

Abstract
Metabolic reprogramming is a hallmark of cancer. Oncogenic growth signaling regulates glucose, glutamine and lipid metabolism to meet the bioenergetics and biosynthetic demands of rapidly proliferating tumor cells. Emerging evidence indicates that sterol regulatory element-binding protein 1 (SREBP-1), a master transcription factor that controls lipid metabolism, is a critical link between oncogenic signaling and tumor metabolism. We recently demonstrated that SREBP-1 is required for the survival of mutant EGFR-containing glioblastoma, and that this pro-survival metabolic pathway is mediated, in part, by SREBP-1-dependent upregulation of the fatty acid synthesis and low density lipoprotein (LDL) receptor (LDLR). These results have identified EGFR/PI3K/Akt/SREBP-1 signaling pathway that promotes growth and survival in glioblastoma, and potentially other cancer types. Here, we summarize recent insights in the understanding of cancer lipid metabolism, and discuss the evidence linking SREBP-1 with PI3K/Akt signaling-controlled glycolysis and with Myc-regulated glutaminolysis to lipid metabolism. We also discuss the development of potential drugs targeting the SREBP-1-driven lipid metabolism as anti-cancer agents.
Keywords: TCA cycle, PI3K/Akt, SREBP-1, ACLY, ACC, FASN, LDLR, LXR, ABCA1, SREBP-2
Introduction
Increased evidence shows that the lipogenic phenotype is a major characteristic of cancer [1-4]. Cancer cells engage in extensive de novo lipogenesis, producing lipids from glucose and glutamine [5, 6]. This ability of cancer cells to engage in cell autonomous generation of lipids provides a critical route by which cancer cells become independent of systemic regulation. It also provides a potential Achilles hell, as cancer cells may develop enhanced dependence on the lipogenic machinery, in an oncogene-dependent fashion. Multiple studies showed that glucose and glutamine both contribute to lipogenesis [6-9], which may open a new avenue to inhibit tumor growth by disrupting the metabolic flux from glucose, glutamine to lipogenesis. However, the detailed molecular mechanism by which the metabolic flux is regulated, and whether oncogenic growth signaling is tightly involved in these processes is poorly understood.
An abundant supply of lipids is required for rapid tumor cell proliferation. Phospholipids and cholesterol form the major structure of cell membranes [10-14]. Fatty acids and triglycerides provide ample energy substrates [15], and lipids are also utilized as precursors to synthesize some hormones and second messengers that mediate signal transduction pathways [10, 16]. Here, we will discuss recent advances in the understanding of lipid metabolism regulation, address how oncogenic growth signaling coordinates glucose, glutamine metabolism to lipid synthesis, and further indicate the potential druggable molecular targets in the regulatory network of lipid metabolism.
Lipogenesis pathway
PI3K/Akt signaling and C-Myc control the metabolic flux from glucose and glutamine to de novo lipid synthesis [5, 7, 9, 17]. Recently, SREBP-1 was revealed to be highly activated in cancers [18-20], and has been shown to play an integral role in connecting oncogenic signaling-regulated glucose and glutamine metabolism to de novo lipogenesis [8, 21]. Genetic and pharmacological targeting of SREBP-1 has been shown to significantly inhibit tumor cell growth, which strongly suggests that SREBP-1 is a novel molecular target in cancer [18].
SREBP-1 regulation by sterol levels
SREBPs are key transcriptional factors that control lipogenesis and lipid uptake [22]. There are two SREBP genes in mammals, SREBP-1 and -2 [23, 24]. SREBP-1 gene transcribes two isoforms SREBP-1a and -1c encoded from different promoters, which are demonstrated to mainly regulate genes that control fatty acid synthesis [22-25]. SREBP-2 is shown to regulate genes involved in cholesterol synthesis [23, 25, 26]. However, the roles of SREBP-1 and -2 significantly overlap in the regulation of lipid metabolism [25-28]. In normal tissues, SREBPs levels and activity are tightly controlled by endogenous sterol levels via negative feedback regulation [22]. SREBPs are located in the endoplasmic reticulum (ER) membrane in association with SREBPs cleave activating protein (SCAP) in which they are retained by Insulin-induced gene (Insig) when cellular sterol levels are sufficient. Once sterol levels drop, SCAP protein dissociates with Insig and escorts SREBPs to the Golgi, in which they are cleaved by site-1 and site-2 proteases (S1P and S2P) sequentially thereby releasing the N-terminus, which then enters into the nucleus to transcribe lipogenesis genes and LDLR (Fig. 1) [22, 23]. In addition, SREBP-1 has also been shown to be involved in other cellular functions, such as regulation of cell cycle and cell proliferation [29, 30]. However, it is still not clear whether the alteration of cell cycle progression when SREBP-1 is activated or inhibited is caused by the change of lipid metabolism or from separate SREBP-1 molecular functions. More detailed studies are essential in order to unravel the complete function of SREBP-1, particularly in cancer cells.
Figure 1. Model for sterols-regulated maturation of SREBPs.
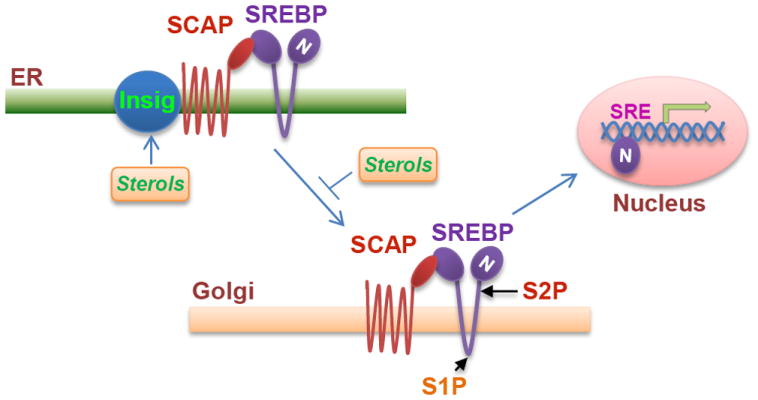
The precursor SREBPs locates in the ER membrane in association with SCAP, and Insig interacts with SCAP and retains the SCAP/SREBP complex in the ER membrane in the condition of high sterol levels. SCAP will dissociate from Insig and escort SREBPs translocation to the Golgi apparatus when sterol levels drop. In the Golgi, SREBPs are cleaved sequentially by the S1P and S2P proteases and then release the mature N-terminal fragment (N), which enters the nucleus to transcribe their target genes expression.
SREBP-1 is activated by PI3K/Akt signaling
In addition to regulation by sterols, SREBP-1 has been shown to be stabilized and activated by the PI3K/Akt oncogenic signaling pathway in cancer [18, 31, 32]. There are two mechanisms that could explain this regulation. The first is that activated Akt can stabilize the SREBP-1 nuclear form and promote its target gene expression through down-regulation of Fwb-7, an ubiquitin enzyme E3 which mediates SREBP-1 N-terminus degradation by inhibiting its regulator, GSK3-β [33-35]. Another mechanism is PI3K/Akt signaling through mTORC1 which regulates Lipin 1, a phosphatidic acid phosphatase, to control SREBP-1 nuclear localization and its transcriptional activity (Fig. 2) [36, 37]. These two distinct observations of regulatory mechanisms for SREBP-1 likely originate from the different organ-derived cell lines. Specifically, cancer cell lines HeLa and HepG2 were compared to the normal tissue cell lines HEK293, retinal pigment epithelial cells (RPE), NIH3T3, hepatocytes and mouse embryonic fibroblasts (MEF). Moreover, SREBP-1 mRNA and protein levels seem to be more sensitive to mTORC1 inhibition in normal tissue cells than cancer cell lines [36-39].
Figure 2. PI3K/Akt signaling regulates SREBP-1-mediated lipid metabolism.
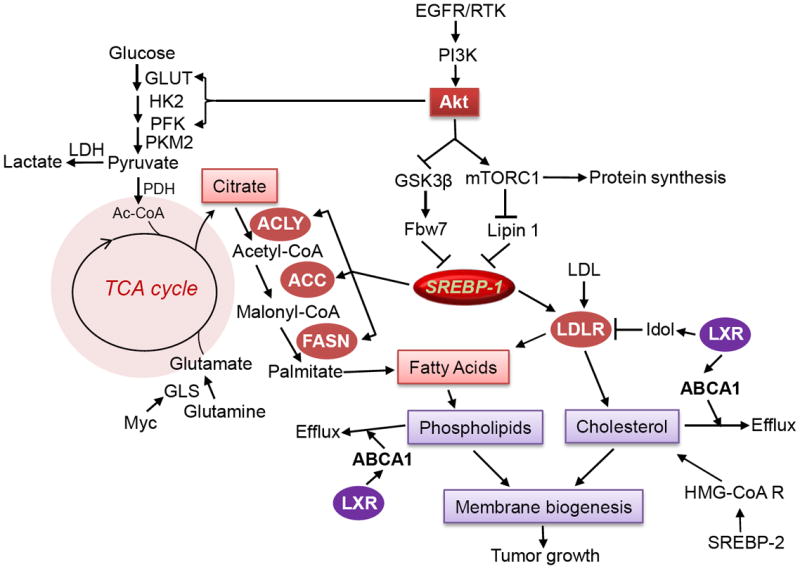
The scheme at the left side shows that glycolysis is promoted by PI3K/Akt signaling via promoting GLUT translocation and activating HK2 and PFK to produce pyruvate. Following, pyruvate enters into the mitochondria and generates citrate, and some portion of citrate are released into cytoplasm and hydrolysed by ACL to produce acyl-CoA, which is the precursor for lipid synthesis. In addition, glutamine incorporates into the TCA cycle through glutaminolysis regulated by the Myc oncogene, to provide an additional energy source for cell growth, and also to produce citrate as a precursor for lipid synthesis through reductive carboxylation. The scheme at right side shows that SREBP-1 plays an integral role in mediating oncogenic signaling RTK/PI3K/Akt to fatty acid synthesis and cholesterol uptake. SREBP-1 upregulates the expression of ACL, ACC and FASN to promote fatty acid synthesis, also promotes the expression of LDLR to enhance cholesterol uptake. Activation of LXR stimulates ABCA1 expression and promotes cholesterol and phospholipids efflux, also reduces LDLR levels via upregulating Idol, a ubiquitin ligase E3. GLUT, glucose transporter; HK2, hexokinase 2; PFK, phosphofructokinase; PKM2, pyruvate kinase M2; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase; GLS, glutaminase; EGFR, epidermal growth factor receptor; RTK, receptor tyrosine kinase.
In cancer cells, SREBP-1 regulation appears to be more complex. A recent study showed that inhibiting mTORC1 using rapamycin has little effect on SREBP-1 nuclear localization and its abundance, but inhibiting its upstream factors like EGFR, PI3K and Akt significantly decreases SREBP-1 N-terminal levels and diminishes its abundance in the nucleus [18]. These data suggest that other oncogenic molecules beyond mTORC1 are involved in SREBP-1 regulation. It is also possible that inhibiting mTORC1 by rapamycin could maintain SREBP-1 levels via feedback activation of Akt [40]. Furthermore, mammalian target of rapamycin complex 2 (mTORC2) is possibly involved in the stability of the nucleus form of SREBP-1 according to the results from the mTOR kinase inhibitor Torin-1 [41], which inhibits both mTORC1 and mTORC2 activity [42], and significantly decreases SREBP-1 abundance in the nucleus compared to the inhibition of mTORC1 alone by rapamycin [37, 43]. Given that lipogensis is a critical source for the formation of new cellular membranes, it is not surprising that cancer cells may develop redundant molecular pathways to control SREBP-1 expression and the stability of its nuclear form to guarantee cells the ability to obtain ample lipids for their rapid growth and division. Additionally, this may partially explain the reason why targeting mTORC1 had an incremental favorable response in the clinic using rapamycin or its derivatives to treat cancer [40]. Taken together, these data suggest that directly targeting SREBP-1 may be one of the most feasible approaches to treat cancer.
SREBP-1 integrates the metabolic flux from PI3K/Akt-regulated glycolysis to lipogenesis
Addictive glycolysis is a main feature of cancers even in an oxygen-rich condition [8, 44, 45]. PI3K/Akt signaling has been demonstrated to activate glycolysis through regulating glucose transport (GLUT) and hexokinase 2 (HK2) [9, 17, 46-48]. In addition to the ATP production, glucose metabolism also contributes to NADPH and macromolecule synthesis, such as nucleic acids, amino acids and lipids [9, 49]. Cancer cells acquire glucose to synthesize fatty acids and support new membranes and lipid raft formation [1, 50, 51]. The glycolytic intermediate pyruvate enters into mitochondria and produce acetyl-CoA, which is then condensed with oxaloacetate to form citrate. To synthesize lipids, a portion of citrate is released into the cytoplasm and then lysed by ATP citrate lyase (ACL) to produce the lipid precursor, acetyl-CoA [52, 53]. Then, acetyl-CoA carboxylase (ACC) catalyzes the irreversible carboxylation of acetyl-CoA to form malonyl-CoA [54, 55], which is then incorporated with acetyl-CoA to synthesize long-chain saturated fatty acid, palmitate, by fatty acid synthase (FASN) (Fig. 2) [1, 56]. Palmitate can be further elongated or de-saturated to produce diverse fatty acids, which can further synthesize phospholipids for new cell membrane formation, store in triglycerides for energy storage, enter into mitochondria for energy production, modify proteins by acetylation, or synthesize hormones [1, 56, 57].
Furthermore, there is a correlation between elevated glycolysis and exacerbated fatty acid synthesis [9, 21]. PI3K/Akt signaling upregulates glucose uptake and glycolysis [17, 49, 58], and also promotes fatty acid synthesis (Fig. 2) [1, 59, 60]. Meanwhile, the fatty acid synthesis pathway is tightly regulated by SREBP-1, which upregulates ACL, ACC and FASN expression [22, 23]. FASN, which controls the terminal step for the synthesis of palmitate, has been shown to be upregulated by PI3K/Akt signaling [1, 60, 61]. Multiple studies have revealed that PI3K/Akt signaling promotes fatty acid synthesis through upregulation of SREBP-1 [18, 27, 36]. Taken together, SREBP-1 has been revealed as a key player that integrates the metabolic flux from PI3K/Akt signaling regulated glycolysis to fatty acid synthesis (Fig. 2).
Glutamine metabolism and lipogenesis
In addition to glycolysis, glutamine also contributes to energy production and lipid synthesis by entering the mitochondria and integrating into the TCA cycle [7, 15, 62, 63]. The oncogene Myc upregulates glutamine transporter and glutaminase to synthesize glutamate, then enters into the mitochondria and is converted into α-ketoglutarate. Then, α-ketoglutarate can either enter into the TCA cycle through oxidative phosphorylation to produce ATP [7, 15, 62, 64, 65], or form citrate through reductive carboxylation under hypoxia or defective mitochondria condition mediated by IDH1 or IDH2, to contribute to SREBP-1-driven fatty acid synthesis (Fig. 2) [5, 6, 18, 66]. Several groups recent reported that cancer cells preferentially utilize glutamine as a precursor to synthesize fatty acid during hypoxia or in an impaired mitochondria condition [5, 6, 65, 66]. SREBP-1 could also be a key regulator of glutamine metabolism in order to synthesize lipids. Therefore, future studies demonstrating the correlation between glutamine metabolism and SREBP-1 activity is required.
Targeting the SREBP-1 regulated lipogenesis pathway is a promising therapeutic strategy to treat cancer
Over the past decade, pharmacological drug development has been moved towards the identification of molecular targets and discovery of specific small molecular compounds to antagonize these targets to treat cancer, such as EGFR, PI3K, Akt, mTOR and B-Raf [67-70]. These explorations have brought significant advances in cancer treatment. Unfortunately, resistance is frequently observed to these targeted therapies and has compromised the efficacy of the molecular targeted drugs in clinic, such as erlotinib, rapamycin, bevacizumab, PLX4032 et al. [40, 68, 71-74]. Therefore, identifying key molecular targets and developing new effective therapeutic strategies to treat cancer is still urgent.
SREBP-1
Targeting metabolic alterations has emerged as a new strategy to treat cancer in the past few years, such as targeting glycolysis using 2-deoxyglucose (2-DG) or an HK2 inhibitor, which has shown promising results in a pre-clinical mouse model [48, 75, 76]. Lipogenesis has been shown to be highly activated in cancer and is integrated with both glucose and glutamine metabolism [6, 9, 17, 77, 78]. Thus, suppression of fatty acid synthesis could be a new approach to block cancer malignant growth [1, 79]. SREBP-1, a central regulator of the integration of PI3K/Akt signaling regulated glucose metabolism and Myc-regulated glutamine metabolism to fatty acid synthesis (Fig. 2) [8, 18, 21, 63], has emerged as a very promising druggable target. In human cancer, SREBP-1 has been shown to be highly present in glioblastoma tissues, and its active N-terminus highly localizes in the tumor cell nucleus. This is accompanied by highly expressed downstream genes ACC and FASN [18]. Genetic and pharmacological inhibition of SREBP-1 has been shown to significantly suppress tumor cell growth and lead to cell death [18]. Given the critical role of SREBP-1 in mediating PI3K/Akt signaling and Myc-regulated metabolic flux and possible other signaling pathways, developing specific inhibitors to target SREBP-1 could become a new therapeutic strategy to treat cancer, particularly in tumors with hyperactivated PI3K/Akt signaling.
ACLY, ACC and FASN
In SREBP-1-regulated fatty acid synthesis pathway, ACL, ACC and FASN have been revealed to be highly expressed in cancer (Fig. 2) [1, 18, 53, 80]. Inhibition of these key factors has shown a significant reduction of cancer cell growth in vitro and in vivo [53, 81-83]. Pharmacological and genetic inhibition of ACL, the enzyme that produces acetyl-CoA in cytoplasm and provides the precursor of lipid synthesis, has been shown to significantly inhibit cancer cell growth in vitro and in vivo [53, 81]. ACC, the enzyme that controls the rate-limiting step of fatty acid synthesis, is also highly expressed in human cancer tissues [18]. Inhibition of ACC was also shown to significantly suppress tumor growth [84]. At the terminal step in the de novo synthesis of fatty acids, FASN has been shown to play a key role in tumor malignant growth. It has been shown to be elevated in most cancers including prostate, breast, glioblastoma and ovarian [1, 18, 80]. Inhibitors that target FASN have been tested in preclinical models and have been shown to exert a significant inhibitory effect on tumor growth [85-87]. In normal tissues, SREBP-1 and its regulated fatty acid synthesis pathway usually display low levels of expression and activity. Therefore, targeting SREBP-1-regulated fatty acid synthesis pathway may be become a very promising strategy for treating cancer.
Inhibiting lipolysis to treat cancer
Fatty acids contribute to cell membrane formation and energy production. When fatty acid levels are excessive in cells, they are converted into triglycerides and deposit into the lipid droplet for temporary storage [88, 89]. Once cells experience a shortage of fatty acids or energy, triglycerides deposited in the lipid droplet will undergo lipolysis via sequential lipases to release free fatty acids and maintain cellular activity [88, 90, 91]. Monoacylglycerol lipase (MAGL), which catalyzes the final step of lipolysis to convert monoacyglycerol into glycerol and one free fatty acid, has been identified to be highly activated in cancer. Pharmacological inhibition of MAGL has been shown to significantly inhibit tumor growth in a xenograft model [92]. In addition, adipose triglyceride lipase (ATGL) which hydrolytes triacylglycerol to diacylglycerol and releases one free fatty acid [90, 93], and hormone-sensitive lipase (HSL) which catalyzes diacylglycerol to release one free fatty acid and form monoacyglycerol (Fig. 3) [94, 95], may also become potential therapeutic targets. Recently, Nieman et al. demonstrated that adipocytes adjacent to metastatic ovarian cancer cells in the omentum through lipolysis provide fatty acid for tumor cell rapid growth [96]. This study demonstrates the importance of lipids in cancer growth. Therefore, further investigation of lipid metabolism within the tumor microenviroment certainly will provide new insights in the understanding of tumorigenesis and further identify effective approaches to treat cancer.
Figure 3. Lipolysis process for triacylglycerol.
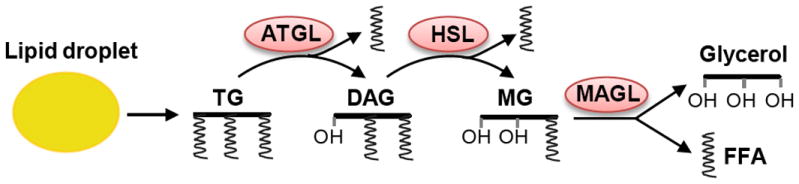
Triacylglycerol (TG) stored in lipid droplet is sequentially hydrolyzed by lipases ATGL, HSL and MAGL to liberate free fatty acid (FFA) and glycerol as substrates for energy production or membrane formation. ATGL, adipose triglyceride lipase; HSL, hormone-sensitive lipase; MAGL, monoacylglycerol lipase.
When free fatty acids are utilized in cells, they will be converted to acyl-CoA by acyl-CoA synthetases (ACSs), which then can either enter into the mitochondria for beta-oxidation to produce ATP, be incorporated into phospholipids or associate with proteins for acyl-modification [97]. ACS has been shown to be elevated in cancer [98]. Additionally, pharmacological and genetic inhibition of ACS has been shown to significantly suppress tumor growth and lead to cell death [99].
Cholesterol metabolism
Cholesterol is an important component of cellular membranes. It also contributes to hormone formation, and participates in important signal transduction pathways [10]. Cholesterol maintains cellular membrane integrity and fluidity, and its homeostasis is tightly regulated by uptake, de novo synthesis and efflux [23, 100, 101]. SREBPs and nuclear receptor Liver X Receptor (LXR) are critical factors that control cellular cholesterol levels [101, 102]. However, it is less known how cholesterol homeostasis is controlled and whether cholesterol metabolism is altered in cancer cells. Our recent study first uncovered that activated EGFR/PI3K signaling promotes cholesterol uptake by upregulating low density lipoprotein (LDL) receptor (LDLR), which was demonstrated to also be mediated by SREBP-1 (Fig. 2) [19]. This study further showed that depriving cells of cholesterol results in cell death through the activation of LXR by its synthetic agonist.
SREBP-1 mediates PI3K/Akt signaling-regulated cholesterol metabolism through upregulating LDLR
LDL is a major cholesterol-carrier in the bloodstream, which transports cholesterol from the liver to tissues in the rest of the body [103, 104]. When cellular cholesterol levels decrease, LDLR will be expressed and then inserted into the clathrin-coated pits on the cell membrane. The extracellular domain of LDLR will bind circulated LDL and promote its uptake through endocytosis. After LDL enters into cells, it will be delivered to the lysosome, in which LDL is then hydrolyzed by lipases and subsequently releases free cholesterol for cell utilization [105, 106].
One of the main features of tumor cells is high proliferation rates. It is predicted that cancer cells should maintain high levels of cholesterol in order to quickly form new membranes for its rapid division [107]. However, how cancer cells maintain its cholesterol levels are less known. Exogenous uptake and de novo synthesis both contribute to cellular cholesterol pools [100], but which route is preferred in cancer cells is unclear. Recently, we revealed that glioblastoma cells prefer to uptake LDL from exogenous media to obtain cholesterol and meet the demand for rapid growth and proliferation, and our data also showed that de novo biosynthesis is a compensatory pathway when exogenous cholesterol is scarce [19]. Our study revealed that EGFR/PI3K/Akt signaling promotes cholesterol uptake through upregulation of LDLR. We further revealed that SREBP-1 mediates this signaling pathway (Fig. 2) [19]. Collectively, the studies demonstrated that the oncogenic signaling PI3K/Akt pathway via SREBP-1 integrates glucose metabolism, lipogenesis and cholesterol uptake to provide sufficient energy and essential building blocks for rapid cancer cell growth (Fig. 2) [18, 19]. Taken together, SREBP-1 has been revealed as a central regulator of oncogenic signaling and metabolic flux. Given LDLR is highly upregulated in cancer, and its critical role for cholesterol uptake [18, 108], it could be a promising molecular target in cancer. Inhibition of LDLR should be tested in cancer cells and tumor xenograft models to determine its function in tumor growth and to provide the rationale for developing specific inhibitors or antibodies to block LDLR-mediated cholesterol uptake in order to treat cancer.
LXR/ABCA1 regulates cholesterol efflux
LXR is a key regulator of cellular cholesterol homeostasis. When endogenous cholesterol levels increase, oxysterol levels will also elevate. They then enter into the nucleus and bind to LXR to activate its transcription activity and promote the expression of its target genes, cholesterol transporter genes ATP-binding cassette, subfamily A (ABCA1) or subfamily G (ABCG1). These target genes are responsible for transporting cholesterol to the outside of cells in order to maintain cellular cholesterol homeostasis [101, 109]. Pharmacologically activating LXR has been shown to significantly reduce atherosclerotic lesions in mouse models via elevating cholesterol reverse transport [109, 110]. In cancer, activating LXR by its agonists has been shown to significantly inhibit tumor cell growth and lead to cell death in vitro and in vivo through elevating ABCA1-mediated cholesterol efflux [19, 111, 112].
Therefore, the LXR/ABCA1 axis has emerged as a promising druggable target in cancer therapy through reducing cells of cholesterol levels [19]. However, activation of LXR by its agonists unfortunately upregulates SREBP-1 gene expression and enhances fatty acid synthesis, which could decrease the efficacy of LXR agonists on tumor growth [113]. To enhance the efficacy of LXR agonists in cancer treatment, LXR agonists could be combined with inhibitors of the fatty acid synthesis pathway to increase the suppression of tumor growth. However, considering the potential toxicity of the combination of drugs, it may be a better strategy to develop a novel LXR agonist with a low effect on SREBP-1 expression in cancer treatment.
LXR and LDLR
LDLR has been shown to be regulated by SREBP-1 or SREBP-2 [26]. A recent study shows LDLR is mainly upregulated by SREBP-1 in cancer cells [19]. Intriguingly, Tontonoz’s group observed that activation of LXR significantly reduces LDLR levels in addition to elevating ABCA1-mediated cholesterol efflux. They further demonstrated that this process is mediated by the inducible degrader of LDLR (Idol), a ubiquitin ligase E3 [114]. In cancers, activation of LXR also elevates Idol expression and significantly reduces LDLR levels [18]. Taken together, these studies demonstrated that reducing cellular cholesterol levels by activation of LXR is caused by elevating ABCA1-mediated cholesterol efflux as well as reducing LDL uptake via degradation of LDLR, which strongly suggests that LXR could be a very promising molecular target in cancer treatment. In addition, development of inhibitors to block LDLR function will be a good approach to decrease cholesterol uptake and therefore treat cancer. Furthermore, cellular cholesterol levels could be significantly reduced by combining an LXR agonist with an LDLR inhibitor and will likely show a significant synergistic effect on tumor growth. In another way, developing a potent agonist of LXR with low SREBP-1 expression activity is desired but may be difficult to achieve. There is recent promising data from Freed-Pastor et al. shows that fatostatin, a novel SREBP-1 inhibitor, significantly suppressed tumor growth in breast cancer xenografts [115]. Combination of an SREBP-1 inhibitor, such as fatostatin, with LXR agonists could overcome the upregulation of SREBP-1 expression and result in complete inhibition of tumor growth.
SREBP-2 and cholesterol synthesis
SREBP-2 regulates de novo cholesterol synthesis by upregulating enzymes participating in this process [23]. Particularly, the rate-limiting enzyme HMG-CoA reductase has been explored as a significant drug target in order to reduce plasma cholesterol levels [116]. Its inhibitor statin has been shown to dramatically improve the treatment of cardiovascular diseases and significantly enhance heart attack prevention [117, 118]. Given statin’s significance in reducing cholesterol levels in plasma, it has been extensively tested in cancer treatment. Unfortunately, the results are controversial for the effects of statin in the inhibition on tumor progression and in the prevention for cancer initiation [119, 120]. Recent meta-analysis of a large population showed that statin treatment does not have a significant effect on tumor growth and in prevention of cancer incidence [121, 122]. These reports are consistent with the recent study in glioblastoma treatment in vitro and in vivo, in which atorvastatin administration did not significantly inhibit tumor growth in a xenograft model [18]. However, the study demonstrated that atorvastain treatment inhibited tumor cell growth when extracellular cholesterol levels are limited [19]. It could be a good strategy to treat cancer with the combination of a statin and an LDLR inhibitor.
Perspectives
In summary, emerging data indicate that SREBP-1 plays a critical role linking oncogenic signaling with lipid metabolism. Further, recent work indicates that genetic context may impose a differential requirement for SREBP-1 in mediating tumor cell survival, hence providing the opportunity for effective, specific and less toxic targeted therapy. Although in its early stages, and with much work to do, SREBP-1 and the lipogenic machinery provide a promising new set of anti-cancer drug targets. Given that SREBP-1 activity is pretty low in normal tissues, designing specific inhibitors and less toxic drugs to inhibit the enzymes in the SREBP-1-driven fatty acid synthesis pathway will be a promising strategy to treat cancer.
Acknowledgments
This work was supported by Rose DiGangi American Brain Tumor Association Translational Grant (DG); NIH NS072838 (DG).
References
- 1.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature reviews Cancer. 2007;7:763–77. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 2.Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer research. 1953;13:27–9. [PubMed] [Google Scholar]
- 3.Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition. 2000;16:202–8. doi: 10.1016/s0899-9007(99)00266-x. [DOI] [PubMed] [Google Scholar]
- 4.Kuhajda FP, Piantadosi S, Pasternack GR. Haptoglobin-related protein (Hpr) epitopes in breast cancer as a predictor of recurrence of the disease. The New England journal of medicine. 1989;321:636–41. doi: 10.1056/NEJM198909073211003. [DOI] [PubMed] [Google Scholar]
- 5.Metallo CM, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–4. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mullen AR, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–8. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:6479–83. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deberardinis RJ, et al. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maxfield FR, van Meer G. Cholesterol, the central lipid of mammalian cells. Current opinion in cell biology. 2010;22:422–9. doi: 10.1016/j.ceb.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Meer G. Lipids of the Golgi membrane. Trends in cell biology. 1998;8:29–33. doi: 10.1016/s0962-8924(97)01196-3. [DOI] [PubMed] [Google Scholar]
- 12.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nature reviews. Molecular cell biology. 2008;9:112–24. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maxfield FR. Plasma membrane microdomains. Current opinion in cell biology. 2002;14:483–7. doi: 10.1016/s0955-0674(02)00351-4. [DOI] [PubMed] [Google Scholar]
- 14.Mukherjee S, Maxfield FR. Membrane domains. Annual review of cell and developmental biology. 2004;20:839–66. doi: 10.1146/annurev.cellbio.20.010403.095451. [DOI] [PubMed] [Google Scholar]
- 15.Barger JF, Plas DR. Balancing biosynthesis and bioenergetics: metabolic programs in oncogenesis. Endocrine-related cancer. 2010;17:R287–304. doi: 10.1677/ERC-10-0106. [DOI] [PubMed] [Google Scholar]
- 16.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–21. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- 17.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nature reviews Cancer. 2011;11:325–37. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 18.Guo D, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Science signaling. 2009;2:ra82. doi: 10.1126/scisignal.2000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo D, et al. An LXR agonist promotes GBM cell death through inhibition of an EGFR/AKT/SREBP- 1/LDLR-dependent pathway. Cancer discovery. 2011;1:442–456. doi: 10.1158/2159-8290.CD-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ettinger SL, et al. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer research. 2004;64:2212–21. doi: 10.1158/0008-5472.can-2148-2. [DOI] [PubMed] [Google Scholar]
- 21.DeBerardinis RJ, et al. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–31. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–40. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 24.Osborne TF, Espenshade PJ. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it’s been. Genes & development. 2009;23:2578–91. doi: 10.1101/gad.1854309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horton JD, et al. Overexpression of sterol regulatory element-binding protein-1a in mouse adipose tissue produces adipocyte hypertrophy, increased fatty acid secretion, and fatty liver. J Biol Chem. 2003;278:36652–60. doi: 10.1074/jbc.M306540200. [DOI] [PubMed] [Google Scholar]
- 26.Horton JD, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027–32. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends in endocrinology and metabolism: TEM. 2011 doi: 10.1016/j.tem.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokoyama C, et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell. 1993;75:187–97. [PubMed] [Google Scholar]
- 29.Motallebipour M, et al. Novel genes in cell cycle control and lipid metabolism with dynamically regulated binding sites for sterol regulatory element-binding protein 1 and RNA polymerase II in HepG2 cells detected by chromatin immunoprecipitation with microarray detection. The FEBS journal. 2009;276:1878–90. doi: 10.1111/j.1742-4658.2009.06914.x. [DOI] [PubMed] [Google Scholar]
- 30.Nakakuki M, et al. A transcription factor of lipid synthesis, sterol regulatory element-binding protein (SREBP)-1a causes G(1) cell-cycle arrest after accumulation of cyclin-dependent kinase (cdk) inhibitors. The FEBS journal. 2007;274:4440–52. doi: 10.1111/j.1742-4658.2007.05973.x. [DOI] [PubMed] [Google Scholar]
- 31.Porstmann T, et al. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24:6465–81. doi: 10.1038/sj.onc.1208802. [DOI] [PubMed] [Google Scholar]
- 32.Yecies JL, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell metabolism. 2011;14:21–32. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sundqvist A, Ericsson J. Transcription-dependent degradation controls the stability of the SREBP family of transcription factors. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13833–8. doi: 10.1073/pnas.2335135100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bengoechea-Alonso MT, Ericsson J. SREBP in signal transduction: cholesterol metabolism and beyond. Current opinion in cell biology. 2007;19:215–22. doi: 10.1016/j.ceb.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 35.Sundqvist A, et al. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7) Cell metabolism. 2005;1:379–91. doi: 10.1016/j.cmet.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 36.Porstmann T, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell metabolism. 2008;8:224–36. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peterson TR, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–20. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:3441–6. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duvel K, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–83. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cloughesy TF, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN- deficient glioblastoma. PLoS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peterson TR, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–86. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 43.Hagiwara A, et al. Hepatic mTORC2 Activates Glycolysis and Lipogenesis through Akt, Glucokinase, and SREBP1c. Cell metabolism. 2012;15:725–38. doi: 10.1016/j.cmet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 44.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 45.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–70. [PubMed] [Google Scholar]
- 46.Katome T, et al. Use of RNA interference-mediated gene silencing and adenoviral overexpression to elucidate the roles of AKT/protein kinase B isoforms in insulin actions. The Journal of biological chemistry. 2003;278:28312–23. doi: 10.1074/jbc.M302094200. [DOI] [PubMed] [Google Scholar]
- 47.Cong LN, et al. Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Molecular endocrinology. 1997;11:1881–90. doi: 10.1210/mend.11.13.0027. [DOI] [PubMed] [Google Scholar]
- 48.Wolf A, et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. The Journal of experimental medicine. 2011;208:313–26. doi: 10.1084/jem.20101470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nature reviews. Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 50.Menendez JA, Vellon L, Lupu R. Targeting fatty acid synthase-driven lipid rafts: a novel strategy to overcome trastuzumab resistance in breast cancer cells. Medical hypotheses. 2005;64:997–1001. doi: 10.1016/j.mehy.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 51.Menendez JA, Colomer R, Lupu R. Why does tumor-associated fatty acid synthase (oncogenic antigen-519) ignore dietary fatty acids? Medical hypotheses. 2005;64:342–9. doi: 10.1016/j.mehy.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 52.Hardwick DC. The fate of acetyl groups derived from glucose in the isolated perfused goat udder. The Biochemical journal. 1966;99:228–31. doi: 10.1042/bj0990228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bauer DE, et al. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24:6314–22. doi: 10.1038/sj.onc.1208773. [DOI] [PubMed] [Google Scholar]
- 54.Ganguly J. Studies on the mechanism of fatty acid synthesis. VII. Biosynthesis of fatty acids from malonyl CoA. Biochimica etbiophysica acta. 1960;40:110–8. doi: 10.1016/0006-3002(60)91320-2. [DOI] [PubMed] [Google Scholar]
- 55.Gregolin C, et al. Molecular characteristics of liver acetyl CoA carboxylase. Proceedings of the National Academy of Sciences of the United States of America. 1966;56:148–55. doi: 10.1073/pnas.56.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Menendez JA, Lupu R, Colomer R. Obesity, fatty acid synthase, and cancer: serendipity or forgotten causal linkage? Molecular genetics and metabolism. 2005;84:293–5. doi: 10.1016/j.ymgme.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 57.Wellen KE, et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–80. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wolf A, et al. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiology of disease. 2011;44:84–91. doi: 10.1016/j.nbd.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 59.Menendez JA, Lupu R, Colomer R. Targeting fatty acid synthase: potential for therapeutic intervention in her-2/neu-overexpressing breast cancer. Drug news & perspectives. 2005;18:375–85. doi: 10.1358/dnp.2005.18.6.927929. [DOI] [PubMed] [Google Scholar]
- 60.Van de Sande T, et al. Role of the phosphatidylinositol 3’-kinase/PTEN/Akt kinase pathway in the overexpression of fatty acid synthase in LNCaP prostate cancer cells. Cancer research. 2002;62:642–6. [PubMed] [Google Scholar]
- 61.Kumar-Sinha C, et al. Transcriptome analysis of HER2 reveals a molecular connection to fatty acid synthesis. Cancer research. 2003;63:132–9. [PubMed] [Google Scholar]
- 62.Dang CV. Therapeutic Targeting of Myc-Reprogrammed Cancer Cell Metabolism. Cold Spring Harbor symposia on quantitative biology. 2011 doi: 10.1101/sqb.2011.76.011296. [DOI] [PubMed] [Google Scholar]
- 63.DeBerardinis RJ, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–50. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Erickson JW, Cerione RA. Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget. 2010;1:734–40. doi: 10.18632/oncotarget.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wise DR, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:19611–6. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Le A, et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell metabolism. 2012;15:110–21. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosell R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. The lancet oncology. 2012 doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 68.Prahallad A, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012 doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 69.Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- 70.Cheng CK, Fan QW, Weiss WA. PI3K signaling in glioma--animal models and therapeutic challenges. Brain pathology. 2009;19:112–20. doi: 10.1111/j.1750-3639.2008.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chinot OL, et al. AVAglio: Phase 3 trial of bevacizumab plus temozolomide and radiotherapy in newlydiagnosed glioblastoma multiforme. Advances in therapy. 2011;28:334–40. doi: 10.1007/s12325-011-0007-3. [DOI] [PubMed] [Google Scholar]
- 72.Atefi M, et al. Reversing Melanoma Cross-Resistance to BRAF and MEK Inhibitors by Co-Targeting the AKT/mTOR Pathway. PLoS One. 2011;6:e28973. doi: 10.1371/journal.pone.0028973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Niehr F, et al. Combination therapy with vemurafenib (PLX4032/RG7204 and metformin in melanoma cell lines with distinct driver mutations. Journal of translational medicine. 2011;9:76. doi: 10.1186/1479-5876-9-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sondergaard JN, et al. Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J Transl Med. 2010;8:39. doi: 10.1186/1479-5876-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maschek G, et al. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer research. 2004;64:31–4. doi: 10.1158/0008-5472.can-03-3294. [DOI] [PubMed] [Google Scholar]
- 76.Dang CV, et al. Therapeutic targeting of cancer cell metabolism. Journal of molecular medicine. 2011;89:205–12. doi: 10.1007/s00109-011-0730-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo D, et al. AMPK: A metabolic checkpoint that regulates the growth of EGFR activated glioblastomas. Cell Cycle. 2010;9:211–2. doi: 10.4161/cc.9.2.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guo D, et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12932–7. doi: 10.1073/pnas.0906606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abramson HN. The lipogenesis pathway as a cancer target. Journal of medicinal chemistry. 2011;54:5615–38. doi: 10.1021/jm2005805. [DOI] [PubMed] [Google Scholar]
- 80.Yoon S, et al. Up-regulation of acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. The Journal of biological chemistry. 2007;282:26122–31. doi: 10.1074/jbc.M702854200. [DOI] [PubMed] [Google Scholar]
- 81.Hatzivassiliou G, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–21. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 82.Vazquez-Martin A, et al. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int J Oncol. 2007;31:769–76. [PubMed] [Google Scholar]
- 83.Chuang HY, Chang YF, Hwang JJ. Antitumor effect of orlistat, a fatty acid synthase inhibitor, is via activation of caspase-3 on human colorectal carcinoma-bearing animal. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2011;65:286–92. doi: 10.1016/j.biopha.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 84.Beckers A, et al. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer research. 2007;67:8180–7. doi: 10.1158/0008-5472.CAN-07-0389. [DOI] [PubMed] [Google Scholar]
- 85.Kuhajda FP, et al. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:3450–4. doi: 10.1073/pnas.050582897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lupu R, Menendez JA. Pharmacological inhibitors of Fatty Acid Synthase (FASN)--catalyzed endogenous fatty acid biogenesis: a new family of anti-cancer agents? Current pharmaceutical biotechnology. 2006;7:483–93. doi: 10.2174/138920106779116928. [DOI] [PubMed] [Google Scholar]
- 87.Menendez JA, et al. Pharmacological inhibition of fatty acid synthase (FAS): a novel therapeutic approach for breast cancer chemoprevention through its ability to suppress Her-2/neu (erbB-2) oncogene-induced malignant transformation. Molecular carcinogenesis. 2004;41:164–78. doi: 10.1002/mc.20054. [DOI] [PubMed] [Google Scholar]
- 88.Karantonis HC, Nomikos T, Demopoulos CA. Triacylglycerol metabolism. Current drug targets. 2009;10:302–19. doi: 10.2174/138945009787846443. [DOI] [PubMed] [Google Scholar]
- 89.Yen CL, et al. Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. Journal of lipid research. 2008;49:2283–301. doi: 10.1194/jlr.R800018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lass A, et al. Lipolysis - a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Progress in lipid research. 2011;50:14–27. doi: 10.1016/j.plipres.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krahmer N, et al. SnapShot: Lipid Droplets. Cell. 2009;139:1024–1024 e1. doi: 10.1016/j.cell.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 92.Nomura DK, et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zechner R, et al. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. Journal of lipid research. 2009;50:3–21. doi: 10.1194/jlr.R800031-JLR200. [DOI] [PubMed] [Google Scholar]
- 94.Holm C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochemical Society transactions. 2003;31:1120–4. doi: 10.1042/bst0311120. [DOI] [PubMed] [Google Scholar]
- 95.Holm C, et al. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Annual review of nutrition. 2000;20:365–93. doi: 10.1146/annurev.nutr.20.1.365. [DOI] [PubMed] [Google Scholar]
- 96.Nieman KM, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nature medicine. 2011;17:1498–503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Coleman RA, et al. Do long-chain acyl-CoA synthetases regulate fatty acid entry into synthetic versus degradative pathways? The Journal of nutrition. 2002;132:2123–6. doi: 10.1093/jn/132.8.2123. [DOI] [PubMed] [Google Scholar]
- 98.Yamashita Y, et al. Fatty acid induced glioma cell growth is mediated by the acyl-CoA synthetase 5 gene located on chromosome 10q25.1-q25.2, a region frequently deleted in malignant gliomas. Oncogene. 2000;19:5919–25. doi: 10.1038/sj.onc.1203981. [DOI] [PubMed] [Google Scholar]
- 99.Mashima T, et al. p53-defective tumors with a functional apoptosome-mediated pathway: a new therapeutic target. Journal of the National Cancer Institute. 2005;97:765–77. doi: 10.1093/jnci/dji133. [DOI] [PubMed] [Google Scholar]
- 100.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 101.Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–14. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Raghow R, et al. SREBPs: the crossroads of physiological and pathological lipid homeostasis. Trends in endocrinology and metabolism: TEM. 2008;19:65–73. doi: 10.1016/j.tem.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 103.Maxfield FR, van Meer G. Cholesterol, the central lipid of mammalian cells. Curr Opin Cell Biol. 2010;22:422–9. doi: 10.1016/j.ceb.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Segrest JP, et al. Structure of apolipoprotein B-100 in low density lipoproteins. J Lipid Res. 2001;42:1346–67. [PubMed] [Google Scholar]
- 105.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–8. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brown MS, Ho YK, Goldstein JL. The low-density lipoprotein pathway in human fibroblasts: relation between cell surface receptor binding and endocytosis of low-density lipoprotein. Ann N Y Acad Sci. 1976;275:244–57. doi: 10.1111/j.1749-6632.1976.tb43358.x. [DOI] [PubMed] [Google Scholar]
- 107.Di Vizio D, Solomon KR, Freeman MR. Cholesterol and cholesterol-rich membranes in prostate cancer: an update. Tumori. 2008;94:633–9. doi: 10.1177/030089160809400501. [DOI] [PubMed] [Google Scholar]
- 108.Rudling MJ, et al. Low density lipoprotein receptor activity in human intracranial tumors and its relation to the cholesterol requirement. Cancer Res. 1990;50:483–7. [PubMed] [Google Scholar]
- 109.Calkin AC, Tontonoz P. Liver x receptor signaling pathways and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:1513–8. doi: 10.1161/ATVBAHA.109.191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Joseph SB, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A. 2002;99:7604–9. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chuu CP, Lin HP. Antiproliferative effect of LXR agonists T0901317 and 22(R)-hydroxycholesterol on multiple human cancer cell lines. Anticancer research. 2010;30:3643–8. [PubMed] [Google Scholar]
- 112.Mehrotra A, Kaul D, Joshi K. LXR-alpha selectively reprogrammes cancer cells to enter into apoptosis. Molecular and cellular biochemistry. 2011;349:41–55. doi: 10.1007/s11010-010-0659-3. [DOI] [PubMed] [Google Scholar]
- 113.Schultz JR, et al. Role of LXRs in control of lipogenesis. Genes & development. 2000;14:2831–8. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zelcer N, et al. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325:100–4. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Freed-Pastor WA, et al. Mutant p53 Disrupts Mammary Tissue Architecture via the Mevalonate Pathway. Cell. 2012;148:244–58. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Goldstein JL, Brown MS. From fatty streak to fatty liver: 33 years of joint publications in the JCI. J Clin Invest. 2008;118:1220–2. doi: 10.1172/JCI34973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rosenson RS, Otvos JD, Hsia J. Effects of rosuvastatin and atorvastatin on LDL and HDL particle concentrations in patients with metabolic syndrome: a randomized, double-blind, controlled study. Diabetes care. 2009;32:1087–91. doi: 10.2337/dc08-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kones R. The Jupiter study, CRP screening, and aggressive statin therapy-implications for the primary prevention of cardiovascular disease. Therapeutic advances in cardiovascular disease. 2009;3:309–15. doi: 10.1177/1753944709337056. [DOI] [PubMed] [Google Scholar]
- 119.Demierre MF, et al. Statins and cancer prevention. Nat Rev Cancer. 2005;5:930–42. doi: 10.1038/nrc1751. [DOI] [PubMed] [Google Scholar]
- 120.Solomon KR, Freeman MR. Do the cholesterol-lowering properties of statins affect cancer risk? Trends Endocrinol Metab. 2008;19:113–21. doi: 10.1016/j.tem.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 121.Bonovas S, Filioussi K, Sitaras NM. Statins are not associated with a reduced risk of pancreatic cancer at the population level, when taken at low doses for managing hypercholesterolemia: evidence from a meta-analysis of 12 studies. The American journal of gastroenterology. 2008;103:2646–51. doi: 10.1111/j.1572-0241.2008.02051.x. [DOI] [PubMed] [Google Scholar]
- 122.Dale KM, et al. Statins and cancer risk: a meta-analysis. JAMA : the journal of the American Medical Association. 2006;295:74–80. doi: 10.1001/jama.295.1.74. [DOI] [PubMed] [Google Scholar]