina V., Kittler J. T., Chen G., Pechstein A., Enders S., Lee S. H., Sheng M., Yan Z., Haucke V. (2007) Molecular determinants for the interaction between AMPA receptors and the clathrin adaptor complex AP-2. Proc. Natl. Acad. Sci. U.S.A. 104, 2991–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chi S., Cao H., Chen J., McNiven M. A. (2008) Eps15 mediates vesicle trafficking from the trans-Golgi network via an interaction with the clathrin adaptor AP-1. Mol. Biol. Cell 19, 3564–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benmerah A., Bégue B., Dautry-Varsat A., Cerf-Bensussan N. (1996) The ear of α-adaptin interacts with the COOH-terminal domain of the Eps 15 protein. J. Biol. Chem. 271, 12111–12116 [DOI] [PubMed] [Google Scholar]
- 20. van Bergen En Henegouwen P. M. (2009) Eps15: a multifunctional adaptor protein regulating intracellular trafficking. Cell Commun. Signal. 7, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Traub L. M. (2009) Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat. Rev. Mol. Cell Biol. 10, 583–596 [DOI] [PubMed] [Google Scholar]
- 22. Piper R. C., Lehner P. J. (2011) Endosomal transport via ubiquitination. Trends Cell Biol. 21, 647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hou Q., Zhang D., Jarzylo L., Huganir R. L., Man H. Y. (2008) Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc. Natl. Acad. Sci. U.S.A. 105, 775–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Man H. Y., Sekine-Aizawa Y., Huganir R. L. (2007) Regulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc. Natl. Acad. Sci. U.S.A. 104, 3579–3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jarzylo L. A., Man H. Y. (2012) Parasynaptic NMDA receptor signaling couples neuronal glutamate transporter function to AMPA receptor synaptic distribution and stability. J. Neurosci. 32, 2552–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sigismund S., Woelk T., Puri C., Maspero E., Tacchetti C., Transidico P., Di Fiore P. P., Polo S. (2005) Clathrin-independent endocytosis of ubiquitinated cargos. Proc. Natl. Acad. Sci. U.S.A. 102, 2760–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen H., De Camilli P. (2005) The association of epsin with ubiquitinated cargo along the endocytic pathway is negatively regulated by its interaction with clathrin. Proc. Natl. Acad. Sci. U.S.A. 102, 2766–2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. von Kleist L., Stahlschmidt W., Bulut H., Gromova K., Puchkov D., Robertson M. J., MacGregor K. A., Tomilin N., Tomlin N., Pechstein A., Chau N., Chircop M., Sakoff J., von Kries J. P., Saenger W., Kräusslich H. G., Shupliakov O., Robinson P. J., McCluskey A., Haucke V. (2011) Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 146, 471–484 [DOI] [PubMed] [Google Scholar]
- 29. Benmerah A., Gagnon J., Bègue B., Mégarbané B., Dautry-Varsat A., Cerf-Bensussan N. (1995) The tyrosine kinase substrate eps15 is constitutively associated with the plasma membrane adaptor AP-2. J. Cell Biol. 131, 1831–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burbea M., Dreier L., Dittman J. S., Grunwald M. E., Kaplan J. M. (2002) Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron 35, 107–120 [DOI] [PubMed] [Google Scholar]
- 31. Kowalski J. R., Dahlberg C. L., Juo P. (2011) The deubiquitinating enzyme USP-46 negatively regulates the degradation of glutamate receptors to control their abundance in the ventral nerve cord of Caenorhabditis elegans. J. Neurosci. 31, 1341–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Madshus I. H. (2006) Ubiquitin binding in endocytosis: how tight should it be and where does it happen? Traffic 7, 258–261 [DOI] [PubMed] [Google Scholar]
- 33. Hawryluk M. J., Keyel P. A., Mishra S. K., Watkins S. C., Heuser J. E., Traub L. M. (2006) Epsin 1 is a polyubiquitin-selective clathrin-associated sorting protein. Traffic 7, 262–281 [DOI] [PubMed] [Google Scholar]
- 34. Sims J. J., Cohen R. E. (2009) Linkage-specific avidity defines the lysine 63-linked polyubiquitin-binding preference of rap80. Mol. Cell 33, 775–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sato Y., Yoshikawa A., Mimura H., Yamashita M., Yamagata A., Fukai S. (2009) Structural basis for specific recognition of Lys 63-linked polyubiquitin chains by tandem UIMs of RAP80. EMBO J. 28, 2461–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim H. C., Huibregtse J. M. (2009) Polyubiquitination by HECT E3s and the determinants of chain type specificity. Mol. Cell Biol. 29, 3307–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sehat B., Andersson S., Vasilcanu R., Girnita L., Larsson O. (2007) Role of ubiquitination in IGF-1 receptor signaling and degradation. PLoS ONE 2, e340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Song I., Huganir R. L. (2002) Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 25, 578–588 [DOI] [PubMed] [Google Scholar]
- 39. Aguilar R. C., Wendland B. (2005) Endocytosis of membrane receptors: two pathways are better than one. Proc. Natl. Acad. Sci. U.S.A. 102, 2679–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Di Guglielmo G. M., Le Roy C., Goodfellow A. F., Wrana J. L. (2003) Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat. Cell Biol. 5, 410–421 [DOI] [PubMed] [Google Scholar]
- 41. Benmerah A., Bayrou M., Cerf-Bensussan N., Dautry-Varsat A. (1999) Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J. Cell Sci. 112, 1303–1311 [DOI] [PubMed] [Google Scholar]
- 42. Benmerah A., Poupon V., Cerf-Bensussan N., Dautry-Varsat A. (2000) Mapping of Eps15 domains involved in its targeting to clathrin-coated pits. J. Biol. Chem. 275, 3288–3295 [DOI] [PubMed] [Google Scholar]