

Abstract
We hypothesized that particular genetic backgrounds enhance rates of colonization, increase severity of enteritis, and allow for extraintestinal spread when inbred IL-10−/− mice are infected with pathogenic C. jejuni. Campylobacter jejuni stably colonized C57BL/6 and NOD mice, while congenic strains lacking IL-10 developed typhlocolitis following colonization that mimicked human campylobacteriosis. However, IL-10 deficiency alone was not necessary for presence of C. jejuni in extraintestinal sites. C3H/HeJtlr4−/− mice that specifically express the Cdcs1 allele showed colonization and limited extraintestinal spread without enteritis implicating this interval in the clinical presentation of C. jejuni infection. Furthermore, when the IL-10 gene is inactivated as in C3Birtlr4−/− IL-10−/− mice, enteritis and intensive extraintestinal spread were observed, suggesting that clinical presentations of C. jejuni infection are controlled by a complex interplay of factors. These data demonstrate that lack of IL-10 had a greater effect on C. jejuni induced colitis than other immune elements such as TLR4 (C3H/HeJ, C3Bir IL-10−/−), MHC H-2g7, diabetogenic genes, and CTLA-4 (NOD) and that host genetic background is in part responsible for disease phenotype. C3Bir IL-10−/− mice where Cdcs1 impairs gut barrier function provide a new murine model of C. jejuni and can serve as surrogates for immunocompromised patients with extraintestinal spread.
Keywords: Campylobacter jejuni, enteritis, murine disease model, IL-10, Toll-like receptor 4, cytokine deficiency–induced colitis susceptibility 1 (Cdcs1) allele, Inflammatory Bowel Disease
1. Introduction
Campylobacter jejuni is a leading cause of bacterial diarrhea with cases in the US exceeding those of Salmonella and Escherichia coli (1). Self-limiting gastroenteritis is the most common clinical syndrome (2), but infected patients show a spectrum of diseases including gastroenteritis, septicemia, meningitis, proctitis, abortion, and autoimmune diseases (3). Immunocompromised patients have both a higher incidence of infection with Campylobacter and more severe disease manifestations with profuse diarrhea and persistence of the organism (4, 5). Less commonly, extremes of age or underlying immune compromise can predispose to bacteremia and subsequent spread to other organs (4, 6). Reactive inflammatory disorders can also occur as sequelae of C. jejuni infection. Transmural inflammatory changes in colon or small intestines sometimes occur that resemble inflammatory bowel disease (IBD)(7, 8). Antecedent C. jejuni has also been linked to Guillain Barré Syndrome (GBS), a debilitating inflammatory polyneuritis that often results in long term disability (9, 10). Reactive arthritis can also follow diarrheic episodes of (11) or asymptomatic exposure to (12) C. jejuni and is attributed to autoimmune response in joints (13, 14). Recently, presence of C. jejuni in tissues was associated with mucosa-associated lymphoid-tissue (MALT) lymphoma, an immunoproliferative small intestinal disease (15).
In order to understand the basis of bacterial strain dependent disease manifestations, studies have been conducted to examine genetic diversity among C. jejuni strains. Microarray analysis of C. jejuni isolates has shown that both highly divergent and highly conserved gene classes exist among isolates from cases (16–20); however, comparison of C. jejuni isolates implicated in GBS to isolates associated with mild enteritis showed that specific GBS genes or regions could not be identified (16, 21). Clinical evidence that C. jejuni strain variation does not account for all disease syndromes is shown by the fact that the same strains producing watery diarrhea in children in developing countries have been isolated from visitors with acute inflammatory disease (22). In another study, there was no difference in serotypes isolated from symptomatic and asymptomatic children (23). Although exploration of the significant diversity of C. jejuni genomes is ongoing, the outcome of infection likely depends on host, bacterial, and epidemiological factors, including bacterial strain variation, dose, number and timing of previous infections, host age and immune status, presence of other pathogens, or variation in the composition of the flora in the gastrointestinal (GI) tract. In this study we consider the allelic composition of the host genome as a determinant of disease after C. jejuni challenge.
Host genetic background plays a substantial role in susceptibility to infectious diseases (24). Use of mice with altered immune systems has been crucial in eliciting specific clinical syndromes to model infection with GI pathogens, including Campylobacter (25) and Helicobacter (26). Each inbred mouse strain has unique background alleles that can interact with and modify the expression of targeted mutations, genetic regions introduced from other strains, and transgenes (27). Inbred mouse strains often vary in normal development and physiology due to the action of modifier genes that suppress or enhance gene expression by altering DNA transcription rates, altering mRNA stability, or inducing epigenetic effects such as DNA methylation (28). Modifier genes can be normal constituents of the genome, arise by spontaneous mutation, or be discovered during construction of congenic mouse strains due to linkage to the locus of interest (28). Modifier genes unique to particular genetic backgrounds were shown to have deleterious effects in exacerbating murine cecal hyperplasia associated with cytokine deficiency–induced colitis susceptibility 1 (Cdcs1) even in previously well-characterized inbred strains (29).
With such a complex interplay of factors, reliable animal infection models that reproduce human disease outcomes would facilitate research into mechanisms of C. jejuni pathogenesis and therapies. Our strategy was to develop murine models of enteritis induced by primary oral C. jejuni challenge using mice with a genetic background and immune bias expected to increase susceptibility and demonstrate the range of clinical syndromes observed in patients. Recently, we showed that C57BL/6 IL-10+/+ and congenic IL-10−/− mice can be used as colonization and disease models, respectively (25). C. jejuni 11168 colonized both C57BL/6J IL-10+/+ and IL-10−/− mice and caused a high rate of typhlocolitis in IL-10−/− mice in time course and dose response experiments. A dose as small as 102 cfu produced clinical disease and histological lesions in the GI tracts of the mice (unpublished results). By 28 days post infection, both C57BL/6 IL-10+/+ and IL-10−/− mice developed robust T helper cell 1 (Th1) associated plasma immunoglobulin responses to C. jejuni. To extend these studies, we considered the possibility that background modifier genes may influence the development of C. jejuni enteritis due to IL-10 deficiency. We sought IL-10−/− mice of different genetic backgrounds that could be obtained and reared free of colitogenic bacteria with background genes, especially immune system defects that might influence susceptibility and disease manifestations.
Our approach was to infect C57BL/6, non-obese diabetic (NOD), and C3H/HeJ wild-type (WT) mice and their corresponding IL-10 knockouts (B6.129P2-IL10tm1Cgn/J [referred to below as C57BL/6 IL-10−/−]; NOD.Cg-Il10tm1Cgn/LtJ [NOD IL-10−/−]; C3Bir.129P2(B6)-Il10tm1Cgn/Lt [C3Bir IL-10−/−]) with a primary oral challenge with C. jejuni 11168 and then to compare the outcomes by quantifying clinical signs, gross pathology, histopathology, immunohistochemical (IHC) staining, and anti-C. jejuni isotype specific antibody response. C57BL/6 mice have MHC H-2b, NOD mice have MHC H-2g7, a range of diabetes linked genes, and Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) that inhibits T cell function, and C3 mice have H-2k, a Toll-like receptor 4 deficiency (TLR4), and the Cdcs1 allele. These experiments demonstrated that IL-10−/− mice of these genetic backgrounds have similar inflammatory disease processes in the colon when infected with virulent C. jejuni. All IL-10−/− and WT mice were colonized by C. jejuni at 35 days after infection. C. jejuni infected IL-10−/− mice of all three genetic backgrounds and a few NOD WT mice developed typhlocolitis that resembled IBD by 35–40 days post infection. For IL-10+/+ mice of the C57BL/6 and NOD backgrounds, C. jejuni colonization was necessary but not sufficient for lesions. However, in the two experiments combined, 86% of C3Bir IL-10−/− uninfected control mice developed some degree of typhlocolitis despite an environment lacking previously identified colitis-causing bacteria compared to 61% in the C. jejuni infected group, although the latter had twice as many mice with the highest grade lesions. These results suggest the hypotheses (1) that TLR4 and IL-10 deficiency along with the Cdcs1 allele (27) disrupt gut barrier function and (2) that C. jejuni is just one of many bacteria that can elicit colitis in these mice. Taken together, these results demonstrate that the lack of the cytokine IL-10 had the greatest impact on enhancing susceptibility to C. jejuni induced enteritis. Furthermore, mice of the C3 genetic background had significant extraintestinal spread of C. jejuni that was exacerbated by IL-10 deficiency. These results implicate TLR4, Cdcs1, and possibly other background modifier genes in extraintestinal spread of C. jejuni and suggest that C3Bir IL-10−/− mice can provide usable models for this aspect of infection.
2. Results
2.1. Monitoring for spontaneous colitis and diabetes in breeding colony mice
We recognized that the responses of IL-10−/− mice to C. jejuni infection could be skewed if environmental factors such as colitis-causing bacteria were present in either the mouse breeding colony or the containment challenge facility. Mice of C3 and NOD genetic backgrounds were most at risk because of their respective additional immune system deficiencies. Therefore, we monitored the background rate of colitis development in mice of the C57BL/6 and C3Bir genetic background and the background rate of both colitis and diabetes development in mice of the NOD genetic background in the breeding colony. Retired breeding mice of all genetic backgrounds and breeding colony mice euthanized due to evidence of spontaneous colitis were necropsied; each mouse was examined for colitis (enlargement and thickening of the wall of the proximal colon), enlargement of the spleen, and, in NOD mice, elevated urine glucose levels.
Breeding colony mice of the C57BL/6 and NOD genetic backgrounds had low rates of spontaneous colitis and splenic enlargement. Four of 88 (4%) C57BL/6 IL-10−/− mice and 0/33 C57BL/6 IL-10+/+ mice developed spontaneous colitis; 2/88 (2%) C57BL/6 IL-10−/− mice and 0/33 C57Bl/6 IL-10+/+ mice developed splenic enlargement, while 1/43 (2%) NOD IL-10+/+ mice and 0/27 (0%) NOD IL-10−/− mice developed spontaneous colitis, and no NOD mice developed splenic enlargement. The single NOD IL-10+/+ mouse that developed spontaneous colitis was also diabetic. Nineteen of 66 (29%) NOD IL-10+/+ and −/− breeding colony mice in the age range of experimental mice developed blood glucose levels ≥100 mg/dL, but only one experimental NOD IL-10+/+ mouse became diabetic during the course of the experiments. In contrast, 44/73 (60%) C3Bir IL-10−/− mice in the breeding colony in the same age range of experimental mice developed spontaneous colitis as evidenced by soft or liquid feces, enlargement of the colon, and thickening of the colon wall; 42/73 (57%) had enlarged spleens; and 48/73 (66%) had enlarged ileocecocolic/mesenteric lymph nodes. Thirty-one of 73 (42%) of these mice exhibited all three kinds of pathological change: colitis, lymph node enlargement, and splenic enlargement. One C3H/HeJ IL-10+/+ mouse necropsied at 22 weeks of age had enlargement of the colon only.
2.2. Development of spontaneous colitis and diabetes in experimental mice
Prior to infection species-specific PCR for known colitogenic bacteria (Helicobacter hepaticus, C. jejuni, Citrobacter rodentium, Enterococcus faecalis) were negative for all experimental mice. However in Experiment 1, one C57BL/6 IL-10−/− control mouse and both C3Bir IL-10−/− control mice developed spontaneous colitis as assessed by clinical observations and gross pathology findings. All three mice had elevated histological scores and IHC staining of tissues at or below the basolateral surface of the intestinal epithelium with a polyclonal anti-C. jejuni antibody. No spiral bacteria were found on exam of immunohistochemically stained tissue sections, although some rod-shaped bacteria in the lumen of the GI tract appeared stained. All of these mice were negative for C. jejuni by culture and PCR gyrA assay. In Experiment 2, no control C3Bir IL-10−/− mice showed clinical or gross pathological signs of spontaneous colitis; however, 4/5 control mice had elevated histological scores falling in grade 1 and IHC staining of tissues at or below the basolateral surface of the intestinal epithelium (Table 1). Once again, all seven of these mice were negative for C. jejuni by culture and PCR gyrA assay.
Table 1.
Gross pathology and C. jejuni specific PCR assays of infected C3Bir IL-10−/− and C3H IL-10+/+ mice in Experiment 2. All uninfected control mice had no pathology and were negative for all values presented.
| Genotype | Day euthanized | Enlarged lymph nodes | Enlarged spleen | PCR+ spleen | PCR+ liver | PCR+ blood (day(s)) |
|---|---|---|---|---|---|---|
| C3Bir IL-10−/− | 7 | + | + | − | − | − |
| 11 | + | + | + | + | − | |
| 13 | + | + | − | + | − | |
| 28 | + | + | − | + | − | |
| 28 | + | − | + | + | − | |
| 28 | − | − | + | − | + (14) | |
| 28 | + | − | + | + | − | |
| 28 | − | − | + | + | − | |
| 28 | − | + | − | − | + (14) | |
| 28 | + | − | + | − | + (14) | |
| 28 | + | + | + | − | − | |
| 28 | − | − | + | + | + (14, 28) | |
| 28 | + | − | + | + | + (14) | |
| C3H IL-10+/+ | 29 | − | − | + | − | − |
| 29 | − | − | − | − | − | |
| 29 | − | − | − | + | − | |
| 29 | − | − | − | − | − | |
| 29 | − | − | − | − | − | |
| 29 | − | − | − | − | − | |
| 29 | − | − | − | − | + (14) | |
| 29 | − | − | − | − | + (14) | |
| 29 | − | − | − | − | + (14) | |
| 29 | − | − | − | − | − | |
| 29 | − | − | − | − | − | |
| 29 | − | − | + | + | − |
Because of the documented association of typhlocolitis with Helicobacter species (30) and the widespread presence of Helicobacter spp. infection in research mouse colonies (31), we tested the single C57BL/6 IL-10−/− control mouse and all six C3Bir IL-10−/− control mice that had spontaneous colitis for the presence of Helicobacter spp. using two PCR assays based on the 16S rRNA gene. The PCR assay of Beckwith et al. (1997) used to screen fecal samples from the breeding colony was performed on fecal samples taken from each cage of mice prior to transport to the University Research Containment Facility; these PCR assays were all negative (32). The same PCR assay was performed on DNA extracted from all tissues (stomach, jejunum, cecum, colon, liver, and spleen), blood, and fecal samples from all of the seven control mice that exhibited elevated histopathology scores. All samples were negative in this assay except for spleen and fecal samples from one C3Bir IL-10−/− mouse and liver and spleen samples from a second C3Bir IL-10−/− mouse both in Experiment 2 that had faint positive bands. Five species-specific PCR assays for murine Helicobacter spp. (H. hepaticus, H. muridarum, and H. typhlonius (33), H. ganmani (34), and H. mastomyrinus (35)) were performed on the four positive DNA samples from the two C3Bir IL-10−/− control mice in Experiment 2; all results were negative (data not shown).
These four DNA samples were then subjected to PCR-RFLP analysis of a 1.2 kbp fragment of the 16S RNA gene digested with the restriction endonuclease HhaI (36); one PCR product was subjected to digestion with BslI. The HhaI 16S PCR-RFLP pattern obtained for all four of these samples consisted of a doublet containing two bands ≤ 400 bp each and two additional fragments of approximately 280 and 180 bp. The BslI pattern included fragments of approximately 550 bp, 200 bp, and a number of small fragments ≤ 100 bp. The combined HhaI and BslI patterns are not consistent with any 16S PCR-RFLP patterns that have been reported in the literature (35, 36) or that can be predicted from published sequences of any Helicobacter species or “Flexispira” taxa that have been reported in the literature as infecting mice (37, 38). Although Helicobacter genus specific primers were applied to all mouse samples, we could not test for H. bilis, H. rappini and H. trogontum by PCR because they are so rare that positive control DNA was not available (31). One C. jejuni infected NOD IL-10+/+ mouse became diabetic during Experiment 1; no NOD background mice became diabetic during Experiment 3. No uninfected NOD mice developed spontaneous colitis.
2.3. Campylobacter jejuni 11168 colonizes the GI tract of all experimentally infected mice
Colonization levels in the ceca and colons of inoculated mice are shown in Figure 1 for all three experiments. No control mice were colonized by C. jejuni in any experiment based on culture or C. jejuni specific PCR of GI tract tissues and feces (Tables 1 and 2). C. jejuni was not cultured from one inoculated C57BL/6 IL-10−/− mouse on the low fat diet but was detected by PCR in DNA extracted from all four GI tract tissues and feces. All other C. jejuni inoculated mice of all three genetic backgrounds and both IL-10 genotypes in all three experiments were colonized based on culture (Figure 1) and PCR (Table 1). As found previously in C57BL/6 IL-10−/− and IL-10+/+ mice, colonization levels were highest in ceca of mice of all genetic backgrounds regardless of presence or absence of IL-10; similarly, levels in stomach and jejunum (data not shown) and in feces of all mice were lower than in ceca and colons (25). All putative C. jejuni isolates were verified by PCR assay (data not shown).
Figure 1. Colonization by Campylobacter jejuni.

Presence of Campylobacter jejuni in cecum and colon of the GI tract in C57BL/6 IL-10−/−, C57BL/6 IL-10+/+, NOD IL-10−/−, NOD IL-10+/+, C3BirIL-10−/−, and C3H/HeJ IL-10+/+ mice inoculated with C. jejuni 11168. Colonization rates were ranked according to numbers of Campylobacter colonies on the plate as 0 (none), 1 (~1–20 cfu), 2 (~20–200 cfu), 3 (>200 cfu), and 4 (confluent growth). All cultures from the uninoculated control mice were negative for Campylobacter jejuni. Panels A, D show C57BL/6 genotype results, Panels B, E show C3BirIL-10−/−, and C3H/HeJ IL-10+/+ genotype results, and Panels C, F show NOD IL-10−/−, NOD IL-10+/+ genotype results. Numbers in parentheses along X-axis show percentage fat in the mouse diets.
Table 2.
C. jejuni gyr A specific PCR on DNA extracted from tissues of infected mice only (# samples positive/# samples tested); ND, not done; no DNA preparations from any tissues or feces of control mice were positive for C. jejuni by this PCR assay.
| Mouse strain | Genotype | % fat in diet | n | stomach | jejunum | cecum | colon | feces | blood | spleen | liver |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A. Experiment 1 | |||||||||||
| C57BL/6 | IL-10−/− | 11 | 10 | 10/10 | 10/10 | 10/10 | 10/10 | 10/10 | 0/10 | -- | -- |
| C57BL/6 | IL-10−/− | 6 | 10 | 10/10 | 9/10 | 9/10 | 10/10 | 10/10 | 1/10 | -- | -- |
| C3Bir | IL-10−/− | 11 | 10 | 10/10 | 10/10 | 10/10 | 10/10 | 8/9 | 2/10 | -- | -- |
| NOD | IL-10−/− | 6 | 10 | 10/10 | 10/10 | 10/10 | 10/10 | 7/9 | 0/10 | -- | -- |
| C57BL/6 | IL-10+/+ | 11 | 10 | 10/10 | 10/10 | 10/10 | 9/9 | 9/10 | 0/10 | -- | -- |
| C3H/HeJ | IL-10+/+ | 11 | 10 | 9/10 | 10/10 | 10/10 | 10/10 | 10/10 | 1/10 | -- | -- |
| NOD | IL-10+/+ | 6 | 10 | 10/10 | 10/10 | 10/10 | 10/10 | 10/10 | 0/10 | -- | -- |
| B. Experiment 2 | |||||||||||
| C3Bir | IL-10−/− | 11 | 13 | 12/13 | 13/13 | 13/13 | 13/13 | 13/13 | 6/33 | 9/13 | 8/13 |
| C3H/HeJ | IL-10+/+ | 11 | 12 | 7/13 | 12/12 | 12/12 | 12/12 | 12/12 | 3/36 | 2/12 | 2/12 |
| C. Experiment 3 | |||||||||||
| NOD | IL-10−/− | 6 | 3 | 1/3 | 2/3 | 3/3 | 3/3 | 3/3 | ND | 0/3 | 1/3 |
| NOD | IL-10+/− | 6 | 4 | 2/4 | 4/4 | 4/4 | 4/4 | 3/4 | ND | 0/4 | 0/4 |
| NOD | IL-10+/+ | 6 | 18 | 5/18 | 11/18 | 18/18 | 18/18 | 13/18 | ND | 0/18 | 0/18 |
2.4. Campylobacter jejuni infected IL10−/− mice experience clinical campylobacteriosis
C. jejuni infected IL-10−/− mice of all three genetic backgrounds exhibited clinical signs and gross pathological changes similar to those reported previously for C57BL/6 IL-10−/− mice (Figure 2)(25). Major clinical signs included ruffled hair coat, hunched posture, lethargy and watery diarrhea. Gross pathological changes in all mice included enlargement of the ileocecocolic lymph node, enlargement of the cecum and colon, and thickening of the GI tract wall. However, unlike C57BL/6 IL-10−/− mice, some control C3Bir IL-10−/− mice, C. jejuni infected C3Bir IL-10−/− mice, and a few (3/13) C. jejuni infected NOD IL-10−/− mice also exhibited enlarged spleens (Figure 3). No C. jejuni infected C57BL/6 IL-10+/+ or C3H/HeJ IL-10+/+ mice exhibited clinical signs or gross pathology; however, 2/10 C. jejuni infected NOD IL-10+/+ mice in Experiment 1 exhibited enlargement of the ileocecocolic lymph node only or enlargement of the lymph node, cecum and colon, but neither mouse exhibited clinical signs.
Figure 2.
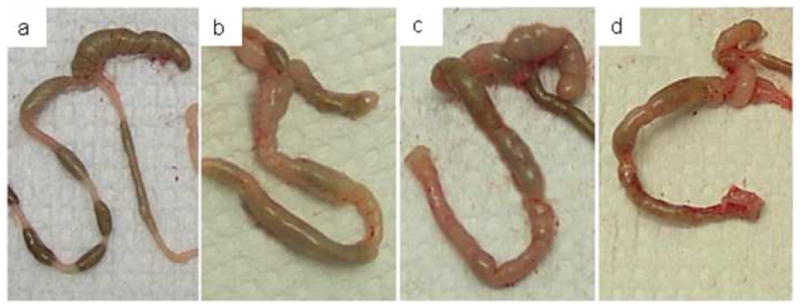
Colon gross pathology in C57BL/6 IL-10−/−, NOD IL-10−/−, and C3BirIL-10−/− mice given oral Campylobacter jejuni and sacrificed 28–35 days after infection. A) Normal colon from an uninfected C57BL/6 IL-10+/+ control mouse 30 days after gavage with tryptone soy broth. Colons from the Campylobacter jejuni infected B) C57BL/6 IL-10−/− mouse, C) NOD IL-10−/− mouse, and D) C3BirIL-10−/− mouse. Ceca and colons in B, C, and D are enlarged and pale with thickened walls and watery contents.
Figure 3.

Spleen gross pathology in a C3BirIL-10−/− mouse given oral Campylobacter jejuni and sacrificed 30 days post infection. A) Shows normal spleen from an uninfected C57BL/6 IL-10+/+ control mouse 30 days after gavage with tryptone soy broth compared to B) greatly enlarged spleen from infected C3BirIL-10−/− mouse. Table shows the means and the standard error of the mean for spleen body weight ratios for mice of C3BirIL-10−/− and C3H/HeJ genotypes either sham inoculated with tryptone soy broth or infected with Campylobacter jejuni. Two way analysis of variance showed there were differences between means based on genotype (P=0.030), but not based on infection status (P=0.269).
C. jejuni infected C57BL/6 IL-10−/− mice in Experiment 1 exhibited enlargement of the colon and thickening of its walls (4/10 infected mice on each of the two diets and one control mouse on the 11% fat diet) and enlargement of lymph nodes draining the colon (2/10 infected mice on the 11% fat diet, 1/10 infected mice on the 6% fat diet, and 1/10 control mice on the 11% fat diet). No infected or control C57BL/6 IL-10−/− mice exhibited enlarged spleens. No infected or control C57BL/6 IL-10+/+ mice exhibited any gross pathological changes.
In the Experiment 1, 6/10 infected and 2/2 control IL-10−/− mice of the C3Bir genetic background exhibited enlargement and thickening of the colon; 3/10 infected and 1/2 control C3Bir IL-10−/− mice exhibited enlarged GI lymph nodes. In addition, 5/10 infected and 1/2 control C3Bir IL-10−/− mice exhibited markedly enlarged spleens. In Experiment 2, 6/10 infected C3Bir IL-10−/− mice exhibited enlargement and thickening of the colon, 9/10 infected C3Bir IL-10−/− mice exhibited enlarged GI lymph nodes, and 6/10 infected C3Bir IL-10−/− mice exhibited enlarged spleens (Table 1). No control C3Bir IL-10−/− mice in the Experiment 2 exhibited any gross pathological changes. No infected or control C3H IL-10+/+ mice in either experiment exhibited any gross pathological changes.
In Experiment 1, 5/10 infected IL-10−/− mice of the NOD genetic background exhibited enlargement and thickening of the colon, and 3/10 infected NOD IL-10−/− mice exhibited enlarged ileocecocolic lymph nodes. In addition, 2/10 infected NOD IL-10−/− mice exhibited markedly enlarged spleens. In Experiment 3, 1/3 infected NOD IL-10−/− mice exhibited enlargement and thickening of the colon, enlarged ileocecocolic lymph nodes, and an enlarged spleen. No control NOD IL-10−/− mice in either experiment exhibited any gross pathological changes. In Experiment 1, one NOD IL-10+/+ mice exhibited enlargement and thickening of the GI tract, and a second mouse exhibited enlarged ileocecocolic lymph nodes. Neither the single NOD IL-10+/+ control mouse in Experiment 1 nor any infected or control NOD IL-10+/+ mice in Experiment 3 exhibited gross pathological changes.
2.5. Detection of C. jejuni DNA in extraintestinal tissues (blood, spleen, and liver)
In Experiment 1, splenic enlargement was noted in several IL-10−/− mice of C3Bir and NOD genetic backgrounds; such enlargement might reflect transport of C. jejuni beyond the GI tract. C. jejuni specific PCR assays were performed on DNA extracted from blood, spleen, and liver samples from selected mice (Table 2). Some samples were positive, but because spleen and liver samples had been removed from the body cavity after excision of the GI tract, results could not be regarded as definitively showing that C. jejuni was present in organs outside the GI tract. Therefore, in Experiments 2 and 3, liver and spleen samples were obtained prior to excision of the GI tract to minimize possible contamination. DNA extracted from 9/13 (70%) spleen, 8/13 (61%) liver, and 6/13 (46%) blood samples from C3Bir IL-10−/− mice was positive for C. jejuni by PCR assay (Table 1). DNA extracted from 2/12 (17%) spleen, 2/12 (17%) liver, and 3/12 (25%) blood samples from C3H/HeJ IL-10+/+ mice was positive for C. jejuni by PCR assay. Blood, spleen, and liver samples in Experiment 2 (C3 genetic background) were all negative by culture. A single liver sample from an infected NOD IL-10−/− mouse in Experiment 3 was positive for C. jejuni DNA by PCR assay, but all spleens were negative. Culturing of blood, spleen, and liver samples was not performed in this experiment because of the negative results in Experiment 2. In another trial not reported here, DNA extracted from spleen tissue of 1/23 (4%) C57BL/6 IL-10−/− mice infected with C. jejuni 11168 was positive for C. jejuni by PCR assay.
2.6. Campylobacter jejuni produces enteric lesions in IL-10−/− mice of various genotypes
Hematoxylin and eosin stained sections of the ileocecocolic junction of all mice were evaluated and scored according to criteria published previously (25). In this scoring system, grade 0 indicates normal histology; grade 1 indicates mild to moderate histopathologic change, and grade 2 indicates severe histopathologic change. Incidence of the individual histological characters that make up the criteria of the scoring system did not differ among C57BL/6 IL-10−/−, C3Bir IL-10−/−, and NOD IL-10−/− mice (data not shown). Distributions of total histological scores of the various mouse IL-10 genotypes and genetic backgrounds are shown in Figure 4.
Figure 4. Histopathology scoring.

Histopathology scores of sections of ileocecocolic junctions from C57BL/6 IL-10−/−, C57BL/6 IL-10+/+, NOD IL-10−/−, NOD IL-10+/+, C3BirIL-10−/−, and C3H/HeJ IL-10+/+ mice inoculated with Campylobacter jejuni 11168 or sham inoculated with tryptone soy broth and sacrificed 30 days after infection. The bar labeled “TSB” shows data for all control mice in each experiment. Panel A, experiment 1 with all three mouse genotypes; Panel B, experiment 2 with C3Bir IL-10−/− and C3H/HeJ IL-10+/+ mice; Panel C, experiment 3 with NOD IL-10−/−, NOD IL-10+/− mice, and NOD IL-10+/+ mice. Boxes enclose data falling within the second and third quartiles of the distributions; the heavy bar within each box indicates the median of the distribution; whisker bars indicate the maximum and minimum values observed.
In the Experiment 1 (Figures 4 - Panel A and 5), at least half of the C. jejuni infected IL-10−/− mice of all three genetic backgrounds had histopathological scores falling into grades 1 or 2. No C. jejuni infected C57BL/6 IL-10+/+ or C3H IL-10+/+ mice had histopathological scores in grades 1 or 2; the C57BL/6 IL-10−/− control mouse and the two C3Bir IL-10−/− that developed spontaneous colitis had scores in grade 2. The two C. jejuni infected NOD IL-10+/+ mice that had gross pathological changes had histopathological scores falling into grades 1 and 2. Nonparametric Kruskal Wallis one-way ANOVA on ranks was used to test the hypothesis that there were differences in histopathological scores among the groups of mice; control groups containing less than 10 animals were excluded from the analysis. This analysis indicated that there were significant differences among the groups of mice (P ≤ 0.001). Post hoc analysis using Tukey’s method showed that the scores of C. jejuni infected C57BL/6 IL-10+/+ mice were significantly different from those of C. jejuni infected C57BL/6 IL-10−/− mice on both diets (P < 0.05 for both comparisons), C. jejuni infected NOD IL-10−/− mice (P < 0.05), and C. jejuni infected C3Bir IL-10−/− mice (P < 0.05). Scores of C. jejuni infected NOD IL-10−/− and NOD IL-10+/+ mice were also significantly different from each other in this analysis (P ≤ 0.05), but scores of C3Bir IL-10−/− and C3H IL-10+/+ mice were not (P >0.05). Scores of C. jejuni infected IL-10−/− mice of the three genetic backgrounds were not significantly different from one another (P > 0.05). Scores of C57BL/6 IL-10−/− sham inoculated control mice on the 11% fat diet were not significantly different (p >0.05) from those of any other groups of mice in this analysis, likely due to the single control C57BL/6 IL-10−/− mouse that developed spontaneous colitis. Scores of C. jejuni infected IL-10+/+ mice of the three genetic backgrounds were not significantly different from each other or from those of C57BL/6 IL-10−/− control mice (P > 0.05).
In Experiment 2 (Figures 4 - Panel B and 5), 69% of C. jejuni infected C3Bir IL-10−/− mice had histological scores falling into grades 1 or 2; however, 80% of uninfected C3Bir IL-10−/− mice had histological scores falling into grade 1. All uninfected and C. jejuni infected C3H IL-10+/+ mice had scores falling in grade 0. Statistically significant differences were detected among the groups of mice using nonparametric Kruskal Wallis ANOVA (P ≤ 0.001). Post hoc analysis by Dunn’s method showed that histopathology scores of the C. jejuni infected C3Bir IL-10−/− mice were significantly different from those of both C. jejuni infected and control C3H IL-10+/+ mice (P < 0.05) but not significantly different from those of control C3Bir IL-10−/− mice (P > 0.05), four of five of the latter had spontaneous colitis. Histopathology scores of control C3H IL-10+/+ and C. jejuni infected C3H IL-10+/+ mice were not significantly different (P > 0.05).
In the NOD genetic background Experiment 3 (Figures 4 - Panel C and 5), a single C. jejuni infected IL-10−/− mouse was scored as grade 2. All other uninfected and C. jejuni infected NOD IL-10−/−, IL10+/−, and IL-10+/+ mice had scores in grade 0. Nonparametric Kruskal Wallis ANOVA revealed significant differences among the groups of mice at the 0.05 level (P = 0.012) and post hoc analysis indicated that C. jejuni infected NOD IL-10−/− mice had significantly different scores from control NOD IL-10−/− mice (P < 0.05). These results were similar to those of Experiment 1; the observation of fewer infected IL-10−/− mice with high histologic scores may have been influenced by the relatively small sample size.
2.7. Splenic enlargement as a marker of extraintestinal spread of Campylobacter jejuni
Spleens were graded based on two characters: presence or absence of marked extra medullary hematopoiesis (EMH) and expansion or lack of expansion of periarteriolar lymphatic sheaths (PALS). All spleens showed at least a moderate degree of EMH. These changes are illustrated in Table 3 and Figure 6.
Table 3.
Histopathological scoring of spleen sections.
| Mouse genotype | Treatment | Number of spleens examined | Number of normal spleens | Number of spleens exhibiting marked extramedullary hematopoeisis (EMH) only | Number of spleens exhibiting expansion of periarteriolar lymphatic sheaths (PALS) only | Number of spleens exhibiting both EMH and PALS |
|---|---|---|---|---|---|---|
| Experiment 1 | ||||||
| C57BL/6 IL-10−/− | control | 10 | 9/10 | 0/10 | 0/10 | 1/10a |
| infectedb | 18 | 14/18 | 1/18 | 1/18 | 2/18 | |
| C57BL/6 IL-10+/+ | infected | 10 | 9/10 | 0/10 | 1/10 | 0/10 |
| NOD IL-10−/− | control | 2 | 2/2 | 0/2 | 0/2 | 0/2 |
| infected | 10 | 3/10 | 1/10 | 1/10 | 5/10 | |
| NOD IL-10+/+ | infected | 10 | 7/10 | 1/10 | 0/10 | 2/10 |
| C3Bir IL-10−/− | control | 2 | 0/2 | 1/2a | 0/2 | 1/2a |
| infected | 9 | 5/9 | 0/9 | 0/9 | 4/9 | |
| C3H IL-10+/+ | control | 2 | 2/2 | 0/2 | 0/2 | 0/2 |
| infected | 10 | 9/10 | 0/10 | 1/10 | 0/10 | |
| Experiment 2 | ||||||
| C3Bir IL-10−/− | control | 5 | 2/5 | 0/5 | 1/5a | 2/5c |
| infected | 13 | 1/13 | 0/13 | 4/13 | 8/13 | |
| C3H IL-10+/+ | control | 5 | 4/5 | 0/5 | 1/5 | 0/5 |
| infected | 12 | 9/12 | 0/12 | 3/12 | 0/12 | |
| Experiment 3 | ||||||
| NOD IL-10−/− | control | 2 | 0/2 | 0/2 | 2/2 | 0/2 |
| infected | 2 | 1/2 | 0/2 | 1/2 | 0/2 | |
| NOD IL-10+/− | control | 2 | 1/2 | 0/2 | 1/2 | 0/2 |
| infected | 4 | 3/4 | 0/4 | 1/4 | 0/4 | |
| NOD IL-10+/+ | control | 7 | 1/7 | 0/7 | 6/7 | 0/7 |
| infected | 18 | 6/18 | 0/18 | 12/18 | 0/18 | |
This mouse had spontaneous colitis.
Includes mice on both 6% fat and 11% fat diets.
One of these mice had spontaneous colitis.
Figure 6.
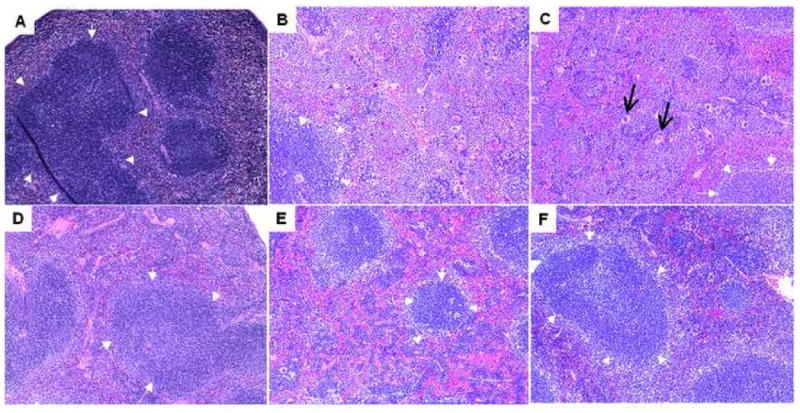
Representative micrographs of spleen at 10X magnification from C57BL/6 IL-10−/−, NOD IL-10−/−, and C3BirIL-10−/− mice given oral Campylobacter jejuni or sham inoculated with tryptone soy broth and sacrificed 28–35 days post infection. Sections were fixed in formalin, sectioned and stained with hematoxylin and eosin. Panels show infected mice of the following genotypes A) C57BL/6 IL-10−/−, B) NOD IL-10−/−, and C) C3BirIL-10−/− versus sham inoculated control mice of the same genotypes D) C57BL/6 IL-10−/−, E) NOD IL-10−/−, and F) C3BirIL-10−/−. In all panels small white arrows indicate the borders of the main periarteriolar lymphatic sheath in the section. Black arrows in panel C show prominent megakaryocytes. Dark staining as in panel A was seen consistently in infected C57BL/6 IL-10−/− mice and is not an artifact.
In Experiment 1, histological changes were found in 4/18 (22%) of spleens from C. jejuni infected C57BL/6 IL-10−/− mice (Table 3). One of ten control C57BL/6 IL-10−/− mice exhibited both expansion of PALS and marked EMH; this mouse also exhibited spontaneous colitis. A single mouse 1/10 (10%) C. jejuni infected C57BL/6 IL-10+/+ exhibited expansion of PALS only. Seven of ten (70%) C. jejuni infected NOD IL-10−/−, 2/10 (20%) C. jejuni infected NOD IL-10+/+, 4/9 (44%) C. jejuni infected C3Bir IL-10−/−, and 1/10 (10%) C. jejuni infected C3H IL-10+/+ mice had histological changes in the spleen. None of the uninfected NOD IL-10−/− and uninfected C3H IL-10+/+ mice had histological changes in the spleen. However, both of the uninfected C3Bir IL-10−/− mice with spontaneous colitis had histological changes in the spleen.
In Experiment 2, 3/5 (60%) of control C3Bir IL-10−/− mice and 12/13 (92%) of C. jejuni infected C3Bir IL-10−/− mice had histological changes in the spleen (Table 3). One of five (20%) control C3H IL-10+/+ and 3/12 (25%) C. jejuni infected C3H IL-10+/+ mice exhibited histological changes in the spleen. All of the mice that exhibited EMH were IL-10−/−; 6 were C. jejuni infected (46% of infected IL-10−/− mice) and 2 were control mice (40% of control IL-10−/− mice). Thus, while the C3Bir IL-10−/− mice had a substantially greater frequency of histological changes in the spleen than the C3H IL-10+/+ mice in this experiment, the presence of C. jejuni in the GI tract did not markedly increase the incidence of histological changes in the spleen in either C3Bir IL-10−/− or C3H IL-10+/+ mice. Results of the Fisher-Freeman-Halton test indicated that there were significant differences among the groups (P = 0.0009); post-hoc analysis indicated that differences in spleen histopathology were significant for comparison of infected C3Bir IL-10−/− mice to infected C3H IL-10+/+ mice (P = 0.006) and to uninfected control C3H IL-10+/+ mice (P = 0.047), but not to uninfected control C3Bir IL-10−/− mice (P = 0.172). In Experiment 2, there was a significant association between the presence of histological changes at the ileocecocolic junction and histological changes in the spleen in all C3Bir IL-10−/− mice, regardless of whether GI tract pathology arose from spontaneous colitis or from C. jejuni infection (P = 0.044).
In Experiment 3 with NOD as the genetic background, a large proportion of mice (23/35; 66%), including both control and infected mice and mice of all three IL-10 genotypes, exhibited PALS; no mice exhibited EMH. Analysis of the entire data set could not be performed because of low expected frequencies in most cells. Comparison of uninfected and C. jejuni-infected NOD IL-10+/+ mice by Fisher’s exact test showed no significance difference between groups(P=0.626).
2.8. Campylobacter jejuni invades enteric tissues as assessed by immunohistochemistry
Sections of the ileocecocolic junction of all mice were immunohistochemically stained with anti-C. jejuni antibody and were evaluated and scored as described previously (25)(Table 4 and Figure 7). Approximately half of C. jejuni infected C57BL/6 IL-10−/− but no C57BL/6 IL-10+/+ mice showed C. jejuni specific IHC staining within the intestinal epithelium and erosive lesions of the epithelium. C. jejuni-specific staining was found both within and between cells within and below the lamina propria in C. jejuni infected C57BL/6 IL-10−/− mice. Staining patterns were similar in Experiments 1–3 for C3Bir/C3H and NOD mice except that in Experiment 3, 3/7 uninfected and 14/18 C. jejuni infected NOD IL-10+/+ mice had staining in the lamina propria, whereas little staining was observed in the lamina propria in NOD IL-10+/+ in Experiment 1. Both C. jejuni infected C3Bir IL-10−/− and C. jejuni infected C3H IL-10+/+ mice showed IHC staining within the intestinal epithelium and erosive lesions of the epithelium; infected C3H IL-10+/+ mice were less likely to have IHC staining in deeper tissues. Four of five control C3Bir IL-10−/− mice but 0/6 control C3H IL-10+/+ mice showed some IHC staining. In Experiment 3, infected but not uninfected IL-10−/− and IL-10+/− NOD mice showed IHC staining in the lamina propria as well as associated with the epithelium. Both control and C. jejuni infected NOD IL-10+/+ mice showed staining in some parts of the lamina propria, although C. jejuni infected NOD IL-10−/− in Experiment 1 did not. Campylobacter jejuni specific IHC staining was performed on spleens from all mice. Some of the mice with positive C. jejuni PCR tests had low numbers of cells within the spleen with intracellular staining and no mice with negative PCR had positive splenic IHC.
Table 4.
Percentage of mice showing IHC staining in particular locations in the wall of the GI tract
| epithelium | lamina propria | submucosa | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| Genotype | Status - % fat in diet (n) |
basolateral surface |
paracellular junctions |
effacing lesion |
between cells |
within all cell types | within PMNs | within MP/DC | within connective tissue |
within all cell types | within PMNs |
within MP/DC |
associated with vascular tissue |
associated with lymphoid tissue |
associated with histiocytes |
| Experiment 1 | |||||||||||||||
| C57BL/6 IL-10−/− | control - 11% (10) | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| infected - 11% (10) | 50 | 60 | 50 | 20 | 50 | 20 | 40 | 0 | 40 | 0 | 20 | 0 | 30 | 10 | |
| infected - 6% (10) | 50 | 50 | 60 | 40 | 50 | 30 | 40 | 10 | 40 | 10 | 30 | 10 | 10 | 50 | |
| C57BL/6 IL-10+/+ | infected - 11% (10) | 0 | 0 | 0 | 0 | 10 | 0 | 10 | 0 | 20 | 10 | 20 | 0 | 0 | 0 |
| C3Bir IL-10−/− | infected - 11% (10) | 60 | 60 | 60 | 30 | 60 | 20 | 30 | 0 | 60 | 10 | 20 | 20 | 20 | 20 |
| C3H IL-10+/+ | infected - 11% (10) | 20 | 20 | 30 | 0 | 50 | 0 | 20 | 0 | 40 | 0 | 30 | 0 | 0 | 0 |
| NOD IL-10−/− | infected - 6% (10) | 60 | 60 | 60 | 20 | 80 | 20 | 40 | 0 | 70 | 0 | 10 | 30 | 40 | 40 |
| NOD IL-10+/+ | infected - 6% (10) | 20 | 30 | 40 | 0 | 20 | 0 | 0 | 0 | 20 | 0 | 0 | 0 | 0 | 0 |
| Experiment 2 | |||||||||||||||
| C3Bir IL-10−/− | control - 11% (5) | 80 | 80 | 80 | 20 | 20 | 20 | 20 | 0 | 40 | 20 | 40 | 20 | 0 | 0 |
| infected - 11% (13) | 61 | 69 | 77 | 23 | 54 | 23 | 54 | 15 | 77 | 8 | 77 | 31 | 31 | 15 | |
| C3H IL-10+/+ | control - 11% (6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| infected - 11% (12) | 33 | 33 | 33 | 8 | 8 | 8 | 8 | 0 | 17 | 8 | 17 | 8 | 0 | 0 | |
| Experiment 3 | |||||||||||||||
| NOD IL-10−/− | control - 6% (2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| infected - 6% (3) | 67 | 67 | 67 | 67 | 67 | 0 | 67 | 0 | 33 | 0 | 33 | 33 | 33 | 0 | |
| NOD IL-10+/− | control - 6% (2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| infected - 6% (4) | 25 | 25 | 25 | 0 | 25 | 0 | 25 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| NOD IL-10+/+ | control - 6% (7) | 0 | 0 | 0 | 0 | 43 | 0 | 43 | 0 | 14 | 0 | 14 | 0 | 0 | 0 |
| infected - 6% (18) | 67 | 67 | 44 | 5 | 78 | 0 | 78 | 0 | 28 | 0 | 28 | 0 | 5 | 0 | |
Figure 7.

Representative sections of the ileocecocolic junction of C57BL/6 IL-10−/−, NOD IL-10−/−, and C3BirIL-10−/− mice given oral Campylobacter jejuni 11168 or sham inoculated with tryptone soy broth and sacrificed 28 days after infection. Sections are stained with an anti-Campylobacter jejuni specific IHC stain. Sections shown in A, B, C, J, K, and L were taken at 20X magnification. Sections shown in D, E, F, G, H, and I were taken at 40X magnification. Sections from an infected A) C57BL/6 IL-10−/− mouse, B) NOD IL-10−/− mouse, and C) C3BirIL-10−/− mouse all show extensive inflammatory response in all segments, compared to their respective uninfected controls in J) C57BL/6 IL-10−/− mouse, K) NOD IL-10−/− mouse, and L) C3BirIL-10−/− mouse. Note crypt hyperplasia, extensive mononuclear cell infiltrates, and neutrophilic exudates (appear at the top of all six upper panels. Black double headed arrows show extent of mucosal thickening in infected versus uninfected control mice. Crypt abscesses occurred in mice of all three genetic backgrounds in response to C. jejuni as shown in inserts from a G) C57BL/6 IL-10−/− mouse H) NOD IL-10−/− mouse, and I) C3BirIL-10−/− mouse. In panels D, E, and F small black arrows show mucosal villus tip lesions. In these cases the villus tip is sloughing and C. jejuni organisms can be found within or amongst cells.
2.9. Infected mice produced Campylobacter jejuni- specific IgG2b immunoglobulin responses
Levels of anti-C. jejuni antibody in mouse plasma are shown in Figures 8 and 9. As observed previously in C57BL/6 IL-10−/− and C57BL/6 IL-10+/+ mice, antibody responses in C. jejuni infected mice were mainly comprised of the IgG2b subtype in all three genetic backgrounds for both IL-10−/− and IL-10+/+ mice, indicating a Th1 response to C. jejuni. Nonparametric ANOVA was conducted on IgG2b data from Experiment 1, in which the three mouse genetic backgrounds were directly compared. The results indicated that there were statistically significant differences among the groups of mice (P ≤ 0.001). Post hoc analysis showed that there were significant differences between both groups of control mice (IL-10−/− and IL-10+/+) and C. jejuni infected C57BL/6 IL-10−/− mice on the 11% fat diet, C. jejuni infected NOD IL-10−/− mice, and C. jejuni infected NOD IL-10+/+ mice (P < 0.05).
Figure 8. Antibody subclasses (OD450/μg plasma protein).

Antibody response as measured by ELISA in C57BL/6 IL-10−/−, C57BL/6 IL-10+/+, NOD IL-10−/−, NOD IL-10+/+, C3BirIL-10−/−, and C3H/HeJ IL-10+/+ mice infected with C. jejuni 11168 or sham inoculated with tryptone soy broth and sacrificed 30 days after infection. Each column indicates the mean absorbance value/μg plasma protein; error bars are ± standard error. Panels A, B and D show results for Experiment 1 for total IgG1, IgG2c, IgG2b, IgG3, and IgA. Results for C57BL/6 IL-10+/+ and C57BL/6 IL-10−/− mice appear in panel A, those for C3H/HeJ IL-10+/+ and C3BirIL-10−/− mice appear in panel B, and those for NOD IL-10+/+ and NOD IL-10−/− mice appear in panel D. Panel C shows result for C3BirIL-10−/−, and C3H/HeJ IL-10+/+ mice and panel D shows results for NOD IL-10−/− and NOD IL-10+/+ mice for total IgG1, IgG2c, IgG2b, IgG3, and IgA in Experiments 2 and 3. Black bars represent values from infected mice and white bars values from uninfected mice.
Figure 9. Time course of total immunoglobulin (IgM, IgA, IgG) production during C.jejuni infection.

Total antibody response as measured by ELISA in C3BirIL-10−/− and C3H/HeJ IL-10+/+ mice infected with C. jejuni 11168 or sham inoculated with tryptone soy broth and sacrificed at 14 or 21 days after infection. Each column indicates the mean absorbance value/μg plasma protein; error bars are ± standard error. Secondary antibody detects total IgM, IgG, and IgA. Gray bars represent values from infected mice and white bars values from uninfected mice. C3BirIL-10−/− mice that also lack functional TLR4 develop a robust antibody response to C. jejuni that does not prevent gastrointestinal tract pathology or systemic spread of the bacteria.
In Experiment 2 (C3 genetic background), Kruskal Wallis ANOVA indicated significant differences among the groups of mice in plasma levels of IgG2b (P = 0.003). Post hoc analysis indicated significant differences between C. jejuni infected and uninfected C3Bir IL-10−/− mice (P < 0.05). Figure 9 shows the development of total IgG + IgM + IgA anti-C. jejuni circulating antibodies during the course of C. jejuni infection of C3Bir IL-10−/− and C3H IL-10+/+ mice in Experiment 2. The anti-C. jejuni antibody response appeared to be delayed in tlr4-deficient C3H IL-10+/+ mice; however, C3Bir IL-10−/− mice, which are deficient in both IL-10 and tlr4, exhibited a prompt, robust antibody response. In Experiment 3 (NOD genetic background), significant differences were detected between the groups of mice in Kruskal Wallis ANOVA (P = 0.001); post hoc analysis indicated that infected NOD IL-10 +/− and IL-10+/+ mice were significantly different from control NOD IL-10+/+ mice (P < 0.05). The single C57 IL-10−/− control mouse that developed spontaneous colitis in Experiment 1 had moderate plasma levels of IgG2c but only background levels of IgG2b reactive with C. jejuni antigen in enzyme linked immunosorbent assay (ELISA). One of the two C3Bir IL-10−/− control mice in Experiment 1 had moderate plasma levels of IgG2a reactive with C. jejuni antigen in ELISA; the other C3Bir IL-10−/− mouse had only background levels of IgG2a reactive with C. jejuni antigen in ELISA. Neither of these mice had more than background levels of IgG2b reactive with C. jejuni antigen and both had severe histological changes. However, none of the four control mice that developed histological lesions in Experiment 2 (C3 genetic background) had more than background plasma levels of IgG1, IgG2b, IgG2c, IgG3, or IgA reactive with C. jejuni antigen in ELISA. Histological changes in mice with spontaneous colitis were not as severe as in C57BL/6 IL-10−/− or the two C3Bir IL10−/− mice in Experiment 1.
2.10. Bacteroides spp. and E. coli DNA were detected in spleen and liver of C3Bir IL-10−/− and C3H/HeJ IL-10+/+ mice
Q-PCR for Bacteroides spp. and E. coli in C. jejuni infected and tryptone soy broth (TSB) sham inoculated mice of the C3 genetic background showed the presence of these organisms in extraintestinal sites in all mice, both IL-10+/+ and IL-10−/−. These data indicate that common members of the colon microbiota are present in spleen and liver and along with the C. jejuni gyrA PCR results show that C. jejuni is just one of the bacteria that escapes the gut and are found in spleen and liver.
3. Discussion
The major goal of these studies was to develop mouse models of human C. jejuni infections that mimic the wide range of clinical syndromes observed in patients. Expected host responses to this pathogen include a spectrum from resistance, to colonization without disease, to enteritis, to dissemination from the GI tract to other organ systems, and rarely systemic collapse and death. The rationale for these studies was that many of the available inbred mouse strains with the IL-10−/− targeted mutation have background genes, including other known immune system defects, which influence susceptibility to pathogens and disease manifestations. We hypothesized that a particular genetic background enhances the rate of colonization, increases the severity of enteritis lesions observed, and allows for extraintestinal spread when inbred IL-10−/− mice are infected with a pathogenic strain of C. jejuni. These results demonstrate the complex nature of interactions among modifier genes in these mouse models, especially those known defects in TLR4, Cdcs1 and IL-10, which did produce a range of C. jejuni disease phenotypes from colonization alone (C57BL/6 IL-10+/+, NOD IL-10+/+), to colonization with enteritis and no extraintestinal spread (C57BL/6 IL-10−/−, NOD IL-10−/−), to colonization with limited extraintestinal spread without enteritis (C3H/HeJ IL-10+/+) and to enteritis with extensive extraintestinal spread (C3Bir IL-10−/−).
All mice regardless of genotype became stably colonized with C. jejuni 11168 after experimental inoculation. Patterns of C. jejuni colonization of compartments of the GI tracts in infected IL-10−/− and IL-10+/+ mice of all three genetic backgrounds were similar to each other and to patterns observed previously (25). Both IL-10−/− and IL-10+/+ mice were colonized, most heavily in the cecum. However, C. jejuni DNA could be detected in at least one extraintestinal location—blood, liver, or spleen—in 12/13 (92%) C3Bir IL-10−/− mice. Although C. jejuni was found occasionally in either blood or spleen of NOD mice (2/38, 5%) and C57BL/6 mice (1/30, 3 %) these low numbers show that bacterial escape from the gut has a low frequency in mice of these genetic backgrounds. These observations suggest that mice of the C57BL/6 and NOD genotypes, both IL-10+/+ and IL10−/−, mainly control C. jejuni infection at the level of the gut mucosa and local lymph nodes, but that C3Bir IL-10−/− and C3H/HeJ IL-10+/+ mice do not. While both C3Bir IL-10−/− and C3H IL-10+/+ mice have demonstrable C. jejuni in blood, spleen and liver, the number of mice with extraintestinal spread is ~4 times higher in those lacking IL-10. Spleen/body weight ratio analysis showed a significant increase in spleen size based on genotype–mice of the C3 genotype had the biggest spleens—, but there was a trend to enlargement in C3Bir IL-10−/− mice infected with C. jejuni. Thus, in hosts with TLR4 and Cdcs1 deficiencies extraintestinal spread was made much worse by lack of IL-10. We expect that C3Bir IL-10−/− mice are candidates for studies of spread of C. jejuni beyond the GI tract, for example, in screening C. jejuni strains carrying targeted mutations in putative virulence determinants. The fraction of mice having detectable C. jejuni in spleen (69%) in Experiment 2 was large enough to allow detection of a decrease in that fraction due to a targeted gene disruption in C. jejuni.
C. jejuni infected IL-10−/− mice of all three genetic backgrounds developed clinical signs and GI lesions both gross and histologic that mimic those seen in human campylobacteriosis. C. jejuni infected C3Bir IL-10−/−, C3H IL-10+/+, NOD IL-10−/−, and NOD IL-10+/+ mice had clinical signs consistent with previous observations in C57BL/6 IL-10−/− and IL-10+/+ mice (25). In infected C3Bir IL-10−/− and NOD IL-10−/− the most severe lesions were observed at the ileocecocolic junction similar in both incidence and pattern to those previously reported in C57BL/6 IL-10−/− mice (25). The only IL-10+/+ mice that developed gross and histologic pathological changes in response to C. jejuni were two NOD IL-10+/+ mice in Experiment 1. All pathological lesions previously observed in the GI tract of C. jejuni infected C57BL/6 IL-10−/− mice were seen in C3Bir IL-10−/− and NOD IL-10−/− mice, but the presence of enlarged spleens in the latter two strains suggested that the additional immune system deficiencies in these mice allowed C. jejuni to escape the GI tract, and, as mentioned above, raised the possibility that these mice could be used as models to explore extraintestinal infection by C. jejuni. However, NOD IL-10−/− mice were more likely to have spleens with marked extramedullary hematopoiesis and expanded periarteriolar lymphatic sheaths with few C. jejuni than in the C3Bir IL-10−/− genotype where most mice had C. jejuni in spleen or liver with and without splenic enlargement. Most C3Bir IL-10−/− and C3H/HeJ IL-10+/+ mice either control or C. jejuni infected with and without spontaneous colitis also had significant amounts of E. coli and Bacteroides in spleen and liver as measured by Q-PCR indicating that this genetic background experiences decreased gut barrier function. C. jejuni infected IL-10−/− mice of all genetic backgrounds exhibited elevated IHC staining for this organism within and below the epithelium, in the lamina propria, and in the submucosa compared to IL-10+/+ mice of the corresponding genetic background. Thus, IL-10 deficiency appears to permit increased invasion of GI tissues by C. jejuni, but is apparently not necessary for the presence of this organism in extraintestinal sites.
Histological lesions at the ileocecocolic junction of infected C57BL/6 IL-10−/− and NOD IL-10−/− were readily attributed to the presence of C. jejuni because uninfected controls had no colitis. However, the situation was more complicated in the C3Bir IL-10−/− mice where both the C. jejuni infected and uninfected mice had high rates of colitis. In general, rates of spontaneous colitis in C57BL/6 and NOD backgrounds were very low in these experiments. However, necropsy results on retired breeding mice and breeding colony mice that had to be euthanized due to the development of spontaneous colitis revealed that almost 60% of C3Bir IL-10−/− mice developed spontaneous colitis and that this colitis was almost always accompanied by splenic enlargement. Control C3Bir IL-10−/− mice in the two experiments reported here also exhibited high rates of spontaneous colitis (100% in the Experiment 1 and 80% in the Experiment 2) and splenic enlargement (50% in Experiment 1 and 60% in Experiment 2). These findings are in strong contrast to both C57BL/6 IL-10−/− and NOD IL-10−/− mice and indicate that the use of C3Bir IL-10−/− mice for some kinds of investigations focusing on enteric pathogens within the GI tract would be problematic, since it would be difficult to distinguish statistically between uninfected and infected animals.
Substantial efforts failed to detect known mouse-associated Helicobacter or “Flexispira” species in these mice. Although we cannot absolutely rule out the possibility that the C3Bir IL-10−/− mice exhibiting spontaneous colitis carried low levels of a previously undescribed Helicobacter or “Flexispira” species, it is unlikely that such an organism was present in these mice and caused the degree of histological change observed. Immunohistochemical staining did not reveal any spiral bacteria in the GI tracts of uninfected mice with spontaneous colitis. Also, all of the PCR tests conducted on C3Bir IL-10−/− control uninfected mouse tissues for known colitogenic bacteria were negative. However, all control mice that had spontaneous colitis also exhibited IHC staining of sections of the ileocecocolic junction with a polyclonal anti-C. jejuni antibody. This staining was associated with various bacteria in the lumen, chiefly large and small rods and cocci, but not with the presence of C. jejuni because culture and PCR tests were negative. Furthermore, all control mice were serologically negative for C. jejuni specific antibodies further confirming that cross contamination did not occur. These rod-shaped and coccal bacteria would therefore appear to possess epitopes that cross-react with C. jejuni and may account for the positive IHC results described here. An alternative explanation is that damage to the gut barrier causes a general stickiness of the tissue sections that increased non-specific binding of the antibodies used in IHC staining.
Enlargement of the spleen with presence of C. jejuni revealed an effect of genetic background on extraintestinal spread of the challenge organism. C. jejuni was detected by culture or PCR in blood and spleens of infected C3Bir IL-10−/−, NOD IL-10−/−, and NOD IL-10+/+ mice, but not mice of the C57BL/6 background, showing effects of genetic background on bacterial escape from the GI tract. In particular, on the C3H/HeJ IL-10+/+ background, deficiency in TLR4 alone was not sufficient for enteritis, but in C3Bir IL-10−/− mice the IL-10 and TLR4 deficiencies and the Cdcs1 allele present in this background enhanced extraintestinal spread of both C. jejuni and members of the normal enteric microbial community. C. jejuni and other bacteria were present in the spleens of these mice suggesting a role for these gene products in limiting disease to the local GI environment. Our data supports the conclusions of Beckwith, Leiter and colleagues that documented a role for Cdcs1 gene product(s) in development of spontaneous colitis (27, 29). These investigators constructed reciprocal Cdcs1 congenic stocks on both C3Bir and C57BL/6 IL-10−/− mouse backgrounds to identify the susceptibility gene and its function (27). In these studies, spontaneous colitis in C3Bir IL-10−/− mice was significantly diminished by the C57BL/6J IL-10−/− 7-Mb genome in this allele and, conversely, C57BL/6J IL-10−/− mice with the 7-Mb genome allele from C3Bir IL-10−/− mice had much more severe disease. Functionally, C3Bir IL-10−/− mice had macrophages that constitutively expressed higher levels of NF-κB p50, leading to reduced innate responsiveness to bacterial ligands and increased CD4 T-cell responses, a compensatory adaptive response. In previous work, C57BL/129 inbred mice deficient in NF-κB subunits (p50−/− p65+/−) developed spontaneous and H. hepaticus- and C. jejuni-induced colitis (39). In those studies, Fox and colleagues showed that persistent colonization of NF-κB -deficient mice with C. jejuni was associated with impaired IgG and IgG2a humoral responses consistent with an innate or adaptive immune system defect(s); they suggested that clearance of C. jejuni is NF-κB dependent. Of the three mouse strains used in our study, the C3H/HeJ and C3Bir IL-10−/− mice have the Cdcs1 allele described by Beckwith and colleagues while the C57BL/6 and C57BL/6 IL-10−/− mice do not; NOD mice remain uncharacterized at this locus. Taken together with our data it is likely that C3Bir IL-10−/− and IL-10+/+ mice with TLR4 deficiencies and the Cdcs1 allele have decreased colonic barrier function that enables pathogens and members of the microbiome to gain access to parenteral sites such as spleen. This leads to enhanced inflammation that goes unchecked in the absence of IL-10. While Beckwith, Leiter and colleagues concluded that the colitogenic Cdcs1 allele containing NF-κB impairs innate immunity to bacterial products and in turn skews the adaptive immune response toward compensatory hyperresponsiveness and chronic intestinal inflammation, we suggest that Cdcs1 has greater effects toward enhancing sensitivity to the microbiota, whereas IL-10 has greater impact on limiting excessive intestinal inflammation.
As observed in C57BL/6 mice (25), infected IL-10−/− and IL-10+/+ mice of all three genetic backgrounds exhibited C. jejuni specific plasma IgG2b antibody responses. This observation is consistent with a Th1 directed immune response and the largely mononuclear cell infiltrates that we observed in histological lesions at the ileocecocolic junction. The additional immune deficiencies of C3Bir/C3H and NOD mice apparently did not affect this aspect of the immune response. Total antibody levels made up largely of IgG2b and IgG2a were somewhat higher in C3Bir IL-10−/− than C3H/HeJ IL-10+/+ mice. It is known that C3H/HeJ IL-10+/+ mice have delayed antibody responses (The Jackson Laboratory web site), but IL-10 deficiency restores a normally-timed response. Generally, IL-10 deficient mice produced more IgG2b antibody than the IL-10 competent C3H/HeJ.
The results of Experiment 1 also raised the possibility that NOD IL-10+/+ mice could be used to study the full effects of C. jejuni infection in IL-10 sufficient mice and thus used in conjunction with NOD IL-10−/− mice to delineate the role of IL-10 in the host response to C. jejuni. Unfortunately, however, the initial observation of marked histological changes in C. jejuni infected NOD IL-10+/+ mice could not be repeated with a larger sample size in Experiment 3. The polygenic defects in NOD mice that lead to autoimmune (type 1) insulin dependent diabetes mellitus (IDDM)(40) had less of an effect on generation of GI pathology after C. jejuni challenge than lack of IL-10; NOD mice given C. jejuni had only mild pathological lesions while those with IL-10 deficiency had severe GI pathology. At The Jackson Laboratories 85% of female and 37% of male specific pathogen free NOD/LtJ IL-10+/+ and IL-10−/− mice housed in barrier facilities develop IDDM by 30 weeks of age and IL-10 deficiency renders them susceptible to mild spontaneous colitis that is not as severe as other IL-10 deficient mice (41). Susceptibility to IDDM in NOD mice is polygenic (42), but the H-2g7 MHC haplotype is the major component controlling susceptibility (43). There are at least four more MHC-linked type 1 diabetogenic genes in the 33.9 Mbp region centromeric to the Lmp2 gene (Idd1a, Idd1b, Idd1c and Idd1d) (44). Proteasome dysfunction occurs due to a tissue- and developmental-stage-specific defect in expression of the MHC-linked Lmp2 gene, resulting in altered activity of the transcription factor NF-κB (45). T cell activation and CTLA-4 up-regulation are also impaired due to low CD86 expression (46). The ligation of CTLA-4 on activated T cells by B7 molecules expressed by antigen presenting cells delivers a negative signal that results in inhibition of IL-2 production and T cell proliferation (47). The presence of I-Ag7 leads to the development of an autoimmune T-cell repertoire, and local costimulation of CD8 T-cells precipitates aggressive diabetes. Our NOD IL-10−/− mice did not express colitis unless they were infected with C. jejuni showing that these immune defects leading to IDDM had little impact on spontaneous enteric disease or in exacerbating systemic spread of this bacterium.
4. Materials and methods
4.1. Specific-pathogen-free mice and experimental infections
Full details of the standard operating procedure developed for study of C. jejuni infection in C57BL/6 IL-10+/+ and IL-10−/− mice has been published (25)(Murine Enteric Diseases Phenome Database (MEDPeD) http://foodsafe.msu.edu/mru_web/MurineEntericDiseasesPhenomeDatabase.htm. All procedures involving animals were approved by the All University Committee on Animal Use and Care of Michigan State University (protocol 02/04-029-00) following National Institutes of Health standards for animal use. All mouse strains were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Thereafter, mice were housed and bred in a closed specific-pathogen-free colony at MSU to produce age-matched offspring for experiments. C57BL/6 IL-10+/+, C57BL/6 IL-10−/−, C3Bir IL-10−/−, and C3H/HeJ IL-10+/+ mice were fed irradiated mouse chow containing 11–12% fat (Mouse Breeder Diet 7904, Harlan Teklad, Indianapolis, IN, USA). NOD IL-10−/− and NOD IL-10+/+ mice were fed irradiated mouse chow containing 6–7% fat (NIH-31 Modified Open Formula Mouse/Rat Diet 7913, Harlan Teklad, Indianapolis, IN, USA).
Mouse husbandry, monitoring, tracking, handling, and genotyping procedures were as described (25). Mice were monitored for C. jejuni, Helicobacter hepaticus, Citrobacter rodentium, and Enterococcus faecalis by PCR assay of DNA extracted from total fecal bacterial populations as described (25). PCR assays of fecal DNA for Helicobacter spp. (48) and Enterococcus spp. (49) were also performed. Four other species-specific PCR assays for murine Helicobacter spp. (H. muridarum, and H. typhlonius (33), H. ganmani (34), and H. mastomyrinus (35)) were performed on any suspect samples. Retired breeding mice and breeding colony mice with any clinical signs were euthanized, necropsied, and examined for evidence of spontaneous colitis or other abnormalities and this data is presented as a basis to interpret the induced C. jejuni infection model. NOD mice were tested for diabetes using Keto-Diastix reagent strips (Bayer, Tarrytown, NY, USA) at necropsy or when excessive drinking or urination was noted. Diabetic mice were euthanized.
Before experimental infections, mice were moved to the University Research Containment Facility in filter top cages inside of sterile dog crates. Mice were randomized to group and housed individually in filter top cages that were randomized for placement on the cage rack. Eight to 12 week old mice were inoculated with approximately 1010 cfu C. jejuni 11168 suspended in 0.2 mL of tryptone soy broth by oral gavage using a 3.5 French red rubber pediatric feeding tube (Kendall Sovereign; Tyco Healthcare Group, Mansfield, MA, USA) and a one mL syringe (Becton Dickinson, Franklin Lakes, NJ, USA). Control mice were inoculated with 0.2 mL TSB. Mice were observed twice daily for adverse clinical signs and euthanized and necropsied promptly if they became ill.
In the C3Bir/C3H genetic background experiment, blood samples were obtained prior to inoculation and on days 14 and 21 by nicking the lateral saphenous vein with a sterile 25 gauge needle and collecting approximately 50–100 μl of blood in a heparinized capillary tube or Microvette tube (Sarstedt, Nümbrecht, Germany) and handled as below.
4.2. Experimental designs
Three experiments were conducted. In Experiment 1, mice of all genetic backgrounds were used with mice of the C57BL/6 background serving as controls. Ten C57BL/6 IL-10−/− mice kept on the 11% fat diet, 10 C57BL/6 IL-10−/− mice adapted to the 6% fat diet prior to the experiment, and 10 C57BL/6 IL-10+/+ mice kept on the 11% fat diet were inoculated with C. jejuni 11168. Ten C57BL/6 IL-10−/− mice kept on the 11% fat diet, two C57BL/6 IL-10−/− mice adapted to the 6% fat diet, and two C57BL/6 IL-10+/+ mice kept on the 11% fat diet were inoculated with TSB to serve as vehicle controls. These two diets were employed because (1) disease in all other mouse strains and genotypes was to be compared to that in C57BL/6 IL-10−/− mice, and (2) mice of the C3 genetic background were kept on the 11% fat diet while mice of the NOD genetic background were kept on the 6% fat diet to decrease the incidence of diabetes. Ten mice of each of the following genotypes were inoculated with C. jejuni: C3H/HeJ IL-10+/+, C3Bir IL-10−/−, NOD IL-10+/+, and NOD IL-10−/−; two mice of each genotype except NOD IL-10+/+ (one mouse) were inoculated with TSB. Results from Experiment 1 prompted two additional experiments, one to repeat C. jejuni 11168 infection of C3H/HeJ IL-10+/+ and C3Bir IL-10−/− mice (Experiment 2) and another to repeat C. jejuni 11168 infection of NOD IL-10+/+ and NOD IL-10−/− mice (Experiment 3). In Experiment 2, thirteen C3Bir IL-10−/− and twelve C3H IL-10+/+ mice were given C. jejuni, five C3Bir IL-10−/− mice and six C3H IL-10+/+ mice were given TSB. In Experiment 3, 3 NOD IL-10−/−, 18 NOD IL-10+/+, and 4 IL-10 +/− mice were given C. jejuni; 2 NOD IL-10−/−, 7 IL-10+/+, and 2 IL-10 +/− mice were given TSB.
4.3. Bacterial strains, media, and growth conditions; inoculum preparation
Campylobacter jejuni 11168 inocula for experiments were prepared and quantified as described (25). The inoculum for Experiment 1 had 8 × 109 cfu/mouse, the inoculum for Experiment 2 (C3Bir and C3H/HeJ mice) had 1.0 × 1010 cfu/mouse, and the inoculum for Experiment 3 (NOD IL-10−/− and NOD IL-10+/+ mice) had 3.8 × 1010 cfu/mouse.
4.4 Necropsy procedures
All mice were humanely euthanized by CO2 overdose; mice that exhibited adverse clinical signs were euthanized promptly to prevent suffering. Mice not requiring early euthanasia were euthanized and necropsied 35–38 days after inoculation in Experiment 1, 28–29 days after inoculation in Experiment 2, and 36–37 days after inoculation in Experiment 3. All post-mortem procedures were as previously described (25). Body weights and spleen weights were taken to calculate spleen/body weight ratios.
4.5 Campylobacter jejuni culture, DNA extraction, and PCR
To detect C. jejuni by culture, a small portion of tissue from stomach, jejunum, cecum, and colon was used to inoculate a separate plate of tryptose soya sheeps’ blood (TSSB) agar containing 20 μg cefoperazone, 10 μg vancomycin, and 2 μg amphotericin B per mL (TSSBA-CVA)(50))(antibiotics from Sigma-Aldrich, St. Louis, MO, USA). Growth was scored as a rank ranging from 0 to 4 as previously described (25). The identity of isolates was verified by C. jejuni gyrA-specific PCR as previously described (51). DNA was extracted from total fecal bacterial populations and tissue samples using Qiagen DNeasy tissue kits (Qiagen, Valencia, CA, USA) as previously described (25) and subjected to C. jejuni gyrA-specific PCR. Blood samples were separated into plasma and packed cell fractions by centrifugation and frozen at minus 80 C. DNA was later extracted from cellular fractions and subjected to C. jejuni specific PCR assay; plasma was used in ELISA to quantify anti-C. jejuni antibodies.
4.6. Histopathology and immunohistochemistry
The body of the cecum with attached distal ileum and proximal colon gently purged of their contents were mounted in a histological cassette along with one-half of the spleen. The cassette was placed in phosphate buffered 10% formalin solution, which was replaced with 60% ethanol after 24 hours. Paraffin embedding of the fixed tissues, sectioning, hematoxylin and eosin staining, and IHC staining with a rabbit polyclonal antibody against C. jejuni (US Biologicals, Swampscott, MA, USA) were performed by the Histology Laboratory, Department of Physiology, Michigan State University as previously described (25). Histological lesions and IHC staining at the ileocecocolic junction were randomized and scored by a single blinded investigator (LSM) using the standardized systems described previously (25)(http://foodsafe.msu.edu/mru_web/MurineEntericDiseasesPhenomeDatabase.htm). Histological lesions in spleens were scored by a single blinded investigator (JSP) using the standardized system described below.
4.7. Establishing criteria for assessing and scoring spleen pathology
Spleen sections from 5 control and 5 C. jejuni infected mice of each genotype were examined by a board certified veterinary pathologist (JSP) to determine the range of cell types and changes present. Two criteria were established based on pathological anatomical descriptions of these sections. Thereafter, all experimental spleen sections were blinded to identity by a separate operator (JAB), randomized and given to this pathologist to score based on the established criteria. The criteria are as follows:
Criterion 1: marked extramedullary hematopoiesis (EMH)
Rodent spleens typically show histological evidence of EMH characterized by dense or loose aggregates of erythroid, granulocytic, lymphoid, and monocytic precursor cells, as well as megakaryocytes. Densely aggregated cells commonly have densely basophilic (hyperchromatic) to moderately basophilic, medium-sized to large, round to oval nuclei and scant pale basophilic cytoplasm; these are interpreted to be primarily lymphoid, monocytic, and immature erythroid precursor cells (indistinguishable in routine hematoxylin-and-eosin-stained sections). The more loosely scattered nucleated cells are typically in the red pulp, and contain smaller, densely basophilic nuclei and a rim of red cytoplasm; they are interpreted to be erythroid precursors, further along in their maturation than erythroid precursors in the denser cell aggregates. Granulocytic precursor cells are identified by their large irregularly shaped, often donut-shaped, relatively pale basophilic (hypochromatic) nuclei and moderate quantity of pale basophilic cytoplasm. Aggregates of these cells were small and inconspicuous in spleens scored as “negative” or “−” for marked EMH, but were large, irregularly shaped, and identifiable even at 10X magnification in spleens scored as “positive” or “+”. The presence of numerous conspicuous aggregates of granulocytic precursor cells was the main feature of spleens scored as “+” for “marked EMH.” In addition, megakaryocyte numbers appeared increased in these spleens.
Criterion 2: prominent periarteriolar lymphatic sheaths (PALS)
Rodent spleens are typically quite cellular, with fairly prominent PALS. PALS are usually round, with visible germinal centers and marginal zones of uniform thickness. With respect to periarteriolar lymphatic sheaths, spleens that were scored negative, or “normal,” generally contained prominent, densely cellular, round to oval or elongate, “individual” PALS. Spleens scored as positive contained PALS that were generally greater in diameter than normal and irregularly shaped, with prominent marginal zones. Lymphoid cells in marginal zones were large, with hypochromatic nuclei (lymphoblasts). Adjacent PALS frequently coalesced. These changes were interpreted as lymphoid hyperplasia.
4.8. Enzyme-linked immunosorbent assay (ELISA) to detect anti-C. jejuni antibodies in sera
A whole-cell C. jejuni antigen preparation was produced as described (25, 39) and used to coat 96-well Nunc-Immuno MaxiSorp plates (Nalgen Nunc Int., Rochester, NY, USA). Secondary antibodies used included biotinylated goat F(ab′)2 anti-mouse IgG (H+L) (Southern Biotech, Birmingham, AL, USA; detects IgA, IgM, and total IgG), biotinylated goat anti-mouse IgG1, IgG2b, IgG2c and IgG3 (Jackson ImmunoResearch, West Grove, PA, USA), and biotinylated goat anti-mouse IgA and IgM (Sigma Aldrich, St. Louis, MO, USA). Detection employed extravidin peroxidase (Sigma Aldrich, St. Louis, MO, USA) and 3, 3′, 5, 5′-tetramethylbenzidine (TMB; Sigma Aldrich, St. Louis, MO, USA) substrate. Plasma protein concentrations were determined by Bradford assay as described (25).
4.9. Quantitative Real-Time PCR method and analyses
DNA extracted from spleen and liver was used as the template in species-specific Q-PCR assays. The Bacteroides assay was performed using primer sequences from Rinttila et al. (52). Escherichia coli Q-PCR assays were performed using primer sequences from Khan et al. (53). In all assays, 25 μl reactions were performed in triplicate for each sample with SYBR® Green PCR mastermix (Applied Biosystems, Foster City, CA), 12.5 pm of bacterial specific primers, and 50 ng of sample DNA. All Q-PCR assays included a 7-point standard curve in triplicate (R2 > 0.90) and three non-template controls containing all other reaction components on a Bio-Rad iQ5 Real-Time PCR Detection System (Bio-Rad, Hercules, USA). To further confirm the quality of PCR amplification the products were confirmed on a 1.2% agarose gel in TAE, stained using ethidium bromide, visualized using UV illumination and photographed (Alpha Innotech Gel Documentation System, Alpha Innotech Corporation, San Leandro, CA). Bio-Rad iQ5 PCR detection system software (Bio-Rad, Hercules, USA) was used to calculate the Ct value for each reaction and the mean Ct value for each set of triplicates. Using the corresponding standard curve, the starting quantity (SQ) of DNA was calculated for each reaction and the mean SQ for each sample’s set of triplicates. The mice were grouped according to genotype (IL-10+/+ or IL-10−/−) and treatment (C. jejuni infected or TSB sham inoculated) and the mean SQs of DNA for each mouse within a specific treatment calculated.
4.10. Statistical analyses
Statistical analyses were performed using SigmaStat version 3.1 (Systat Software Inc., Point Richmond, CA, USA). The nonparametric Kruskal Wallis ANOVA on ranks was used to test for differences among the groups of infected and uninfected mice in histological score at the ileocecocolic junction. If results of the ANOVA were significant, pairwise comparisons were performed by Dunn’s method with correction for multiple comparisons that was used because of unequal group sizes. For analysis of Experiment 1, uninfected IL-10−/− mice of all genetic backgrounds were grouped together as were uninfected IL-10+/+ mice. Spleen histopathology scores were analyzed using the Fisher-Freeman-Halton test for R X C tables for Experiment 2 (http://faculty.vassar.edu/lowry/VassarStats.html). For Experiment 1, all uninfected mice were combined in a single control group. For all groups, mice were divided into two groups: those having normal spleens versus those having marked extramedullary hematopoiesis, expansion of periarteriolar sheaths, or both. Post-hoc analyses were performed using Fisher’s exact test; probability levels were corrected for multiple comparisons using the Holm-Šidák procedure (54).
ELISA data was transformed with the relation X′ = log10(X+1) due to the presence of zero values, skewness, and kurtosis. Even after transformation, the data for all three experiments examined individually failed either the normality and or the equal variance test and was therefore subjected to nonparametric Kruskal Wallis ANOVA on ranks; if the results of the ANOVA were significant, pairwise comparisons were performed by Dunn’s method with correction for multiple comparisons. For analysis of Experiment 1, uninfected IL-10−/− mice of all genetic backgrounds were grouped together as were uninfected IL-10+/+ mice.
Figure 5.
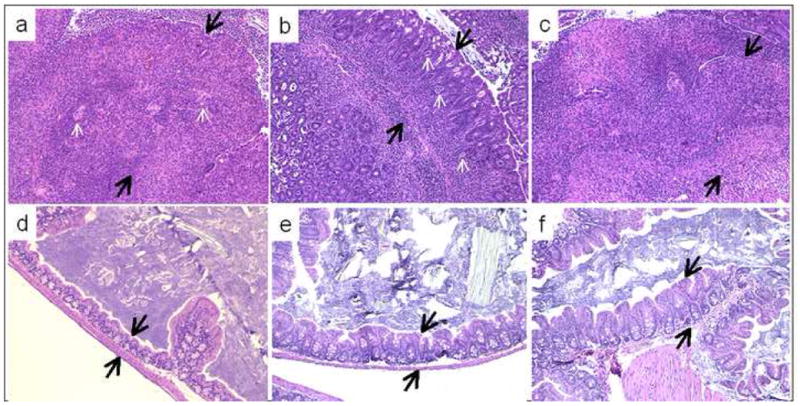
Representative sections of the ileocecocolic junction of C57BL/6 IL-10−/−, NOD IL-10−/−, and C3BirIL-10−/− mice given oral Campylobacter jejuni 11168 or sham inoculated with tryptone soy broth and sacrificed 30 days after infection. Sections are stained either with hematoxylin and eosin. A) Low power view (10X magnification) of ileocecocolic junction of C57BL/6 IL-10−/− mouse given oral Campylobacter jejuni compared to D) a similar section from an uninfected C57BL/6 IL-10−/− control mouse. B) Low power view (10X magnification) of ileocecocolic junction of NOD IL-10−/− mouse given oral Campylobacter jejuni compared to E) a similar section from an uninfected NOD IL-10−/− control mouse. C) Low power view (10X magnification) of ileocecocolic junction of C3BirIL-10−/− mouse given oral Campylobacter jejuni compared to D) a similar section from an uninfected C3BirIL-10−/− control mouse. Note extensive inflammatory response in all segments of sections A, B, and C. Note crypt hyperplasia, extensive mononuclear cell infiltrates, crypt abscesses (small white arrows) and neutrophilic exudates. Black arrows indicate the tip and base of the mucosa and show the extent of mucosal thickening of sections from infected mice compared to their controls.
Table 5.
Bacteroides spp. and Escherichia coli Q-PCR results in spleen, and liver of Campylobacter jejuni infected and trypticase soya broth sham inoculated mice of the C3 genetic background (# samples positive/# samples tested with percentages in parentheses) in Experiment 2.
| Bacteroides spp. | Escherichia coli | |||
|---|---|---|---|---|
|
| ||||
| Mouse Group | Spleen | Liver | Spleen | Liver |
| C3Bir IL-10−/− control | 5/5 (100%) | 5/5 (100%) | 2/15 (16%) | 4/5 (80%) |
| C3Bir IL-10−/− infected | 9/13 (69%) | 9/13 (69%) | 5/13 (38%) | 2/13 (15%) |
| C3H/HeJ IL-10+/+ control | 2/6 (33%) | 4/6 (66%) | 5/6 (83%) | 6/6 (100%) |
| C3H/HeJ IL-10+/+ infected | 9/12 (75%) | 10/12 (83%) | 2/12 (16%) | 7/12 (58%) |
Acknowledgments
These studies were funded in whole with federal funds from NIAID, NIH, Department of Health and Human Services, under Contract No. N01-AI-30058. We thank members of the MSU Investigative Histology Laboratory Kathy Campbell, Rick Rosebury, and Amy Porter for superlative work in processing, sectioning, and staining difficult samples. We thank Tom Whittam for productive discussions of these experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.CDC. PulseNet Pathogens - Campylobacter jejuni. National Center for Infectious Diseases website. 2005 Aug 1;2005:1. http://www.cdc.gov/pulsenet/pathogens_pages/Campylobacter_jejuni.htm(2005) [Google Scholar]
- 2.Blaser MJ. Epidemiologic and clinical features of Campylobacter jejuni infections. Journal of Infectious Diseases. 1997;176(Suppl 2):S103–5. doi: 10.1086/513780. [DOI] [PubMed] [Google Scholar]
- 3.Mansfield LS, Abner SR. Molecular Mechanisms Governing Campylobacter Pathogenicity. In: Carey J, Linz J, editors. Microbial Foodborne Diseases: Mechanisms of Pathogenesis and Toxin Synthesis. 2000. pp. 191–243. [Google Scholar]
- 4.Tee W, Mijch A. Campylobacter jejuni bacteremia in human immunodeficiency virus (HIV)-infected and non-HIV-infected patients: comparison of clinical features and review. Clin Infect Dis. 1998;26(1):91–6. doi: 10.1086/516263. [DOI] [PubMed] [Google Scholar]
- 5.Snijders F, Kuijper EJ, de Wever B, van der Hoek L, Danner SA, Dankert J. Prevalence of Campylobacter-associated diarrhea among patients infected with human immunodeficiency virus. Clin Infect Dis. 1997;24(6):1107–13. doi: 10.1086/513643. [DOI] [PubMed] [Google Scholar]
- 6.Skirrow MB, Jones DM, Sutcliffe E, Benjamin J. Campylobacter bacteraemia in England and Wales, 1981–91. Epidemiol Infect. 1993 Jun;110(3):567–73. doi: 10.1017/s0950268800050986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perkins DJ, Newstead GL. Campylobacter jejuni enterocolitis causing peritonitis, ileitis and intestinal obstruction. Aust N Z J Surg. 1994 Jan;64(1):55–8. doi: 10.1111/j.1445-2197.1994.tb02137.x. [DOI] [PubMed] [Google Scholar]
- 8.Farmer RG. Infectious causes of diarrhea in the differential diagnosis of inflammatory bowel disease. Med Clin North Am. 1990 Jan;74(1):29–38. doi: 10.1016/s0025-7125(16)30584-3. [DOI] [PubMed] [Google Scholar]
- 9.Kuroki S, Haruta T, Yoshioka M, Kobayashi Y, Nukina M, Nakanishi H. Guillain-Barré syndrome associated with Campylobacter infection. Ped Inf Dis J. 1991;10:149–51. doi: 10.1097/00006454-199102000-00014. [DOI] [PubMed] [Google Scholar]
- 10.Fujimoto S, Yuki N, Itoh T, Amako K. Specific serotype of Campylobacter jejuni associated with Guillain-Barré syndrome. J Inf Dis. 1992;165:183. doi: 10.1093/infdis/165.1.183. [DOI] [PubMed] [Google Scholar]
- 11.Nickerson CL, Luthra HS, David CS. Role of enterobacteria and HLA-B27 in spondyloarthropathies: studies with transgenic mice. Ann Rheum Dis. 1990 Jun;49(1):426–33. [PubMed] [Google Scholar]
- 12.Maki-Ikola O, Viljanen MK, Tiitinen S, Toivanen P, Granfors K. Antibodies to arthritis-associated microbes in inflammatory joint diseases. Rheumatol Int. 1991;10(6):231–4. doi: 10.1007/BF02274884. [DOI] [PubMed] [Google Scholar]
- 13.Gerloni V, Fantini F. Reactive arthritis. Pediatr Med Chir. 1990 Sep-Oct;12(5):447–51. [PubMed] [Google Scholar]
- 14.Salyers AA, Whitt DD. Bacterial Pathogenesis: a Molecular Approach. 1994;418:4. [Google Scholar]
- 15.Lecuit M, Abachin E, Martin A, Poyart C, Pochart P, Suarez F, et al. Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N Engl J Med. 2004 Jan 15;350(3):239–48. doi: 10.1056/NEJMoa031887. [DOI] [PubMed] [Google Scholar]
- 16.Leonard EE, 2nd, Tompkins LS, Falkow S, Nachamkin I. Comparison of Campylobacter jejuni isolates implicated in Guillain-Barre syndrome and strains that cause enteritis by a DNA microarray. Infect Immun. 2004 Feb;72(2):1199–203. doi: 10.1128/IAI.72.2.1199-1203.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.On SL, Dorrell N, Petersen L, Bang DD, Morris S, Forsythe SJ, et al. Numerical analysis of DNA microarray data of Campylobacter jejuni strains correlated with survival, cytolethal distending toxin and haemolysin analyses. Int J Med Microbiol. 2006 Oct;296(6):353–63. doi: 10.1016/j.ijmm.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Parker CT, Quinones B, Miller WG, Horn ST, Mandrell RE. Comparative genomic analysis of Campylobacter jejuni strains reveals diversity due to genomic elements similar to those present in C. jejuni strain RM1221. J Clin Microbiol. 2006 Nov;44(11):4125–35. doi: 10.1128/JCM.01231-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pearson BM, Pin C, Wright J, I’Anson K, Humphrey T, Wells JM. Comparative genome analysis of Campylobacter jejuni using whole genome DNA microarrays. FEBS Lett. 2003 Nov 6;554(1–2):224–30. doi: 10.1016/s0014-5793(03)01164-5. [DOI] [PubMed] [Google Scholar]
- 20.Poly F, Threadgill D, Stintzi A. Genomic diversity in Campylobacter jejuni: identification of C. jejuni 81–176-specific genes. J Clin Microbiol. 2005 May;43(5):2330–8. doi: 10.1128/JCM.43.5.2330-2338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taboada EN, van Belkum AF, Yuki N, Acedillo RR, Godschalk PC, Koga M, et al. Comparative genomic analysis of Campylobacter jejuni associated with Guillain-Barre and Miller Fisher syndromes: neuropathogenic and enteritis-associated isolates can share high levels of genomic similarity. BMC Genomics. 2007 Oct 5;8(1):359. doi: 10.1186/1471-2164-8-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blaser MJ. Epidemiologic and clinical features of Campylobacter jejuni infections. J Infect Dis. 1997 Dec;176:S103–S5. doi: 10.1086/513780. [DOI] [PubMed] [Google Scholar]
- 23.Calva JJ, Ruiz-Palacios GM, Lopez-Vidal AB, Ramos A, Bojalil R. Cohort study of intestinal infection with Campylobacter in Mexican children. Lancet. 1988;1(8584):503–6. doi: 10.1016/s0140-6736(88)91297-4. [DOI] [PubMed] [Google Scholar]
- 24.Yoshiki A, Moriwaki K. Mouse phenome research: implications of genetic background. ILAR J. 2006;47:94–102. doi: 10.1093/ilar.47.2.94. [DOI] [PubMed] [Google Scholar]
- 25.Mansfield LS, Bell JA, Wilson DL, Murphy AJ, Elsheikha HM, Rathinam VA, et al. C57BL/6 and congenic interleukin-10-deficient mice can serve as models of Campylobacter jejuni colonization and enteritis. Infect Immun. 2007 Mar;75(3):1099–115. doi: 10.1128/IAI.00833-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Young VB, Schauer DB, Fox JG. Animal models of Campylobacter infections. In: Nachamkin I, Blaser MJ, editors. Campylobacter. 2. Washington, D.C: ASM Press; 2000. [Google Scholar]
- 27.Beckwith J, Cong Y, Sundberg JP, Elson CO, Leiter EH. Cdcs1, a major colitogenic locus in mice, regulates innate and adaptive immune response to enteric bacterial antigens. Gastroenterology. 2005 Nov;129(5):1473–84. doi: 10.1053/j.gastro.2005.07.057. [DOI] [PubMed] [Google Scholar]
- 28.The Jackson Laboratory. The importance of genetic background in mouse-based biomedical research. JAX Notes. 2006 Summer;2006(502):1–4. [Google Scholar]
- 29.Farmer MA, Sundberg JP, Bristol IJ, Churchill GA, Li R, Elson CO, et al. A major quantitative trait locus on chromosome 3 controls colitis severity in IL-10-deficient mice. Proc Natl Acad Sci U S A. 2001 Nov 20;98(24):13820–5. doi: 10.1073/pnas.241258698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCathey SN, Shomer NH, Schrenzel MD, Whary MT, Taylor NS, Fox JG. Colonization and tissue tropism of Helicobacter pylori and a novel urease-negative Helicobacter species in ICR mice are independent of route of exposure. Helicobacter. 1999 Dec;4(4):249–59. doi: 10.1046/j.1523-5378.1999.99291.x. [DOI] [PubMed] [Google Scholar]
- 31.Taylor NS, Xu S, Nambiar P, Dewhirst FE, Fox JG. Enterohepatic Helicobacter species are prevalent in mice from commercial and academic institutions in Asia, Europe, and North America. J Clin Microbiol. 2007 Jul;45(7):2166–72. doi: 10.1128/JCM.00137-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beckwith CS, Franklin CL, Hook RR, Jr, Besch-Williford CL, Riley LK. Fecal PCR assay for diagnosis of Helicobacter infection in laboratory rodents. J Clin Microbiol. 1997 Jun;35(6):1620–3. doi: 10.1128/jcm.35.6.1620-1623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng S, Ku K, Hodzic E, Lorenzana E, Freet K, Barthold SW. Differential detection of five mouse-infecting helicobacter species by multiplex PCR. Clin Diagn Lab Immunol. 2005 Apr;12(4):531–6. doi: 10.1128/CDLI.12.4.531-536.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tolia V, Nilsson HO, Boyer K, Wuerth A, Al-Soud WA, Rabah R, et al. Detection of Helicobacter ganmani-like 16S rDNA in pediatric liver tissue. Helicobacter. 2004 Oct;9(5):460–8. doi: 10.1111/j.1083-4389.2004.00266.x. [DOI] [PubMed] [Google Scholar]
- 35.Shen Z, Xu S, Dewhirst FE, Paster BJ, Pena JA, Modlin IM, et al. A novel enterohepatic Helicobacter species ‘Helicobacter mastomyrinus’ isolated from the liver and intestine of rodents. Helicobacter. 2005 Feb;10(1):59–70. doi: 10.1111/j.1523-5378.2005.00292.x. [DOI] [PubMed] [Google Scholar]
- 36.Shen ZL, Feng Y, Fox JG. Identif ication of enterohepatic Helicobacter species by restriction fragment-length polymorphism analysis of the 16S rRNA gene. Helicobacter. 2000 Sep;5(3):121–8. doi: 10.1046/j.1523-5378.2000.00019.x. [DOI] [PubMed] [Google Scholar]
- 37.Whary MT, Danon SJ, Feng Y, Ge Z, Sundina N, Ng V, et al. Rapid onset of ulcerative typhlocolitis in B6.129P2-IL10tm1Cgn (IL-10−/−) mice infected with Helicobacter trogontum is associated with decreased colonization by altered Schaedler’s flora. Infect Immun. 2006 Dec;74(12):6615–23. doi: 10.1128/IAI.01091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dewhirst FE, Fox JG, Mendes EN, Paster BJ, Gates CE, Kirkbride CA, et al. ‘Flexispira rappini’ strains represent at least 10 Helicobacter taxa. Int J Syst Evol Microbiol. 2000 Sep;50:1781–7. doi: 10.1099/00207713-50-5-1781. [DOI] [PubMed] [Google Scholar]
- 39.Fox JG, Rogers AB, Whary MT, Ge Z, Taylor NS, Xu S, et al. Gastroenteritis in NF-kappaB-deficient mice is produced with wild-type Camplyobacter jejuni but not with C. jejuni lacking cytolethal distending toxin despite persistent colonization with both strains. Infect Immun. 2004 Feb;72(2):1116–25. doi: 10.1128/IAI.72.2.1116-1125.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makino S, Muraoka Y, Kishimoto Y, Hayashi Y. Genetic analysis for insulitis in NOD mice. Jikken Dobutsu. 1985 Oct;34(4):425–31. doi: 10.1538/expanim1978.34.4_425. [DOI] [PubMed] [Google Scholar]
- 41.Gaskins HR, Prochazka M, Hamaguchi K, Serreze DV, Leiter EH. Beta cell expression of endogenous xenotropic retrovirus distinguishes diabetes-susceptible NOD/Lt from resistant NON/Lt mice. J Clin Invest. 1992 Dec;90(6):2220–7. doi: 10.1172/JCI116107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghosh S, Palmer SM, Rodrigues NR, Cordell HJ, Hearne CM, Cornall RJ, et al. Polygenic control of autoimmune diabetes in nonobese diabetic mice. Nat Genet. 1993 Aug;4(4):404–9. doi: 10.1038/ng0893-404. [DOI] [PubMed] [Google Scholar]
- 43.Wong FS, Du W, Thomas IJ, Wen L. The influence of the major histocompatibility complex on development of autoimmune diabetes in RIP-B7.1 mice. Diabetes. 2005 Jul;54(7):2032–40. doi: 10.2337/diabetes.54.7.2032. [DOI] [PubMed] [Google Scholar]
- 44.Hattori M, Yamato E, Itoh N, Senpuku H, Fujisawa T, Yoshino M, et al. Cutting edge: homologous recombination of the MHC class I K region defines new MHC-linked diabetogenic susceptibility gene(s) in nonobese diabetic mice. J Immunol. 1999 Aug 15;163(4):1721–4. [PubMed] [Google Scholar]
- 45.Hayashi T, Faustman D. NOD mice are defective in proteasome production and activation of NF-kappaB. Mol Cell Biol. 1999 Dec;19(12):8646–59. doi: 10.1128/mcb.19.12.8646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dahlen E, Hedlund G, Dawe K. Low CD86 expression in the nonobese diabetic mouse results in the impairment of both T cell activation and CTLA-4 up-regulation. J Immunol. 2000 Mar 1;164(5):2444–56. doi: 10.4049/jimmunol.164.5.2444. [DOI] [PubMed] [Google Scholar]
- 47.Janeway CA, Jr, Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994 Jan 28;76(2):275–85. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 48.Riley LK, Franklin CL, Hook J, RR, Besch-Williford C. Identification of murine helicobacters by PCR and restriction enzyme analyses. J Clin Microbiol. 1996;34:942–6. doi: 10.1128/jcm.34.4.942-946.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deasy BM, Rea MC, Fitzgerald GF, Cogan TM, Beresford TP. A rapid PCR based method to distinguish between Lactococcus and Enterococcus. Syst Appl Microbiol. 2000 Dec;23(4):510–22. doi: 10.1016/S0723-2020(00)80025-9. [DOI] [PubMed] [Google Scholar]
- 50.Young VB, Knox MA, Pratt JS, Cortez JS, Mansfield LS, Rogers AB, et al. In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect Immun. 2004 May;72(5):2521–7. doi: 10.1128/IAI.72.5.2521-2527.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson DL, Abner SR, Newman TC, Mansfield LS, Linz JE. Identification of ciprofloxacin-resistant Campylobacter jejuni by use of a fluorogenic PCR assay. J Clin Microbiol. 2000 Nov;38(11):3971–8. doi: 10.1128/jcm.38.11.3971-3978.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rinttila T, Kassinen A, Malinen E, Krogius L, Palva A. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol. 2004;97(6):1166–77. doi: 10.1111/j.1365-2672.2004.02409.x. [DOI] [PubMed] [Google Scholar]
- 53.Khan IU, Gannon V, Kent R, Koning W, Lapen DR, Miller J, et al. Development of a rapid quantitative PCR assay for direct detection and quantification of culturable and non-culturable Escherichia coli from agriculture watersheds. J Microbiol Methods. 2007 Jun;69(3):480–8. doi: 10.1016/j.mimet.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 54.Ludbrook J. Multiple comparison procedures updated. Clin Exp Pharmacol Physiol. 1998;25:1032–7. doi: 10.1111/j.1440-1681.1998.tb02179.x. [DOI] [PubMed] [Google Scholar]