

Background: Klotho is an age suppressor protein whose brain function is unknown.
Results: Klotho protects hippocampal neurons from glutamate and amyloid β-induced oxidative damage through the induction of the thioredoxin/peroxiredoxin system.
Conclusion: Klotho is neuroprotective via the regulation of the redox system.
Significance: Understanding the mechanism underlying Klotho-induced neuroprotection may lead to the development of novel therapeutic approaches against neurodegeneration.
Keywords: Alzheimer Disease, Amyloid, Glutamate, Neuroprotection, Oxidative Stress
Abstract
Generation of reactive oxygen species (ROS), leading to oxidative damage and neuronal cell death, plays an important role in the pathogenesis of neurodegenerative disorders, including Alzheimer disease. The present study aimed to examine the mechanism by which the anti-aging protein Klotho exerts neuroprotective effects against neuronal damage associated with neurodegeneration and oxidative stress. Pretreatment of rat primary hippocampal neurons and mouse hippocampal neuronal cell line HT22 with recombinant Klotho protected these cells from glutamate and oligomeric amyloid β (oAβ)-induced cytotoxicity. In addition, primary hippocampal neurons obtained from Klotho-overexpressing mouse embryos were more resistant to both cytotoxic insults, glutamate and oAβ, compared with neurons from wild-type littermates. An antioxidative stress array analysis of neurons treated with Klotho revealed that Klotho significantly enhances the expression of the thioredoxin/peroxiredoxin (Trx/Prx) system with the greatest effect on the induction of Prx-2, an antioxidant enzyme, whose increase was confirmed at the mRNA and protein levels. Klotho-induced phosphorylation of the PI3K/Akt pathway, a pathway important in apoptosis and longevity, was associated with sustained inhibitory phosphorylation of the transcription factor forkhead box O3a (FoxO3a) and was essential for the induction of Prx-2. Down-regulation of Prx-2 expression using a lentivirus harboring shRNA almost completely abolished the ability of Klotho to rescue neurons from glutamate-induced death and significantly, but not completely, inhibited cell death mediated by oAβ, suggesting that Prx-2 is a key modulator of neuroprotection. Thus, our results demonstrate, for the first time, the neuroprotective role of Klotho and reveal a novel mechanism underlying this effect.
Introduction
Neuronal cell death, mediated by elevated ROS,2 has been implicated as a contributing cause for neurodegenerative diseases, including Alzheimer disease (AD), Parkinson disease, Huntington disease, and amyotrophic lateral sclerosis (ALS) (1–3), suggesting that antioxidant defense mechanisms are overwhelmed in these conditions (2). l-Glutamate and Aβ are two oxidative stressors widely used as in vitro models for studying mechanisms of neuronal damage associated with neurodegeneration. l-Glutamate, the major excitatory neurotransmitter in the central nervous system (CNS) (4), is neurotoxic at high concentrations due to increases in intracellular calcium levels and enhanced formation of ROS, causing the oxidative stress that contributes to neurodegeneration (5, 6). The neurotoxicity of Aβ(1–42), a primary component of AD pathogenesis, is also associated with cellular injury following ROS exposure (7).
Klotho was identified in 1997 as a gene mutated in the klotho mouse and named after the Greek goddess who spins the thread of life (8). The absence of Klotho in mice leads to an extremely shortened life span and the display of multiple phenotypes resembling human premature aging, including vascular calcification, infertility, emphysema, osteoporosis, skin atrophy and hair loss, thymic involution, osteopenia, motor neuron and hippocampal degeneration, and cognitive impairment (8–10). In contrast, overexpression of Klotho in mice extends life span ∼20–30%, suppresses insulin signaling, and confers resistance against oxidative stress (11).
Klotho is mainly expressed in the brain, kidney, and reproductive organs (12). In the brain, Klotho is expressed by the choroid plexus and by neurons, especially in the hippocampus and pituitary, and by cerebellar Purkinje cells (8, 13, 14). Behavioral studies done in Klotho knock-out mice show memory retention deficits when compared with wild-type mice, probably due to an increase in oxidative stress in the brain (15). Klotho-deficient mice also exhibit hippocampal neurodegeneration with a reduction in synapse numbers (16), perturbations in axonal transport, and a neurodegenerative phenotype in hippocampal pyramidal cells (13). When transgenic Klotho expression was limited to the brain and testes in otherwise Klotho null mice, all of the systemic phenotypes associated with Klotho null mice were improved (8), suggesting that brain expression of Klotho is important to CNS aging. Another correlation linking Klotho and aging comes from a microarray analysis comparing young and aged brains. In experiments designed to take an unbiased look at age-related changes in the monkey corpus callosum, we previously determined that Klotho is decreased in the white matter of the aged brain in non-human primates, mice, and rats (17) and that this is probably due to hypermethylation of its promoter (18). Furthermore, we have shown that Klotho enhances oligodendrocyte differentiation and myelin protein production (19). Most recently, we have reported that increased levels of Klotho are associated with enhanced cognition in humans and mice (20). Mice overexpressing Klotho exhibit enhanced long term potentiation, a form of synaptic plasticity, and enriched synaptic GluN2B, an N-methyl-d-aspartate receptor (NMDAR) subunit with key functions in learning and memory.
The Klotho gene encodes a single-pass type I transmembrane protein. Transmembrane Klotho functions as the FGF-23 co-receptor with FGF-R and is critical in maintaining proper calcium, phosphate, and vitamin D homeostasis (8). We discovered that the extracellular domain of Klotho protein can be clipped at the plasma membrane by membrane-anchored proteases ADAM10 and ADAM17 to generate a secreted form of Klotho protein (21), which is detectable in serum and cerebrospinal fluid (22). Secreted Klotho possesses sialidase activity that modifies glycans on the cell surface of cells and regulates the activity of multiple ion channels, such as TRPV5/6 and ROMK1 (23, 24). The secreted form of Klotho also possesses anti-cancer and antioxidative properties through the inhibition of insulin/IGF-1, Wnt (23), and transforming growth factor-β1 (TGF-β1) signaling (25), the activation of FoxOs, and induction of manganese superoxide dismutase (Mn-SOD), also known as SOD2, expression (26, 27). In line with these properties, Klotho-deficient mice have impaired cognition, which is associated with an increased number of apoptotic pyramidal neurons and levels of oxidized lipid and DNA in the hippocampus (15). In contrast, Klotho-overexpressing (KL-OE) mice have higher Mn-SOD expression in muscles, less phosphorylated FoxOs, and lower levels of urinary 8-oxo-2′-deoxyguanosine, a marker of oxidative damage to DNA, than wild-type mice (26), all findings associated with reduced oxidative stress. KL-OE mice also survive significantly longer than wild-type mice after a sublethal dose of paraquat, a herbicide that generates superoxide, further indicating that Klotho overexpression enhances resistance to oxidative stress (11). Furthermore, treatment of cultured cells with secreted Klotho protein inhibits insulin/IGF-1 signaling, activates FoxOs, induces Mn-SOD expression, and reduces oxidative damage and apoptosis in HeLa cells (26) and in vascular endothelial cells (27). Taken together, the ability of Klotho to suppress oxidative damage observed in different tissues and its important role in cognitive function (20) led us to hypothesize that Klotho could protect the brain, and, specifically the hippocampus, from oxidative damage.
The current work examines the neuroprotective effect of Klotho on hippocampal neurons and the underlying mechanism of this neuroprotection. We show that Klotho protects hippocampal neurons from glutamate and oligomeric Aβ-induced cytotoxicity when added exogenously or when hippocampal neurons are exposed to increased endogenous levels of Klotho in KL-OE mice. We also provide evidence that Klotho acts through the Akt-dependent modulation of the thioredoxin/peroxiredoxin system, an important contributor to neuronal antioxidant defense (28, 29). Finally, our data suggest that the neuroprotective effect of Klotho is relevant to neurodegenerative diseases.
EXPERIMENTAL PROCEDURES
Animals
Pregnant Sprague-Dawley rats and C57BL mice were obtained from Charles River Laboratories and housed in the AAALAC-accredited Boston University School of Medicine animal facility. All studies were conducted under the regulations required by the National Institutes of Health and the Boston University School of Medicine Institutional Animal Care and Use Committee. Klotho knock-out (KL-KO) (8) and KL-OE mice (11) were a kind gift from Dr. M. Kuro-o (University of Texas Southwestern Medical Center). KL-OE mice were generated by transgenic insertion of a construct containing the membrane form of mouse Klotho driven by human elongation factor 1a promoter (11) thought to be ubiquitously active at all developmental stages (30–32) For RNA isolation, KL-KO (8), KL-OE, and control mice (WT) were perfused with 0.1 m phosphate-buffered saline, pH 7.4, at 4 °C, and hemibrains were homogenized in lysis buffer provided by the PerfectPure RNA cultured cell kit (5 Prime, Gaithersburg, MD). RNA was isolated according to the manufacturer's instructions.
Reagents
For all experiments where Klotho was added exogenously to cells, we used recombinant mouse Klotho containing the shed ectodomain of Klotho (Ala35–Lys982, recombinant mouse Klotho, catalog no. 1819-KL-050 from R&D Systems, Minneapolis, MN). Akt inhibitor LY294002 (catalog no. 9901) and ERK inhibitor UO126 (catalog no. 9903) were from Cell Signaling (Danvers, MA). All cell culture reagents were from Invitrogen, and all other chemicals were from Sigma-Aldrich unless otherwise indicated.
Cell Preparation
Hippocampi were obtained from E18 Sprague-Dawley rats and E18 C57BL mouse embryos and incubated in dissociation medium (90 mmol/liter Na2SO4, 30 mmol/liter K2SO4, 5.8 mmol/liter MgCl2, 0.25 mmol/liter CaCl2, 10 mmol/liter kynurenic acid, and 1 mmol/liter HEPES, pH 7.4) containing papain (10 units/ml) and cysteine (3 mmol/liter) for a 20-min period, as described (33). The hippocampi were then rinsed in dissociation medium and incubated in dissociation medium containing trypsin inhibitor (10–20 units/ml) for three 5-min periods. After centrifugation, cells were resuspended in neurobasal medium containing 2% B27, 2 mm l-glutamine, 100 units/ml penicillin, 100 mg/ml streptomycin and plated at a cell density of ∼1500 cells/mm2 in 96-, 12-, or 6-well polylysine/laminin-coated plates (Falcon Labware, Lincoln Park, NJ). Neurons were maintained in growth medium at 37 °C in a humidified atmosphere of 5% CO2 and 95% room air for 2 weeks to reach maturation. The medium was half-replaced at an interval of every 2 days with a maintenance medium containing neurobasal medium and 5% B27 supplement. Nonneuronal cells were negligible. Mouse hippocampal neurons were used mainly for the experiments testing the role of the endogenously overexpressed Klotho isolated from the KL-OE mice. HT22 cells were maintained in Dulbecco's modified Eagle's medium (DMEM, without l-glutamine), supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. For the assessment of cytotoxicity, HT22 cells were seeded onto 96-well plates at 5000 cells/100 μl of growth medium in each well and grown overnight prior to initiation of any experimental treatments. For other experiments, such as protein and RNA analysis, cells were seeded in 12-well plates at 150,000 cells/well. Cells were treated with recombinant mouse Klotho, subsequently referred to as Klotho, at 0.4 μg/ml (19), and glutamate at millimolar concentrations, as described in each experiment.
Oligomeric Aβ(1–42) Preparation and Characterization
Soluble amyloid β oligomers (also known as Aβ-derived diffusible ligands) were prepared using Aβ(1–42) peptide (which was synthesized and purified by Dr. James I. Elliott (Yale University, New Haven, CT) as described (34–36), with minor modifications. One microgram of the peptide was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol, sonicated in an ice water bath for 10 min, and incubated for 1 h at room temperature. The 1,1,1,3,3,3-hexafluoro-2-propanol was evaporated under a steady stream of nitrogen gas to form a clear peptide film, which was stored at −80 °C until use. The peptide film was dissolved in DMSO to a concentration of 5 mm and further diluted in phenol red-free F-12 medium (Caisson, North Logan, UT) to make a 100 μm solution. This preparation was incubated at room temperature for 16 h. In the last step of Aβ oligomer (oAβ) preparation, we used centrifugation to remove large aggregates (at 14,000 × g for 15 min), which was followed by collecting the top 90% of the total volume. Although no visible pellet was observed after centrifugation, the oAβ fraction that was retained in the supernatant was 71.8 ± 14% of the initial concentration of 100 μm as evaluated by Western blotting (WB). The collected supernatant was stored on ice until use or flash frozen in liquid nitrogen and stored at −80 °C. oAβ was used at a 0–10 μm final concentration (37).
Routine characterization of oAβ was performed using size exclusion chromatography (36). To analyze oligomer content, 500 μl of a 100 μm Aβ(1–42) preparation (fresh or thawed) was injected onto a Superdex 75 10/300 column (GE Healthcare). The sample was eluted at a flow rate of 0.5 ml/min in PBS, pH 7.4, using an ÄKTApurifier 10. Blank control injections contained 2% DMSO in F-12 phenol red-free medium. Eluted fractions were monitored by measuring absorbance at 280 and 220 nm.
For the assessment of oAβ concentration before and after centrifugation, the oAβ aliquots were diluted to 1 μm in sample buffer (catalog no. 161-0739), loaded on 16.5% Tris-Tricine gels (catalog no. 456-3064), and electrophoresed in Tris-Tricine SDS-PAGE running buffer (catalog no. 161-0744) from Bio-Rad. Proteins were transferred to PVDF membranes (catalog no. ISEQ15150) (Millipore, Billerica, MA) and immunostained with mouse monoclonal 6E10 antibody against amino acids 1–17 of Aβ peptide (catalog no. SIG-39300) (1:2000) (Covance, Dedham, MA).
Cell Viability and Cytotoxicity
For cell viability assays, cells were plated in 96-well plates in five repeats and allowed to attach for 24 h. The next day, the cells were pretreated with Klotho for 4 h before the addition of different concentrations of glutamate or Aβ. Twenty-four hours later, cell death was assessed using the CellTiter-Glo luminescent cell viability assay (Promega), which measures the production of ATP, according to the manufacturer's instructions. Briefly, 100 μl of CellTiter Glo reagent was added to an equal volume of cell culture medium present in each well. The contents were mixed for 2 min, and after stabilization of the signal at room temperature for 10 min, the luminescence was recorded on a microplate reader (Glomax Multi Detection System, Promega). Cytotoxicity was calculated by the following formula, as described previously (38).
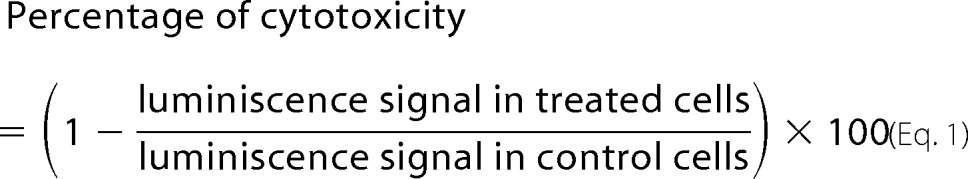 |
In addition, cell death was assessed directly using the CytoTox 96® non-radioactive cytotoxicity assay, Promega), which is a colorimetric alternative to the 51Cr release cytotoxicity assays. The CytoTox 96® assay quantitatively measures lactate dehydrogenase, a stable cytosolic enzyme that is released upon cell lysis. The intensity of the red color formed in the assay was measured at a wavelength of 490 nm and was proportional to lactate dehydrogenase activity and to the number of damaged cells. The data were normalized to the activity of lactate dehydrogenase released from glutamate-treated culture medium (100%) and expressed as a percentage of this control.
ROS Production
ROS production was measured in 96-well plates after staining with the fluorescent dye 2′,7′-dichlorofluorescein diacetate (catalog no. D6883) (5 μm, at 37 °C for 30 min) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm, as described (39).
Western Blotting
Protein concentrations were measured using the Micro BCA protein assay reagent kit (Pierce) according to the manufacturer's protocol. For SDS-PAGE, cell lysates containing the same amount of total protein were boiled for 5 min with sample buffer (250 mm Tris, pH 6.8, 10% SDS, 30% glycerol, 5% β-mercaptoethanol, 0.02% bromphenol blue) and loaded on gels. Proteins were transferred to nitrocellulose membranes (Millipore, Billerica, MA).
All antibodies were diluted in TBST (50 mm Tris, pH 8.0, 150 mm NaCl, and 0.1% (v/v) Tween 20) containing 1% BSA. Secondary antibodies were horseradish peroxidase-conjugated goat anti-mouse, anti-rat, or anti-rabbit (1:5000, Kirkegaard & Perry Laboratories, Gaithersburg, MD). Enhanced chemiluminescence (ECL) was detected using Immobilon Western Chemiluminescent Substrate (Millipore, Billerica, MA). Autoradiography was done using Kodak Scientific Imaging Film X-OMATTM AR (Eastman Kodak Co.). The primary antibodies used were as follows: anti-Prx-2 rabbit antibody (catalog no. R8656) (1:20,000), anti-phospho-ERK mouse monoclonal antibody (catalog no. M8159) (1:5000) from Sigma-Aldrich, anti-β-tubulin monoclonal antibody (catalog no. 322600) (1:1000) from Invitrogen, anti-Trxrd-1 rabbit monoclonal antibody (catalog no. ab124954) (1:2000) from Abcam (Cambridge, MA), anti-Klotho rat monoclonal antibody (clone KM2076 (catalog no. KAL-KO603) (1:2000) from Cosmo BIO USA (Carlsbad, CA), mouse monoclonal 6E10 antibody (see above), mouse monoclonal antibody CP-13 (1:1000) directed against Tau protein phosphorylated at Ser202 and Thr205 (P. Davies, Albert Einstein College of Medicine, Bronx, NY), and mouse monoclonal antibody Tau 5 (1:500) directed against phosphorylation-independent Tau protein (L. Binder, Northwestern University Medical School, Chicago, IL). The antibodies for phospho-Akt (Thr308) (catalog no. 2965), phospho-Akt (Ser473) (catalog no. 9271), total Akt (catalog no. 4691), total ERK (catalog no. 9102), and phospho-GSK3β (catalog no. 9323), FoxO3a (catalog no. 2497) and an anti-phospho-forkhead antibody that recognizes the phosphorylated form of FoxO1a (Thr24), FoxO3a (Thr32), and FoxO4 (Thr28) (catalog no. 2599) were from Cell Signaling (Danvers, MA) and were used according to the manufacturer's protocol.
qRT-PCR
Rat and mouse primary hippocampal neurons were isolated and plated in 6-well plates and maintained for 2 weeks. Cells were then treated as described in each experiment, and total RNA was isolated using PerfectPure RNA cultured cell kit (5 Prime, Gaithersburg, MD). A reverse transcription was performed using the iScript cDNA synthesis kit (Bio-Rad) with 2 μg of total RNA from each sample. The rat oxidative stress primer library array (catalog no. ROSL-I) and all of the primer sets were purchased from RealTimePrimers (Elkins Park, PA). For the confirmation of the array results, the same array primer sets were used and normalized to β-actin, β2-microglobulin, phosphoglycerate kinase 1, hypoxanthine phosphoribosyltransferase 1, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and peptidylprolyl isomerase A, which were provided in the array kit. Specific mouse gene primer sets included sets for Prx-2 (catalog no. VMPS-5017), Trxrd-1 (catalog no. VMPS-6936), Klotho (customized; forward (5′-CAGACCCAAGTGAACCACAC-3′) and reverse (5′-GACCCAGTGACCCAACTCTT-3′)) and housekeeping gene primers (β-actin, β2-microglobulin, hypoxanthine phosphoribosyltransferase 1, GAPDH, and peptidylprolyl isomerase A) from the Mouse Housekeeping Gene Primer Set (catalog no. MHK-1). The qRT-PCR experiments were performed with iQTM SYBR® Green Supermix (Bio-Rad) with detection on a Bio-Rad C1000 thermal cycler. The whole cDNA from 2 μg of total RNA was used for one 96-well plate. Five replicates of 20-μl reactions containing primer and cDNA template were used for quantitation. A PCR was carried out as follows: 1 cycle of 95 °C for 3 min followed by 40 cycles of 95 °C for 10 s, 55 °C for 20 s, and 72 °C for 30 s. This was followed by a dissociation curve beginning at 55 °C and increasing by 0.2 °C every 3 s, with SYBR Green fluorescence measured at every interval. Relative quantitation of the difference between the samples was done using the RT2 Profiler PCR Array Data Analysis Program from Qiagen (Gaithersburg, MD). Genes were tested for statistical significance (p < 0.05), relative to the control, by Student's t test.
Plasmids and shRNAs
Lentiviruses were produced by transfection of HEK 293T cells with plasmids psPAX2 (catalog no. 12260), pMD2.G (catalog no. 12259), GFP (catalog no. 25999), and control empty vector pLKO (catalog no. 10878), purchased from Addgene (Cambridge, MA), or shRNAs for Prx-2 (catalog no. EG21672), obtained from Open Biosystems (Huntsville, AL). 48 h after transfection, supernatants containing lentivirus were collected, and the cells were infected with twice diluted supernatant and 10 μg/ml Polybrene (AmericanBio, Inc., Natick, MA) overnight, washed, and selected with puromycin (10 μg/ml) 48 h after infection. Lentiviral vectors expressing GFP were used as an infection efficiency indicator: usually ∼75% of cells were fluorescent 2 days after infection. For transfections, construct DNA was mixed with Lipofectamine 2000 in Opti-MEM and added directly to cell medium. After 6 h, cells were washed once with medium, medium were replaced, and cells were incubated overnight at 37 °C. GFP was used as a control for transfections.
For shPrx-2, a pool of four target-specific shRNAs was screened for complete silencing. One shRNA with the sequence 5′-GCATTAAAGCAGCGTATC-3′ (shRNA-3) was chosen for further experiments. The cells were infected with GIPZ lentiviral shRNA as described above. Prx-2-deficient cells had a similar morphology as control cells.
Genotyping of Mouse Embryos
Male KL-OE mice were mated with wild type C57BL females; thus, theoretically, only 50% of embryos were KL-OE. At the same time the hippocampus was harvested, the tail from each individual embryo was clipped into a microcentrifuge tube, and genomic DNA was isolated according to the manufacturer's protocol using DirectAmpTM tissue genomic DNA amplification kit (Denville Scientific, Metuchen, NJ). Briefly, 100 μl of extraction solution was added to the tail, followed by 25 μl of preparation solution. The samples were incubated at room temperature for 10 min and then at 95 °C for 3 min. 100 μl of neutralization solution was added to samples and mixed by vortexing. Four microliters of the tissue extract was used for PCR amplification using Choic Taq Blue master mix (Denville Scientific, Metuchen, NJ) in a 20-μl final volume. The Klotho primers used were as follows: forward, 5′-TCGCGCCCTGGCCGACCATTTCAGG-3′; reverse, 5′-AGCACAAAGTCAAGAGACTTCTGGC-3′. PCR was carried out using the following cycling conditions: 94 °C for 3 min and 25 cycles at 94 °C for 30 s, 58 °C for 30 s, and 68 °C for 30 s, followed by 68 °C for 2 min. The PCR products were analyzed by 1% agarose gel electrophoresis for 15 min. Samples from Klotho-overexpressing mice produce a 339-bp band, whereas the wild type samples produced no band. The genotyping of mouse embryos was typically done within 90 min. Once the genotype results were obtained, the hippocampal tissues from WT embryos and Klotho-overexpressing embryos were pooled separately.
Statistical Analysis
Quantitative data are expressed as the means ± S.D. Statistical comparisons between experimental groups were made using the two-tailed, unpaired Student's t test. Probability values of p < 0.05 were considered significant.
RESULTS
Klotho Protects Hippocampal Neurons against Glutamate-induced Cytotoxicity
To determine whether Klotho can counteract oxidative stress induced in brain-derived cells in vitro, we used glutamate cytotoxicity in rat primary hippocampal neurons as a model for neuronal death by oxidative damage. Glutamate is an endogenous excitatory neurotransmitter, which is neurotoxic at high concentrations (6). To induce oxidative stress in hippocampal neurons, we used glutamate at concentrations of 2–5 mm, which is in line with the published data demonstrated in primary neurons and HT22 cells (5, 40–42). Glutamate concentrations lower than 2 mm had no significant cytotoxic effect (data not shown). When primary neurons were cultured in the presence of increasing concentrations of glutamate from 0 to 4 mm for 24 h, cell viability decreased in a concentration-dependent manner (Fig. 1A). Pretreatment of primary neurons for 4 h with Klotho, which did not have any effect on cell cytotoxicity by itself, significantly diminished the extent of cytotoxicity from 65% to 40% and from 80% to 60%, when exposed to 3 and 4 mm glutamate, respectively, as measured by CellTiter Glo. As a second measure of cytotoxicity, we quantified lactate dehydrogenase release into the medium to monitor cell death in the late apoptotic process, when membrane integrity is impaired. As seen in Fig. 1C, the release of lactate dehydrogenase into the medium, which reflects glutamate-induced cell death, was significantly decreased by pretreatment with Klotho.
FIGURE 1.

Klotho protects hippocampal neurons from glutamate-induced cytotoxicity. Rat primary hippocampal neurons (A–D) and immortalized mouse hippocampal neurons (HT22 cells) (E and F) were pretreated without or with Klotho for 4 h and exposed to glutamate at concentration of 3–4 mm. Cell death was assessed 24 h later by CellTiter Glo (A, D, and E) and by lactate dehydrogenase (LDH) release to the medium (C). The accumulation of intracellular ROS was assessed using 2′,7′-dichlorofluorescein diacetate, 8 h after the glutamate addition, and presented as -fold increase in fluorescence units normalized to untreated control (B and F). Asterisks indicate statistical significance of Klotho-treated versus Klotho-untreated cells: *, p < 0.05; **, p < 0.01 by Student's t test. Results represent the average of 3–4 independent experiments. Error bars, S.D.
Intracellular ROS accumulation in hippocampal neurons following glutamate treatment has been reported previously (43). Here we show that exposure of hippocampal neurons to increasing concentrations of glutamate leads to increased ROS accumulation, whereas pretreatment with Klotho significantly decreased ROS production (Fig. 1B). Pretreatment with Klotho for 4–6 h significantly decreased the extent of cell death induced by glutamate (Fig. 1D). Interestingly, when added simultaneously with or 1 h before glutamate, Klotho did not protect hippocampal neurons from glutamate-induced cytotoxicity. Thus, a 4-h preincubation with Klotho was employed in all subsequent experiments.
Results similar to those obtained in hippocampal neurons were also observed in the immortalized murine hippocampal cell line, HT22. Subsequent experiments in HT22 cells confirmed the results obtained in primary neurons: 4-h pretreatment with Klotho before exposure to glutamate markedly reduced cell death (Fig. 1E) and accumulation of intracellular ROS (Fig. 1F).
Klotho Induces Antioxidative Enzymes
As a first step toward understanding the mechanism of Klotho-induced neuroprotection, we used an antioxidative stress qRT-PCR array, which profiles the expression of 84 genes related to oxidative stress and defense. We examined RNA isolated from rat primary hippocampal neurons treated with or without Klotho for 24 h. The qRT-PCR analysis revealed that most genes were highly detectable, suggesting that the primer sets were working appropriately. The enhancing effect of Klotho was detected on numerous factors involved in oxidative stress defense, such as CuZn-SOD and Mn-SOD, glutathione reductase, glutathione synthetase, and sirtuin-1, but the results did not reach statistical significance (data not shown). In contrast, treatment with Klotho significantly increased antioxidative factors of the Trx/Prx redox control system peroxiredoxin 2 and 3 (Prx-2 and Prx-3) and thioredoxin reductase 1 (Trxrd-1) (Fig. 2A). Prx refers to a family of six small non-seleno peroxidases, which are widely distributed in mammalian tissues. Prxs detoxify peroxides using thioredoxin as an electron donor, whereas thioredoxin is then reduced back by Trxrd (44). Next, we performed qRT-PCR for the genes whose expression was statistically different following Klotho treatment to confirm the results of the array. The enhancing effect of Klotho on the induction of Prx-2 and Trxrd-1, under naive (non-stressed) conditions, was confirmed by qRT-PCR analysis (Fig. 2B, top). The induction of Prx-3 was not confirmed by qRT-PCR, and we eliminated this factor from our subsequent investigation.
FIGURE 2.

Klotho induces the expression of Trx/Prx family members in the absence or presence of glutamate. Rat primary hippocampal neurons (A and B) and mouse HT22 cells (C) were pretreated with Klotho (KL) for 4 h, followed by an additional 24 h of treatment in the absence (A) or presence (B and C) of glutamate (Glu) (3 mm). RNA was collected for qRT-PCR, and the results were normalized to endogenous controls, as described under “Experimental Procedures.” A, antioxidative stress array (n = 5 for each group) revealed genes significantly enhanced by treatment with Klotho. B and C, the results of the array were confirmed using specific gene primer sets. The effect of Klotho was assessed in the absence or presence of glutamate. Results are representative of three independent experiments. D, rat primary hippocampal neurons were pretreated with Klotho for 4 h, followed by an additional 24 h of treatment in the absence or presence of glutamate (3 mm), as indicated, and the lysates were collected for WB analysis. Statistical significance for treated versus untreated cells was indicated as follows. *, p < 0.05; **, p < 0.01 by Student's t test; &, p < 0.05 by Student's t test, for cells treated with Klotho and glutamate versus cells treated with glutamate only. Error bars, S.D. Each experiment was repeated at least three times, and representative results are shown.
The results of the array and the confirmation by qRT-PCR analysis led us to focus our research on the profound effect of Klotho on Prx-2 (∼3.7-fold up-regulation) and, to a lesser extent, Trxrd-1 (∼2-fold up-regulation) (Fig. 2, A and B, top). Therefore, we next questioned whether the presence of oxidative conditions, induced by low concentrations of glutamate (3 mm), would result in a greater effect of Klotho on the induction of those antioxidative factors. We found that in hippocampal neurons, the induction of Prx-2 and Trxrd-1 was slightly but not dramatically increased by the presence of glutamate (2.56- and 1.77-fold, respectively), whereas the combination of Klotho pretreatment and exposure to glutamate resulted in a higher increase in the expression of Prx-2 (4.29-fold up-regulation (treated versus untreated cells)) and Trxrd-1 (3.58-fold up-regulation (treated versus untreated cells)) (Fig. 2B). The enhancing effect of Klotho on Prx-2 and Trxrd-1 was confirmed also in HT22 cells in further experiments (Fig. 2C). On the protein level, we found a significant increase in Prx-2, induced by the presence of Klotho alone (under non-oxidative conditions). The treatment of the cells with glutamate alone led also to an enhanced expression of Prx-2 protein, whereas the pretreatment with Klotho increased it further (Fig. 2D, first panel). Trxrd-1 protein was induced mainly in the presence of oxidative conditions at low concentrations of glutamate (3 mm). This induction of Trxrd-1 protein expression by glutamate was significantly increased by pretreatment with Klotho (Fig. 2D, second panel). These results are in line with the qRT-PCR results and confirm the regulatory effect of Klotho on Trx/Prx family members in hippocampal neurons. Because the expression of Prx-2 was induced by Klotho under naive conditions and 4-h pretreatment was essential for the achievement of neuroprotection, we next investigated the role of Prx-2 in the protective effect of Klotho.
The Activation of the Trx/Prx System Is Crucial for the Neuroprotective Effect of Klotho
Based on the results obtained in rat primary hippocampal neurons and the mouse HT22 hippocampal cell line, we next tested whether the induction of Prx-2 is crucial for the neuroprotective effect of Klotho, using the HT22 cells. In this cell line, the response to Klotho in the presence of glutamate (Fig. 1, E and F) and the induction of Prx-2 were similar to what we observed in primary hippocampal neurons. shRNA constructs targeted to Prx-2 were used to achieve stable knockdown of Prx-2 in HT22 cells, using lentivirus infections. As shown in Fig. 3A, Prx-2 protein expression was completely knocked down by shRNA3 and to a lesser extent by shRNA4, as compared with the cells stably infected with control vector. Based on this result and a better conserved cell morphology following the selection process, shRNA3 Prx-2-stably transduced HT22 cells (passages 3–8) were chosen for subsequent experiments. To determine the role of Prx-2, Prx-2-deficient and control cells were pretreated with Klotho and then incubated with glutamate for an additional 24 h. As shown in Fig. 3B, concentration-dependent cell death was observed in all the cells in response to glutamate, whereas the extent of cytotoxicity was slightly, but non-significantly, higher in the Prx-2 knockdown cells than in cells infected with the control vector. In contrast, the protective effect of Klotho against glutamate cytotoxicity was observed only in control cells and was almost completely abolished in the cells lacking Prx-2, suggesting that Prx-2 plays a critical role in the neuroprotective effect of Klotho.
FIGURE 3.

The induction of Prx-2 is essential for the neuroprotective effect of Klotho. HT22 cells were infected with the control vector and four different constructs of shRNA. The Prx-2 expression was assessed by WB (A). HT22 cells stably infected with control vector or Prx-2 shRNA3 were pretreated with Klotho (KL) for 4 h and challenged with glutamate at the indicated concentrations. The extent of cytotoxicity was assessed 24 h later using CellTiter Glo (B). Asterisks indicate statistical significance of Klotho-treated versus Klotho-untreated samples: *, p < 0.05; **, p < 0.01 by Student's t test. Error bars, S.D. Results are representative of three independent experiments. C, qRT-PCR analysis of RNA obtained from whole brain samples of WT, KL-KO, and KL-OE mice. RNA was collected, and the mRNA analysis was performed using selective gene sets and normalized to endogenous controls. Each experiment was repeated at least two times, and representative results are shown.
In line with those results, we have found that brain lysates of KL-KO mice contain significantly lower levels of Prx-2 mRNA as compared with WT, whereas no alteration was detected in the brain lysates of KL-OE mice versus WT. No significant change was observed in the expression of mRNA of Trxrd-1 in KL-KO versus WT or KL-OE versus WT (Fig. 3C).
The Involvement of Akt/PI3K- and ERK-mediated Signaling in Klotho-induced Neuroprotection
To investigate the upstream molecular events that lead to the induction of Prx-2 by Klotho and the subsequent increase in resistance to oxidative stress, we assessed the activation of Akt/PI3K- and ERK-mediated pathways in rat primary hippocampal neurons. As we reported recently, these pathways are activated by Klotho and are essential for the Klotho-induced maturation of oligodendrocytes (19). Results in Fig. 4, A and B, show that exogenous Klotho increases phosphorylation of both kinases as soon as 15 min following Klotho treatment. Importantly, two essential sites of Akt phosphorylation (Thr308 and Ser473) were found to be activated by Klotho (Fig. 4A). Because the induction of Prx-2 is crucial for the neuroprotective effect of Klotho, we examined next the role of Akt and ERK activation in the up-regulation of Prx-2 as a biological end point using specific pharmacological inhibitors. In the presence of the Akt inhibitor LY294002 (10 μm), applied to neurons 1 h prior to Klotho treatment, the induction of Prx-2 was significantly reduced and almost reached the control level (Fig. 4C). These results strongly support the crucial role of Akt in the Klotho-induced up-regulation of Prx-2. However, despite the clear activation of the ERK pathway, the role of ERK in the neuroprotective effect of Klotho is less profound. In the presence of U0126 (5 μm), an inhibitor of the ERK-mediated pathway (MEK kinase 1/2), the induction of Prx-2 by Klotho was only partially and insignificantly inhibited (Fig. 4D). Our results suggest that ERK may play a role in Klotho-induced neuroprotection, but this role is less prominent than that of Akt and needs to be further explored.
FIGURE 4.

The neuroprotective effect of Klotho is mediated mainly through the activation of Akt and partially through the activation of ERK. Rat primary hippocampal neurons were treated with Klotho for the indicated times, and the activation of Akt (A) and ERK (B) was assessed by WB. Cells were treated with 10 μm LY294002 (LY) (C) and U0126 5 μm (D) for 1 h before the treatment with Klotho (KL). Twenty-four hours following the treatment with Klotho, the expression of Prx-2 was assessed by WB in the absence or presence of glutamate (Glu) (3 mm). Akt and ERK phosphorylated proteins are normalized to total Akt or ERK control and presented as a -fold increase normalized to untreated control. Statistical significance for Klotho-treated versus Klotho-untreated samples was indicated as follows. *, p < 0.05; **, p < 0.01 by Student's t test; &, p < 0.05 for cells treated with Klotho in the presence of the inhibitor versus cells treated with Klotho only. Error bars, S.D. Results represent the average of three independent experiments, and representative blots are shown.
Because the Akt-mediated pathway is involved in the rescue of primary neurons by Klotho, we next examined in depth the activation of this pathway and its downstream targets in the presence of oxidative stress induced by glutamate. Exposure of primary neurons to glutamate for 1 h, but not for 24 h, slightly increased Akt phosphorylation (Fig. 5A), whereas the pretreatment with Klotho induced Akt phosphorylation at Thr308 and Ser473 in the absence as well as in the presence of glutamate 1 and 24 h following treatment (Fig. 5, A and B). Forty-eight hours after the treatment, no significant activation of Akt was detected (Fig. 5C).
FIGURE 5.
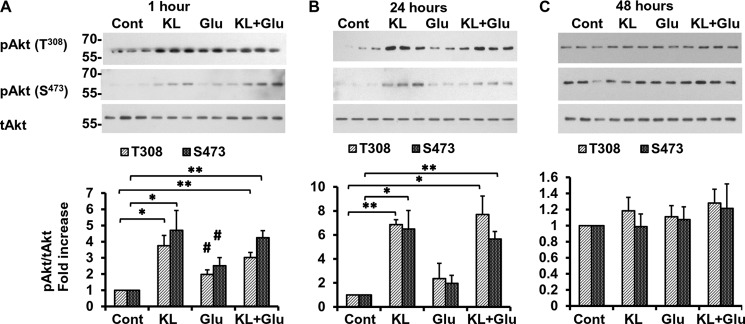
Klotho induces the phosphorylation of Akt. Rat primary hippocampal neurons were pretreated with Klotho (KL) for 4 h followed by additional treatment for 1 h (A), 24 h (B), and 48 h (C), in the absence or presence of glutamate (Glu) (3 mm). The lysates were collected for WB analysis using primary antibody recognizing the Thr308 phosphorylated site (top panels) and Ser473 phosphorylated site (bottom panels). Phosphorylated Akt proteins are normalized to total Akt (tAkt) control and presented as -fold increase normalized to untreated control. Asterisks indicate statistical significance of Klotho-treated versus Klotho-untreated samples. *, p < 0.05; **, p < 0.01; #, p < 0.05 for cells treated with glutamate only versus untreated cells by Student's t test. Error bars, S.D. The average of three independent experiments and representative blots are shown.
Recent studies showed that Klotho alleviated oxidative stress by activating the Forkhead box class O (FoxO) transcription factors (26, 45). Three of the four FoxO isoforms are negatively regulated by Akt through phosphorylation at three specific sites (Thr32, Ser253, and Ser315 for human FoxO3) (reviewed in Ref. 46), which, in turn, prevents their translocation to the nucleus and induction of the apoptotic cascade. Pretreatment with Klotho led to a significant increase in FoxO3 phosphorylation on Thr32 in the absence as well as in the presence of glutamate (Fig. 6A). Interestingly, FoxO3 remained phosphorylated at Thr32 for at least 24 h following the exposure.
FIGURE 6.

Klotho induces the phosphorylation of Akt and FoxO3a. Rat primary hippocampal neurons were pretreated with Klotho (KL) for 4 h followed by additional treatment for 1 and 24 h, as indicated, in the absence or presence of glutamate (Glu) (3 mm). The lysates were collected for WB analysis of the effect of Klotho on phosphorylation of FoxO3a (A), Tau (B), and GSK3β (C). All proteins are normalized to the total control (A and B) or tubulin (C) and presented as a -fold increase normalized to untreated control. Asterisks indicate statistical significance of Klotho-treated versus Klotho-untreated samples: *, p < 0.05; **, p < 0.01 by Student's t test. Error bars, S.D. The average of three independent experiments and representative blots are shown.
Glycogen synthase kinase 3β (GSK3β)/β-catenin signaling is another target of Akt negative regulation. In the non-phosphorylated state, GSK3β constitutively phosphorylates β-catenin, which is then ubiquitinated and degraded by the proteasome (47). Upon phosphorylation, GSK3β is inactivated, which permits β-catenin to translocate into the nucleus to activate transcription factors for a number of genes, including Mn-SOD. Pretreatment of the hippocampal neurons with Klotho in the presence or absence of glutamate did not affect GSK3β phosphorylation after 1 or 24 h (Fig. 6B). This result confirms that the effect of Klotho on neurons does not involve GSK3β regulation.
Neurofibrillary tangles are one of the two neuropathological hallmarks of the AD brain and consist mainly of hyperphosphorylated Tau deposits. Hyperphosphorylation of Tau is regulated by several kinases that phosphorylate specific sites of Tau in vitro. Because of the activation of Akt and ERK by Klotho in our experimental system, we assessed the possible effect of Klotho on Tau phosphorylation in the absence or presence of glutamate. As shown in Fig. 6C, no changes were observed in Tau phosphorylation in the presence of Klotho or glutamate after 1 or 24 h.
The Hippocampal Neurons Obtained from KL-OE Embryos Are More Resistant to Glutamate Cytotoxicity than Neurons from WT Embryos
In order to confirm the neuroprotective effects of Klotho, instead of using exogenous Klotho, we performed experiments using hippocampal neurons isolated from KL-OE mouse embryos. We have developed a completely novel way of obtaining hippocampal neurons separately from WT and KL-OE mouse E18 embryos, as described under “Experimental Procedures.” The protein levels of Klotho in the whole brain lysates obtained from KL-OE E18 embryos were 2.5 times higher than the levels in the brain lysates from WT embryos (Fig. 7A, top). The levels of Klotho mRNA in hippocampal neurons from KL-OE embryos were about 40 times higher (p value <0.05) than in the neurons obtained from WT embryos, as assessed by qRT-PCR. The level of endogenous Klotho protein in hippocampal neurons from KL-OE embryos was quantified by WB by comparison with a standard curve of known concentrations of recombinant mouse Klotho starting from 0.4 μg/ml (which corresponds to 59.2 × 10−6 nmol). We can then conclude that the amount of endogenous Klotho in hippocampal neuronal lysates from KL-OE embryos was about 7.4–14.8 × 10−6 nmol in 25 μg of total protein loaded on the gel. In the medium, the levels of shed endogenous Klotho were below the levels of detection by WB (Fig. 7A, bottom). Note that the transmembrane and shed forms of Klotho from the lysate migrate together at 130 and 135 kDa and cannot be visualized separately on the WB.
FIGURE 7.

Hippocampal neurons obtained from KL-OE embryos are more resistant to glutamate cytotoxicity than neurons from WT embryos. A, top, WB analysis of Klotho expression in 50 μg of whole brain lysate from WT and KL-OE E18 embryos (n = 4 for each group). Bottom panel, WB analysis of Klotho expression in 20 μl of medium containing serial dilutions of recombinant mouse Klotho (× 10−6 nmol), starting at a concentration of 0.4 μg/ml, which corresponds to 59.2 × 10−6 nmol of Klotho (lanes a–d), 25 μg of cell lysate (lanes e and f), and 20 μl of medium (lanes g and h) collected from hippocampal neurons from WT and KL-OE embryos. B and C, mouse primary hippocampal neurons from WT and KL-OE embryos were treated with glutamate at the indicated concentrations. Twenty-four (B) or eight (C) hours later, the percentage cytotoxicity was assessed using CellTiter Glo, and the accumulation of ROS was measured by 2′,7′-dichlorofluorescein diacetate (B and C, respectively). Error bars, S.D. Each experiment was repeated at least three times, and representative results are shown. D, qRT-PCR analysis of the expression of Prx-2 and Trxrd-1 in the presence or absence of glutamate (3 mm), 24 h after the treatment. The results are presented as -fold increase of groups. First panel, neurons from KL-OE embryos versus neurons from WT embryos in the absence of oxidative conditions; second panel, neurons from WT embryos treated with glutamate (3 mm) versus untreated; third panel, neurons from KL-OE embryos treated with glutamate (3 mm) versus untreated. E, WB analysis of the expression of Prx-2 and Trxrd-1 in the presence or absence of glutamate (3 mm), 24 h after the treatment of hippocampal neurons obtained from WT and KL-OE embryos. Asterisks indicate statistical significance: *, p < 0.05; **, p < 0.01 by t test. Error bars, S.D. Each experiment was repeated three times, and representative results are shown.
Hippocampal neurons obtained from KL-OE mice were more resistant to glutamate-induced cytotoxicity than neurons obtained from their WT littermates (Fig. 7B); however, at high glutamate concentrations, Klotho overexpression was not protective. This effect was also observed at the level of ROS accumulation. Whereas the accumulation of ROS significantly increased upon exposure to glutamate, the hippocampal neurons obtained from KL-OE embryos generated significantly lesser amounts of ROS (Fig. 7C).
Notably, consistent with the data obtained in primary neurons treated with exogenous Klotho, the mRNA of Prx-2 in neurons obtained from KL-OE mouse embryos was significantly higher (3-fold up-regulation) as compared with neurons obtained from WT embryos under naive conditions. The presence of oxidative conditions, induced by glutamate (3 mm), increased the levels of Prx-2 in the neurons from WT embryos and increased Prx-2 even further in neurons from KL-OE embryos (Fig. 7D). In contrast to the expression of Prx-2, which was up-regulated in both non-oxidative and oxidative conditions, no significant change in the mRNA expression of Trxrd-1 under naive conditions was detected between the neurons from KL-OE and WT mice, confirming minimal/no induction of Trxrd-1 by Klotho itself. Remarkably, the mRNA expression of Trxrd-1 was significantly increased by the presence of glutamate (3.2-fold) in the neurons obtained from WT embryos, whereas in neurons obtained from KL-OE embryos, the effect of oxidative conditions induced by glutamate was even more profound (5.76-fold) (see Fig. 7D). Similar results were observed at the protein level: Prx-2 was increased in the neurons obtained from KL-OE embryos under the naive conditions and was increased further under the oxidative conditions. In neurons obtained from WT embryos, the exposure to glutamate resulted in slightly increased expression of Prx-2, but this increase was significantly greater in the neurons obtained from KL-OE embryos. Trxrd-1 was only increased under the oxidative conditions in both types of neurons, with more dramatic and significant effects in the primary neurons obtained from KL-OE embryos (Fig. 7E). Taken together, these results confirm the involvement of Prx/Trx system and especially Prx-2 in the protective effect of Klotho under oxidative conditions.
Klotho Protects Hippocampal Neurons against Oligomeric Aβ-induced Cytotoxicity
Although the precise mechanisms leading to neurodegeneration in AD are not entirely clear, neurotoxic oAβs have emerged as a major contributor (48). Having characterized in depth the neuroprotective effect of Klotho against glutamate-induced oxidative stress, we next tested whether Klotho protects against oAβ-induced cytotoxicity, which has also been associated with neuronal injury following ROS exposure (48–50) in primary hippocampal neurons as well as in HT22 cells (50–52). Exposure of rat primary hippocampal neurons to oAβ induced cell death in a concentration-dependent manner, whereas pretreatment with Klotho for 4 h significantly inhibited oAβ-induced cell death (Fig. 8A). Furthermore, hippocampal neurons obtained from KL-OE mice were more resistant to oAβ-induced cytotoxicity than neurons obtained from their WT littermates (Fig. 8B), which is in line with the results obtained with glutamate-induced neurotoxicity.
FIGURE 8.

Klotho protects hippocampal neurons from oligomeric Aβ-induced cytotoxicity. A–C, cytotoxicity was assessed by CellTiter Glo, 24 h after exposure to oAβ. Rat primary hippocampal neurons pretreated with or without Klotho (KL) for 4 h (A), mouse primary hippocampal neurons obtained from WT and KL-OE embryos (B), and HT22 cells stably transduced with control vector or Prx-2 shRNA pretreated with or without Klotho for 4 h (C) were exposed to oAβ (0–10 μm). Cell death was assessed after 24 h by CellTiter Glo. Asterisks indicate statistical significance of Klotho-exposed versus Klotho-unexposed samples: *, p < 0.05; **, p < 0.01 by Student's t test. Error bars, S.D. Each experiment was repeated at least three times, and representative results are shown. D, size exclusion chromatography for oAβ preparation used for the toxicity assays. The far right peaks indicate the background UV absorbance of the buffer; the peaks on the left indicate soluble aggregates of Aβ(1–42).
Each oAβ preparation was evaluated for the presence of soluble oligomers by size exclusion chromatography (Fig. 8D). The oligomeric fraction is detected as a clear peak in the void volume of the column. As shown in Fig. 8D, the fresh preparation of oAβ contained a larger portion of oAβ than the one that was previously frozen and thawed. Both preparations were used to induce cytotoxicity in hippocampal neurons, whereas the cytotoxic effect induced by fresh preparation was higher than the effect of the preparation that was frozen and thawed once.
To examine whether the neuroprotective effect of Klotho against oAβ cytotoxicity is mediated through the same molecular mechanism involving the induction of Prx-2, Prx-2-deficient and control immortalized mouse hippocampal neurons (HT22 cells) were pretreated with Klotho and then incubated with different concentrations of oAβ for an additional 24 h. As shown in Fig. 8C, a concentration-dependent cell death was observed in all of the cells in response to the oAβ preparation (the extent of cell death induced by the once frozen oAβ preparation was mild). Importantly, the protective effect of Klotho against oAβ-induced cytotoxicity that was observed in control cells was considerably but not completely diminished in the cells lacking Prx-2 and was especially decreased at higher concentrations of oAβ, suggesting that Prx-2 plays an important role in the neuroprotective effect of Klotho against Aβ cytotoxicity, but because the protective effect was not completely inhibited, other factors induced by Klotho might be involved.
DISCUSSION
Oxidative stress is a main contributor to both acute (cerebral ischemia and traumatic brain injury) and chronic conditions (AD, Parkinson disease, Huntington disease, ALS, and others) (7, 49). Because hippocampal neurons are among the most vulnerable to oxidative stress (53), we used these cells to investigate the neuroprotective role of Klotho by exposing them to oxidative stress induced by glutamate and the oligomeric form of Aβ(1–42). Here we demonstrate the neuroprotective effect of Klotho on rat and mouse primary hippocampal neurons as well as on the immortalized mouse hippocampal cell line, HT22, as evidenced by inhibition of accumulation of ROS; inhibition of cell death, as reflected by diminished cellular ATP production; and inhibition of the release of lactate dehydrogenase to the medium. We show that Klotho fosters neuronal survival when added exogenously to WT hippocampal neurons and when hippocampal neurons are exposed to increased endogenous levels of Klotho in KL-OE mice. We developed a novel approach to separately isolate hippocampi from KL-OE and WT embryos from a single pregnant mouse carrying embryos of both genotypes. Using this approach, we genotyped DNA obtained from embryo tails while performing individual isolation of hippocampi. Once genotype results were obtained, hippocampi from KL-OE or WT were separately pooled, and neurons cultured to examine the effect of endogenous Klotho overexpression on neuronal survival. Klotho protein was detected on postnatal day 1 in the developing WT brain (54), and here we have detected Klotho at E18 with an increase in Klotho protein levels in whole brain lysates from KL-OE embryos as compared with WT embryos (Fig. 7A). Although the levels of endogenously overexpressed Klotho in KL-OE hippocampal neurons were lower by 75–87.5% than the levels of exogenously added Klotho, it is important to mention that the OE neurons are constantly exposed to enhanced levels of Klotho compared with WT hippocampal neurons exposed to the Klotho for limited time intervals. We were unable to detect the endogenously shed Klotho in the medium of KL-OE hippocampal neurons by WB, and no reliable ELISA is available for quantifying mouse Klotho.3 In the case of the exogenous addition of Klotho, the protective effect was achieved by the secreted form of Klotho. However, the hippocampal neurons obtained from KL-OE embryos produce both transmembrane and, possibly, shed Klotho, although we were unable to detect the shed form in the conditioned medium. Thus, at this time, we do not know which form is inducing the observed effect.
The antioxidative properties of Klotho in non-brain-derived cells were reported previously (26), but only recent reports suggested an anti-inflammatory and antioxidative role for Klotho in the brain. Klotho was found down-regulated in PC12 cells exposed to glutamate (55) and in hippocampal samples from epilepsy patients (56). Other reports suggested the important role of Klotho in the regulation of endoplasmic reticulum stress in SH-SY5Y cells (57) and in the neuroprotective effect of a plant antioxidant, ligustilide, in an AD mouse model (45). In addition, Klotho concentrations were found to be lower in CSF of patients with AD when compared with age-matched controls (58).
Using a qRT-PCR antioxidative stress array, we discovered that Klotho promotes the induction of several members of the Trx/Prx system. Prxs are a newly characterized family of peroxide-scavenging enzymes. Prxs, together with thioredoxin and thioredoxin reductase represent an antioxidant enzymatic system of growing significance in the context of neuronal physiology and pathology (59). Altered expression of Prx-2 (a neuronal specific type of Prx) and Prx-3 has been shown in the pathology of cerebral ischemia (60), AD (61), and Parkinson disease (29). Enhanced neuronal expression and activity of Prx-2 was reported to protect neurons from ischemic injury (62), whereas Trxrd deficiency was shown to potentiate oxidative stress in dopaminergic neurons (63). The most profound effect of treating primary neurons with Klotho was the induction of Prx-2 under naive conditions, and this effect was confirmed both at the mRNA and protein levels. Intriguingly, Klotho only slightly, but significantly, increased the level of Trxrd-1 in the absence of glutamate, as detected by the antioxidative stress array. However, the levels of Trxrd-1 were increased during oxidative conditions in the presence of low concentrations of glutamate (3 mm), and the pretreatment with Klotho further induced the increase. Notably, the induction of the Prx-2 and Trxrd-1 was observed not only after exposing neuronal cells to Klotho but also in the neurons obtained from KL-OE embryos, which represents exposure to endogenously overproduced Klotho, as compared with neurons from WT controls. In line with the results obtained from neurons treated with Klotho, the increased levels of Prx-2 were detected in neurons obtained from KL-OE embryos untreated with glutamate and enhanced further in the presence of glutamate, whereas the induction of Trxrd-1 was observed only following the exposure to glutamate.
To test the concept that Klotho fosters the neuronal survival via modulation of the Trx/Prx redox system, we used specific shRNA-mediated down-regulation of Prx-2 in hippocampal HT22 cells. Knockdown of Prx-2 leads to a slight increase in the levels of cytotoxicity induced by glutamate, but even at low concentrations of glutamate, Klotho failed to rescue cells deficient in Prx-2 in contrast to cells infected with control shRNA. These results are in line with the finding that neurons require pretreatment with Klotho for a minimum of 4 h in naive conditions in order to prompt its neuroprotective effect. Thus, we suggest that induction of Prx-2 in cells pretreated with Klotho renders cells more resistant to the oxidative damage induced by exposure to glutamate, although treatment of neurons with glutamate itself also leads to an enhanced expression of Prx-2. We speculate that this time period of 4 h is required to prepare the neurons to cope with the death stimuli, through the initial induction of Prx-2, followed by the increase in Trxrd-1, which occurs mainly in the presence of glutamate.
Our finding that Klotho regulates the expression of Prx-2 is supported further by qRT-PCR analysis of KL-KO mouse whole brain homogenates that revealed a significant decrease in the levels of Prx-2 (2.6-fold) as compared with WT. The levels of Trxrd-1 were only insignificantly decreased in KL-KO mouse brains as compared with WT. When we performed the comparison of KL-OE mouse brains with WT, no significant alteration in the expression of both factors (Prx-2 and Trxrd-1) was detected between the groups.
Klotho is an anti-aging protein down-regulated with age. Chen et al. reported decreased Prx-2 protein levels in old individuals in a proteomic comparison of old versus young human brain samples (64), further supporting the regulatory connection between Klotho and Prx-2.
It is widely accepted that various forms of oAβ may be key initiators in the cell death cascade in AD brains (65, 66). Oligomeric Aβ(1–42) is known to be toxic to neurons and was found to induce free radical production, alter mitochondrial function, and decrease ATP synthesis (65, 67). Furthermore, cumulative evidence suggests that cellular injury during AD may result from ROS accumulation (reviewed in Ref. 49). Here, we demonstrate the neuroprotective effect of Klotho against glutamate-induced and oAβ(1–42)-induced cytotoxicity. However, because Klotho does not provide complete protection, although statistically significant, there must be other factors that contribute to the neuroprotection. Several studies have shown the connection between high levels of Prxs and increased neuronal resistance to Aβ cytotoxicity (68–70). Aβ-resistant PC12 cells were shown to display increased Prx, Trx, and Trxrd levels (68), and an increase in Prx-2 and Prx-6 expression in AD post-mortem cortical tissue relative to controls (70) was suggested as evidence for the existence of compensatory responses to increased cell loss.
Pretreatment of Prx-2 knockdown cells with Klotho resulted in partial inhibition of the neuroprotective effect of Klotho, suggesting that Prx-2 is essential for Klotho-induced neuroprotection against Aβ cytotoxicity, and other mechanisms, possibly also induced by Klotho, may be involved. The Bcl-2 family of proteins, including Bax and Bim, was implicated in the execution machinery of oAβ-induced neuronal death (71–73) as well as the alteration of mitochondrial functions (52). The possible regulation of these mechanisms by Klotho requires further investigation.
To investigate the upstream events mediating the induction of Prx-2 by Klotho and subsequent neuronal rescue from death, we examined the role of Akt- and ERK-mediating pathways. Akt is a critical survival factor that can modulate cellular pathways in both central and peripheral nervous system (47, 49). Akt was found to be both necessary and sufficient to protect neurons from injury associated with oxidative stress (33, 74–76). It has been reported that phosphorylation of both sites of Akt (Thr308 and Ser473) is required for its full activation (77). In hippocampal neurons, Klotho induced phosphorylation of both sites of Akt as early as 15 min after the treatment, confirming full activation of the Akt pathway. Notably, following longer treatment (1 and 24 h following the addition of glutamate), Klotho-induced phosphorylation of Akt was detected not only in the absence of glutamate but also in its presence, suggesting that this phosphorylation contributes to the protective effect of Klotho. Although the phosphorylation of Akt is considered to be neuroprotective (33, 74–76), in some studies, the increased continuous phosphorylation of Akt in AD was found to be associated with the development of the neurofibrillary pathology (78). We have found no significant increase in Akt phosphorylation 48 h after treatment with Klotho and no changes in Tau phosphorylation in the Klotho-treated neurons, suggesting that Klotho does not affect Tau phosphorylation.
Furthermore, we have shown that the activation of Akt has an essential role in neuronal rescue by Klotho because the application of the specific Akt inhibitor LY294002 significantly abolished the Klotho-induced induction of Prx-2. We have also recently shown the essential role of Klotho-induced activation of the Akt and ERK pathways in the maturation of primary oligodendrocyte progenitor cells (19). In contrast, it was shown previously in HeLa and Chinese hamster ovary (CHO) cells that Klotho increases the resistance to oxidative stress through the down-regulation of IGF-1/Akt signaling and reduced phosphorylation of FoxO1 (26). The discrepancy in the regulation of the Akt pathway by Klotho between previous reports and our findings could be explained by the difference in signaling mechanisms in brain-derived and non-brain-derived cells or by the possibility that the observed activation of Akt by Klotho in brain-derived cells is initiated through a different receptor than the insulin-like growth factor receptor. One strong candidate could be the FGF receptor, which has been shown to interact with Klotho in several mammalian organs, such as the kidney (79) and stomach (80), and in Caenorhabditis elegans (81). In light of our recent report that the beneficial effect of Klotho on learning and memory is mediated through the modulation of synaptic NMDAR in the hippocampus and frontal cortex (20), one can envision the involvement of NMDAR in the neuroprotective effect of Klotho on CNS neurons. The evidence that synaptic NMDA activity boosts antioxidant defenses through changes to the Trx/Prx system (82) supports this hypothesis. Furthermore, synaptic NMDA receptor activity promotes sustained activation of the Akt pathway, leading to the phosphorylation and nuclear export of FoxO and subsequent inactivation of FoxO target genes, such as thioredoxin-interacting protein (Txnip) (82, 83). Phosphorylated Akt inhibits FoxO activity through its phosphorylation that turns off Txnip transcription by inducing dissociation from the Txnip promoter and export from the nucleus (82). Here we show that Klotho induces sustained Akt activation and inhibitory phosphorylation of FoxO3 in hippocampal neurons; thus, it is reasonable to speculate that the antiapoptotic effect of Klotho is mediated through the above-mentioned signaling events, following the enhanced synaptic NMDAR activity. Furthermore, other known prodeath FoxO potential target genes include the proapoptotic Bcl-2 family proteins, such as Noxa, Bim, and Bad, and inactivation of those genes has been implicated in NMDAR-dependent neuroprotection (83, 84). In line with our hypothesis, Shiozaki et al. (13) reported previously that in the CNS tissues of Klotho knock-out mice, the antiapoptotic Bcl-xL and proapoptotic Bax were reduced and enhanced, respectively, which further implicates the involvement of these proteins in Klotho-regulated neuronal function.
The connection between ERK activation and Prx-2 in the hippocampus has been shown previously (85), and we report here that Klotho induces ERK phosphorylation in hippocampal neurons. Using a specific pharmacological ERK inhibitor, U0126, we have shown that the presence of the inhibitor slightly but non-significantly inhibited Klotho-induced induction of Prx-2, suggesting that the ERK-mediated pathway was not critical in the effect of Klotho on neuronal survival.
One of the targets lying downstream of Akt signaling is the class O of forkhead box (FoxO) transcription factors, which have been implicated in the protection against apoptotic injury (33). Akt-phosphorylated FoxO proteins remain in the cytoplasm and thus are unable to regulate gene expression. FoxO3a phosphorylation inhibits the apoptotic process through several mechanisms, such as modulation of mitochondrial membrane depolarization and cytochrome c release, as demonstrated in lung adenocarcinoma cells (86) and primary hippocampal neurons (33). Moreover, FoxO3a was shown to be activated through the inhibition of Akt-mediated phosphorylation and was reported to be a key event mediating neuronal death in response to Aβ (73). Klotho-induced activation of Akt maintains an inhibitory phosphorylation of FoxO3a (on Thr32), which is sustained up to 24 h after the treatment in the absence and presence of glutamate. Klotho-induced sustained inhibitory phosphorylation of FoxO3a may contribute to the antiapoptotic effect of Klotho and is also consistent with our observation that Klotho did not induce Mn-SOD in hippocampal neurons (data not shown). The induction of Mn-SOD through the reduced phosphorylation of FoxO1 was proposed to be important for the extended longevity and resistance to oxidative stress of the KL-OE mice (11), for the antioxidative effect of Klotho in HeLa and CHO cells (26, 87), and for the Klotho-mediated neuroprotection by ligustilide (3-butylidene-4,5-dihydrophthalide), which exerts marked neuroprotective effects through its antioxidant and antiapoptotic properties (45). Lack of Mn-SOD induction and transient phosphorylation of FoxO3 in our experimental paradigm further support the premise that Klotho-induced neuroprotection is associated with inhibitory phosphorylation of FoxO3a, followed by inhibition of its degradation, thus preventing the induction of the apoptotic cascade. Another possible downstream target for Akt inhibition by phosphorylation is GSK3β. GSK3β was shown to be involved in neuronal death, and cumulative evidence demonstrates the role of GSK3β inhibitors in neuroprotection (47, 88). GSK3 activation leads to Tau hyperphosphorylation, microtubule destabilization, and apoptosis in cultured primary neurons. GSK3β and ERK1/2 are among the best characterized kinases able to phosphorylate Tau (89). In rat primary hippocampal neurons, Klotho did not affect the phosphorylation of GSK3β in the absence or presence of glutamate. Consequently, no change in Tau hyperphosphorylation on Ser202 and Thr205 was detected following treatment with Klotho. Thus, Klotho does not protect neurons by inhibition of Tau phosphorylation. Fig. 9 presents a summary of major findings presented in this study and illustrates the mechanism by which Klotho is hypothesized to exert its neuroprotective effect.
FIGURE 9.
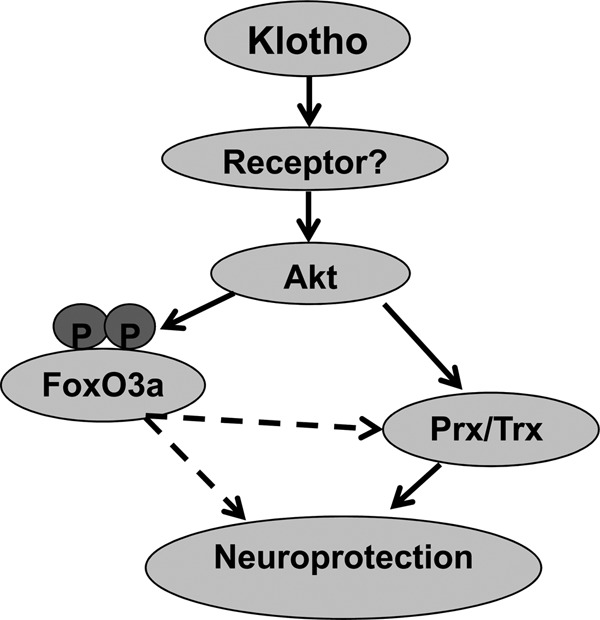
Diagram of the Klotho mechanism of neuroprotection. The neuroprotective effect of Klotho on hippocampal neurons is mediated via Akt-dependent induction of Prx-2/Trxrd-1 and is associated with inhibitory phosphorylation of FoxO3a.
Neuronal damage mediated via oxidative stress plays an important role in neurodegenerative diseases, such as AD, Parkinson disease, Huntington disease, and ALS. Our work demonstrates the neuroprotective properties of Klotho and its ability to counteract neuronal cell loss associated with neurodegenerative diseases. Furthermore, our results reveal that Klotho exerts its neuroprotective effect via the Trx/Prx antioxidative system, which is important for regulating the redox balance in the brain. Finally, our studies suggest that Klotho-enhancing compounds may provide future therapeutic approaches for neurodegenerative disorders (90, 91).
Acknowledgments
We thank Dr. Christina Khodr for help with the mouse colony, Dr. Douglas Rosene for reading the manuscript, Dr. Michael Sherman for helpful discussions, and Drs. David Shubert and Joseph Burdo for the HT22 cells.
This work was supported by a grant from the Alzheimer's Drug Discovery Foundation (to C. R. A.).
M. Kuro-o, personal communication.
- ROS
- reactive oxygen species
- AD
- Alzheimer disease
- Aβ
- amyloid β
- oAβ
- oligomeric Aβ
- NMDAR
- NMDA receptor
- KL-KO
- Klotho knock-out
- KL-OE
- KL-overexpressing
- WB
- Western blotting
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- qRT-PCR
- quantitative RT-PCR
- Prx
- peroxiredoxin
- Trx
- thioredoxin
- GSK3β
- glycogen synthase kinase 3β
- SOD
- superoxide dismutase
- Mn-SOD
- manganese superoxide dismutase
- Trxrd
- thioredoxin reductase
- E18
- embryonic day 18.
REFERENCES
- 1. Armstrong J. S., Khdour O., Hecht S. M. (2010) Does oxidative stress contribute to the pathology of Friedreich's ataxia? A radical question. FASEB J. 24, 2152–2163 [DOI] [PubMed] [Google Scholar]
- 2. Melo A., Monteiro L., Lima R. M., Oliveira D. M., Cerqueira M. D., El-Bachá R. S. (2011) Oxidative stress in neurodegenerative diseases: mechanisms and therapeutic perspectives. Oxid. Med. Cell Longev. 2011, 467180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jin Y. N., Johnson G. V. (2010) The interrelationship between mitochondrial dysfunction and transcriptional dysregulation in Huntington disease. J. Bioenerg. Biomembr. 42, 199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baranzini S. E., Srinivasan R., Khankhanian P., Okuda D. T., Nelson S. J., Matthews P. M., Hauser S. L., Oksenberg J. R., Pelletier D. (2010) Genetic variation influences glutamate concentrations in brains of patients with multiple sclerosis. Brain 133, 2603–2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fukui M., Choi H. J., Zhu B. T. (2010) Mechanism for the protective effect of resveratrol against oxidative stress-induced neuronal death. Free Radic. Biol. Med. 49, 800–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coyle J. T., Puttfarcken P. (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262, 689–695 [DOI] [PubMed] [Google Scholar]
- 7. Shi Q., Gibson G. E. (2007) Oxidative stress and transcriptional regulation in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 21, 276–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuro-o M., Matsumura Y., Aizawa H., Kawaguchi H., Suga T., Utsugi T., Ohyama Y., Kurabayashi M., Kaname T., Kume E., Iwasaki H., Iida A., Shiraki-Iida T., Nishikawa S., Nagai R., Nabeshima Y. I. (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51 [DOI] [PubMed] [Google Scholar]
- 9. Wang Y. A. (2006) Klotho, the long sought-after elixir and a novel tumor suppressor? Cancer Biol. Ther. 5, 20–21 [DOI] [PubMed] [Google Scholar]
- 10. Kuro-o M. (2009) Klotho and aging. Biochim. Biophys. Acta 1790, 1049–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kurosu H., Yamamoto M., Clark J. D., Pastor J. V., Nandi A., Gurnani P., McGuinness O. P., Chikuda H., Yamaguchi M., Kawaguchi H., Shimomura I., Takayama Y., Herz J., Kahn C. R., Rosenblatt K. P., Kuro-o M. (2005) Suppression of aging in mice by the hormone Klotho. Science 309, 1829–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Masuda H., Chikuda H., Suga T., Kawaguchi H., Kuro-o M. (2005) Regulation of multiple ageing-like phenotypes by inducible klotho gene expression in klotho mutant mice. Mech. Ageing Dev. 126, 1274–1283 [DOI] [PubMed] [Google Scholar]
- 13. Shiozaki M., Yoshimura K., Shibata M., Koike M., Matsuura N., Uchiyama Y., Gotow T. (2008) Morphological and biochemical signs of age-related neurodegenerative changes in klotho mutant mice. Neuroscience 152, 924–941 [DOI] [PubMed] [Google Scholar]
- 14. German D. C., Khobahy I., Pastor J., Kuro O. M., Liu X. (2012) Nuclear localization of Klotho in brain: an anti-aging protein. Neurobiol. Aging 33, 1483.e25–1483.e30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nagai T., Yamada K., Kim H. C., Kim Y. S., Noda Y., Imura A., Nabeshima Y., Nabeshima T. (2003) Cognition impairment in the genetic model of aging klotho gene mutant mice: a role of oxidative stress. FASEB J. 17, 50–52 [DOI] [PubMed] [Google Scholar]
- 16. Li S. A., Watanabe M., Yamada H., Nagai A., Kinuta M., Takei K. (2004) Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct. Funct. 29, 91–99 [DOI] [PubMed] [Google Scholar]
- 17. Duce J. A., Podvin S., Hollander W., Kipling D., Rosene D. L., Abraham C. R. (2008) Gene profile analysis implicates Klotho as an important contributor to aging changes in brain white matter of the rhesus monkey. Glia 56, 106–117 [DOI] [PubMed] [Google Scholar]
- 18. King G. D., Rosene D. L., Abraham C. R. (2012) Promoter methylation and age-related downregulation of Klotho in rhesus monkey. Age 34, 1405–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen C. D., Sloane J. A., Li H., Aytan N., Giannaris E. L., Zeldich E., Hinman J. D., Dedeoglu A., Rosene D. L., Bansal R., Luebke J. I., Kuro-o M., Abraham C. R. (2013) The antiaging protein Klotho enhances oligodendrocyte maturation and myelination of the CNS. J. Neurosci. 33, 1927–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dubal D. B., Yokoyama J. S., Zhu L., Broestl L., Worden K., Wang D., Sturm V. E., Kim D., Klein E., Yu G. Q., Ho K., Eilertson K. E., Yu L., Kuro-o M., De Jager P. L., Coppola G., Small G. W., Bennett D. A., Kramer J. H., Abraham C. R., Miller B. L., Mucke L. (2014) Life extension factor klotho enhances cognition. Cell Rep. 7, 1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen C. D., Podvin S., Gillespie E., Leeman S. E., Abraham C. R. (2007) Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc. Natl. Acad. Sci. U.S.A. 104, 19796–19801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matsumura Y., Aizawa H., Shiraki-Iida T., Nagai R., Kuro-o M., Nabeshima Y. (1998) Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem. Biophys. Res. Commun. 242, 626–630 [DOI] [PubMed] [Google Scholar]
- 23. Cha S. K., Ortega B., Kurosu H., Rosenblatt K. P., Kuro-O M., Huang C. L. (2008) Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc. Natl. Acad. Sci. U.S.A. 105, 9805–9810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cha S. K., Hu M. C., Kurosu H., Kuro-o M., Moe O., Huang C. L. (2009) Regulation of renal outer medullary potassium channel and renal K+ excretion by Klotho. Mol. Pharmacol. 76, 38–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doi S., Zou Y., Togao O., Pastor J. V., John G. B., Wang L., Shiizaki K., Gotschall R., Schiavi S., Yorioka N., Takahashi M., Boothman D. A., Kuro-o M. (2011) Klotho inhibits transforming growth factor-β1 (TGF-β1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J. Biol. Chem. 286, 8655–8665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamamoto M., Clark J. D., Pastor J. V., Gurnani P., Nandi A., Kurosu H., Miyoshi M., Ogawa Y., Castrillon D. H., Rosenblatt K. P., Kuro-o M. (2005) Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 280, 38029–38034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ikushima M., Rakugi H., Ishikawa K., Maekawa Y., Yamamoto K., Ohta J., Chihara Y., Kida I., Ogihara T. (2006) Anti-apoptotic and anti-senescence effects of Klotho on vascular endothelial cells. Biochem. Biophys. Res. Commun. 339, 827–832 [DOI] [PubMed] [Google Scholar]
- 28. Patenaude A., Murthy M. R., Mirault M. E. (2005) Emerging roles of thioredoxin cycle enzymes in the central nervous system. Cell Mol. Life Sci. 62, 1063–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhu H., Santo A., Li Y. (2012) The antioxidant enzyme peroxiredoxin and its protective role in neurological disorders. Exp. Biol. Med. (Maywood) 237, 143–149 [DOI] [PubMed] [Google Scholar]
- 30. Uetsuki T., Naito A., Nagata S., Kaziro Y. (1989) Isolation and characterization of the human chromosomal gene for polypeptide chain elongation factor-1 α. J. Biol. Chem. 264, 5791–5798 [PubMed] [Google Scholar]
- 31. Kim D. W., Uetsuki T., Kaziro Y., Yamaguchi N., Sugano S. (1990) Use of the human elongation factor 1 α promoter as a versatile and efficient expression system. Gene 91, 217–223 [DOI] [PubMed] [Google Scholar]
- 32. Hanaoka K., Hayasaka M., Uetsuki T., Fujisawa-Sehara A., Nabeshima Y. (1991) A stable cellular marker for the analysis of mouse chimeras: the bacterial chloramphenicol acetyltransferase gene driven by the human elongation factor 1 α promoter. Differentiation 48, 183–189 [DOI] [PubMed] [Google Scholar]
- 33. Chong Z. Z., Lin S. H., Maiese K. (2004) The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J. Cereb. Blood Flow Metab. 24, 728–743 [DOI] [PubMed] [Google Scholar]
- 34. De Felice F. G., Wu D., Lambert M. P., Fernandez S. J., Velasco P. T., Lacor P. N., Bigio E. H., Jerecic J., Acton P. J., Shughrue P. J., Chen-Dodson E., Kinney G. G., Klein W. L. (2008) Alzheimer's disease-type neuronal tau hyperphosphorylation induced by A β oligomers. Neurobiol. Aging 29, 1334–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stine W. B., Jungbauer L., Yu C., LaDu M. J. (2011) Preparing synthetic Aβ in different aggregation states. Methods Mol. Biol. 670, 13–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Freir D. B., Nicoll A. J., Klyubin I., Panico S., Mc Donald J. M., Risse E., Asante E. A., Farrow M. A., Sessions R. B., Saibil H. R., Clarke A. R., Rowan M. J., Walsh D. M., Collinge J. (2011) Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mishra S., Mishra M., Seth P., Sharma S. K. (2011) Tetrahydrocurcumin confers protection against amyloid β-induced toxicity. Neuroreport 22, 23–27 [DOI] [PubMed] [Google Scholar]
- 38. Zeldich E., Koren R., Dard M., Nemcovsky C., Weinreb M. (2007) Enamel matrix derivative protects human gingival fibroblasts from TNF-induced apoptosis by inhibiting caspase activation. J. Cell Physiol. 213, 750–758 [DOI] [PubMed] [Google Scholar]
- 39. Fukui M., Yamabe N., Kang K. S., Zhu B. T. (2010) Growth-stimulatory effect of resveratrol in human cancer cells. Mol. Carcinog. 49, 750–759 [DOI] [PubMed] [Google Scholar]
- 40. Stanciu M., Wang Y., Kentor R., Burke N., Watkins S., Kress G., Reynolds I., Klann E., Angiolieri M. R., Johnson J. W., DeFranco D. B. (2000) Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 275, 12200–12206 [DOI] [PubMed] [Google Scholar]
- 41. Tobaben S., Grohm J., Seiler A., Conrad M., Plesnila N., Culmsee C. (2011) Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell Death Differ. 18, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kumari S., Mehta S. L., Li P. A. (2012) Glutamate induces mitochondrial dynamic imbalance and autophagy activation: preventive effects of selenium. PLoS One 7, e39382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Das A., McDowell M., O'Dell C. M., Busch M. E., Smith J. A., Ray S. K., Banik N. L. (2010) Post-treatment with voltage-gated Na+ channel blocker attenuates kainic acid-induced apoptosis in rat primary hippocampal neurons. Neurochem. Res. 35, 2175–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arner E. S., Holmgren A. (2001) Measurement of thioredoxin and thioredoxin reductase. Curr. Protoc. Toxicol., Chapter 7, Unit 7.4 [DOI] [PubMed] [Google Scholar]
- 45. Kuang X., Chen Y. S., Wang L. F., Li Y. J., Liu K., Zhang M. X., Li L. J., Chen C., He Q., Wang Y., Du J. R. (2014) Klotho upregulation contributes to the neuroprotection of ligustilide in an Alzheimer's disease mouse model. Neurobiol. Aging 35, 169–178 [DOI] [PubMed] [Google Scholar]
- 46. Salih D. A., Brunet A. (2008) FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol. 20, 126–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Uranga R. M., Katz S., Salvador G. A. (2013) Enhanced phosphatidylinositol 3-kinase (PI3K)/Akt signaling has pleiotropic targets in hippocampal neurons exposed to iron-induced oxidative stress. J. Biol. Chem. 288, 19773–19784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Crews L., Masliah E. (2010) Molecular mechanisms of neurodegeneration in Alzheimer's disease. Hum. Mol. Genet. 19, R12–R20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chong Z. Z., Li F., Maiese K. (2005) Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Prog. Neurobiol. 75, 207–246 [DOI] [PubMed] [Google Scholar]
- 50. Kwon K. J., Kim H. J., Shin C. Y., Han S. H. (2010) Melatonin potentiates the neuroprotective properties of resveratrol against β-amyloid-induced neurodegeneration by modulating AMP-activated protein kinase pathways. J. Clin. Neurol. 6, 127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Behl C., Lezoualc'h F., Trapp T., Widmann M., Skutella T., Holsboer F. (1997) Glucocorticoids enhance oxidative stress-induced cell death in hippocampal neurons in vitro. Endocrinology 138, 101–106 [DOI] [PubMed] [Google Scholar]
- 52. Cha M. Y., Han S. H., Son S. M., Hong H. S., Choi Y. J., Byun J., Mook-Jung I. (2012) Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS One 7, e34929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang X., Michaelis E. K. (2010) Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Clinton S. M., Glover M. E., Maltare A., Laszczyk A. M., Mehi S. J., Simmons R. K., King G. D. (2013) Expression of klotho mRNA and protein in rat brain parenchyma from early postnatal development into adulthood. Brain Res. 1527, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang C. J., Hu C. P., Xu K. P., Yuan Q., Li F. S., Zou H., Tan G. S., Li Y. J. (2010) Protective effect of selaginellin on glutamate-induced cytotoxicity and apoptosis in differentiated PC12 cells. Naunyn Schmiedebergs Arch. Pharmacol. 381, 73–81 [DOI] [PubMed] [Google Scholar]
- 56. Teocchi M. A., Ferreira A. É., da Luz de Oliveira E. P., Tedeschi H., D'Souza-Li L. (2013) Hippocampal gene expression dysregulation of Klotho, nuclear factor κB and tumor necrosis factor in temporal lobe epilepsy patients. J. Neuroinflammation 10, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Banerjee S., Zhao Y., Sarkar P. S., Rosenblatt K. P., Tilton R. G., Choudhary S. (2013) Klotho ameliorates chemically induced endoplasmic reticulum (ER) stress signaling. Cell Physiol. Biochem. 31, 659–672 [DOI] [PubMed] [Google Scholar]
- 58. Semba R. D., Moghekar A. R., Hu J., Sun K., Turner R., Ferrucci L., O'Brien R. (2014) Klotho in the cerebrospinal fluid of adults with and without Alzheimer's disease. Neurosci. Lett. 558, 37–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bell K. F., Hardingham G. E. (2011) CNS peroxiredoxins and their regulation in health and disease. Antioxid. Redox Signal. 14, 1467–1477 [DOI] [PubMed] [Google Scholar]
- 60. Hwang I. K., Yoo K. Y., Kim D. W., Lee C. H., Choi J. H., Kwon Y. G., Kim Y. M., Choi S. Y., Won M. H. (2010) Changes in the expression of mitochondrial peroxiredoxin and thioredoxin in neurons and glia and their protective effects in experimental cerebral ischemic damage. Free Radic. Biol. Med. 48, 1242–1251 [DOI] [PubMed] [Google Scholar]
- 61. Lee Y. J., Goo J. S., Kim J. E., Nam S. H., Hwang I. S., Choi S. I., Lee H. R., Lee E. P., Choi H. W., Kim H. S., Lee J. H., Jung Y. J., Kim H. J., Hwang D. Y. (2011) Peroxiredoxin I regulates the component expression of γ-secretase complex causing the Alzheimer's disease. Lab. Anim. Res. 27, 293–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gan Y., Ji X., Hu X., Luo Y., Zhang L., Li P., Liu X., Yan F., Vosler P., Gao Y., Stetler R. A., Chen J. (2012) Transgenic overexpression of peroxiredoxin-2 attenuates ischemic neuronal injury via suppression of a redox-sensitive pro-death signaling pathway. Antioxid. Redox Signal. 17, 719–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lopert P., Day B. J., Patel M. (2012) Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. PLoS One 7, e50683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen W., Ji J., Xu X., He S., Ru B. (2003) Proteomic comparison between human young and old brains by two-dimensional gel electrophoresis and identification of proteins. Int. J. Dev. Neurosci. 21, 209–216 [DOI] [PubMed] [Google Scholar]
- 65. Calkins M. J., Manczak M., Mao P., Shirendeb U., Reddy P. H. (2011) Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum. Mol. Genet. 20, 4515–4529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shankar G. M., Bloodgood B. L., Townsend M., Walsh D. M., Selkoe D. J., Sabatini B. L. (2007) Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Reddy P. H. (2009) Role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer's disease. CNS Spectr. 14, 8–13; discussion 16–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cumming R. C., Dargusch R., Fischer W. H., Schubert D. (2007) Increase in expression levels and resistance to sulfhydryl oxidation of peroxiredoxin isoforms in amyloid β-resistant nerve cells. J. Biol. Chem. 282, 30523–30534 [DOI] [PubMed] [Google Scholar]
- 69. Cimini A., Gentile R., Angelucci F., Benedetti E., Pitari G., Giordano A., Ippoliti R. (2013) Neuroprotective effects of PrxI over-expression in an in vitro human Alzheimer's disease model. J. Cell Biochem. 114, 708–715 [DOI] [PubMed] [Google Scholar]
- 70. Krapfenbauer K., Engidawork E., Cairns N., Fountoulakis M., Lubec G. (2003) Aberrant expression of peroxiredoxin subtypes in neurodegenerative disorders. Brain Res. 967, 152–160 [DOI] [PubMed] [Google Scholar]
- 71. Kudo W., Lee H. P., Smith M. A., Zhu X., Matsuyama S., Lee H. G. (2012) Inhibition of Bax protects neuronal cells from oligomeric Aβ neurotoxicity. Cell Death Dis. 3, e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Luo H., Yang Y., Duan J., Wu P., Jiang Q., Xu C. (2013) PTEN-regulated AKT/FoxO3a/Bim signaling contributes to reactive oxygen species-mediated apoptosis in selenite-treated colorectal cancer cells. Cell Death Dis. 4, e481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sanphui P., Biswas S. C. (2013) FoxO3a is activated and executes neuron death via Bim in response to β-amyloid. Cell Death Dis. 4, e625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chong Z. Z., Kang J. Q., Maiese K. (2004) AKT1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-xL and caspase 1, 3, and 9. Exp. Cell Res. 296, 196–207 [DOI] [PubMed] [Google Scholar]
- 75. Kang J. Q., Chong Z. Z., Maiese K. (2003) Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol. Pharmacol. 64, 557–569 [DOI] [PubMed] [Google Scholar]
- 76. Kang J. Q., Chong Z. Z., Maiese K. (2003) Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J. Neurosci. Res. 74, 37–51 [DOI] [PubMed] [Google Scholar]
- 77. Alessi D. R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., Hemmings B. A. (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15, 6541–6551 [PMC free article] [PubMed] [Google Scholar]
- 78. Pei J. J., Khatoon S., An W. L., Nordlinder M., Tanaka T., Braak H., Tsujio I., Takeda M., Alafuzoff I., Winblad B., Cowburn R. F., Grundke-Iqbal I., Iqbal K. (2003) Role of protein kinase B in Alzheimer's neurofibrillary pathology. Acta Neuropathol. 105, 381–392 [DOI] [PubMed] [Google Scholar]
- 79. Wöhrle S., Bonny O., Beluch N., Gaulis S., Stamm C., Scheibler M., Müller M., Kinzel B., Thuery A., Brueggen J., Hynes N. E., Sellers W. R., Hofmann F., Graus-Porta D. (2011) FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J. Bone Miner. Res. 26, 2486–2497 [DOI] [PubMed] [Google Scholar]
- 80. Izbeki F., Asuzu D. T., Lorincz A., Bardsley M. R., Popko L. N., Choi K. M., Young D. L., Hayashi Y., Linden D. R., Kuro-o M., Farrugia G., Ordog T. (2010) Loss of Kitlow progenitors, reduced stem cell factor and high oxidative stress underlie gastric dysfunction in progeric mice. J. Physiol. 588, 3101–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Château M. T., Araiz C., Descamps S., Galas S. (2010) Klotho interferes with a novel FGF-signalling pathway and insulin/Igf-like signalling to improve longevity and stress resistance in Caenorhabditis elegans. Aging 2, 567–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Papadia S., Soriano F. X., Léveillé F., Martel M. A., Dakin K. A., Hansen H. H., Kaindl A., Sifringer M., Fowler J., Stefovska V., McKenzie G., Craigon M., Corriveau R., Ghazal P., Horsburgh K., Yankner B. A., Wyllie D. J., Ikonomidou C., Hardingham G. E. (2008) Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 11, 476–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hardingham G. E., Bading H. (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hetman M., Kharebava G. (2006) Survival signaling pathways activated by NMDA receptors. Curr. Top. Med. Chem. 6, 787–799 [DOI] [PubMed] [Google Scholar]
- 85. Kim S. U., Jin M. H., Kim Y. S., Lee S. H., Cho Y. S., Cho K. J., Lee K. S., Kim Y. I., Kim G. W., Kim J. M., Lee T. H., Lee Y. H., Shong M., Kim H. C., Chang K. T., Yu D. Y., Lee D. S. (2011) Peroxiredoxin II preserves cognitive function against age-linked hippocampal oxidative damage. Neurobiol. Aging 32, 1054–1068 [DOI] [PubMed] [Google Scholar]
- 86. Zhong D., Liu X., Khuri F. R., Sun S. Y., Vertino P. M., Zhou W. (2008) LKB1 is necessary for Akt-mediated phosphorylation of proapoptotic proteins. Cancer Res. 68, 7270–7277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Murakami S. (2006) Stress resistance in long-lived mouse models. Exp. Gerontol. 41, 1014–1019 [DOI] [PubMed] [Google Scholar]
- 88. Meijer L., Flajolet M., Greengard P. (2004) Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol. Sci. 25, 471–480 [DOI] [PubMed] [Google Scholar]
- 89. Medina M. G., Ledesma M. D., Domínguez J. E., Medina M., Zafra D., Alameda F., Dotti C. G., Navarro P. (2005) Tissue plasminogen activator mediates amyloid-induced neurotoxicity via Erk1/2 activation. EMBO J. 24, 1706–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. King G. D., Chen C., Huang M. M., Zeldich E., Brazee P. L., Schuman E. R., Robin M., Cuny G. D., Glicksman M. A., Abraham C. R. (2012) Identification of novel small molecules that elevate Klotho expression. Biochem. J. 441, 453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Abraham C. R., Chen C., Cuny G. D., Glicksman M. A., Zeldich E. (2012) Small-molecule Klotho enhancers as novel treatment of neurodegeneration. Future Med. Chem. 4, 1671–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]