

Abstract
Background
Infection with pandemic (pdm) A/H1N1 virus induces high levels of pro-inflammatory mediators in blood and lungs of experimental animals and humans.
Methods
To compare the involvement of seasonal A/PR/8/34 and pdm A/H1N1 virus strains in the regulation of inflammatory responses, we analyzed the changes in the whole-genome expression induced by these strains in macrophages and A549 epithelial cells. We also focused on the functional implications (cytokine production) of the differential induction of suppressors of cytokine signaling (SOCS)-1, SOCS-3, retinoid-inducible gene (RIG)-I and interferon receptor 1 (IFNAR1) genes by these viral strains in early stages of the infection.
Results
We identified 130 genes differentially expressed by pdm A/H1N1 and A/PR/8/34 infections in macrophages. mRNA levels of SOCS-1 and RIG-I were up-regulated in macrophages infected with the A/PR/8/34 but not with pdm A/H1N1 virus. mRNA levels of SOCS-3 and IFNAR1 induced by A/PR/8/34 and pdm A/H1N1 strains in macrophages, as well as in A549 cells were similar. We found higher levels of IL-6, TNF-α, IL-10, CCL3, CCL5, CCL4 and CXCL8 (p<0.05) in supernatants from cultures of macrophages infected with the pdm A/H1N1 virus compared to those infected with the A/PR/8/34 strain, coincident with the lack of SOCS-1 and RIG-I expression. In contrast, levels of INF-α were higher in cultures of macrophages 48 h after infection with the A/PR/8/34 strain than with the pdm A/H1N1 virus.
Conclusions
These findings suggest that factors inherent to the pdm A/H1N1 viral strain may increase the production of inflammatory mediators by inhibiting SOCS-1 and modifying the expression of antiviral immunity-related genes, including RIG-I, in human macrophages.
Keywords: SOCS-1, A/H1N1, Influenza, Cytokines, Inflammation
1. Introduction
The 2009 outbreak of swine-origin influenza A/H1N1 [1,2] continues to affect many countries, having caused over 18,000 deaths worldwide [3]. A growing body of evidence supports the hypothesis that the development of severe pneumonia in patients with pandemic (pdm) A/H1N1 infection is associated with increased immune activation and immune complex deposition [4,5]. Therefore, it is of great importance to understand the factors that determine the development of severe disease. In this regard, high levels of pro-inflammatory cytokines and chemokines have been detected in peripheral blood and lung tissue from patients with severe pneumonia associated to the pdm A/H1N1 infection [4,6]. Even though cytokines, chemokines, and growth factors are required to control influenza virus infection, their overproduction in an uncontrolled inflammatory response can lead to lung tissue damage [4,6,7]. The suppressors of cytokine signaling (SOCS) are a family of proteins that down-regulate cytokine signaling [8-11] by negatively regulating JAK/STAT-mediated signal transduction [911]. Experimental infection of A549 epithelial cells with seasonal influenza A virus up-regulates the expression of SOCS-1 and SOCS-3. These proteins regulate the immune response against influenza A viruses through a retinoid-inducible gene (RIG)-I/mitochondrial antiviral signaling protein (MAVS)/interferon (alpha and beta) receptor 1 (IFNAR1)-dependent pathway [12].
On the other hand, the combination of gene segments from North American and Eurasian swine lineages of the pdm A/H1N1 virus [13] create unique molecular structures [14,15] that could regulate host immune responses differently than seasonal influenza strains, and contribute to the particular clinical presentation of pandemic influenza infections.
We therefore hypothesized that immune feedback or regulatory mechanisms that normally control host inflammatory responses to pathogens may be absent or impaired in severe pdm A/H1N1 infection, due to the virus itself.
To establish if particular patterns of gene expression and alterations of immune regulation are attributable to specific viral factors, we analyzed the whole genome expression patterns induced by the pdm A/H1N1 and A/PR/8/34 strains through microarray analysis of infected human macrophages and A549 cells. In addition, we performed in vitro assays of macrophages and A549 cells in order to evaluate the differences between the pdm A/H1N1 and A/PR/8/34 in their capacity to induce SOCS-1, SOCS-3, and the antiviral response molecule RIG-I, as well as the production of pro-inflammatory cytokines, chemokines and growth factors.
2. Materials and methods
2.1. Ethics statement
The Institutional Review Board of the National Institute of Respiratory Diseases (INER) reviewed and approved this protocol (protocol number B27-10), under which all subjects were recruited. All subjects provided written informed consent, and authorized the storage of their samples at INER repositories for this and future studies.
2.2. Seasonal and pandemic A/H1N1 influenza virus isolation, identification, and propagation
Influenza pdm A/H1N1 virus isolates were obtained from patients with severe pneumonia, who signed an informed consent letter, during the 2009 outbreak in Mexico City, at the National Institute for Respiratory Diseases. Detection of pdm A/H1N1 viral RNA from the respiratory specimens was assessed by real time RT-PCR according with CDC and WHO guidelines. Live influenza pdm A/H1N1 and seasonal A/PR/8/34 viruses were isolated in Madin-Darby canine kidney cells (MDCK). Virus infectivity was assessed by determination of tissue culture infection dose 50% (TCID50) in MDCK cells. The titers of virus stocks were adjusted to 1 × 106 TCID50/mL The H1N1 strain (A/PR/8/34) was obtained from the American Type Culture Collection (ATCC) and titrated to the same concentration as pdm A/H1N1.
2.3. PBMC isolation, monocyte isolation and macrophage differentiation
Buffy coats from five healthy blood donors, who signed an informed consent letter, were obtained from the Blood Bank of the INER. Total peripheral blood mononuclear cells (PBMCs) were obtained by density gradient centrifugation using Lymphoprep (Axis-Shield, Oslo, Norway). CD14+ monocytes were purified using magnetic beads (Miltenyi, Auburn, CA, USA). Purity of isolated monocytes was assessed by flow cytometry using anti-human monoclonal antibodies: CD14-FITC and CD3-PE (BioLegend, San Diego, CA, USA), obtaining a 99% purity. Isolated monocytes were seeded at a concentration of 5×105 cells per well onto 24-well low-adherence culture plates (Corning Life Sciences, Corning, NY) in 10% FBS, 1% L-glutamine (Gibco BRL, Life Technologies, Gaithersburg, MD) supplemented RPMI-1640 culture medium (Sigma Chemical Co., St. Louis, MO, USA) with penicillin (0.6 mg/mL), and streptomycin (60 mg/mL) (Gibco BRL, Life Technologies), and were incubated at 37 °C and 5% CO2 during 14 days. At day 14, 98% of macrophage differentiation was obtained, as assessed by flow cytometric analysis of CD11b, HLA-DR and CD14 expression (BD Biosciences, San José, CA, USA).
2.4. In vitro infection of macrophages and epithelial cells with seasonal A/PR/8/34 or pdm A/H1N1 influenza viruses
Macrophages were infected with 5 × 105 TCID50 of the pdm A/H1N1 or seasonal A/PR/8/34 strains. Mock-treated cells received virus-free culture medium. Culture supernatants were collected 30 min, 1 h, 2 h, 5 h, 10 h, 15 h, 24 h, and 48 h later for cytokine, chemokine, and growth factor measurements. Macrophages were harvested for RNA isolation. All assays were performed by triplicate. A549 epithelial cells were infected with pandemic or A/PR/8/34 virus under the same conditions used for human macrophages. The infection of macrophages was confirmed using monoclonal antibody anti-influenza A virus hemagglutinin (HA) (Light Diagnostics, Millipore, Billerica, MA, USA), after 6 and 48 h of infection (Supplementary Fig. 1A and B). In addition, we analyzed the viral titers using the haemagglutination inhibition (HAI) assay. Briefly, two fold dilutions of supernatants from infected macrophages or A549 cells were prepared and mixed with chicken red blood cells and incubated at 37 °C during 90 min. A significant rise of the viral titers after 5 h of infection of macrophages and A549 cells was detected. However, higher titers of pdm A/H1N1 in cultures of macrophages were detected earlier (Supplementary Fig. 1C).
2.5. Microarray gene expression analysis
Total RNA was obtained from macrophages and A549 epithelial cell cultures 10 h after infection with either the A/PR/8/34 or pdm A/H1N1 strains and from uninfected cells (Mock). Equimolar concentrations of total RNA from five independent in vitro experiments were pooled for microarray gene expression analysis. Each RNA pool was processed in duplicate. cDNA synthesis, amplification, and gene expression profiling were done according to the manufacturers instructions (Affymetrix WT Sense Target labeling assay manual). Labeled DNA was added to hybridization cocktail and the sample was injected into the array, (GeneChip Human Gene 1.0 ST Array). Wash and stain processes were performed in the GeneChip Fluidics Station 450. The probe arrays were scanned using The GeneChip® Scanner 3000 7G (Affmetrix, Santa Clara CA, USA).
2.6. SOCS-1, SOCS-3, RIG-I and IFNAR1 mRNA expression
Total RNA was isolated from macrophages and A549 epithelial cells using the RNA easy isolation Kit (Qiagen, Valencia CA, USA). SOCS-1, SOCS-3, RIG-I and, IFNAR1 mRNA expression levels were measured by real time RT-PCR using validated TaqMan assays from Applied Biosystems (SOCS-1: hs00864158_g1, SOCS-3: hs01000485_g1, RIG-I: hs00204833 and IFNAR1: hs01066116). β-actin expression was used as an endogenous control (Life Technologies/Applied Biosystems, Foster City, CA). qRT-PCR was performed for each target gene and β-actin as house-keeping gene. Triplicate cycle threshold CT values were analyzed using the comparative CT (ΔΔCT) and then presented as relative quantification (RQ) units.
2.7. Western blot assays for SOC-1 and SOCS-3 protein detection
Cells were lysed in RIPA buffer M-PER (Pierce, Cheshire, UK) containing a protease inhibitor cocktail (P8340; Sigma Aldrich, St. Louis MO, USA). Protein concentrations were determined by the Bradford method. Equal amounts of proteins (30 μg) were resolved on SDS-PAGE in 12.5% acrylamide gels and transferred to nitrocellulose membranes (Bio-Rad Laboratories, Inc., Hercules, CA, USA). After blocking with non-fat dried milk, the membranes were incubated with primary anti-SOCS-1 (1:500 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-SOCS-3 (1:500 dilution; Santa Cruz Biotechnology) and anti-β-tubulin antibodies (1:200 dilution; Santa Cruz Biotechnology) followed by horseradish peroxidase-labeled antibody (Santa Cruz Biotechnology). Immunodetection was performed by chemiluminescence using the Molecular Imager ChemiDoc XR + System and Quantity One software version 4.6.9 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
2.8. Cytokine and chemokine quantitation in culture supernatants by Luminex and ELISA
Macrophages from five healthy donors were cultured with either pdm A/H1N1 or seasonal A/PR/8/34 strain as described above. Culture supernatants, collected at 30 min, 1 h, 2 h, 5 h, 10 h, 15 h, 24 h and 48 h, were assayed in triplicate. The levels of IL-1β, IL-1RA, IL-2, IL-4, IL-5, IL-6, IL-7, CXCL8, IL-9, IL-10, IL-12 (p70), IL-13, IL-15, IL-17, FGF, Eotaxin, G-CSF, GM-CSF, IFN-γ, CXCL10, CCL2, CCL3, CCL4, PDGF-BB, CCL5, TNF-α and VEGF were determined in a Bio-Plex Luminex 200 instrument (Bio-Rad Laboratories, Inc., Hercules, CA, USA) as previously described [4].
Levels of IFN-α and IFN-β in supernatants from infected macrophages were measured by ELISA (VeriKine ELISA kit; Piscataway Township, NJ, USA). Briefly, for IFN-α, 50 μL of sample or standards were incubated with equal volumes of sample diluent whereas for IFN-β 100 μL of undiluted sample or standards (standard curve range 12.5–5000 pg/mL for IFN-α and 25–2000 pg/mL for IFN-β, respectively) were used and incubated during 1 h. After three washes, samples and standards were incubated with 100 μL of diluted antibody during 1 h. After three new washes, 100 μL of diluted HRP solution was added to each well and incubated by 1 h. A volume of 100 μL of TMB substrate were added to each well and the reaction was stopped after 15 min by adding 100 μL of stop solution. Absorbance at 450 nm was measured with a microplate ELISA reader Bio-Rad model iMark (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
2.9. Statistical analysis
Microarray background correction and normalization were done using the Oligo package in R Bioconductor. Differentially expressed genes comparing the infections with the pandemic and seasonal viruses were determined using the Limma package, considering a B value threshold of ≥3 and a fold change (logFC) value of at least 1.4. Unsupervised hierarchical clustering analysis with the differentially expressed genes was used to identify clusters of genes with differential expression between the two infection conditions. The microarray data were deposited in the Geo-Omnibus database with the MIAME guidelines under the number GSE36012. To identify the biological processes in which the differentially expressed genes are involved, an enrichment analysis was done using the DAVID (http://david.abcc.ncifcrf.gov/) and Reactome databases (http://www.reactome.org/ReactomeGWT/entry-point.html). For pathway visualization we used the Pathvision software obtaining the cellular pathways from KEGG and WikiPathways.
Differences in mRNA expression and in the levels of cytokines/chemokines/growth factors between cells infected with pandemic A/H1N1 versus seasonal A/PR/8/34 strains were evaluated by the Wilcoxon signed rank test or the Friedman's test using the Graph Pad Prism software version 5.04 (GraphPad Software, La Jolla, CA). p values < 0.05 were considered significant.
3. Results
3.1. Differential expression of antiviral immune response-related genes is induced by pandemic A/H1N1 and A/PR/8/34 strains
In order to compare the transcriptional profiles induced by different influenza A virus strains, we compared genome-wide expression induced by infection (10 h) with the pdm A/H1N1 and A/PR/8/34 viruses in macrophages and A549 cells using the exon array 1.0 ST. We also analyzed the gene expression profile of macrophages without virus infection (Mock at 10 h of infection) and the results were used as basal expression to determine the differences induced in the gene expression provoked by the infection of macrophages with the seasonal and pandemic virus. Considering a B value threshold of ≥3 and a fold change value of at least 1.4, the comparison between macrophages infected with the seasonal A/PR/8/34 and the pdm A/H1N1 virus revealed 130 genes with statistically significant differential expression (seven over-expressed genes and 123 down-regulated genes in the pdm A/H1N1 compared with the A/PR/8/34 infection), (see online Supplementary list). Unsupervised clustering analysis using the 130 differentially expressed genes showed that this set of genes is capable of distinguishing macrophages infected with pdm A/H1N1 from those infected with A/PR/8/34, Fig. 1. Enrichment analysis of the differentially expressed genes identified several pathways including antiviral response, cytokine signaling regulation, innate response and proliferation that are significantly enriched in our dataset, Supplementary Fig. 2. Several genes encoding immune response proteins involved in antiviral and inflammatory immune responses against pathogens, including SOCS-1, RIG-I (DDX58), CXCL11, CXCL10, CXCL9, CCL8, CD69, TLR3, JAK2, IFIT3, IFITM3, TRIM5, DHX58, DDX60 and IRF7 amongst others, were significantly down-regulated (considering a B value threshold of ≥3 and a fold change value ≤−1.4) in macrophages infected with pdm A/H1N1 virus. In contrast, CCL1 and other genes including RASGRP1, ETS2, EBI3, ASAP2, ITGB8, and THBS1, were significantly up-regulated in macrophages infected with the pdm A/H1N1 strain (see online Supplementary list).
Fig. 1.
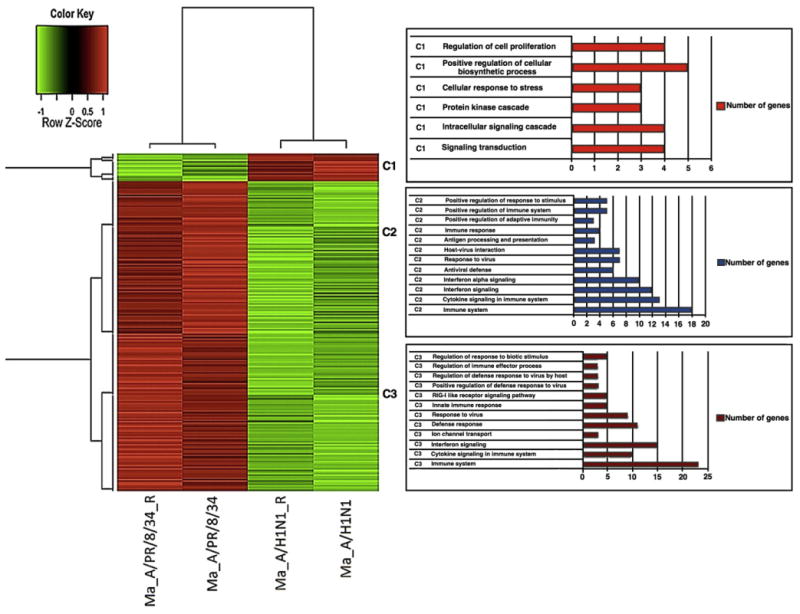
Heatmap showing differences in gene expression by macrophages infected with pdm A/H1N1 compared to seasonal A/PR/8/34 influenza viruses. Samples and replicates are listed in columns; red highlighting indicates high expression and green highlighting indicates low expression. Dendograms indicating the similarity of gene expression between the macrophages infected with different viral strains are shown (left). The numbers of genes in each cluster involved with specific pathways are shown in the right panel.
Furthermore, in correlation with the high levels of IL-6, TNF-α, and CXCL8 proteins observed in 10 h cultures of pdm A/H1N1 infected macrophages, these genes were up-regulated in the micro array analysis, although they did not reach statistical significance.
In agreement to previous studies, the gene expression characteristics of A549 cells induced by the pdm A/H1N1 strain were similar to those induced by a H1N1 seasonal strain [16]. In our analysis, we did not detect significant differential expression of genes in A549 cells infected with the pdm A/H1N1 compared to A/PR/8/34 after 10 h of infection.
3.2. Induction of SOCS-1 and RIG-I mRNA by pdm A/H1N1 and A/PR/8/34 viral strains
In order to validate the microarray expression profiles of genes involved in the regulation of the production of cytokines and chemokines and antiviral response, we analyzed the kinetics of the mRNA expression levels of SOCS-1, SOCS-3, RIG-I and IFNAR1 by qRT-PCR in uninfected human macrophages and A549 epithelial cells and those in the early stages of infection. We found significantly greater SOCS-1 mRNA expression in macrophages at 10 h, 15 h, 24 h and 48 h of infection with the A/PR/8/34 virus in comparison with those infected with pdm A/H1N1 virus. At these times, the A/PR/8/34 virus induced a 25-fold or greater increase in SOCS-1 expression compared to mock-treated macrophages, whereas pdm A/H1N1 virus induced less than a 5-fold increase (p < 0.05, Fig. 2A).
Fig. 2.
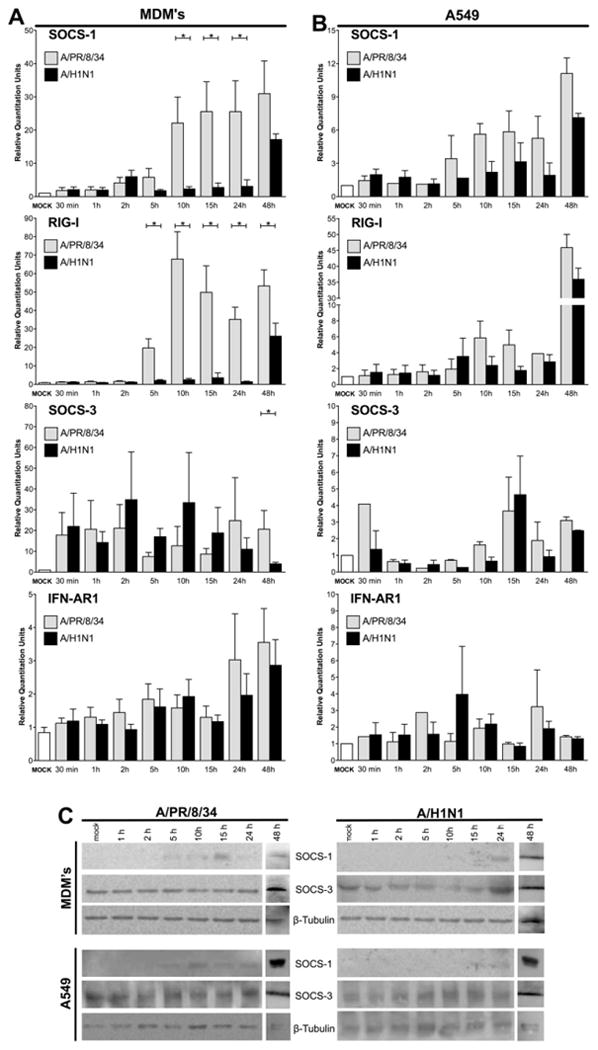
mRNA levels of SOCS-1, SOCS-3, RIG-I and IFNAR1 induced by the infection with seasonal (A/PR/8/34) or pdm A/H1N1 influenza strains. Levels of SOCS-1, SOCS-3, RIG-I and IFNAR1 mRNA induced by the seasonal or pdm A/H1N1 influenza strains in macrophages (A) and A549 cells (B) were determined by qRT-PCR. Significantly higher levels of SOCS-1 and RIG-I mRNA were detected in macrophages at 5 h, 10 h, 15 h, 24 h and 48 h of infection with the seasonal virus compared to those infected with pdm A/H1N1 virus. SOCS-1 and SOCS3 protein production by macrophages and A549 cells after infection with A/PR/8/34 and A/H1N1, as determined by Western blotting (C). Differences in the gene expression were analyzed by Wilcoxon signed-rank test. p values < 0.05 (*) were considered statistically significant.
Expression of RIG-I mRNA was also significantly up-regulated in macrophages infected with A/PR/8/34 virus at 5, 10, 15 and 24 h (p < 0.05), compared to those infected with the pdm A/H1N1 virus, which expressed RIG-I mRNA levels only slightly greater than those observed in mock-treated macrophages. Importantly, the levels of RIG-I mRNA after 48 h of infection were also significantly higher in macrophages infected with the seasonal A/PR/8/34 strain. mRNA levels of SOCS-3 and IFNAR1 induced by APR/8/34 and the pdm A/H1N1 strains in macrophages were similar. However, levels of SOCS-3 mRNA were substantially higher in macrophages infected with either virus compared with mock-treated macrophages, consistent with the known induction of SOCS-3 by seasonal influenza A strains [12], while IFNAR1 mRNA was only slightly increased after infection with either viral strain, Fig. 2A. mRNA expression levels of SOCS-1 and RIG-I in A/PR/8/34- or pdm A/H1N1-infected A549 cells were not statistically different. However, these genes were slightly up-regulated in the A549 cells infected with the A/PR/8/34 virus compared with the pdm A/H1N1 virus, Fig. 2B.
The production of SOCS-1 protein was assessed by Western blot assays in whole-cell lysates of macrophages and A549 cells. In line with the qRT-PCR results, this protein was detected in lysates of macrophages infected with A/PR/8/34 after 5 h, 10 h, 24 h and 48 h, yet in macrophages infected with the pdm A/H1N1 virus, SOCS-1 was detected only in the 24 h and 48 h post-infection samples. The same pattern was observed in lysates of A549 infected cells. SOCS-3 was detected in all lysates from macrophages and A549 cells infected with both viral strains, Fig. 2C. As expected, the levels of the protein RIG-I were decreased in the lysates of macrophages infected with the pdm A/H1N1 virus (data not shown).
3.3. The infection of macrophages with the pdm A/H1N1 or A/PR/8/34 strains induces different levels of inflammatory mediators
The levels of cytokines, chemokines and growth factors in supernatants of macrophages infected with A/PR/8/34 or pdm A/H1N1 strains are shown in Fig. 3. Higher levels (p < 0.05) of the cytokines IL-6, TNF-α and IL-10 were detected in supernatants from macrophage cultures infected with pdm A/H1N1 virus compared with those infected with the A/PR/8/34 virus. Both strains induced similar kinetics and levels of IFN-γ production. Regarding chemokines, levels of CCL3, CCL4, CCL5 and CXCL8 were considerably higher (p < 0.05) in cultures treated with the pdm A/H1N1 virus than those treated with A/PR8/34. In contrast, high levels of CXCL10 were induced by A/PR8/34, while this cytokine was almost undetectable in pdm A/H1N1-infected cultures. Significantly higher levels of G-CSF, particularly at 10 h post-infection, were induced by the pdm A/H1N1 strain. Levels of IFN-α and IFN-β were also measured in cultures from infected macrophages. Here, higher production of IFN-α was detected at 48 h in the cultures infected by the A/PR/8/34 strain. IFN-β was undetectable, Fig. 3. In contrast to the pattern seen with infected macrophages, no significant differences in the levels of TNF-α, IL-10, CCL3, and CCL5 in supernatants of A549 cells infected with pdm A/H1N1 or seasonal virus were observed. However, a slight increase in the levels of IL-6, IFN-γ, CCL4, CXCL8, CXCL10 and G-CSF was observed, particularly in 10 h supernatants from A549 cultures, infected with the A/PR/8/34 strain (Supplementary Fig. 3).
Fig. 3.

Cytokine/chemokine/growth factor levels in culture supernatants of macrophages infected with seasonal or A/H1N1 influenza strains. Results are shown as means ± SEM of 3 independent experiments per strain. Significantly higher levels of IL-6, TNF-α and IL-10 in supernatants from macrophage cultures infected with pdm A/H1N1 strain were observed compared to those infected with the seasonal (A/PR/8/34) strain. Both strains induced similar production of IFN-γ. Levels of CCL3, CCL4, CCL5 and CXCL8 were considerably higher in cultures treated with the pdm A/H1N1 virus. Differences in the levels of the cytokines/chemokines and growth factors induced by the different viral strains on macrophages from the same donors at each time point (1 h, 2 h, 5 h, 10 h, 15 h, 24 h and 48 h) were analyzed by Wilcoxon signed-rank test (*). The differences including all time points in seasonal and pdm A/H1N1 strains treatment were evaluated by using the Friedman's test. p values < 0.05 were considered statistically significant.
4. Discussion
In previous studies, elevated levels of pro-inflammatory cytokines and chemokines in blood and lung from patients with severe pneumonia associated with pdm A/H1N1 virus have been identified, suggesting that hypercytokinemia may contribute to the severity of the disease [4,6].
Here, we analyzed the ability of pdm A/H1N1 and A/PR8/34 viruses to induce specific gene expression patterns in infected macrophages and A549 epithelial cells. These analyses allowed us to identify 130 genes differentially induced (B value threshold of ≥ 3 and a fold change value P1.4) by these strains, particularly in macrophages. These genes are mainly involved in the antiviral response, cytokine signaling regulation, innate immune responses against pathogens, and in cell proliferation pathways (see Supplementary list and the enriched pathways in Supplementary Fig. 2). Findings from experimental in vitro infection assays provide evidence that in macrophage cultures, the pdm A/H1N1 strain induces a significant attenuation of SOCS-1 and RIG-1 mRNA expression compared to the A/PR/8/34 strain. Moreover, we detected that the lack of expression of SOCS-1 induced by the pdm A/H1N1 strain in early phases of infection (particularly between 10 and 15 h) is associated with higher production of immune mediators, including IL-6, TNF-α, IL-10, CCL3, CCL4, CCL5, and CXCL8.
Changes in the expression of SOCS proteins play a critical role in the regulation of cytokine/chemokine production in both innate and adaptive immune responses [8–11], and in the regulation of immunity against the influenza A virus [12].
Previous studies have demonstrated that seasonal influenza A viruses are able to induce the expression of both SOCS-1 and SOCS-3 mRNA in human respiratory epithelial cells [12]. However, our findings suggest that the pdm A/H1N1 virus efficiently induces SOCS-3 but not SOCS-1 in early stages of infection. Furthermore, we find that the pdm A/H1N1 virus induces lower SOCS-1 mRNA levels than the A/PR/8/34 virus after 24 h of infection, as well as a delay in the production of the SOCS-1 protein.
SOCS-1 is the best studied of the SOCS family members. Its expression is induced by many cytokines, in particular by IFN-γ [17], and is critical in the activation of macrophages through TLR signaling [18,19]. Functional studies have revealed that SOCS-1 interacts with JAK kinases, inhibiting their tyrosine kinase activity [20]. SOCS-1 negatively regulates the production of multiple cytokines, primarily IFN-α, IFN-γ, IL-2, IL-3, IL-4, IL-6, IL-7, IL-12, IL-13, IL-15 and TNF-α [10].
Monocyte/macrophage lineage cells are abundantly recruited to the lungs during the initial stages of influenza virus infection [21]. Our findings are relevant because the delayed induction of SOCS-1, through a deficient regulation of the JAK/STAT pathway, may allow an over-production of inflammatory mediators by infected infiltrating macrophages, and may thus be deleterious.
While macrophages are critical to the defense against lung pathogens as revealed by infection assays in macrophage-deficient experimental animals [22], the over-activation and augmented recruitment of circulating monocytes/macrophages to the virus-infected lung may in fact promote progressive tissue damage due to the production of inflammatory mediators [23]. In this context, we believe that studies of circulating monocyte/macrophages isolated after their activation, possibly secondary to the initial in vivo infection of the respiratory epithelium, might help to understand the role of infiltrating macrophages in the development of exuberant inflammatory responses and pathogenesis of severe pneumonia.
Importantly, the exclusive induction of SOCS-3, in absence of SOCS-1, in initial phases of the pdm A/H1N1 virus infection, may be consistent with classical macrophage activation or M1 polarization [24], which promotes tissue inflammation and destruction [25]. In accordance with this possible profile of M1 polarization, we found that macrophages stimulated with pdm A/H1N1 characteristically produced G-CSF, TNF-α, and IL-6 [26,27].
Among the in vitro elicited molecules, IL-6 and CXCL8 stand out, since they have been detected in blood of pdm A/H1N1 influenzainfected subjects with severe pneumonia in concentrations significantly higher than in subjects exposed to the virus but not developing severe disease [4]. Our findings suggest that IL-6 and CXCL8 induction appears to be stimulated, at least partially, by viral factors, and these cytokines, therefore, could serve as biomarkers of severe manifestations of pdm A/H1N1 infection. The over-production of pro-inflammatory mediators induced by the pdm A/H1N1 strain in circulating macrophages suggest that this strain could elicit the same phenotype in alveolar macrophages, inducing a local and systemic inflammatory state. Our results also suggest that over-activated lung macrophages might be critical in the development of pathogenic responses, contributing in a greater extent to immune dysregulation and lung damage than respiratory epithelial cells.
Concerning regulatory cytokines, high levels of IL-10 have been consistently detected in experimental models [28] and in patients with severe pneumonia caused by pdm A/H1N1 infection [6]. In our study, high levels of IL-10 were induced by the pdm A/H1N1, but not by the A/PR/8/34 strain. However the relevance of IL-10 in the severe forms of pneumonia associated to the pdm A/H1N1 remains unclear.
Interestingly, significant amounts of CXCL10 were selectively induced by the A/PR/8/34 seasonal strain. Microarray analysis also revealed that mRNAs of CXCR3 ligands (CXCL10, CXCL11 and CXCL9), as well as CCL8, were significantly up-regulated by A/PR/8/34 virus, and, in contrast, they were down-regulated during infection by the pdm A/H1N1 strain. Although IFN-γ is a major inducer of CXCL10, we did not detect significant differences in the induction of IFN-γ between the two viral strains, consistent with previous reports [4,28]. The expression of CXCL10 may be enhanced directly by pathogens and other pro-inflammatory cytokines other than IFN-γ [29]. Further functional studies are required to determine the mechanisms underlying the failure of pdm A/H1N1-infected macrophages to express CXCL10, as reported in our findings.
Innate immunity to viral infections is induced by pattern recognition receptors such as toll-like receptors (TLR's) and the retinoic acid-inducible gene I (RIG)-like helicase (RLH) receptors [30]. Our results show that, in contrast to the A/PR/8/34 strain, pdm A/H1N1 virus failed to induce RIG-I mRNA. In concordance with this result, the microarray analysis of gene expression demonstrates that several antiviral proteins normally induced by the RIG-I activity, such as OAS, MX1, RIG-I itself, IFIT2, IFIT3, TLR3, TRIM25, DHX58, DDX60 and ISG15, among others, were considerably down-modulated in pdm A/H1N1 infected macrophages.
RIG-I is critical in the detection of and response to viral infection in host cells. Upon influenza virus infection, the recognition of viral ssRNA by RIG-I induces the TRIM25-dependent ubiquitination of RIG-I and promotes its interaction with MAVS/IPS-1. This triggers the activation of IRF3, an NFkB transcription factor that in turn induces IFN production and other innate response genes [31]. The importance of RIG-I has been demonstrated in experimental models of infection with respiratory viruses in RIG-I-deficient cells [32]. Among other immune escape strategies so far identified, the influenza virus A NS1 protein inhibits the TRIM25 dependent ubiquitination of RIG-I, and in turn the production of type I IFN [33]. Therefore, NS1 is considered a major virulence factor, and there has been great interest in studying the possible correlation of virulence with strain-specific differences in the NS1 protein [34,35]. Interestingly, gene profile analysis of human cells infected with genetically engineered influenza A virus has demonstrated that strain specific differences in the NS1 protein correlate with different expression patterns of type I IFN-related genes [36]. In this context, it has been shown that A/PR/8/34 NS1 protein induces strong expression of SOCS-1 and RIG-I compared with NS1 protein of the 1918 pandemic virus [37]. The NS1 gene of the pdm A/H1N1 virus is of swine origin [13–15], and was not previously found in human seasonal viruses. Keeping this in mind, we cannot rule out that strain-specific differences in the NS1 protein of seasonal A/PR/8/34 versus pdm A/H1N1 could in part explain the different responses we found in macrophages infected with these viruses.
Our study has some limitations, particularly regarding the selection of the seasonal H1N1 viral strain (A/PR/8/34) used in the comparative assays with pdm A/H1N1. The A/PR/8/34 strain has been laboratory adapted by several passages in mice and chick-en embryos, but has nevertheless been widely used for direct comparisons of pathogenic characteristics with different influenza A strains. In this study, we did not use a contemporary non-pdm influenza A H1N1 strain to perform the functional assays and com pare its effects with the pdm A/H1N1 virus. However, preliminary data generated in our laboratory using a circulating H3N2 strain, contemporary to the pdm A/H1N1 strain, suggest that similar to the pdm A/H1N1 virus, the H3N2 strain failed to induce SOCS-1. Interestingly, infection with the seasonal H3N2 virus induces a high expression of RIG-1 in macrophages, of the same magnitude as the A/PR/8/34 (Cruz-Lagunas A, in preparation), contrasting the lack of induction of RIG-I by the pdm A/H1N1 strain.
In conclusion, our results suggest that factors inherent to pdm A/H1N1 influenza strain may in part incite the development of severe inflammatory disease through inhibiting SOCS-1 and RIG-1 expression, resulting in the increased production of inflammatory mediators (cytokine storm) and the inhibition of the antiviral response. These effects could, along with host-related factors, explain the associated poor clinical outcomes observed in some patients. Further work to identify the responsible virulence factors of pdm A/H1N1 is warranted.
Supplementary Material
Acknowledgments
This work was supported by The Mexican National Council of Science and Technology (CONACYT) Grants [127002, 142364 CB-2010-155382 and E-1105].
Footnotes
Appendix A. Supplementary material: Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cyto.2013.01.018.
References
- 1.Lapinsky SE. Epidemic viral pneumonia. Curr Opin Infect Dis. 2010;23:139–44. doi: 10.1097/QCO.0b013e328336eaae. [DOI] [PubMed] [Google Scholar]
- 2.Perez-Padilla R, de la Rosa-Zamboni D, Ponce de Leon S, Hernandez M, Quiñones-Falconi F, Bautista E, et al. Pneumonia and respiratory failure from swine-origin influenza A (H1N1) in Mexico. N Eng J Med. 2009;361:680–9. doi: 10.1056/NEJMoa0904252. [DOI] [PubMed] [Google Scholar]
- 3.Writing Committee of the WHO Consultation on Clinical Aspects of Pandemic (H1N1) 2009. Influenza E, Bautista T, Chotpitayasunondh TZ, Gao SA, Harper M, Shaw, et al. Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N Eng J Med. 2010;362:1708–19. doi: 10.1056/NEJMra1000449. [DOI] [PubMed] [Google Scholar]
- 4.Zúñiga J, Torres M, Romo J, Torres D, Jiménez L, Ramírez G, et al. Inflammatory profiles in severe pneumonia associated to the pandemic influenza A/H1N1 virus isolated in Mexico City. Autoimmunity. 2011;44:562–70. doi: 10.3109/08916934.2011.592885. [DOI] [PubMed] [Google Scholar]
- 5.Monsalvo AC, Batalle JP, Lopez MF, Krause JC, Klemenc J, Hernández JZ, et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat Med. 2011;17:195–9. doi: 10.1038/nm.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bermejo-Martin JF, Martin-Loeches I, Rello J, Antón A, Almansa R, Xu L, et al. Host adaptive immunity deficiency in severe pandemic influenza. Crit Care. 2010;14:R167. doi: 10.1186/cc9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:267–8. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- 8.Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 2005;387:921–4. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 9.Naka T, Fujimoto M, Tsutsui H, Yoshimura A. Negative regulation of cytokine and TLR signalings by SOCS and others. Adv Immunol. 2005;87:61–122. doi: 10.1016/S0065-2776(05)87003-8. [DOI] [PubMed] [Google Scholar]
- 10.Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–87. doi: 10.1016/s1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- 11.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–65. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 12.Pothlichet J, Chignard M, Si-Tahar M. Cutting edge: innate immune response triggered by influenza A virus is negatively regulated by SOCS1 and SOCS3 through a RIG-I/IFNAR1-dependent pathway. J Immunol. 2008;180:2034–8. doi: 10.4049/jimmunol.180.4.2034. [DOI] [PubMed] [Google Scholar]
- 13.Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature. 2009;459:1122–5. doi: 10.1038/nature08182. [DOI] [PubMed] [Google Scholar]
- 14.Saxena SK, Mishra N, Saxena R, Swamy ML, Sahgal P, Saxena S, et al. Structural and antigenic variance between novel influenza A/H1N1/2009 and influenza A/H1N1/2008 viruses. J Infect Dev Ctries. 2009;4:1–6. doi: 10.3855/jidc.546. [DOI] [PubMed] [Google Scholar]
- 15.Sinha NK, Roy A, Das B, Das S, Basak S. Evolutionary complexities of swine flu H1N1 gene sequences of 2009. Biochem Biophys Res Commun. 2009;390:349–51. doi: 10.1016/j.bbrc.2009.09.060. [DOI] [PubMed] [Google Scholar]
- 16.Yang XX, Du N, Zhou JF, Li Z, Wang M, Guo JF, et al. Gene expression profiles comparison between 2009 pandemic and seasonal H1N1 influenza viruses in A549 cells. Biomed Environ Sci. 2010;23:259–66. doi: 10.1016/S0895-3988(10)60061-X. [DOI] [PubMed] [Google Scholar]
- 17.Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, et al. A family of cytokine inducible inhibitors of signaling. Nature. 1997;387:917–21. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 18.Dalpke AH, Opper S, Zimmermann S, Heeg K. Suppressors of cytokine signaling (SOCS)-1 and SOCS-3 are induced by CpG-DNA and modulate cytokine responses in APCs. J Immunol. 2001;166:7082–9. doi: 10.4049/jimmunol.166.12.7082. [DOI] [PubMed] [Google Scholar]
- 19.Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, et al. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17:583–91. doi: 10.1016/s1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- 20.Kimura A, Naka T, Muta T, Takeuchi O, Akira S, Kawase I, et al. Suppressor of cytokine signaling-1 selectively inhibits LPS-induced IL-6 production by regulating JAK-STAT. Proc Natl Acad Sci USA. 2005;102:17089–94. doi: 10.1073/pnas.0508517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perrone LA, Plowden JK, García-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008;4:e1000115. doi: 10.1371/journal.ppat.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters W, Cyster JG, Mack M, Schlöndorff D, Wolf AJ, Ernst JD, et al. CCR2-dependent trafficking of F4/80dim macrophages and CD11cdim/intermediate dendritic cells is crucial for T cell recruitment to lungs infected with Mycobacterium tuberculosis. J Immunol. 2004;172:7647–53. doi: 10.4049/jimmunol.172.12.7647. [DOI] [PubMed] [Google Scholar]
- 23.La Gruta NL, Kedzierska K, Stambas J, Doherty PC. A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol. 2007;85:85–92. doi: 10.1038/sj.icb.7100026. [DOI] [PubMed] [Google Scholar]
- 24.Martínez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 25.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 27.Whyte CS, Bishop ET, Rückerl D, Gaspar-Pereira S, Barker RN, Allen JL, et al. Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J Leukoc Biol. 2011;90:845–54. doi: 10.1189/jlb.1110644. [DOI] [PubMed] [Google Scholar]
- 28.Itoh Y, Shinya K, Kiso M, Watanabe T, Sakoda Y, Hatta M, et al. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature. 2009;460:1021–5. doi: 10.1038/nature08260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu M, Guo S, Hibbert JM, Jain V, Singh N, Wilson NO, et al. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011;22:121–30. doi: 10.1016/j.cytogfr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Basler CF, Garcia-Sastre A. Sensing RNA virus infections. Nat Chem Biol. 2007;3:20–1. doi: 10.1038/nchembio0107-20. [DOI] [PubMed] [Google Scholar]
- 31.Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell. 2010;140:397–408. doi: 10.1016/j.cell.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 32.Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–45. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microb. 2009;5:439–49. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol. 2008;89:2359–76. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- 35.Hale BG, Steel J, Manicassamy B, Medina RA, Ye J, Hickman D, et al. Mutations in the NS1 C-terminal tail do not enhance replication or virulence of the 2009 pandemic H1N1 influenza A virus. J Gen Virol. 2010;91:1737–42. doi: 10.1099/vir.0.020925-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geiss GK, Salvatore M, Tumpey TM, Carter VS, Wang X, Basler CF, et al. Cellular transcriptional profiling in influenza A virus infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc Natl Acad Sci USA. 2002;99:10736–41. doi: 10.1073/pnas.112338099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Billharz R, Zeng H, Proll SC, Korth MJ, Lederer S, Albrecht R. The NS1 protein of the 1918 pandemic influenza virus blocks host interferon and lipid metabolism pathways. J Virol. 2009;83:10557–70. doi: 10.1128/JVI.00330-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.