

Background: Recent research has uncovered tumor suppressive and oncogenic potential of miRNAs.
Results: miR-941 expression is regulated by DNA methylation, and is an important regulator of cell proliferation, migration, and invasion by directly targeting KDM6B in hepatocellular carcinoma (HCC).
Conclusion: miR-941 may play an important role in suppressing tumor growth and metastasis in HCC.
Significance: miR-941 may provide a potential biomarker for the diagnosis and treatment of HCC.
Keywords: DNA Methylation, Epigenetics, Epithelial-Mesenchymal Transition (EMT), Hepatocellular Carcinoma, Invasion, Migration, Proliferation, KDM6B, miR-941, miRNAs
Abstract
MicroRNAs (miRNAs) have been shown to play important roles in carcinogenesis. However, their underlying mechanisms of action in hepatocellular carcinoma (HCC) are poorly understood. Recent evidence suggests that epigenetic silencing of miRNAs through tumor suppression by CpG island hypermethylation may be a common hallmark of human tumors. Here, we demonstrated that miR-941 was significantly down-regulated in HCC tissues and cell lines and was generally hypermethylated in HCC. The overexpression of miR-941 suppressed in vitro cell proliferation, migration, and invasion and inhibited the metastasis of HCC cells in vivo. Furthermore, the histone demethylase KDM6B (lysine (K)-specific demethylase 6B) was identified as a direct target of miR-941 and was negatively regulated by miR-941. The ectopic expression of KDM6B abrogated the phenotypic changes induced by miR-941 in HCC cells. We demonstrated that miR-941 and KDM6B regulated the epithelial-mesenchymal transition process and affected cell migratory/invasive properties.
Introduction
MicroRNAs (miRNA)4 are small, non-coding RNAs of ∼22 nucleotides in length that are endogenously expressed and regulate gene expression by binding to the 3′ untranslated regions (UTRs) of target genes. miRNA binding leads to translational repression or mRNA degradation (1). miRNAs can function as tumor suppressors or oncogenes by targeting oncogenes or tumor suppressor genes, respectively (2). There is increasing evidence indicating that miRNAs exist in many types of cancers (3–5), including hepatocellular carcinoma (HCC) (6, 7). HCC is one of the most common malignant tumors with a poor prognosis and is the third leading cause of cancer-related death in the world (8). The rapid proliferation of cancer cells and their ability to migrate to other tissue sites leads to poor clinical outcomes (9). Our understanding of the clinical pathogenesis of HCC is limited, which hinders the development of effective treatment methods. Research has focused on elucidating the molecular pathogenesis of HCC, which is critical for indentifying novel molecular targets. Recent studies have reported that miRNAs such as miR-490–3p (9), miR-21 (10), miR-10a (6), and miR-519d (11) are associated with HCC. The epithelial to mesenchymal transition (EMT) is an important cellular phenotypic change in which polarized, immotile epithelial cells acquire the motile mesenchymal phenotype. EMT is regulated by miRNAs (12, 13) that promote tumor invasion and metastasis and allow tumor cells to escape apoptosis (14).
Aberrant miRNA expression can be regulated by many factors, including chromosome modification. DNA methylation is one of the most important chromosome modifications involved in the aberrant epigenetic profile in cancer. Recent evidence indicates that methylation may play a role in the regulation of tumor malignancy (15, 16) and the silencing of several miRNAs is intimately linked to the epigenetic mechanisms of DNA methylation. Furthermore, the aberrant methylation of CpG islands is an event frequently observed in the process of tumorigenesis (17–19).
Our previous work indicated that miR-941 in HepG2 cells treated with 5-azacytidine (an inhibitor of DNA methyltransferase) was up-regulated compared with control cells by miRNA microarray analysis (data not shown), thus, miR-941 was chosen for further study. Here, we determined that miR-941 was significantly down-regulated in HCC tissues and influenced the phenotypes of QGY-7703 and HepG2 cells. CpG islands are present in the upstream region of the miR-941 gene, therefore, we performed bisulfite sequencing analysis and demonstrated that miR-941 down-regulation may be caused by epigenetic inactivation. Furthermore, we also identified that miR-941 regulated the expression of KDM6B via directly binding to its 3′ UTR. KDM6B belongs to a small family of JmjC domain containing enzymes that mediate the demethylation of H3K27 trimethyl groups (20–23). Many miRNAs, including miR-138, miR-148a, miR-185, and miR-339-5p, had been reported to decrease KDM6B expression by targeting the 3′ UTR and increasing H3K27 methylation (24). Our findings indicated that miR-941 was influenced by methylation and functioned as a tumor suppressor by regulating KDM6B. Both miR-941 and KDM6B were involved in the EMT process.
EXPERIMENTAL PROCEDURES
Vector Constructions
Pri-miR-941 (the fragment containing the precursor of the miR-941) was amplified using the primers shown in Table 1 and then cloned into the BamHI/EcoRI restriction sites of pcDNA3. The resulting construct was confirmed by DNA sequencing. miR-941 antisense oligonucleotide (ASO-miR-941) was used as the inhibitor of miR-941 and ASO-NC was used as its control.
TABLE 1.
The oligonucleotides used in vector constructions
| Name | Sequence (5′-3′) |
|---|---|
| Pri-941-S | 5′-GGAGGATCCGCACCGACGTGTCCGGGG-3′ |
| Pri-941-As | 5′-GGAGAGAATTCAGCCTGAGGCCCCCGCATG-3′ |
| ASO-miR-941 | 5′-GCACAUGUGCACACAGCCGGGUG-3′ |
| ASO-NC | 5′-CAGUACUUUUGUGUAGUACAA-3′ |
| EGFP-S | 5′-GCAGCCAAGCTTGCCACCATGTGTAGCAAGGGC-3′ |
| EGFP-AS | 5′-CGCGGATCCTTTACTTGTACAGCTCGTCC-3′ |
| KDM6B-3′UTR-S | 5′-CGCGGATCCTCTATTTATTCATTTTTGG-3′ |
| KDM6B-3′UTR-AS | 5′-CGGAATTCGAGAAAGTGCAGGACGTAG-3′ |
| KDM6B-3′UTR-MS | 5′-CGCGGCTCCACGCCGCATCTCATGGTG-3′ |
| KDM6B-3′UTR-MA | 5′-CACCATGAGATGCGGCGTGGAGCCGCG-3′ |
| KDM6B-shR-Top | 5′-GATCCGCAGCTTGGGCAACTGTACGTTCAAGAGACGTACAGTTGCCCAAGCTGTTTTTTGGAAGAATTCA-3′ |
| KDM6B-shR-Bot | 5′-AGCTTGAATTCTTCCAAAAAACAGCTTGGGCAACTGTACGTCTCTTGAACGTACAGTTGCCCAAGCTGCG-3′ |
To construct the enhanced green fluorescent protein (EGFP) reporter plasmid, the EGFP coding region from the pEGFP-N1 vector was subcloned into pcDNA3 using HindIII and BamHI. Wild-type or mutant forms of the KDM6B mRNA 3′ UTR were inserted into the downstream region of the pcDNA3/EGFP vector.
The pSilencer/shR-KDM6B vector was constructed by annealing the top and bottom strands of hairpin RNA and inserting them into the pSilencer2.1 neo vector (Ambion). All the oligonucleotides used are shown in Table 1.
Cell Culture and Treatment
The HepG2, Hep3B, SK-Hep-1, PLC-PRF-5, and LM6 HCC cell lines were grown in α-minimal essential medium (Invitrogen), QGY-7703 cells were cultured in RPMI 1640 medium (Invitrogen) supplemented with 10–20% FBS, 100 IU/ml of penicillin, and 100 mg/ml of streptomycin. All the cells were incubated in a humidified atmosphere with 5% CO2 at 37°C. The transient transfections were performed using LipofectamineTM 2000 (Invitrogen).
For the 5-aza-CdR (Sigma) treatments, the cells were seeded into 25-cm2 culture flasks at a density of 5 × 105 cells/flask and cultured with 5 (QGY-7703) or 10 μmol (HepG2) of 5-aza-CdR. The drug-containing medium was changed every 24 h. After 72 h of treatment, the cells were harvested for genomic DNA and total RNA extraction for use in the subsequent experiments.
To select a pool of QGY-7703 clones, cells were placed into cell culture flanks (250 ml) containing G418 (Geneticin, 1 mg/ml or 500 g/ml) for 24 h after transient transfection with the miR-941 or pcDNA3 vectors. The medium was changed every 3 days. On day 12, there were numerous G418-resistant colonies. Total RNA was extracted and used for qRT-PCR analysis. We obtained transformed cells that were resistant to G418.
Human Tissue Samples and RNA Isolation
Human HCC and adjacent non-tumor liver tissues were collected from the Tumor Bank Facility of Cancer Center, Sun Yat-sen University. All samples were obtained with informed consent from the patients, and this study was approved by the Ethics Committee of Tianjin Medical University. RNA was isolated from tissue samples with the mirVana miRNA Isolation Kit (Ambion, Austin, TX) according to the manufacturer's protocol.
Genomic DNA Extraction and Bisulfite Treatment
Genomic DNA was extracted from the HCC cell lines or clinical HCC tissue specimens according to the Kit instructions (Biotete Corp.). The bisulfite treatment was performed according to the EZ DNA Methylation-Direct Kit protocol (Zymo Research, Orange, CA).
DNA Methylation Analysis
The miRNA sequences were analyzed using miRBase and the University of California at Santa Cruz Human Genome Browser. The bisulfate primers used in this study were designed using MethPrimer (25).
Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
Stem-loop quantitative RT-PCR was performed to detect miR-941. Briefly, 2 μg of small RNA extracted from cells or tissue samples was reverse transcribed to cDNA using a stem-loop RT primer with Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI). cDNA was used to amplify miR-941, and endogenous U6 snRNA was used as the internal control. The PCR cycling parameters were as follows: 94 °C for 3 min, followed by 40 cycles of 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 30 s.
To quantitate KDM6B expression, 5 μg of large RNA extracted from cells or tissue samples was reverse transcribed to cDNA using Molony murine leukemia virus reverse transcriptase. The cDNA was used to amplify KDM6B as well as the β-actin endogenous control. The PCR cycling parameters were as follows: 94 °C for 3 min followed by 40 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s. The PCRs were performed using SYBR Premix Ex Mix TaqTM (TaKaRa, Dalian, China) on an iQ5 Real-Time PCR Detection System (Bio-Rad). The relative gene expression levels were calculated with the 2−ΔΔCt method
Western Blot Analysis
Cells were transfected and then lysed with RIPA lysis buffer. Protein lysates were resolved by SDS-PAGE (polyacrylamide) and then transferred onto a nitrocellulose membrane. The membranes were incubated with blocking buffer (Blotto) for 90 min at room temperature and then with antibodies against KDM6B, E-cadherin, vimentin, or GAPDH overnight at 4 °C. Subsequently, the membranes were washed and incubated with a HRP-conjugated secondary antibody. Protein expression was analyzed by enhanced chemiluminescence and exposure to chemiluminescent film. The band intensities were quantified using Lab Works Image Acquisition and Analysis Software (UVP). The E-cadherin and vimentin antibodies were purchased from Tianjin Saier Biotechnology (Tianjin, China), and the KDM6B antibody was purchased from Millipore Biotechnology (Millipore).
Fluorescent Reporter Assay
Cells were co-transfected with pri-miR-941 or pcDNA3, and ASO-miR-941 or control oligonucleotides in a 48-well plate along with the reporter vector pcDNA3/EGFP-KDM6B 3′ UTR or the mutated KDM6B 3′ UTR. The RFP expression vector pDsRed2-N1 (Clontech) was used for normalization. The EGFP and RFP fluorescent intensities were detected with Fluorescence Spectrophotometer F-4500 (HITACHI, Tokyo, Japan).
MTT and Colony Formation Assays
Twenty-four hours after transfection, the cells were seeded into 96-well plates at a density of either 7,000 cells/well (QGY-7703) or 10,000 cells/well (HepG2). An MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was used to determine cell viability 48 h after transfection. The absorbance at 570 nm was measured using a μQuant Universal Microplate Spectrophotometer (BioTek, Winooski, VT).
For colony formation assays, the cells were counted and seeded at a density of either 150 cells/well (QGY-7703) or 700 cells/well (HepG2) in 12-well plates in triplicate. The culture medium was changed every 3 days. Colonies were counted only if they contained more than 50 cells, and the number of colonies was counted at either 6 (QGY-7703) or 15 (HepG2) days after seeding. The colonies were stained using crystal violet, and the colony formation rate was calculated using the following equation: colony formation rate (%) = (number of colonies/number of seeded cells) × 100%.
Migration and Invasion Assays
For the transwell migration assays, 6 × 104 QGY-7703 or 1.5 × 105 HepG2 cells in 0.2 ml of RPMI 1640 or α-minimal essential medium without FBS were seeded into the upper part of each Transwell chamber (Transwell filter inserts with a pore size of 8 μm; Corning) with a non-coated membrane. For the invasion assays, 6 × 104 QGY-7703 or 1.5 × 105 HepG2 cells were placed on the upper chamber of each insert coated with 40 μl of 2 mg/ml of Matrigel (growth factor-reduced BD MatrigelTM matrix). In the lower part of the chamber, 0.8 ml of RPMI 1640 or α-minimal essential medium with 20% FBS was added. After incubating for several hours (12 h for QGY-7703 and 48 h for HepG2 in the migration assays and 24 h for QGY-7703 and 72 h for HepG2 in the invasion assays), the upper surface of the membrane was wiped with a cotton tip, and the cells that were attached to the lower surface were stained for 20 min with crystal violet. Images were acquired from three different regions. The mean of triplicate assays for each experimental condition was reported.
In Vivo Metastasis Assays
To assess in vivo metastasis, 3 × 106 QGY-7703 cells (stably transfected with miR-941 and pcDNA3) were suspended in 40 μl of serum-free RPMI 1640/Matrigel (1:1) for each mouse. Each nude mouse was inoculated in the upper pole of the spleen with a microsyringe under anesthesia. After 24 days, the mice were sacrificed, and their spleens and livers were harvested. The tissues were fixed with phosphate-buffered neutral formalin. All the studies were performed according to the American Association for the Accreditation of Laboratory Animal Care guidelines for the humane treatment of animals and adhered to national and international standards.
Immunofluorescence Staining
Transfected QGY-7703 cells were seeded into 14-well plates. After 24 h, the cells were rinsed with 1 × PBS, fixed with paraformaldehyde, and blocked with 10% normal donkey serum. The cells were then incubated with a primary antibody (KDM6B, rabbit). A FITC-conjugated goat anti-rabbit IgG secondary antibody was used to detect the primary antibody. The cell nuclei were labeled with DAPI. All the images were captured using a fluorescence microscope (model Eclipse 660, Nikon, Japan) and a digital camera (ACT-2U, Nikon, Japan) with the NIS Element FW software package.
Immunohistochemical Staining
All of the tissue samples were fixed in phosphate-buffered neutral formalin and embedded in paraffin, finally cut into 5-μm thick sections. Sections were soaked in hydrogen peroxide/phosphate-buffered saline for 30 min and incubated with a primary antibody against KDM6B, E-cadherin, and vimentin at a 1:100 dilution overnight at 4 °C. Detection of the primary antibody was performed by goat anti-rabbit HRP for 1 h at room temperature and visualized with DAB substrate.
Statistical Analysis
The data are presented as the mean ± S.D. The statistical analyses were performed using a paired Student's t test. A p < 0.05 was considered statistically significant. One representative experiment is presented for experiments performed in duplicate or triplicate.
RESULTS
miR-941 was Down-regulated in Human HCC Tissues and Cells
We used qRT-PCR to determine the miR-941 expression level in 15 pairs of HCC and adjacent non-cancerous tissues, miR-941 was significantly down-regulated in HCC tissues compared with the adjacent normal tissues (Fig. 1A). We detected miR-941 expression in the LO2 immortalized normal liver cell line and HCC cell lines (QGY-7703, HepG2, Hep3B, SK-Hep-1, PLC-PRF-5, and LM6), miR-941 was down-regulated in HCC cells compared with LO2 cells (Fig. 1B).
FIGURE 1.
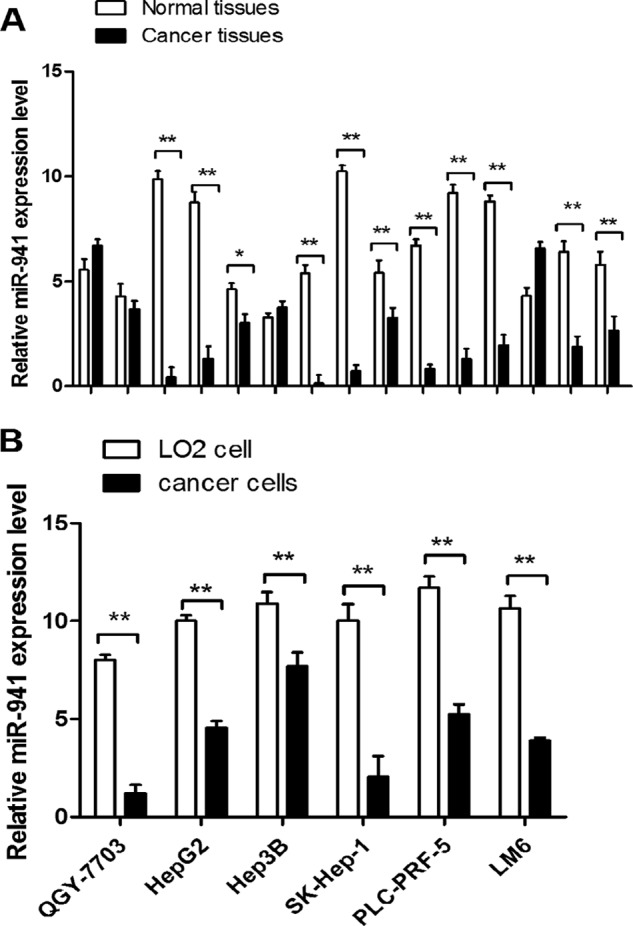
miR-941 is down-regulated in HCC tissues and cell lines. A, differential expression of miR-941 in 15 pairs of HCC tissues compared with adjacent normal tissues. B, differential expression of miR-941 in six different HCC cell lines, including QGY-7703, HepG2, Hep3B, SK-Hep-1, PLC-PRF-5, and LM6 compared with a non-cancerous cell (LO2). The relative expression of miR-941 was detected by quantitative RT-PCR. *, p < 0.05; **, p < 0.01). The primers for quantitative RT-PCR are shown in Table 2.
miR-941 Expression Was Regulated by DNA Methylation in HCC
We investigated whether the down-regulation of miR-941 expression in human HCC tissues and cell lines was caused by an epigenetic mechanism such as DNA methylation. CpG islands are located in the promoter region of miR-941, which is embedded in intron-1 of DNAJC5 at chromosome 20q13.3. One CpG-rich island is within 500 bp of miR-941, and its upstream region was found using MethPrimer with the default criteria (Fig. 2A). First, we analyzed the DNA methylation status of the miR-941 gene in 10 pairs of HCC tumor and adjacent non-cancerous tissues, the DNA methylation status of each of the 10 CpGs was determined by bisulfate analysis. Data indicated that the unmethylated CG sites in adjacent non-tumor tissues were higher than HCC tissues (Fig. 2B). Then, we examined methylation in the HCC cell lines. As shown in Fig. 2C, the CpG island was highly methylated in QGY-7703 and HepG2 cells, but poorly methylated in LO2 cells. The expression of miR-941 was determined by qRT-PCR after the cells were treated with 5-aza-CdR. Compared with the controls, miR-941 expression was significantly up-regulated in QGY-7703 (19.33-fold) and HepG2 (7-fold) cells (Fig. 2E). The methylation frequency decreased from 98.3 to 68.3% in QGY-7703 cells and from 96.7 to 71.7% in HepG2 cells. The methylation frequency in LO2 cells was 36.7% (Fig. 2D). The primers for genomic bisulfite sequencing are shown in Table 2. These results indicated that miR-941 down-regulation in HCC cells and tissues may be regulated by DNA methylation.
FIGURE 2.

miR-941 is regulated by DNA methylation in QGY-7703 and HepG2 cell lines. A, genomic representation of Chr20, which includes the DNAJC5 gene and miR-941 is embedded in intron-1 of DNAJC5. Relative location of CpG sites (the red vertical bars) are indicated upstream of miR-941 and the start site is designated as +1. The CpG island (blue box) and primers for bisulfite PCR (black box) are also indicated. B, we carried out a bisulfite sequencing analysis to determine the DNA methylation status of each of the 10 CpGs in HCC tumor tissues and their adjacent non-tumor tissues. Open and filled squares denote unmethylated and methylated CpG sites, respectively. Each row represents a single clone. C, the methylation status of the CpG island is assessed before and after 5-aza-CdR treatment in HCC cell lines or LO2 cells. D, the data in C was presented as percentages of the methylated/total CpGs in the plot. E, qRT-PCR detected the level of miR-941 expression in QGY-7703 and HepG2 cell lines after treatment with 5-aza-CdR compared with the control of treatment with DMSO (*, p < 0.05; **, p < 0.01).
TABLE 2.
The primers in quantitative RT-PCR and genomic bisulfite sequencing (BS)
| Name | Sequence (5′-3′) |
|---|---|
| β-Actin-S | 5′-CGTGACATTAAGGAGAAGCTG-3′ |
| β-Actin-AS | 5′-CTAGAAGCATTTGCGGTGGAC-3′ |
| miR-941 RT primer | 5′-GTCGTATCCAGTGCAGGGTCCGAGGTGCACTGGATACGACGCACATG-3′ |
| miR-941 forward primer | 5′-TGCGGCACCCGGCTGTGTGCACA-3′ |
| U6 RT primer | 5′-GTCGTATCCAGTGCAGGGTCCGAGGTGCACTGGATACGACAAAATATGG-3′ |
| U6 forward primer | 5′-TGCGGGTGCTCGCTTCGGCAGC-3′ |
| U6 reverse primer | 5′-CCAGTGCAGGGTCCGAGGT-3′ |
| SNAIL1-S | 5′-TTCTCTAGGCCCTGGCTGC-3′ |
| SNAIL1-AS | 5′-TACTTCTTGACATCTGAGTGGGTCTG-3′ |
| SLUG-S | 5′-CCTCCATCTGACACCTCC-3′ |
| SLUG-AS | 5′-CCCAGGCTCACATATTCC-3′ |
| BS-941-S | 5′-TTGAGGAGTAGGGATTTGTTAAATT-3′ |
| BS-941-AS | 5′-TAAACCCTAAACACATATCCACACA-3′ |
miR-941 Suppressed in Vitro Cell Proliferation, Migration, and Invasion and in Vivo HCC Metastasis
We constructed the miR-941 expression plasmid (pcDNA3/pri-miR-941) and used antisense oligonucleotides (ASO) to inhibit miR-941 to investigate the in vitro roles of this particular miRNA. As shown in Fig. 3A, miR-941 expression in cells transfected with pri-miR-941 was up-regulated 8.9-fold, whereas transfecting with ASO-miR-941 decreased the expression by almost 65%. We performed MTT, colony formation, cell migration, and invasion assays using QGY-7703 and HepG2 cells. The MTT assays demonstrated that cell viability was significantly inhibited by 22 and 20% in pri-miR-941-transfected QGY-7703 and HepG2 cells, respectively. However, compared with control cells, ASO-miR-941 increased cell viability (Fig. 3B). The colony formation assays showed that pri-miR-941 suppressed colony development and that ASO-miR-941 increased colony formation compared with the respective control groups in QGY-7703 and HepG2 cells (Fig. 3C).
FIGURE 3.

miR-941 represses cell proliferation, migration, and invasion in vitro and represses HCC metastasis in vivo. A, the relative level of miR-941 in QGY-7703 cells after transfection with pri-miR-941 or ASO-miR-941. B, MTT assays in 96-well plates after transfected with pri-miR-941 or ASO-miR-941. C, colony formation assays after transfection, the representative image is shown. D and E, transwell migration and invasion assay of QGY-7703 and HepG2 cells. Representative images are shown below. Cells in five random fields of view were counted and expressed the average number of cells per field. F, qRT-PCR was used to detect the mRNA level of miR-941 in QGY-7703 pooled clones. G, a representative picture of tumor nodules in primary sites (spleen) and metastatic sites (liver) after spleen transplantation is shown on the left. Quantification of the metastatic ability of the miR-941 vector or control vector is shown on the right. The numbers of intrahepatic metastatic nodules in each mouse were counted. Black arrows indicate the location of primary tumors and metastatic nodules. H, the miR-941 level of metastatic nodules in liver. I, hematoxylin-eosin-stained sections of primary spleen tumors and intrahepatic metastatic nodules. Red arrows indicate the HE-stained location of primary and metastatic tumors. Black arrows indicate the normal liver and spleen tissues. J, immunohistochemical staining of KDM6B in miR-941 transplants of HCC tissue and its control were shown (*, p < 0.05; **, p < 0.01).
The migration of pri-miR-941 cells was decreased by ∼42 and 56%, conversely, the migration of ASO-miR-941 cells was increased by ∼1.8- or 1.7-fold in QGY-7703 and HepG2 cells, respectively (Fig. 3D). The invasion capability of ASO-miR-941 cells increased 1.9- or 1.8-fold, whereas invasion was decreased by 49% in pri-miR-941 QGY-7703 cells and 58% in pri-miR-941 HepG2 cells (Fig. 3E).
We next explored the role of miR-941 in HCC metastasis in vivo. Using G418 screening, we obtained pooled QGY-7703 cells (QGY-7703/miR-941 cells) that stably expressed higher levels of miR-941 based on qRT-PCR (Fig. 3F). QGY-07703/miR-941 cells or empty vector control pooled clones of QGY-7703/pcDNA3 cells were transplanted into the upper pole of the spleen of nude mice. After 24 days, the spleens and livers were harvested. The average number of metastatic nodules in the liver was dramatically decreased by 62.6% in mice with tumors ectopically expressing miR-941, compared with the control group (Fig. 3G). The miR-941 level of metastatic nodules in liver was detected in Fig. 3H. Representative primary splenic tumors and intrahepatic metastatic nodules were stained with hematoxylin-eosin, the normal liver and spleen tissues were also shown as controls (Fig. 3I). Additionally, the protein levels of KDM6B in HCC tissues were analyzed using immunohistochemical staining. Strong staining of tissues with controls rather than overexpression of miR-941 was observed (Fig. 2J).
miR-941 Bound Directly to the KDM6B 3′ UTR and Negatively Regulated KDM6B Expression at the Post-transcriptional Level in HCC Cells
To identify specific targets of miR-941, we used Target-Scan, PicTar, and miRBase to predict potential target genes. We chose KDM6B as a candidate gene and identified miR-941 complementary binding sites in the 3′ UTR of KDM6B mRNA (Fig. 4A). The KDM6B 3′ UTR fragment was cloned, and another vector was constructed with 5 mutations in the seed sequence of the KDM6B 3′ UTR. As shown in Fig. 4B, the EGFP intensity was reduced by pri-miR-941 and enhanced by ASO-miR-941 when the wild-type KDM6B 3′ UTR was present. However, neither overexpressed nor down-regulated miR-941 had an effect on EGFP fluorescence in cells transfected with the mutant 3′ UTR vector (Fig. 4C). Additionally, qRT-PCR (Fig. 4D), immunofluorescence staining (Fig. 4E), and Western blot (Fig. 4F) revealed that miR-941 overexpression markedly inhibited endogenous KDM6B mRNA and protein expression, and inhibiting miR-941 expression increased KDM6B mRNA and protein expression. These results demonstrated that miR-941 bound directly to the 3′ UTR of KDM6B and negatively regulated endogenous KDM6B expression at both the mRNA and protein levels.
FIGURE 4.

miR-941 targets KDM6B and negatively regulates KDM6B at the post-transcriptional level. A, the predicted binding sites for miR-941 on the 3′ UTR of KDM6B mRNA are shown. The mutant 3′ UTR contains 5 mutated nucleotides in the complementary seed sequences. B and C, relative EGFP activity was analyzed after the wild-type or mutant 3′ UTR reporter plasmids were cotransfected with miR-941 or ASO-miR-941 in QGY-7703 cells. The histogram shows the mean ± S.D. of the normalized EGFP intensity from three independent experiments. D, qRT-PCR shows the mRNA level of KDM6B after the cells were transfected with pri-miR-941 and ASO-miR-941 compared with the control group. E, KDM6B immunostaining (green) and DAPI staining (blue) in QGY-7703 cells after transfection. F, Western blot shows the protein level of KDM6B after the cells were transfected with pri-miR-941 and ASO-miR-941. G, different expression of KDM6B in 15 pairs of HCC tissues compared with adjacent normal tissues. H, different expression of KDM6B in six different HCC cell lines compared with the non-cancerous cell (LO2) (*, p < 0.05; **, p < 0.01).
KDM6B mRNA levels were measured in 15 paired clinical specimens, and up-regulated in diseased tissue compared with the corresponding normal tissue (Fig. 4G). Consistently, KDM6B was more highly expressed in human HCC cell lines (QGY-7703, HepG2, Hep3B, SK-Hep-1, PLC-PRF-5, and LM6) than in the LO2 non-cancerous liver cell line (Fig. 4H).
KDM6B Affected Cell Viability, Migration, and Invasion of HCC Cells
The pSilencer/shR-KDM6B (shR-KDM6B) plasmid was constructed to knockdown KDM6B expression. The immunofluorescence staining (Fig. 5A) and Western blot (Fig. 5B) analysis indicated that KDM6B protein expression was reduced in shR-KDM6B-transfected HCC cells. Furthermore, the qRT-PCR analysis demonstrated that KDM6B was down-regulated by 60% in QGY-7703 cells and 55% in HepG2 cells (Fig. 5C). The MTT and colony formation assays indicated that inhibition of KDM6B decreased cell viability and growth but overexpression of KDM6B improved cell viability (Fig. 5D) and colony formation (Fig. 5E), compared with the controls. Furthermore, inhibiting KDM6B significantly decreased cell migration (Fig. 5F) and invasion (Fig. 5G). KDM6B overexpression significantly increased migration and invasion in QGY-7703 and HepG2 cells. Cumulatively, the results suggested that KDM6B plays a role in cell proliferation, migration, and invasion and that its functions are opposite those of miR-941.
FIGURE 5.

Knockdown of KDM6B suppresses proliferation, migration, and invasion in HCC cells. A and B, KDM6B protein level was measured by immunofluorescence and Western blot analysis after transfection with shR-KDM6B. C, the KDM6B mRNA expression level was determined by quantitative RT-PCR after transfection with shR-KDM6B. D, the effects of KDM6B knockdown on cell viability were detected by a MTT assay. E, cell proliferation capacity was detected through a colony formation assay and the representative image is shown. F and G, cell migration and invasion ability induced by shR-KDM6B were determined by transwell migration and invasion assays. Representative images are shown below. Cells in five random fields of view were counted and expressed the average number of cells per field (*, p < 0.05; **, p < 0.01).
miR-941 Repressed HCC Cell Migration and Invasion through a KDM6B-dependent EMT Process
Because miR-941 and KDM6B affected cell migration and invasion, we aimed to determine which pathway mediated the metastatic properties of HCC cells. In this study, we noticed a striking change in cellular shape. An initial spindle and fibroblast-like spindle were observed to switch to the cobblestone appearance of epithelial cells when overexpression of miR-941 and the opposite changes were found with overexpression of KDM6B (Fig. 6A). We examined localization of the adherent and tight junction marker E-cadherin and the mesenchymal marker vimentin in transfected QGY-7703 cells. Western blotting indicated that E-cadherin was up-regulated (Fig. 6B), and vimentin was down-regulated when miR-941 was overexpressed. The opposite results were observed when KDM6B was overexpressed (Fig. 6C). qRT-PCR was performed to quantitate expression of transcription factors SNAIL1 and SLUG, which promote EMT. The results indicated that overexpression of miR-941 reduced SNAIL1 expression by 67% and SLUG expression by ∼50%. Conversely, overexpression of KDM6B elicited the opposite results (Fig. 6, D and E). Additionally, the protein levels of E-cadherin and vimentin in tissues were analyzed using immunohistochemical staining. Overexpression of miR-941 showed strong staining of E-cadherin and weak staining of vimentin compared with control groups (Fig. 6F). Therefore, these results suggested that miR-941 and KDM6B regulated the migratory and invasive behavior of HCC cells via the EMT process.
FIGURE 6.

miR-941 and KDM6B regulated the EMT process. A, QGY-7703 cells were transiently transfected with miR-941 and KDM6B, bright field morphology of transfected cells were shown. B and C, the protein expression levels of E-cadherin and vimentin when miR-941 and KDM6B was over-expressioned. D and E, the qRT-PCR analysis for the expression of EMT transcription factors, SNAIL1 and SLUG, in cells transfected with miR-941 and KDM6B with their control construct. F, immunohistochemical staining of E-cadherin and vimentin in miR-941 transplants of HCC tissues and its controls (*, p < 0.05; **, p < 0.01).
KDM6B Restoration Counteracted the Effects of miR-941 in HCC Cells
We used the KDM6B overexpression vector without the 3′ UTR to avoid miRNA interference and determine whether miR-941 regulated cell proliferation, migration, and invasion by targeting KDM6B. We observed that the reductions in cell viability (Fig. 7A), colony formation (Fig. 7B), migration (Fig. 7C), and invasion (Fig. 7D) resulting from miR-941 overexpression were rescued by restoring KDM6B expression in QGY-7703 and HepG2 cells. Taken together, our results suggested that KDM6B overexpression counteracted the effects of miR-941 on cell proliferation, migration, and invasion. These results suggested that miR-941 exerts its activity by regulating KDM6B expression.
FIGURE 7.

Restoration of KDM6B rescues the miR-941-induced cellular phenotypes in HCC cells. A and B, cell viability was measured by a MTT assay and colony formation assay with or without KDM6B restoration. C and D, transwell migration or invasion assay was measured when ectopic expression of KDM6B compared with controls. Representative images are shown below. Cells in five random fields of view were counted and expressed the average number of cells per field (*, p < 0.05; **, p < 0.01).
DISCUSSION
miRNAs are dysregulated in various cancers and are defined as tumor suppressors or oncogenes (2). Numerous factors, including transcriptional regulation and chromosome modification, lead to the dysregulation of miRNAs. DNA methylation is one form of chromosome modification that stably maintains diverse cell characteristics in different tissues but does not alter the primary DNA sequence (26). An increasing number of reports have shown that aberrant methylation of promoter CpG islands inactivates tumor suppressor genes, this has been reported to occur in HCC (27, 28). Although miR-941 was identified several years ago, there are no literature reports investigating the underlying mechanisms of action of this miRNA or its role in HCC. In this study, we demonstrated that the suppression of miR-941 expression in HCC is at least partly mediated by DNA hypermethylation. We discovered that miR-941 was significantly down-regulated in HCC tissues and cell lines and that the CpG island upstream of miR-941 was highly methylated in cancer cells. Additionally, the methylation frequency decreased and the expression of miR-941 increased after the cells were treated with a DNA demethylating agent (5-aza-CdR). The miR-941 methylation levels were higher in tumor tissues than in adjacent normal tissues. Collectively, the results suggested that methylation was an important mechanism for miR-941 down-regulation in HCC.
miRNAs affect cell processes by regulating target gene expression. Approximately 60% of human genes are believed to be miRNA targets (29). Combining bioinformatics-based predictions and the resulting candidate functions of miR-941 in HCC cells, we found that KDM6B had the highest recurrence rate and contained a miR-941 binding site in its 3′ UTR. Furthermore, miR-941 directly and negatively regulated KDM6B gene expression at both the mRNA and protein levels via binding sites in the 3′ UTR.
KDM6B (formerly JMJD3) belongs to the histone lysine demethylase (KDM) family. Research investigating the function of KDMs, including LSD1 and the family of JmjC domain-containing enzymes, has become popular. Several KDMs have been implicated in development, differentiation, and stem cell renewal and have been intimately linked to cancers (30–32). KDM6B specifically demethylates Lys-27 of histone H3 tri- and dimethyl (me3/2) repressive chromatin marks (20–23) and has been implicated in the regulation of several 1,25(OH)2D3 target genes, such as CDH1/E-cadherin and CST5/cystatin D, in human colorectal carcinoma (33). Moreover, KDM6B plays a critical role in the terminal differentiation of neural stem cells, dermal keratinocytes, and antigen-driven B cells. It also plays a central role in the regulation of posterior development by modulating HOX gene expression and is involved in inflammatory responses via its participation in macrophage differentiation (20, 23, 34). KDM6B is induced upon activation of the RAS-RAF signaling pathway in diploid fibroblasts (35, 36).
Previous studies have shown that KDM6B is a potential tumor suppressor. The KDM6B gene is located at chromosome 17p13.1 within 0.15 Mb of TP53, The genetic loss of TP53 and KDM6B occurs in various human cancers. As a result, it is difficult to distinguish whether these deletions confer an advantage to the tumor or are a by-product of TP53 deletions. However, the genetic lesion is rather poorly correlated with mutations in TP53 (35). Deregulation of JMJD3 may contribute to gliomagenesis by inhibiting the p53 pathway with obstruction by terminal differentiation (37). Thus, KDM6B may be a tumor suppressor gene. Conversely, KDM6B overexpression may have oncogenic activity. The H3K27-specific demethylase KDM6B is overexpressed in HPV16 E7-expressing primary human epithelial cells, and the depletion of KDM6B inhibits the proliferation of CaSki cervical cancer cells (38). Moreover, KDM6B expression is low in benign prostate but is up-regulated in prostate cancer and is highly expressed in metastatic prostate cancer (39). Certain reports have demonstrated that KDM6B expression can be modulated by Epstein-Barr virus in Hodgkin lymphoma (40). KDM6B is highly expressed in ER-dependent breast cancer cells (41) and in primary renal cell carcinoma (42). Here, we determined that KDM6B depletion suppressed cell proliferation, migration, and invasion in HCC cells (Fig. 5, D–G), which is consistent with the oncogenic model.
The EMT is important for tumor invasion and metastasis. Several recent studies have identified the miR-200 family and miR-205 as key regulators of EMT (43). We speculated that miR-941 and KDM6B were involved in the EMT process in HCC. In this study, the induction of EMT was evidenced by the increased expression of the mesenchymal marker vimentin and decreased expression of the epithelial marker E-cadherin (44). The SNAIL1 and SLUG transcription factors that are associated with EMT (45–47) contribute to regulation of vimentin expression and inhibiting E-cadherin either directly or indirectly (48). As expected, the up-regulation of miR-941 suppressed transition from an epithelial to a mesenchymal phenotype, and KDM6B increased cell migration and invasion by mediating the EMT process. It has been reported that KDM6B inhibits EMT inducers SNAIL1 and ZEB1 and promotes E-cadherin expression in colon cancer by regulating vitamin D signaling (33). Conversely, another report demonstrated that KDM6B increased the expression of mesenchymal genes during TGF-β-induced EMT in mammary epithelial cells by stimulating SNAIL1 expression (49). This result is consistent with our findings in HCC. Therefore, the discrepancies between these two studies might be due to the differential action of KDM6B in various cancer types and/or signaling mechanisms.
In summary, we identified miR-941 down-regulation in HCC tissues and cell lines and determined that miR-941 expression was inversely correlated with the level of miR-941 methylation. miR-941 functioned as a tumor suppressor gene that inhibited cell proliferation, migration, and invasion in vitro and in vivo and suppressed EMT in HCC cell lines. The functions of miR-941 were at least partially caused by down-regulation of KDM6B expression. Understanding the relationship between aberrant miR-941 methylation and its target gene KDM6B will provide insight into the molecular mechanism of HCC carcinogenesis and may be valuable for the development of future HCC diagnostics and therapeutics.
This work was supported by the National Natural Science Foundation of China Grants 31270818, 91029714, and 31071191 and Natural Science Foundation of Tianjin Grant 12JCZDJC25100.
- miRNA
- microRNA
- HCC
- hepatocellular carcinoma
- KDM6B
- lysine-specific demethylase 6B
- EMT
- epithelial-mesenchymal transition
- EGFP
- enhanced green fluorescent protein
- ASO
- antisense oligomer
- qRT-PCR
- quantitative reverse transcription PCR
- MTT
- (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
REFERENCES
- 1. Bartel D. P. (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 [DOI] [PubMed] [Google Scholar]
- 2. Esquela-Kerscher A., Slack F. J. (2006) Oncomirs: microRNAs with a role in cancer. Nat. Rev. Cancer 6, 259–269 [DOI] [PubMed] [Google Scholar]
- 3. Song S. J., Poliseno L., Song M. S., Ala U., Webster K., Ng C., Beringer G., Brikbak N. J., Yuan X., Cantley L. C., Richardson A. L., Pandolfi P. P. (2013) MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET family-dependent chromatin remodeling. Cell 154, 311–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Calin G. A., Liu C. G., Sevignani C., Ferracin M., Felli N., Dumitru C. D., Shimizu M., Cimmino A., Zupo S., Dono M., Dell'Aquila M. L., Alder H., Rassenti L., Kipps T. J., Bullrich F., Negrini M., Croce C. M. (2004) MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc. Natl. Acad. Sci. U.S.A. 101, 11755–11760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pallante P., Visone R., Ferracin M., Ferraro A., Berlingieri M. T., Troncone G., Chiappetta G., Liu C. G., Santoro M., Negrini M., Croce C. M., Fusco A. (2006) MicroRNA deregulation in human thyroid papillary carcinomas. Endocr. Relat. Cancer 13, 497–508 [DOI] [PubMed] [Google Scholar]
- 6. Yan Y., Luo Y. C., Wan H. Y., Wang J., Zhang P. P., Liu M., Li X., Li S., Tang H. (2013. miR-10a is involved in metastatic process by regulating EphA4-mediated epithelial-mesenchymal transition and adhesion in hepatoma cells. Hepatology 57, 667–677 [DOI] [PubMed] [Google Scholar]
- 7. Zhang J. F., He M. L., Fu W. M., Wang H., Chen L. Z., Zhu X., Chen Y., Xie D., Lai P., Chen G., Lu G., Lin M. C., Kung H. F. (2011) Primate-specific microRNA-637 inhibits tumorigenesis in hepatocellular carcinoma by disrupting signal transducer and activator of transcription 3 signaling. Hepatology 54, 2137–2148 [DOI] [PubMed] [Google Scholar]
- 8. El-Serag H. B., Rudolph K. L. (2007) Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 132, 2557–2576 [DOI] [PubMed] [Google Scholar]
- 9. Zhang L. Y., Liu M., Li X., Tang H. (2013) MiR-490–3p modulates cell growth and epithelial to mesenchymal transition of hepatocellular carcinoma cells by targeting endoplasmic reticulum-Golgi intermediate compartment protein 3*(ERGIC3). J. Biol. Chem. 288, 4035–4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meng F., Henson R., Wehbe-Janek H., Ghoshal K., Jacob S. T., Patel T. (2007) MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133, 647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hou Y. Y., Cao W. W., Li L., Li S. P., Liu T., Wan H. Y., Liu M., Li X., Tang H. (2011) MicroRNA-519d targets MKi67 and suppresses cell growth in the hepatocellular carcinoma cell line QGY-7703. Cancer Lett. 307, 182–190 [DOI] [PubMed] [Google Scholar]
- 12. Díaz-Martín J., Díaz-López A., Moreno-Bueno G., Castilla M. Á., Rosa-Rosa J. M., Cano A., Palacios J. (2014) A core microRNA signature associated with inducers of the epithelial-to-mesenchymal transition. J. Pathol. 232, 319–329 [DOI] [PubMed] [Google Scholar]
- 13. Ding X. M. (2014) MicroRNAs: regulators of cancer metastasis and epithelial-mesenchymal transition (EMT). Chin. J. Cancer 33, 140–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thiery J. P., Acloque H., Huang R. Y., Nieto M. A. (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 [DOI] [PubMed] [Google Scholar]
- 15. Watts G. S., Futscher B. W., Holtan N., Degeest K., Domann F. E., Rose S. L. (2008) DNA methylation changes in ovarian cancer are cumulative with disease progression and identify tumor stage. BMC Med. Genomics 1, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aran D., Hellman A. (2013) DNA methylation of transcriptional enhancers and cancer predisposition. Cell 154, 11–13 [DOI] [PubMed] [Google Scholar]
- 17. Saito Y., Liang G., Egger G., Friedman J. M., Chuang J. C., Coetzee G. A., Jones P. A. (2006) Specific activation of microRNA-127 with down-regulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 9, 435–443 [DOI] [PubMed] [Google Scholar]
- 18. Lujambio A., Ropero S., Ballestar E., Fraga M. F., Cerrato C., Setién F., Casado S., Suarez-Gauthier A., Sanchez-Cespedes M., Git A., Gitt A., Spiteri I., Das P. P., Caldas C., Miska E., Esteller M. (2007) Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 67, 1424–1429 [DOI] [PubMed] [Google Scholar]
- 19. Han L., Witmer P. D., Casey E., Valle D., Sukumar S. (2007) DNA methylation regulates microRNA expression. Cancer Biol. Ther. 6, 1284–1288 [DOI] [PubMed] [Google Scholar]
- 20. Agger K., Cloos P. A., Christensen J., Pasini D., Rose S., Rappsilber J., Issaeva I., Canaani E., Salcini A. E., Helin K. (2007) UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449, 731–734 [DOI] [PubMed] [Google Scholar]
- 21. Hong S., Cho Y. W., Yu L. R., Yu H., Veenstra T. D., Ge K. (2007) Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. U.S.A. 104, 18439–18444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sen G. L., Webster D. E., Barragan D. I., Chang H. Y., Khavari P. A. (2008) Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes Dev. 22, 1865–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lan F., Bayliss P. E., Rinn J. L., Whetstine J. R., Wang J. K., Chen S., Iwase S., Alpatov R., Issaeva I., Canaani E., Roberts T. M., Chang H. Y., Shi Y. (2007) A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 449, 689–694 [DOI] [PubMed] [Google Scholar]
- 24. Ichi S., Costa F. F., Bischof J. M., Nakazaki H., Shen Y. W., Boshnjaku V., Sharma S., Mania-Farnell B., McLone D. G., Tomita T., Soares M. B., Mayanil C. S. (2010) Folic acid remodels chromatin on Hes1 and Neurog2 promoters during caudal neural tube development. J. Biol. Chem. 285, 36922–36932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li L. C., Dahiya R. (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics 18, 1427–1431 [DOI] [PubMed] [Google Scholar]
- 26. Jones P. A., Takai D. (2001) The role of DNA methylation in mammalian epigenetics. Science 293, 1068–1070 [DOI] [PubMed] [Google Scholar]
- 27. Saito Y., Hibino S., Saito H. (2014) Alterations of epigenetics and microRNA in hepatocellular carcinoma. Hepatol. Res. 44, 31–42 [DOI] [PubMed] [Google Scholar]
- 28. Goeppert B., Schmezer P., Dutruel C., Oakes C., Renner M., Breinig M., Warth A., Vogel M. N., Mittelbronn M., Mehrabi A., Gdynia G., Penzel R., Longerich T., Breuhahn K., Popanda O., Plass C., Schirmacher P., Kern M. A. (2010) Down-regulation of tumor suppressor A kinase anchor protein 12 in human hepatocarcinogenesis by epigenetic mechanisms. Hepatology 52, 2023–2033 [DOI] [PubMed] [Google Scholar]
- 29. Xu N., Zhang L., Meisgen F., Harada M., Heilborn J., Homey B., Grandér D., Ståhle M., Sonkoly E., Pivarcsi A. (2012) MicroRNA-125b down-regulates matrix metallopeptidase 13 and inhibits cutaneous squamous cell carcinoma cell proliferation, migration, and invasion. J. Biol. Chem. 287, 29899–29908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamane K., Tateishi K., Klose R. J., Fang J., Fabrizio L. A., Erdjument-Bromage H., Taylor-Papadimitriou J., Tempst P., Zhang Y. (2007) PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol. Cell 25, 801–812 [DOI] [PubMed] [Google Scholar]
- 31. van Haaften G., Dalgliesh G. L., Davies H., Chen L., Bignell G., Greenman C., Edkins S., Hardy C., O'Meara S., Teague J., Butler A., Hinton J., Latimer C., Andrews J., Barthorpe S., Beare D., Buck G., Campbell P. J., Cole J., Forbes S., Jia M., Jones D., Kok C. Y., Leroy C., Lin M. L., McBride D. J., Maddison M., Maquire S., McLay K., Menzies A., Mironenko T., Mulderrig L., Mudie L., Pleasance E., Shepherd R., Smith R., Stebbings L., Stephens P., Tang G., Tarpey P. S., Turner R., Turrell K., Varian J., West S., Widaa S., Wray P., Collins V. P., Ichimura K., Law S., Wong J., Yuen S. T., Leung S. Y., Tonon G., DePinho R. A., Tai Y. T., Anderson K. C., Kahnoski R. J., Massie A., Khoo S. K., Teh B. T., Stratton M. R., Futreal P. A. (2009) Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 41, 521–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dalgliesh G. L., Furge K., Greenman C., Chen L., Bignell G., Butler A., Davies H., Edkins S., Hardy C., Latimer C., Teague J., Andrews J., Barthorpe S., Beare D., Buck G., Campbell P. J., Forbes S., Jia M., Jones D., Knott H., Kok C. Y., Lau K. W., Leroy C., Lin M. L., McBride D. J., Maddison M., Maguire S., McLay K., Menzies A., Mironenko T., Mulderrig L., Mudie L., O'Meara S., Pleasance E., Rajasingham A., Shepherd R., Smith R., Stebbings L., Stephens P., Tang G., Tarpey P. S., Turrell K., Dykema K. J., Khoo S. K., Petillo D., Wondergem B., Anema J., Kahnoski R. J., Teh B. T., Stratton M. R., Futreal P. A. (2010) Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463, 360–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pereira F., Barbáchano A., Silva J., Bonilla F., Campbell M. J., Muñoz A., Larriba M. J. (2011) KDM6B/JMJD3 histone demethylase is induced by vitamin D and modulates its effects in colon cancer cells. Hum. Mol. Genet. 20, 4655–4665 [DOI] [PubMed] [Google Scholar]
- 34. De Santa F., Totaro M. G., Prosperini E., Notarbartolo S., Testa G., Natoli G. (2007) The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130, 1083–1094 [DOI] [PubMed] [Google Scholar]
- 35. Barradas M., Anderton E., Acosta J. C., Li S., Banito A., Rodriguez-Niedenführ M., Maertens G., Banck M., Zhou M. M., Walsh M. J., Peters G., Gil J. (2009) Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 23, 1177–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Agger K., Cloos P. A., Rudkjaer L., Williams K., Andersen G., Christensen J., Helin K. (2009) The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 23, 1171–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ene C. I., Edwards L., Riddick G., Baysan M., Woolard K., Kotliarova S., Lai C., Belova G., Cam M., Walling J., Zhou M., Stevenson H., Kim H. S., Killian K., Veenstra T., Bailey R., Song H., Zhang W., Fine H. A. (2012) Histone demethylase Jumonji D3 (JMJD3) as a tumor suppressor by regulating p53 protein nuclear stabilization. PLoS One 7, e51407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McLaughlin-Drubin M. E., Crum C. P., Münger K. (2011) Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. U.S.A. 108, 2130–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xiang Y., Zhu Z., Han G., Lin H., Xu L., Chen C. D. (2007) JMJD3 is a histone H3K27 demethylase. Cell Res. 17, 850–857 [DOI] [PubMed] [Google Scholar]
- 40. Anderton J. A., Bose S., Vockerodt M., Vrzalikova K., Wei W., Kuo M., Helin K., Christensen J., Rowe M., Murray P. G., Woodman C. B. (2011) The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin's lymphoma. Oncogene 30, 2037–2043 [DOI] [PubMed] [Google Scholar]
- 41. Svotelis A., Bianco S., Madore J., Huppé G., Nordell-Markovits A., Mes-Masson A. M., Gévry N. (2011) H3K27 demethylation by JMJD3 at a poised enhancer of anti-apoptotic gene BCL2 determines ERα ligand dependency. EMBO J. 30, 3947–3961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shen Y., Guo X., Wang Y., Qiu W., Chang Y., Zhang A., Duan X. (2012) Expression and significance of histone H3K27 demethylases in renal cell carcinoma. BMC Cancer 12, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gregory P. A., Bracken C. P., Bert A. G., Goodall G. J. (2008) MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 7, 3112–3118 [DOI] [PubMed] [Google Scholar]
- 44. Kraljevic Pavelic S., Sedic M., Bosnjak H., Spaventi S., Pavelic K. (2011) Metastasis: new perspectives on an old problem. Mol. Cancer 10, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kajita M., McClinic K. N., Wade P. A. (2004) Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol. Cell. Biol. 24, 7559–7566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vega S., Morales A. V., Ocaña O. H., Valdés F., Fabregat I., Nieto M. A. (2004) Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 18, 1131–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang J. P., Zeng C., Xu L., Gong J., Fang J. H., Zhuang S. M. (2013) MicroRNA-148a suppresses the epithelial-mesenchymal transition and metastasis of hepatoma cells by targeting Met/Snail signaling. Oncogene 33, 4069–4076 [DOI] [PubMed] [Google Scholar]
- 48. Kroepil F., Fluegen G., Totikov Z., Baldus S. E., Vay C., Schauer M., Topp S. A., Esch J. S., Knoefel W. T., Stoecklein N. H. (2012) Down-regulation of CDH1 is associated with expression of SNAI1 in colorectal adenomas. PLoS One 7, e46665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ramadoss S., Chen X., Wang C. Y. (2012) Histone demethylase KDM6B promotes epithelial-mesenchymal transition. J. Biol. Chem. 287, 44508–44517 [DOI] [PMC free article] [PubMed] [Google Scholar]