

Background: We examined alternative mechanisms by which fumarate levels contribute to hypoxia inducible factor (HIF)-1α accumulation and fumarate hydratase (FH)-deficient renal carcinogenesis.
Results: Fumarate promotes HIF-1α transcription through Tank binding kinase 1 (TBK1)-dependent noncannonical activation of NF-κB signaling.
Conclusion: Fumarate-mediated, TBK-dependent accumulation of HIF-1α mediates cell invasion in FH-deficient RCC.
Significance: TBK is a novel putative therapeutic target for the treatment of aggressive fumarate-driven tumors.
Keywords: Cancer Biology, Cell Metabolism, Hypoxia-inducible Factor (HIF), NF-κB (NF-KB), Small Molecule, Small
Abstract
Inactivating mutations of the gene encoding the tricarboxylic acid cycle enzyme fumarate hydratase (FH) have been linked to an aggressive variant of hereditary kidney cancer (hereditary leiomyomatosis and renal cell cancer). These tumors accumulate markedly elevated levels of fumarate. Fumarate is among a growing list of oncometabolites identified in cancers with mutations of genes involved in intermediary metabolism. FH-deficient tumors are notable for their pronounced accumulation of the transcription factor hypoxia inducible factor-1α (HIF-1α) and aggressive behavior. To date, HIF-1α accumulation in hereditary leiomyomatosis and renal cell cancer tumors is thought to result from fumarate-dependent inhibition of prolyl hydroxylases and subsequent evasion from von Hippel-Lindau-dependent degradation. Here, we demonstrate a novel mechanism by which fumarate promotes HIF-1α mRNA and protein accumulation independent of the von Hippel-Lindau pathway. Here we demonstrate that fumarate promotes p65 phosphorylation and p65 accumulation at the HIF-1α promoter through non-canonical signaling via the upstream Tank binding kinase 1 (TBK1). Consistent with these data, inhibition of the TBK1/p65 axis blocks HIF-1α accumulation in cellular models of FH loss and markedly reduces cell invasion of FH-deficient RCC cancer cells. Collectively, our data demonstrate a novel mechanism by which pseudohypoxia is promoted in FH-deficient tumors and identifies TBK1 as a novel putative therapeutic target for the treatment of aggressive fumarate-driven tumors.
Introduction
Alterations in cellular metabolism are now considered a key phenotype of cancer. One of the most notable examples of the connection between metabolism and cancer is through the identification of oncometabolites. Oncometabolites represent small molecules, which can accumulate secondary to enzymatic alterations in cancer with putative transforming properties. To date, the most common oncometabolites include fumarate, succinate, and 2-hydroxyglutarate, which have been linked with mutations of genes encoding fumarate hydratase (FH),3 succinate dehydrogenase (SDH), and isocitrate dehydrogenase (IDH1/2), respectively (1, 2). Fumarate accumulation has been identified principally in the context of a hereditary cancer syndrome referred to as hereditary leiomyomatosis and renal cell cancer (HLRCC). Patients with HLRCC harbor germline mutations of the FH gene and are at risk for the development of aggressive renal cell carcinoma (RCC) (3, 4). FH is an enzyme of the tricarboxylic acid (TCA) cycle that converts fumarate to malate. As such, inactivating mutations of FH result in elevated cellular levels of fumarate (1). Fumarate shares structural similarity with another TCA cycle intermediate α-ketoglutarate, also referred to as 2-oxglutarate (2-OG). 2-OG is a required cofactor for a family of enzymes called 2-OG-dependent dioxygenases (5). Among the enzymes that belong to this enzyme family are the prolyl hydroxylases. The most well established substrates of the prolyl hydroxylases are the α subunits of hypoxia-inducible factor (HIF) (6–8). HIF is comprised of α subunits (either HIF-1α or HIF-2α), which are labile under normoxic conditions and a constitutively expressed β subunit (HIF-1β; also referred to as ARNT). Proline hydroxylation of HIF-α by prolyl hydroxylases facilitates recognition by the E3 ubiquitin ligase pVHL (9–13). Ubiquitination by the von Hippel-Lindau (VHL) complex targets HIF-α for proteosomal mediated degradation (11). Disruptions of this response lead to aberrant expression of HIF-α. Current models for HLRCC indicate elevated fumarate and reactive oxygen species lead to stabilization of HIF-1α through inhibition of prolyl hydroxylation therefore preventing VHL-mediated degradation (14, 15). A notable observation in FH-deficient tissues and cell lines is the increased expression of HIF-1α (14, 16). Although prevention of degradation is a mechanism by which HIF can accumulate, HIF-1α is also subject to regulation at the level of synthesis, including transcriptional regulation (17). Given the robust expression of HIF-1α in HLRCC renal tumors, we set out to examine the contribution of fumarate in the transcriptional regulation of HIF-1α.
EXPERIMENTAL PROCEDURES
Chemicals
Diethyl fumarate (DEF) and dimethyl fumarate (DMF), dimethyl sulfoxide and all other chemicals were purchased from Sigma. BX-795 (TBK1 inhibitor) was purchased from Axon Medchem.
Cells
HK-2 and HEK-293 cells were obtained from the American Type Culture Collection. RCC4 cells were kindly provided by P. Ratcliffe (Oxford). Paired mouse embryonic fibroblast (MEF) lines (WT, FH−/−, FH−/−, and FH) were kindly provided by P. Pollard (Oxford) and have previously been described (18). IKKα/β null MEFs (double knock-out; DKO) were kindly provided by Inder Verma (Salk Institute). UOK262 cells were acquired from WM Linehan (National Institutes of Health, NCI) and have previously been reported (15). UOK262 cells were stably transfected utilizing retrovirus with a control vector (pBabePuro) or a vector containing wild-type FH with a C-terminal FLAG tag. Puromycin-resistant clones were selected and screened for transgene expression via immunoblotting. All cell lines except UOK262, MEFS, and HK-2 were cultured in low glucose (1 g/liter) Dulbecco's modified Eagle's medium (DMEM) supplemented with penicillin (100 units/ml), streptomycin (100 mg/ml), 10% heat-inactivated fetal bovine serum, and HEPES (10 mm). HK-2 cells were purchased from ATCC and cultured in DMEM/Ham's F-12 media with l-glutamine supplemented with penicillin (100 units/ml), streptomycin (100 mg/ml), 10% heat-inactivated fetal bovine serum, and HEPES (10 m). MEFs were cultured in DMEM containing 4.5 g/liter of glucose supplemented with 10% (v/v) fetal bovine serum (Sigma), 2 mm glutamine (Sigma) and maintained in a humidified atmosphere of 5% CO2 and 21% O2. UOK262 cells were grown similar to MEFs along with addition of 100 μm sodium pyruvate.
Fumarate Treatment
All cell lines were treated with cell permeable esterified derivatives of fumarate, DEF, and DMF at various concentrations (0 to 150 μm) in serum-free media.
Immunoblotting
Nuclear extracts were prepared utilizing differential salt lysis buffers as previously described (14). All other immunoblot analyses were performed on whole-cell lysates prepared with the use of radioimmunoprecipitation assay buffer (50 mm Tris-HCl, 150 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) supplemented with protease inhibitor mixture (Roche Applied Science). Protein (either nuclear or whole cell) was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Membranes were blocked with 5% nonfat milk in TBST (10 mm Tris-HCl, 100 mm NaCl, 0.1% Tween 20). Membranes were then incubated with antibodies from the following sources: HIF-1α (BD Biosciences (mouse monoclonal) or Cayman (rabbit polyclonal)), total p65, p65 Ser536, IKKα, IKKβ, total TBK1, TBK1 Ser172, total IκB, Tubulin, GAPDH, CREB, LaminB, and Actin (Cell Signaling), FH (Genetex), and FLAG (Sigma). Membranes were washed with TBST followed by incubation with the indicated secondary antibody (horseradish peroxidase conjugated). Proteins were visualized with enhanced chemiluminescence.
RNA Interference
For FH, p65, and TBK1 knockdown, cells were transfected with pooled siRNA reagent (Dharmacon) with the Amaxa Nucleofector system according to the manufacturer's protocol. Cells were harvested at 48–72 h following transfection. A non-targeting scramble siRNA pool was used as a negative control (Dharmacon).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) assay kit (Millipore) was utilized to examine the binding of p65 (RelA) to the HIF-1α promoter. RCC4 cells and UOK262 FH overexpression along with control cells were fixed with 1% formaldehyde on ice to cross-link the proteins bound to the chromatin DNA. After washing, the tissue was homogenized and the chromatin DNA was sheared by sonication to produce DNA fragments of around 500–1,000 base pairs. The same amounts of sheared DNA were used for immunoprecipitation with antibody against p65 antibody (Cell Signaling Technologies) or an equal amount of preimmune rabbit IgG (Millipore). The immunoprecipitate then was incubated with protein A-agarose/salmon sperm DNA (Millipore), and the antibody-protein-DNA-agarose complex was collected for subsequent reverse cross-linking. The same amount of sheared DNA without antibody precipitation was processed for reverse cross-linking and served as input control. DNA recovered from reverse cross-linking was used for PCR. PCR was performed with primers for the HIF-1α promoter (forward, 5′-GAACAGAGAGCCCAGCAGAG-3′ and reverse, 5′-TGTGCACTGAGGAGCTGAGG-3′) flanking the NF-κB binding site (−197/188 base pairs) at 55 °C for 35 cycles (19).
Isolation of RNA and Real-time RT-PCR
Total RNA was isolated from appropriate treatment conditions according to each experiment using the RNAeasy kit (Qiagen). The RNA concentration was measured using UV spectroscopy (Nanodrop 2000). cDNA was synthesized using 1 μg of total RNA (High capacity cDNA reverse transcription kit, ABI). Expression of the HIF-1α gene was determined (TaqMan probe Hs00936368_m1 HIF1A) using RT-PCR analysis, quantitated by Δ-Δ Ct analysis, and normalized to 18 S (Hs99999901_s1 18 S) RNA. Differences in gene expression between the various treatment conditions were expressed relative to their appropriate control cells.
RESULTS
Fumarate Induces HIF-1α mRNA Levels
We initially compared HIF-1α mRNA expression in normal immortalized renal epithelial HK2 cells and FHs-deficient UOK262 renal cancer cells. We find UOK262 cells expressed higher mRNA levels of HIF-1α as examined by real-time RT-PCR, and protein levels as examined by Western blot analysis compared with HK2 cells (Fig. 1A, top and bottom panels, respectively). To determine whether loss of FH contributes to enhanced HIF-1α mRNA expression, we re-introduced wild-type (WT) FH, which harbors a FLAG tag (FH-FLAG), back to FH-deficient cells. Re-introduction of FH-FLAG into UOK262 reduced HIF-1α mRNA expression, and HIF-1α protein levels compared with UOK262 control vector-transfected cells (Fig. 1B, top and bottom panel). FH-FLAG expression was confirmed by Western blot analysis using FLAG antibody (Fig. 1B, bottom panel).
FIGURE 1.
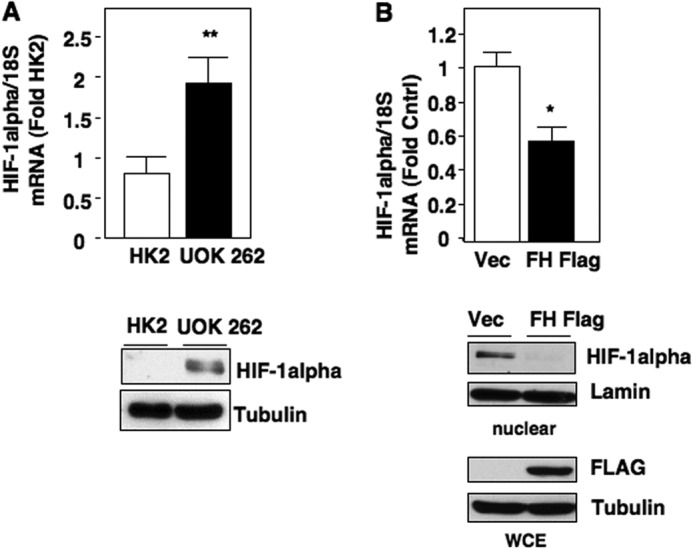
Cell models of high fumarate state demonstrate increased HIF-1α mRNA levels. A, HIF-1α mRNA expression was assessed in HK2- and FH-deficient UOK 262 cultured cells and normalized to 18 S RNA as described under “Experimental Procedures.” In parallel, HIF-1α protein expression was analyzed by Western blot. Tubulin was used as a loading control. The data were quantitated and the results are expressed as mean ± S.E., **, p < 0.01. B, HIF-1α mRNA levels were examined in FH-deficient UOK 262 stably transfected with FH FLAG or vector control (Vec). The data are expressed as mean ± S.E., *, p < 0.05. HIF-1α expression was analyzed in the nuclear extract and Lamin was used as a loading control. FLAG antibodies were used to detect overexpression of FH in whole cell lysate (WCE). Tubulin was used as a loading control.
As reintroduction of WT FH reduces fumarate levels in UOK262 cells (16), we considered whether elevated fumarate levels promote HIF-1α transcription. Fumarate has previously been shown to block HIF-α degradation via inhibition of prolyl hydroxylase hydroxylation/VHL-dependent degradation (14). Therefore to determine the specific effects of fumarate on HIF-1α synthesis, we used cultured cells, which do not express VHL as a model system. Endogenous levels of intracellular fumarate have been reported at ≈700 mm in FH-deficient UOK262 cells (20). We initially utilized two esterified forms of fumarate, DEF and DMF, to permit cell entry at concentrations of 0–150 and 0–200 μm, respectively, as previously reported for exogenous treatment (21, 22). We find that VHL-deficient RCC4 cells exposed to DEF or DMF resulted in a dose-dependent increase in both HIF-1α mRNA and protein levels (Fig. 2, A and B, upper and lower panels, respectively). Similar effects of DEF and DMF on HIF-1α mRNA and protein levels were observed in VHL-deficient RCC10 cells (data not shown). Next, we utilized a genetic approach to examine the effects of FH loss on HIF-1α mRNA. siRNA-mediated knockdown of FH in RCC4 cells resulted in an increase in both HIF-1α mRNA and protein levels (Fig. 2, C and D, respectively). Collectively, these data indicate that fumarate enhances mRNA expression of HIF-1α.
FIGURE 2.

Effects of fumarate on HIF-1α expression in VHL-deficient cells. VHL-deficient RCC4 cells were treated with increasing concentrations of DEF or DMF in serum-free media for 8 h. A, upper panel, HIF-1α mRNA was assessed in DEF-treated cells as described in the legend to Fig. 1 and expressed relative to dimethyl sulfoxide-treated control. Lower panel, HIF-1α protein levels were examined by Western blot in parallel. Tubulin was used as a loading control. The data were quantitated and the results are expressed as mean ± S.E. *, p < 0.05 and **, p < 0.01. B, upper panel, HIF-1α mRNA was assessed in DMF-treated cells as described in the legend to Fig. 1 and expressed relative to dimethyl sulfoxide-treated control. Lower panel, HIF-1α protein levels were examined by Western blot in parallel. GAPDH was used as a loading control. The data were quantitated and the results are expressed as mean ± S.E. *, p < 0.05. C, HIF-1α mRNA expression was analyzed in RCC4 cells silenced of fumarate hydratase using siRNA (siFH) or scrambled control (scr). The data were quantitated and the results are expressed as mean ± S.E. ***, p < 0.001. D, HIF-1α and FH expression was assessed in total cell lysates prepared from RCC4 cells transfected with siFH or scrambled control from C. Actin was used as loading control.
Fumarate Induces HIF-1α mRNA Expression through an NF-κB-dependent Mechanism
Multiple studies have established the NF-κB transcription factor complex as a promoter of HIF-1α transcription (17, 23). The NF-κB complex consists of RelA (p65), p50, and IκB. Upon stimulation, IκB is phosphorylated and degraded by the 26 S proteosome(24). p65 is activated via phosphorylation on Ser536 and translocates to the nucleus to up-regulate target genes (24–27). We examined p65 phosphorylation at Ser536, a marker of NF-κB activation, following treatment with esterified fumarate. Both DEF and DMF promote p65 Ser536 phosphorylation in RCC4 cells, which was concomitant with a decrease in IκB levels (Fig. 3A, left and right panels). Similar findings of p65 activation were obtained in RCC10 cells exposed to DEF and DMF (data not shown).
FIGURE 3.

Fumarate-induces HIF-1α mRNA expression through an NF-κB-dependent mechanism. A, RCC4 cells were treated with increasing concentrations of DEF and DMF (left and right panels, respectively) in serum-free media for 8 h. Cell lysates were prepared and phospho-p65 Ser536 (p-p65 S536), total p65, and IκB expression were analyzed by Western blot. Actin was used as loading control. B, RCC4 cells were transiently transfected with siRNA oligos for fumarate hydratase (siFH) or scrambled control (scr). Total protein was analyzed for phospho-p65 Ser536, total p65, and FH. Actin was used as loading control. C, p65 was transiently down-regulated in RCC4 cells using small inhibitory RNA (siRNA). Scrambled oligos were transfected as a negative control. Cell lysates were prepared and expression of HIF-1α, phospho-p65 Ser536, and total p65 were assessed. GAPDH was used as loading control. D, HIF-1α DNA associated with p65 was assessed in nuclear extracts of RCC4 cells treated with 100 μm DEF for 8 h using ChIP as described under “Experimental Procedures.” Input DNA from RCC4 cells treated or not with DEF was used as a positive control for HIF-1α PCR. Immunoprecipitation using nonspecific IgG was used as a negative control in RCC4 cells treated or not with DEF. HIF-1α is detected at 518 bp. Nonspecific (NS) bands and primer dimer bands (PD) are indicated by arrows. The data were quantitated from two independent experiments and the results are expressed as mean ± S.E. **, p < 0.01 (lower panel).
We next examined the effects of FH knockdown on p65 phosphorylation. siRNA-mediated knockdown of FH in RCC4 cells resulted in increased p65 Ser536 phosphorylation (Fig. 3B). Based on these results, we examined whether inhibition of p65 could interfere with the effect of fumarate on HIF-1α levels. RCC4 cells were transfected with scrambled control or specific siRNA for p65 and subsequently treated with DEF. Knockdown of p65, but not scrambled control, inhibited DEF-induced HIF-1α accumulation (Fig. 3C). Successful knockdown of p65 was confirmed by Western blot analysis (Fig. 3C). To further define the relationship between fumarate-dependent, p65-mediated HIF-1α mRNA accumulation, we assessed p65 binding at the HIF-1α promoter via ChIP utilizing primers directed to a previously reported NF-κB binding element within the HIF-1α promoter (19). Treatment of RCC4 cells with DEF resulted in a marked increase in p65 binding to the HIF-1α promoter relative to control treated cells (Fig. 3D, upper panel). Quantitation is presented (Fig. 3D, lower panel).
Fumarate Promotes p65 Phosphorylation in an IKK-independent, TBK1-dependent Manner
To further characterize the mechanism by which fumarate enhances mRNA expression of HIF-1α, we utilized FH−/− MEFs. We find that phosphorylation of p65 at Ser536 is enhanced in FH−/− MEFs compared with wild-type MEFs, whereas total levels of p65 did not change (Fig. 4A). Importantly, re-introducing wild-type FH cDNA into FH−/− MEFs (FH−/− + FH) reduced endogenous phosphorylation of p65 Ser536 and HIF-1α expression (Fig. 4A). Similarly, treatment of WT MEFs with increasing concentrations of DMF stimulated p65 Ser536 phosphorylation similar to the endogenous levels found in FH null (−/−) MEFs (Fig. 4B). The effects observed in our models of high fumarate states on p65 phosphorylation prompted us to examine known kinases that phosphorylate p65 at Ser536. The most well characterized upstream kinases of p65 at Ser536 include IKKα and IKKβ. These upstream p65 kinases constitute part of the canonical NF-κB signaling pathway. We therefore wanted to determine whether the effect of fumarate on p65 phosphorylation was IKK dependent. As fumarate promotes p65 phosphorylation in cells of murine origin, we tested the effect of fumarate on p65 in IKK α/β DKO MEFs. Unexpectedly, we find that p65 was readily phosphorylated at Ser536 and IκB levels were reduced in DKO cells treated with DEF, whereas not affecting total p65 levels (Fig. 4C). Together, this suggests fumarate activates p65 independent of IKKα and IKKβ and suggests fumarate mediates HIF-1α mRNA expression through a non-canonical NF-κB signaling pathway.
FIGURE 4.

Fumarate promotes p65 phosphorylation in an IKK-independent manner. A, HIF-1α phospho-p65 Ser536 (p-p65 S536) and total p65 were analyzed by Western blot analysis using cell lysates prepared from FH null MEFs or FH-deficient MEFs stably expressing FH (FH−/− + FH). Tubulin expression was used as loading control. B, HIF-1α, phospho-p65 Ser536, and total p65 expression was examined in FH-null MEFs and wild-type (WT) MEFs treated with increasing concentrations of DMF in serum-free media for 6 h. Actin was used as loading control. C, DKO IKKα and IKKβ MEFs were treated with increasing concentrations of DEF in serum-free media for 6 h. Whole cell lysates were analyzed for phospho-p65 Ser536 and total p65, and IκB. Actin was used as loading control.
We therefore considered alternate kinases that have previously been reported to phosphorylate p65 at Ser536. Tank binding kinase 1 (TBK1) is among the known upstream kinases of p65 Ser536 (28). TBK1 is a member of the IKK-related kinases and is a known upstream mediator of non-canonical NF-κB activation (29). TBK1 contains an autophosphorylation site at Ser172, which is indicative of TBK1 kinase activation (30). To determine the effects of fumarate on TBK1 activation, we treated DKO IKKα/β MEFs with increasing concentrations of DEF. Importantly, we find TBK1 is readily phosphorylated at Ser172 in IKK α/β null cells exposed to DEF (Fig. 5A). In a similar manner, DEF and DMF resulted in dose-dependent TBK1 activation in RCC4 and RCC10 cells (Fig. 5B, upper and lower panels, respectively, and data not shown). Next we transiently silenced FH using siRNA in RCC4 cells and found that knockdown of FH, but not scrambled control, lead to enhanced phosphorylation of TBK1 while not changing the overall TBK1 levels (Fig. 5C). In support of these findings, we find that FH-deficient MEFs have enhanced basal phosphorylation of TBK1 compared with WT MEFs (Fig. 5D). Re-introduction of FH to FH-null MEFs resulted in a decrease of TBK1 phosphorylation (Fig. 5D). Together, these data suggest fumarate plays a novel role in the activation of TBK1.
FIGURE 5.

Fumarate mediates HIF-1α mRNA expression through non-canonical NF-κB signaling. A, phospho-TBK Ser172 (p-TBK S172) and total TBK expression was examined in DKO (IKKα and IKKβ) MEFs treated with increasing concentrations of DEF in serum-free media for 6 h. B, phospho-TBK Ser172 and total TBK expression was assessed in RCC4 cells treated with increasing concentrations of DEF (upper panel) and DMF (lower panel) for 8 h in serum-free media. C, RCC4 cells were transiently transfected with siFH or scrambled control and total protein was analyzed for phospho-TBK Ser172, and total TBK. Actin was used as loading control. D, total TBK and phospho-TBK Ser172 expression was assessed in wild-type (WT), FH-deficient (FH−/−), or FH-deficient with FH stably added back (FH−/− + FH) MEFs.
To define the putative FH/TBK1/p65 signaling axis, we silenced FH in DKO IKKα/β MEFs. siRNA-mediated knockdown of FH in IKK DKO MEFs resulted in enhanced phosphorylation of p65 Ser536 compared with scrambled control. However, concurrent knockdown of TBK1 blocked siFH-mediated p65 phosphorylation (Fig. 6A). In support of these findings, pretreatment of HK2 cells with the TBK1 pharmacological inhibitor, BX795, blocked DEF-induced p65 Ser536 phosphorylation and HIF-1α expression (Fig. 6B).
FIGURE 6.
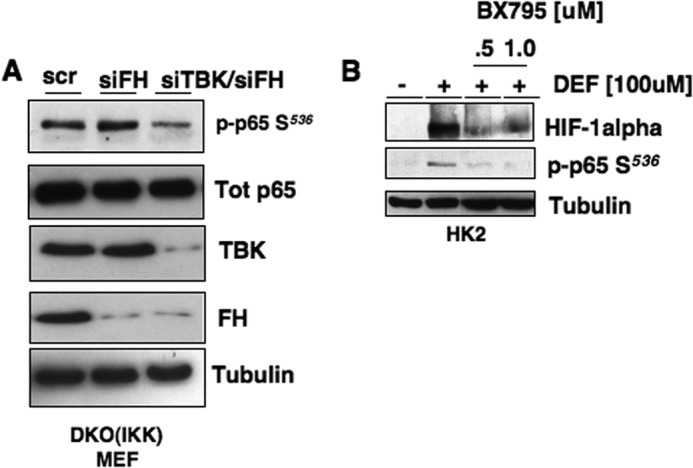
Fumarate mediates HIF-1α mRNA expression through TBK1. A, FH or fumarate hydratase + Tank-binding kinase-1 (FH + TBK) were transiently silenced (siFH and siTBK/siFH, respectively) in DKO (IKKα and IKKβ) MEFs. Scrambled oligos were transfected as a negative control. After 48 h, cell lysates were prepared and analyzed for phospho-p65 Ser536 (p-p65 S536), total p65, total TBK and FH. Tubulin was used as loading control. B, HK2 cells were treated with DEF simultaneously with the Tank-binding kinase inhibitor BX795 at the indicated concentrations for 8 h. Total protein was used to assess HIF-1α, phospho-p65 Ser536 expression. Tubulin was used as loading control.
Fumarate Mediates p65-dependent HIF-1α Accumulation in a TBK1-dependent Manner in FH-deficient Cells
We next examined the status of TBK1 activation in FH-deficient UOK262 cells. We find that basal phosphorylation of TBK1 on its activation site (Ser172) is enhanced in FH-deficient RCC cells compared with the normal immortalized renal cell line, HK2 (Fig. 7A). Reintroduction of FH resulted in reduced TBK1 phosphorylation compared with vector control (Fig. 7A). No changes in total TBK1 were observed. Similarly, p65 phosphorylation at Ser536 was markedly reduced in UOK262 cells reconstituted with FH compared with vector control as examined by Western blot analysis (Fig. 7B). Next we examined specific p65 binding at the HIF-1α promoter using ChIP analysis in the aforementioned cells. Fig. 7C shows p65 bound to the HIF-1α promoter in UOK262 cells transfected with empty vector when immunoprecipitated with p65 antibodies (EV, lane 4), but not IgG alone (EV, lane 3). In contrast, p65 binding to the HIF-1α promoter was not identified in UOK262 cells stably transfected with FH (FH, lane 5). Input DNA was used as positive control (lanes 1 and 2).
FIGURE 7.

Activated p65/TBK axis in FH-deficient tumor cells promotes HIF-1α expression. A, phospho-TBK Ser172 and total TBK expression was analyzed in cell extracts prepared from HK2, FH-deficient UOK 262 stably expressing vector control, or FH-FLAG. B, phospho-p65 Ser536 (p-p65 S536) and total p65 expression was assessed in FH-deficient UOK 262 stably expressing vector control or FH-FLAG. Tubulin was used as loading control. C, HIF-1α DNA associated with p65 was assessed in nuclear extracts of FH-deficient UOK 262 cells stably expressing vector control or FH using ChIP as outlined under “Experimental Procedures.” Input DNA was used as a positive control for HIF-1α PCR. Immunoprecipitation using nonspecific IgG was used as a negative control. HIF-1α is detected at 518 bp. The data were quantitated from two independent experiments and the results are expressed as mean ± S.E. *, p < 0.05 (lower panel). D, UOK 262 cells were transiently transfected with siTBK or scramble control. After 48 h nuclear and cytoplasmic extracts were prepared. HIF-1α, phospho-p65 Ser536 (p-p65 S536), and total p65 expression were analyzed in the nuclear fraction by Western blot analysis. CREB was used as loading control. Total TBK expression was assessed in the cytosolic lysate. Tubulin was used as loading control. E, UOK 262 cells was transiently transfected with siTBK or scramble control. HIF-1α mRNA expression was analyzed as described in the legend to Fig. 1. The data were quantitated and the results are expressed mean ± S.E., **, p < 0.01.
To determine the effects of TBK1 on p65 phosphorylation in FH-deficient UOK262 cells, we silenced TBK1 using siRNA. Scrambled RNA was used as a control. Silencing of TBK1, but not scrambled control, reduced phosphorylation of p65 Ser536 concomitant with reduced levels of HIF-1α expression in nuclear fractions (Fig. 7D). CREB was used as a loading control. Successful knockdown of TBK1 in the cytosolic fraction was confirmed by Western blot analysis where tubulin was used as a loading control for whole cell lysate (Fig. 7D, lower panel). Importantly, we find that transient knockdown of TBK1 using small inhibitory RNA (siRNA) in UOK262 cells reduced HIF-1α mRNA expression (Fig. 7E). Together, our data demonstrate that fumarate mediates HIF-1α mRNA expression through a novel TBK1-dependent, p65-mediated signaling pathway.
FH-deficient renal carcinoma cells are highly invasive. HIF-1α contributes to cell invasion in FH-deficient cells (16). To determine whether TBK1 plays a role in cell invasion in FH-deficient RCC, we transfected UOK262 cells with scrambled control or siRNA for TBK1 (siTBK1). Invasion was examined using a Matrigel Boyden chamber assay. Fig. 8A shows FH-deficient UOK262 silenced of TBK1 significantly reduces cell invasion compared with scrambled transfected control cells (Fig. 8A). TBK1 knockdown was verified by Western blot analysis (Fig. 8B).
FIGURE 8.
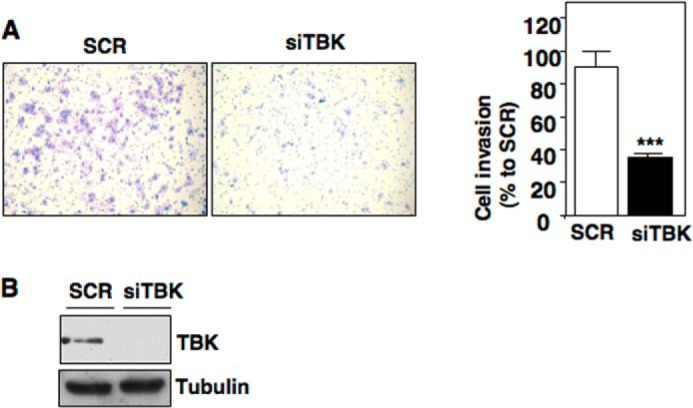
TBK1 silencing reduces cell invasion in FH-deficient RCC cells. UOK 262 cells were transiently transfected with siTBK or scramble control (scr). A, left panel, transfected cells were seeded in a Matrigel invasion chamber. After 72 h of incubation, inserts were subjected for staining. Right panel, invaded cells were quantitated by counting and the results are expressed as mean ± S.E. ***, p < 0.0001. B, total protein was analyzed for total TBK and Tubulin as loading control.
DISCUSSION
In this report, we present the first evidence that the oncometabolite fumarate promotes mRNA expression of HIF-1α through a non-canonical NF-κB-dependent pathway. In particular, we have identified a novel mechanism whereby fumarate promotes p65 activation through the upstream activator TBK1. In accord with these data, inhibition of the TBK1/p65 axis blocks HIF-1α accumulation and cell invasion in cellular models of FH loss (Fig. 9).
FIGURE 9.
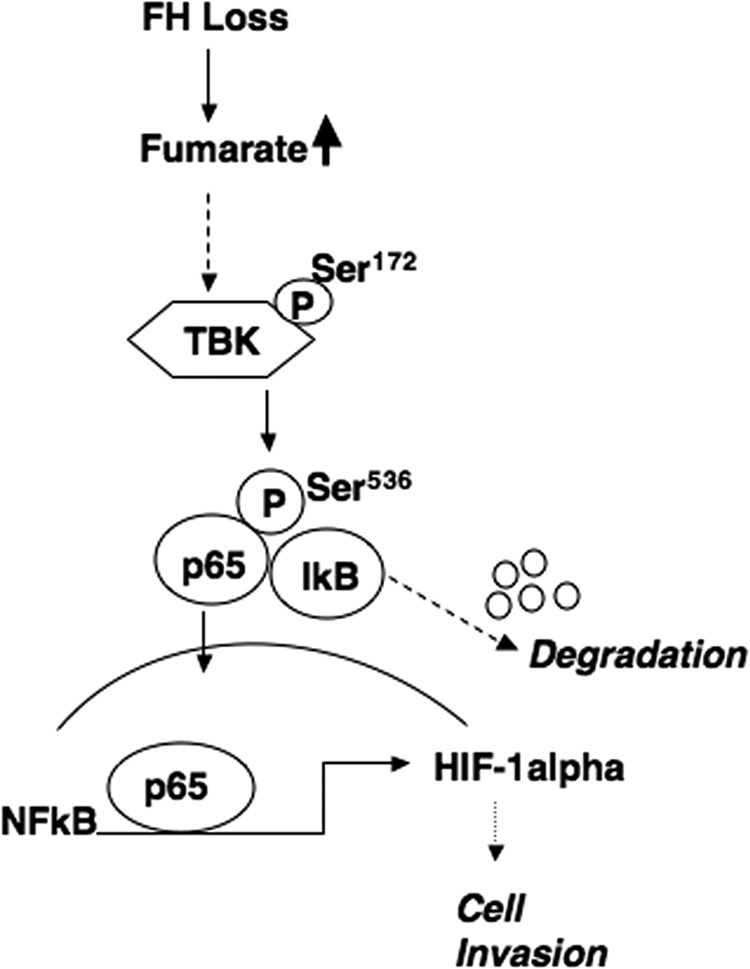
Model of fumarate-mediated HIF-1α mRNA expression through TBK1.
Our studies are the first to identify a role for fumarate in the promotion of HIF-1α synthesis through effects at the transcriptional level. The initial link between FH and HIF was reported by Isaacs et al. (14) who demonstrated that fumarate acts as a competitive inhibitor of prolyl hydroxylases (due to structural similarity with the cofactor 2-OG), which normally targets HIF α subunits for VHL complex-mediated proteosomal degradation. Isaacs et al. (14) reported exogenous administration of monoethyl fumarate in combination with a pharmacologic inhibitor of FH did not increase HIF-1α mRNA in cultured lung carcinoma A549 cells. However, we find that fumarate promotes HIF-1α transcription in multiple VHL-deficient RCC cell lines. Moreover, we find HIF-1α mRNA is enhanced in FH-deficient cells, which is reduced when FH is reintroduced. These data may indicate cell type-specific responses to fumarate treatment. Additionally, potential differences in experimental design including the type of fumarate ester may account for the differing results. Notably, Koivunen et al. (21) found that DMF and DEF were far more potent (∼2 orders of magnitude) at increasing HIF-1α protein levels in cells than the monoethyl ester although HIF-1α mRNA levels were not examined. More recent studies have demonstrated a role for reactive oxygen species signaling in the accumulation of HIF-1α through protein stabilization (15, 31). The collective data support that transcriptional as well as post-transcriptional mechanisms are relevant and contributory to FH null pseudohypoxia given that targeting either mechanism can successfully reduce HIF-1α accumulation.
Another novel aspect of the current study is the link between non-canonical NF-κB signaling and HIF-1α. It is well established that IKK α/β kinases mediate NF-κB activation through phosphorylation of p65 at Ser536 (known as NF-κB canonical signaling) and can promote the transcription of HIF-1α (17, 19). Canonical NF-κB signaling has primarily been examined in the context of cytokine-induced activation. However, it is now well established that NF-κB can be activated via non-canonical means. Non-canonical signaling has also been shown to promote the transcription of HIF-1α. Kumar and Mehta (32) recently demonstrated that the protein tissue transglutaminase TG2 promotes transcription of HIF-1α through interaction with p65. Similar to these data, we have found that fumarate promotes HIF-1α transcription in an IKK-independent fashion.
Prior studies have demonstrated a role for HIF-1α in cell invasion of FH-deficient RCC (16). Our studies demonstrate a novel role for TBK1 in RCC cell invasion in FH-deficient RCC cells. To our knowledge, this is the first study demonstrating a role for TBK1 in cancer cell invasion. It is noteworthy that TBK1 is overexpressed in several malignancies including breast, colon, and lung cancers (33–35). In summary, our studies uncover a novel signaling node activated in high fumarate states, which has the potential to lead to rationale therapeutic approaches with emphasis on cell invasion.
This work was supported, in whole or in part, by National Institutes of Health Grant K08 CA138774 (to S. S.), Urology Care Foundation and Astellas Pharma Rising Star Award, and America Cancer Society RSG-12-127-01 CNE (to S. S.) and Veterans Affairs Merit Grants CPRIT RP120190 and P30CA054174 (to K. B).
- FH
- fumarate hydratase
- HLRCC
- hereditary leiomyomatosis and renal cell cancer
- RCC
- renal cell carcinoma
- 2-OG
- 2-oxglutarate
- HIF
- hypoxia inducible factor
- VHL
- von Hippel-Lindau
- DEF
- diethyl fumarate
- DMF
- dimethyl fumarate
- MEF
- mouse embryonic fibroblasts
- DKO
- double knock-out
- TBK1
- Tank binding kinase 1
- CREB
- cAMP-response element-binding protein.
REFERENCES
- 1. Pollard P. J., Brière J. J., Alam N. A., Barwell J., Barclay E., Wortham N. C., Hunt T., Mitchell M., Olpin S., Moat S. J., Hargreaves I. P., Heales S. J., Chung Y. L., Griffiths J. R., Dalgleish A., McGrath J. A., Gleeson M. J., Hodgson S. V., Poulsom R., Rustin P., Tomlinson I. P. (2005) Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 14, 2231–2239 [DOI] [PubMed] [Google Scholar]
- 2. Ward P. S., Patel J., Wise D. R., Abdel-Wahab O., Bennett B. D., Coller H. A., Cross J. R., Fantin V. R., Hedvat C. V., Perl A. E., Rabinowitz J. D., Carroll M., Su S. M., Sharp K. A., Levine R. L., Thompson C. B. (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grubb R. L., 3rd, Franks M. E., Toro J., Middelton L., Choyke L., Fowler S., Torres-Cabala C., Glenn G. M., Choyke P., Merino M. J., Zbar B., Pinto P. A., Srinivasan R., Coleman J. A., Linehan W. M. (2007) Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J. Urol. 177, 2074–2079; discussion 2079–2080 [DOI] [PubMed] [Google Scholar]
- 4. Tomlinson I. P., Alam N. A., Rowan A. J., Barclay E., Jaeger E. E., Kelsell D., Leigh I., Gorman P., Lamlum H., Rahman S., Roylance R. R., Olpin S., Bevan S., Barker K., Hearle N., Houlston R. S., Kiuru M., Lehtonen R., Karhu A., Vilkki S., Laiho P., Eklund C., Vierimaa O., Aittomäki K., Hietala M., Sistonen P., Paetau A., Salovaara R., Herva R., Launonen V., Aaltonen L. A., and Multiple Leiomyoma Consortium (2002) Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 30, 406–410 [DOI] [PubMed] [Google Scholar]
- 5. Schofield C. J., Ratcliffe P. J. (2004) Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5, 343–354 [DOI] [PubMed] [Google Scholar]
- 6. Kaelin W. G., Jr. (2002) Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2, 673–682 [DOI] [PubMed] [Google Scholar]
- 7. Kaelin W. G., Jr. (2003) The von Hippel-Lindau gene, kidney cancer, and oxygen sensing. J. Am. Soc. Nephrol. 14, 2703–2711 [DOI] [PubMed] [Google Scholar]
- 8. Pugh C. W., Ratcliffe P. J. (2003) Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9, 677–684 [DOI] [PubMed] [Google Scholar]
- 9. Bruick R. K., McKnight S. L. (2001) A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337–1340 [DOI] [PubMed] [Google Scholar]
- 10. Epstein A. C., Gleadle J. M., McNeill L. A., Hewitson K. S., O'Rourke J., Mole D. R., Mukherji M., Metzen E., Wilson M. I., Dhanda A., Tian Y. M., Masson N., Hamilton D. L., Jaakkola P., Barstead R., Hodgkin J., Maxwell P. H., Pugh C. W., Schofield C. J., Ratcliffe P. J. (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54 [DOI] [PubMed] [Google Scholar]
- 11. Ivan M., Kondo K., Yang H., Kim W., Valiando J., Ohh M., Salic A., Asara J. M., Lane W. S., Kaelin W. G., Jr. (2001) HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468 [DOI] [PubMed] [Google Scholar]
- 12. Jaakkola P., Mole D. R., Tian Y. M., Wilson M. I., Gielbert J., Gaskell S. J., von Kriegsheim A., Hebestreit H. F., Mukherji M., Schofield C. J., Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001) Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472 [DOI] [PubMed] [Google Scholar]
- 13. Yu F., White S. B., Zhao Q., Lee F. S. (2001) HIF-1α binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. U.S.A. 98, 9630–9635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Isaacs J. S., Jung Y. J., Mole D. R., Lee S., Torres-Cabala C., Chung Y. L., Merino M., Trepel J., Zbar B., Toro J., Ratcliffe P. J., Linehan W. M., Neckers L. (2005) HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8, 143–153 [DOI] [PubMed] [Google Scholar]
- 15. Sudarshan S., Sourbier C., Kong H. S., Block K., Valera Romero V. A., Yang Y., Galindo C., Mollapour M., Scroggins B., Goode N., Lee M. J., Gourlay C. W., Trepel J., Linehan W. M., Neckers L. (2009) Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1α stabilization by glucose-dependent generation of reactive oxygen species. Mol. Cell. Biol. 29, 4080–4090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tong W. H., Sourbier C., Kovtunovych G., Jeong S. Y., Vira M., Ghosh M., Romero V. V., Sougrat R., Vaulont S., Viollet B., Kim Y. S., Lee S., Trepel J., Srinivasan R., Bratslavsky G., Yang Y., Linehan W. M., Rouault T. A. (2011) The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell 20, 315–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Uden P., Kenneth N. S., Rocha S. (2008) Regulation of hypoxia-inducible factor-1α by NF-κB. Biochem. J. 412, 477–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. O'Flaherty L., Adam J., Heather L. C., Zhdanov A. V., Chung Y. L., Miranda M. X., Croft J., Olpin S., Clarke K., Pugh C. W., Griffiths J., Papkovsky D., Ashrafian H., Ratcliffe P. J., Pollard P. J. (2010) Dysregulation of hypoxia pathways in fumarate hydratase-deficient cells is independent of defective mitochondrial metabolism. Hum. Mol. Genet. 19, 3844–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Belaiba R. S., Bonello S., Zähringer C., Schmidt S., Hess J., Kietzmann T., Görlach A. (2007) Hypoxia up-regulates hypoxia-inducible factor-1α transcription by involving phosphatidylinositol 3-kinase and nuclear factor κB in pulmonary artery smooth muscle cells. Mol. Biol. Cell 18, 4691–4697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zheng L., Mackenzie E. D., Karim S. A., Hedley A., Blyth K., Kalna G., Watson D. G., Szlosarek P., Frezza C., Gottlieb E. (2013) Reversed argininosuccinate lyase activity in fumarate hydratase-deficient cancer cells. Cancer Metab. 1, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koivunen P., Hirsilä M., Remes A. M., Hassinen I. E., Kivirikko K. I., Myllyharju J. (2007) Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 282, 4524–4532 [DOI] [PubMed] [Google Scholar]
- 22. Ooi A., Wong J. C., Petillo D., Roossien D., Perrier-Trudova V., Whitten D., Min B. W., Tan M. H., Zhang Z., Yang X. J., Zhou M., Gardie B., Molinié V., Richard S., Tan P. H., Teh B. T., Furge K. A. (2011) An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer cell 20, 511–523 [DOI] [PubMed] [Google Scholar]
- 23. Rius J., Guma M., Schachtrup C., Akassoglou K., Zinkernagel A. S., Nizet V., Johnson R. S., Haddad G. G., Karin M. (2008) NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature 453, 807–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mercurio F., Zhu H., Murray B. W., Shevchenko A., Bennett B. L., Li J., Young D. B., Barbosa M., Mann M., Manning A., Rao A. (1997) IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science 278, 860–866 [DOI] [PubMed] [Google Scholar]
- 25. Spencer E., Jiang J., Chen Z. J. (1999) Signal-induced ubiquitination of IκBα by the F-box protein Slimb/β-TrCP. Genes Dev. 13, 284–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Mahony A. M., Montano M., Van Beneden K., Chen L. F., Greene W. C. (2004) Human T-cell lymphotropic virus type 1 tax induction of biologically active NF-κB requires IκB kinase-1-mediated phosphorylation of RelA/p65. J. Biol. Chem. 279, 18137–18145 [DOI] [PubMed] [Google Scholar]
- 27. Sakurai H., Chiba H., Miyoshi H., Sugita T., Toriumi W. (1999) IκB kinases phosphorylate NF-κB p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem. 274, 30353–30356 [DOI] [PubMed] [Google Scholar]
- 28. Fujita F., Taniguchi Y., Kato T., Narita Y., Furuya A., Ogawa T., Sakurai H., Joh T., Itoh M., Delhase M., Karin M., Nakanishi M. (2003) Identification of NAP1, a regulatory subunit of IκB kinase-related kinases that potentiates NF-κB signaling. Mol. Cell. Biol. 23, 7780–7793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hutti J. E., Porter M. A., Cheely A. W., Cantley L. C., Wang X., Kireev D., Baldwin A. S., Janzen W. P. (2012) Development of a high-throughput assay for identifying inhibitors of TBK1 and IKKϵ. PLoS One 7, e41494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shu C., Sankaran B., Chaton C. T., Herr A. B., Mishra A., Peng J., Li P. (2013) Structural insights into the functions of TBK1 in innate antimicrobial immunity. Structure 21, 1137–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sullivan L. B., Martinez-Garcia E., Nguyen H., Mullen A. R., Dufour E., Sudarshan S., Licht J. D., Deberardinis R. J., Chandel N. S. (2013) The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 51, 236–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kumar S., Mehta K. (2012) Tissue transglutaminase constitutively activates HIF-1α promoter and nuclear factor-κB via a non-canonical pathway. PLoS One 7, e49321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barbie D. A., Tamayo P., Boehm J. S., Kim S. Y., Moody S. E., Dunn I. F., Schinzel A. C., Sandy P., Meylan E., Scholl C., Fröhling S., Chan E. M., Sos M. L., Michel K., Mermel C., Silver S. J., Weir B. A., Reiling J. H., Sheng Q., Gupta P. B., Wadlow R. C., Le H., Hoersch S., Wittner B. S., Ramaswamy S., Livingston D. M., Sabatini D. M., Meyerson M., Thomas R. K., Lander E. S., Mesirov J. P., Root D. E., Gilliland D. G., Jacks T., Hahn W. C. (2009) Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kan Z., Jaiswal B. S., Stinson J., Janakiraman V., Bhatt D., Stern H. M., Yue P., Haverty P. M., Bourgon R., Zheng J., Moorhead M., Chaudhuri S., Tomsho L. P., Peters B. A., Pujara K., Cordes S., Davis D. P., Carlton V. E., Yuan W., Li L., Wang W., Eigenbrot C., Kaminker J. S., Eberhard D. A., Waring P., Schuster S. C., Modrusan Z., Zhang Z., Stokoe D., de Sauvage F. J., Faham M., Seshagiri S. (2010) Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466, 869–873 [DOI] [PubMed] [Google Scholar]
- 35. Korherr C., Gille H., Schäfer R., Koenig-Hoffmann K., Dixelius J., Egland K. A., Pastan I., Brinkmann U. (2006) Identification of proangiogenic genes and pathways by high-throughput functional genomics: TBK1 and the IRF3 pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 4240–4245 [DOI] [PMC free article] [PubMed] [Google Scholar]