

Abstract
Desulfotomaculum gibsoniae is a mesophilic member of the polyphyletic spore-forming genus Desulfotomaculum within the family Peptococcaceae. This bacterium was isolated from a freshwater ditch and is of interest because it can grow with a large variety of organic substrates, in particular several aromatic compounds, short-chain and medium-chain fatty acids, which are degraded completely to carbon dioxide coupled to the reduction of sulfate. It can grow autotrophically with H2 + CO2 and sulfate and slowly acetogenically with H2 + CO2, formate or methoxylated aromatic compounds in the absence of sulfate. It does not require any vitamins for growth. Here, we describe the features of D. gibsoniae strain GrollT together with the genome sequence and annotation. The chromosome has 4,855,529 bp organized in one circular contig and is the largest genome of all sequenced Desulfotomaculum spp. to date. A total of 4,666 candidate protein-encoding genes and 96 RNA genes were identified. Genes of the acetyl-CoA pathway, possibly involved in heterotrophic growth and in CO2 fixation during autotrophic growth, are present. The genome contains a large set of genes for the anaerobic transformation and degradation of aromatic compounds, which are lacking in the other sequenced Desulfotomaculum genomes.
Keywords: spore-forming anaerobes, sulfate reduction, autotrophic, anaerobic degradation of aromatic compounds, complete oxidizer, Peptococcaceae, Clostridiales
Introduction
Desulfotomaculum gibsoniae strain GrollT (DSM 7213) is a mesophilic sulfate-reducing bacterium isolated from a freshwater ditch in Bremen, Northern Germany [1,2]. It grows with a wide range of substrates, including organic acids, such as medium-chain fatty acids, short-chain fatty acids, and several aromatic compounds [1]. These substrates are degraded to CO2 coupled to sulfate reduction. The strain is also able to grow autotrophically with H2/CO2 and sulfate, and is able to ferment pyruvate and crotonate. In the absence of sulfate, it grows slowly on H2/CO2, formate, and methoxylated aromatic compounds. D. gibsoniae does not require vitamins for growth.
The genus Desulfotomaculum is a heterogeneous group of anaerobic spore-forming sulfate-reducing bacteria, with thermophilic, mesophilic, and psychrophilic members that grow at neutral or alkaline pH values [3]. Their cell wall stains Gram-negative, but the ultrastructure of the cell wall is characteristic of Gram-positive bacteria [4]. They are physiologically very diverse. In contrast to Gram-negative sulfate-reducing bacteria and closely related Clostridia, very little is known about their physiology, but members of this genus are known to play an important role in the carbon and sulfur cycle in diverse habitats.
The Desulfotomaculum genus is divided phylogenetically into different subgroups [1]. To get a thorough understanding of the evolutionary relationships of the different Desulfotomaculum subgroups and the physiology of the individual species, it is important to have genome sequence information. Here, we present a summary of the features of D. gibsoniae strain GrollT, together with the description of the complete genomic sequencing and annotation. A special emphasis is put on the ability of this strain to grow on a large variety of aromatic compounds and the responsible genes, and its capacity for acetogenic growth in the absence of sulfate.
Classification and features
D. gibsoniae is a member of the phylum Firmicutes. Phylogenetic analysis of the 16S rRNA genes of D. gibsoniae shows that it clusters in Desulfotomaculum cluster 1, subgroup b. (Figure 1 [1]). Other species in this subgroup are D. geothermicum, D. arcticum, D. alcoholivorax, D. thermosapovorans, D. sapomandens and the non-Desulfotomaculum species Sporotomaculum hydroxybenzoicum and S. syntrophicum.
Figure 1.

Neighbor joining tree based on 16S rRNA gene sequences showing the phylogenetic affiliations of Desulfotomaculum and related species and highlighted to show the subgroups of Desulfotomaculum cluster 1. D. gibsoniae is printed in bold type. The recently described Desulfotomaculum defluvii (cluster 1a), Desulfotomaculum intricatum (cluster 1f), Desulfotomaculum peckii (cluster 1e), and Desulfotomaculum varum (cluster 1a) and the entire cluster 1g are not included in the tree. A set of Thermotogales species were used as outgroup, but were pruned from the tree. Closed circles represent bootstrap values between 75 and 100%. The scale bar represents 10% sequence difference.
D. gibsoniae is a mesophilic sulfate reducer, with an optimum growth temperature between 35-37°C [1,2]. Fermentative and acetogenic growth was shown with pyruvate, crotonate, formate, H2 + CO2, and methoxylated aromatic compounds as substrates. In the presence of an electron acceptor it can completely oxidize substrates to CO2. Suitable electron acceptors are sulfate, thiosulfate and sulfite. The cells of D. gibsoniae are straight or slightly curved rods (1.0-2.5 × 4-7 μm) with pointed ends (Figure 2). Spores of D. gibsoniae are spherical and located in the center of the cells, causing swelling. A summary of the classification and general features of D. gibsoniae is presented in Table 1.
Figure 2.

Scanning electron micrograph of D. gibsoniae strain GrollT.
Table 1. Classification and general features of D. gibsoniae strain GrollT (DSM 7213) according to the MIGS recommendations [5].
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [6] | |
| Phylum Firmicutes | TAS [6-8] | ||
| Class Clostridia | TAS [9,10] | ||
| Order Clostridiales | TAS [11,12] | ||
| Family Peptococcaceae | TAS [11,13] | ||
| Genus Desulfotomaculum | TAS [11,14,15] | ||
| Species Desulfotomaculum gibsoniae | TAS [1] | ||
| Type strain Groll | |||
| Gram stain | Negative with a Gram-positive cell wall structure | TAS [1,2] | |
| Cell shape | Straight or slightly curved rods with pointed ends | TAS [1,2] | |
| Motility | Motile, but motility was lost during cultivation | TAS [1,2] | |
| Sporulation | Spherical and central, slightly swelling the cell | TAS [1,2] | |
| Temperature range | 20-40°C | TAS [1,2] | |
| Optimum temperature | 35-37°C | TAS [1,2] | |
| Carbon source | CO2 (autotrophic) and many organic compounds including aromatic compounds | TAS [1,2] | |
| Energy source | Sulfate-dependent growth and fermentative growth with pyruvate, crotonate, formate, H2 + CO2, and methoxylated aromatic compounds |
TAS [1,2] | |
| Electron acceptor | Sulfate, thiosulfate and sulfite | TAS [1,2] | |
| MIGS-6 | Habitat | Fresh water, mud, soil | TAS [1,2] |
| MIGS-6.2 | pH range Optimum pH |
6.0-8.0 6.9-7.2 |
TAS [1,2] |
| MIGS-6.3 | Salinity | 0-35 g l-1, no addition of NaCl necessary | TAS [1,2] |
| MIGS-22 | Oxygen | Obligate anaerobe | TAS [1,2] |
| MIGS-15 | Biotic relationship | Free living | TAS [1,2] |
| MIGS-14 | Pathogenicity | BSF 1 [16] | |
| MIGS-4 | Geographic location | Grolland, Bremen, Germany | TAS [1,2] |
| MIGS-5 | Sample collection time | Spring 1989 | |
| MIGS-4.1 | Latitude | 53.058 N | |
| MIGS-4.2 | Longitude | 8.762 E | |
| MIGS-4.3 | Depth | 60 cm (water), 1 cm sediment |
Evidence codes - TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). Evidence codes are from the Gene Ontology project [17].
Genome sequencing and annotation
Genome project history
D. gibsoniae was selected for sequencing in the DOE Joint Genome Institute Community Sequencing Program 2009, proposal 300132_795700 'Exploring the genetic and physiological diversity of Desulfotomaculum species', because of its phylogenetic position in one of the Desulfotomaculum subgroups and its ability to use aromatic compounds for growth. The genome project is listed in the Genome OnLine Database (GOLD) [18] as project Gi07572, and the complete genome sequence is deposited in Genbank. Sequencing, finishing and annotation of the D. gibsoniae genome were performed by the DOE Joint Genome Institute (JGI) using state of the art sequencing technology [19]. A summary of the project information is shown in Table 2.
Table 2. Genome sequencing project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Three genomic libraries: one Illumina shotgun library, one 454 standard library, and one paired end 454 library |
| MIGS-29 | Sequencing platforms | Illumina GAii, 454 Titanium |
| MIGS-31.2 | Genome coverage | 479 × Illumina; 27.2 × pyrosequence |
| MIGS-30 | Assemblers | Newbler v. 2.3 |
| MIGS-32 | Gene calling method | Prodigal, GenePRIMP |
| INSDC ID | CP003273 | |
| Genbank Date of Release | May 13, 2013 | |
| MIGS-13 | GOLD ID NCBI project ID Database: IMG Source material identifier |
Gc0017752 59873 2508501002 DSM 7213T |
| Project relevance | Obtain insight into the phylogenetic and physiological diversity of Desulfotomacum species, and genes for anaerobic degradation of aromatic compounds |
Growth conditions and DNA isolation
D. gibsoniae strain GrollT, DSM 7213, was grown anaerobically in DSMZ medium 124a (Desulfotomaculum Groll Medium) [2,20] at 35°C. DNA was isolated from 0.5-1 g of cell paste using Jetflex Genomic DNA Purification kit (GENOMED 600100) following the standard protocol as recommended by the manufacturer. DNA quality was inspected according the guidelines of the genome sequence laboratory.
Genome sequencing and assembly
The genome was sequenced using a combination of Illumina and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website [21]. Pyrosequencing reads were assembled using the Newbler assembler (Roche). The initial Newbler assembly consisting of 139 contigs in one scaffold was converted into a phrap [22] assembly by making fake reads from the consensus, to collect the read pairs in the 454 paired end library. Illumina GAii sequencing data (2,432 Mb) was assembled with Velvet [23] and the consensus sequences were shredded into 1.5 kb overlapped fake reads and assembled together with the 454 data. The 454 draft assembly was based on 220 Mb 454 draft data and all of the 454 paired end data. Newbler parameters are -consed -a 50 -l 350 -g -m -ml 21. The Phred/Phrap/Consed software package [22] was used for sequence assembly and quality assessment in the subsequent finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution [22], Dupfinisher [24], or sequencing cloned bridging PCR fragments with subcloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J.-F. Chang, unpublished). A total of 132 additional reactions were necessary to close some gaps and to raise the quality of the final contigs. Illumina reads were also used to correct potential base errors and increase consensus quality using a software Polisher developed at JGI [25]. The error rate of the final genome sequence is less than 1 in 100,000. Together, the combination of the Illumina and 454 sequencing platforms provided 506.2 × coverage of the genome. The final assembly is based on 2,347 Mb of Illumina draft data and 133 Mb of pyrosequence draft data.
Genome annotation
Genes were identified using Prodigal [26] as part of the DOE-JGI genome annotation pipeline [27], followed by a round of manual curation using the JGI GenePRIMP pipeline [28]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [29].
Genome properties
The genome consists of one circular chromosome of 4,855,529 bp (45.49% GC content) and includes no plasmids. A total of 4,762 genes were predicted, of which 4,666 are protein-coding genes. In addition, 3,464 of protein coding genes (72.7%) were assigned to a putative function with the remaining annotated as hypothetical proteins. The statistics of the genome are summarized in Table 3. 70.24% of the total genes were assigned to the COG functional categories (Table 4 and Figure 3).
Table 3. Genome statistics.
| Attribute | Value | % of total |
|---|---|---|
| Genome size (bp) | 4,855,529 | 100.00 |
| DNA coding region (bp) | 3,949,133 | 81.33 |
| DNA G+C content (bp) | 2,208,827 | 45.49 |
| Total genes | 4,762 | 100.00 |
| RNA genes | 96 | 2.02 |
| Protein-coding genes | 4,666 | 97.98 |
| Genes in paralog clusters | 2,789 | 58.57 |
| Genes assigned to COGs | 3,345 | 70.24 |
| Pseudo genes | 314 | 6.59 |
| Genes with signal peptides | 737 | 15.48 |
| Genes with transmembrane helices | 966 | 20.29 |
Table 4. Number of genes associated with the general COG functional categories.
| Code | Value | %age | Description |
|---|---|---|---|
| J | 157 | 4.26 | Translation |
| A | 1 | 0.03 | RNA processing and modification |
| K | 331 | 8.98 | Transcription |
| L | 276 | 7.49 | Replication, recombination and repair |
| B | 1 | 0.03 | Chromatin structure and dynamics |
| D | 51 | 1.38 | Cell cycle control, mitosis and meiosis |
| Y | 0 | 0.00 | Nuclear structure |
| V | 68 | 1.84 | Defense mechanisms |
| T | 237 | 6.43 | Signal transduction mechanisms |
| M | 169 | 4.58 | Cell wall/membrane biogenesis |
| N | 81 | 2.20 | Cell motility |
| Z | 0 | 0.00 | Cytoskeleton |
| W | 0 | 0.00 | Extracellular structures |
| U | 74 | 2.01 | Intracellular trafficking and secretion |
| O | 103 | 1.79 | Posttranslational modification, protein turnover, chaperones |
| C | 365 | 9.90 | Energy production and conversion |
| G | 104 | 2.82 | Carbohydrate transport and metabolism |
| E | 255 | 6.92 | Amino acid transport and metabolism |
| F | 73 | 1.98 | Nucleotide transport and metabolism |
| H | 188 | 5.10 | Coenzyme transport and metabolism |
| I | 158 | 4.29 | Lipid transport and metabolism |
| P | 151 | 4.10 | Inorganic ion transport and metabolism |
| Q | 75 | 2.03 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 459 | 12.45 | General function prediction only |
| S | 309 | 8.38 | Function unknown |
| - | 1417 | 29.76 | Not in COGs |
Figure 3.

Graphical map of the chromosome. From outside to the center: Genes on forward strand (color by COG categories), genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content (black), GC skew (purple/olive).
Insights into the genome
Degradation of aromatic compounds
D. gibsoniae can grow on a large variety of aromatic compounds (Figure 4) [1,2]. Other bacteria capable of growth via anaerobic degradation of aromatic compounds linked to nitrate reduction, Fe(III) reduction, or sulfate reduction are much more restricted [30,31].
Figure 4.

Degradation of aromatic compounds in D. gibsoniae based on genomic data. Enzymes are highlighted in red color. Possible encoding genes and their locus tags are shown below. Abbreviations: BamBCDEFGHI, class II benzoyl-CoA reductase; BCL, benzoate CoA-ligase; BCT, succinyl-CoA:benzoate CoA-transferase; BzdNOPQ, class I benzoyl-CoA reductase; BhsABCDEFGH, beta-oxidation of hydroxybenzylsuccinate; DCH, cyclohexa-1,5-diene-CoA hydratase; HAD, 6-OH-cyclohex-1-ene-1-carbonyl-CoA dehydrogenase; HbsABC, hydroxybenzylsuccinate synthase; OAH, 6-oxo-cyclohex-1-ene-1-carbonyl-CoA hydrolase; PadBCD, phenylacetyl-CoA acceptor oxidoreductase; padEGHI, phenylglyoxylate oxidoreductase; PadJ, phenylacetate CoA-ligase; PcmRST, 4-OH-benzoyl-CoA reductase; PPC, phenylphosphate carboxylase; PpsAB, phenylphosphate synthase.
In sulfate-reducing bacteria (e.g. Desulfobacula toluolica) methylated aromatic compounds such as toluenes, xylenes or cresols are thought to be degraded via an initial fumarate addition to the methyl group followed by β-oxidation-like reactions [32-34]. The genes putatively coding for the enzyme catalyzing the fumarate addition reaction (hbsABC) are present in two copies in the genome of D. gibsoniae. They might have different substrate specificities for the growth substrates m- and p-cresol since the genome of D. toluolica possesses one set of these genes and can only grow with p-cresol [34,35]. In D. gibsoniae m- and p-cresol are expected to be converted to 3- or 4-hydroxybenzylsuccinate. The genes coding for enzymes involved in the subsequent β-oxidation (bhsABCDEFGH), yielding 3- or 4-hydroxybenzoyl-CoA, are also present in two copies. In growth experiments toluene degradation was not observed for D. gibsoniae [1,2]. The genome provides no opposing information. All genes for the degradation of the growth substrates phenylacetate and phenol are present including the type of phenylphosphate carboxylase typically found in strict anaerobes [36].
All genes encoding enzymes of the upper benzoyl-CoA degradation pathway were identified in D. gibsoniae. The growth substrate benzoate is activated to benzoyl-CoA either via ATP-dependent CoA-ligase (bcl) or succinyl-CoA dependent CoA-transferase (bct) [37,38]. There are two classes of dearomatizing benzoyl-CoA reductases (BCRs) [39]. Class I are ATP-dependent FeS enzymes composed of four different subunits [40]. There are two subclasses of ATP-dependent BCRs of the Thauera- and the Azoarcus-type. ATP-independent class II BCRs contain eight subunits and harbor a tungsten-containing cofactor in the active site [41]. The ATP-independent class II BCR is characteristic of strictly anaerobic aromatic compound degrading bacteria [42]. In D. gibsoniae the genes of the catalytic subunit (bamB) of the class II BCR are present in six copies. All of the predicted seven genes for the putative electron activating subunits of class II BCR (bamCDEFGHI) were identified in at least two copies and arranged next to each other. Surprisingly, genes of a class I BCR with high similarity (47-68% amino acid identity) to class I BCRs of the Azoarcus-type (bzdNOPQ) were found, but these were not located in a single transcriptional unit. It is unclear which of the putative BCR-encoding genes is used for benzoyl-CoA and/or 3-OH-benzoyl-CoA reduction. The genes necessary to convert the product of BCRs, a cyclic conjugated dienoyl-CoA, to 3-OH-pimelyl-CoA via modified β-oxidation (dch, had, oah) are present in one copy each. It is unclear whether these genes are also involved in 3-OH-benzoyl-CoA degradation.
One of the more unusual growth substrates of D. gibsoniae is catechol, a substrate metabolized only by a very limited number of anaerobic bacteria. The pathway of catechol metabolism via protocatechuate was outlined 20 years ago [2] and is now confirmed by the genome analysis. For the degradation of lignin monomers, the side chains will be degraded and the methoxy-group will be removed by o-demethylation. The genes responsible for this mechanism are present in the genome (Desgi_0674 to Desgi_0676). The resulting compounds can then be degraded by the pathways outlined in Figure 4.
Phylogenetic trees based on hbsA which is a homolog to bssA (Figure 5A) and hbsC which is a homolog tobssC (Figure 5B) show deeply branching lineages for the Desulfotomaculum gibsoniae genes and no clear affiliation to other sulfate-reducing bacteria except, in the case of the hbsC gene to alkane-oxidizing species. Interestingly, similar genes were also found in the genomes of Desulfotignum balticum and Desulfotignum phosphitoxidans. Both are only known to use benzoate or its hydroxyl derivatives, whereas the only other species of this genus, Desulfotignum toluenicum can grow very well on toluene [44-46]. Using the bamB and bamC genes for phylogenetic tree construction (Figure 6A and Figure 6B), the picture is even more heterogenous. The different genes are affiliated with genes found in sulfate-reducing and other bacteria, hence a clear clustering cannot be seen. Again, genome data provides some interesting insights. Desulfospira joergensenii is not described as a benzoate utilizing bacterium, but seems to have some similar genes [47].
Figure 5A.
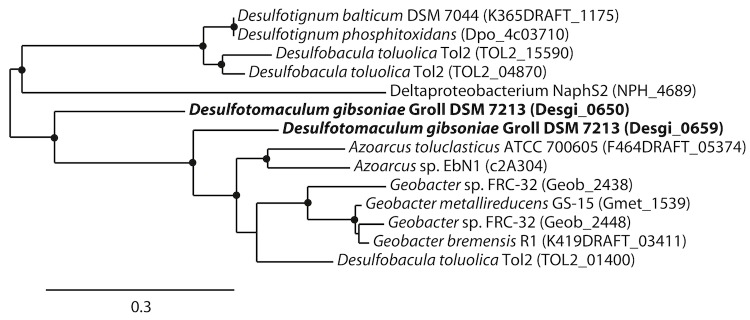
Phylogenetic tree based on amino acid sequences of bssA and hbsA. The trees were calculated with the "One-Click" mode of the online phylogenetic analysis program Phylogeny.fr [43]. Dots represent bootstrap values between 75 and 100%. The sequences of Desulfotomaculum gibsoniae are printed in bold. Azoarcus sp. EbN1 and Geobacter sp. FRC-32 are identical to Aromatoleum aromaticum and Geobacter daltonii respectively.
Figure 5B.
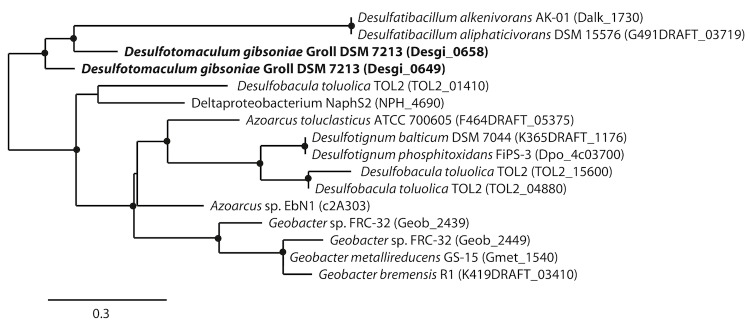
Phylogenetic tree based on amino acid sequences of bssC and hbsC. The trees were calculated with the "One-Click" mode of the online phylogenetic analysis program Phylogeny.fr [43]. Dots represent bootstrap values between 75 and 100%. The sequences of Desulfotomaculum gibsoniae are printed in bold. Azoarcus sp. EbN1 and Geobacter sp. FRC-32 are identical to Aromatoleum aromaticum and Geobacter daltonii respectively.
Figure 6A.

Phylogenetic tree based on amino acid sequences of five of the six homologs of bamB. The trees were calculated with the "One-Click" mode of the online phylogenetic analysis program Phylogeny.fr [43]. Dots represent bootstrap values between 75 and 100%. The sequences of Desulfotomaculum gibsoniae are printed in bold, Geobacter sp. FRC-32 is identical to Geobacter daltonii.
Figure 6B.

Phylogenetic tree based on amino acid sequences of six homologs of bamC. The trees were calculated with the "One-Click" mode of the online phylogenetic analysis program Phylogeny.fr [43]. Dots represent bootstrap values between 75 and 100%. The sequences of Desulfotomaculum gibsoniae are printed in bold, Geobacter sp. FRC-32 is identical to Geobacter daltonii.
Complete substrate oxidation, autotrophic growth and homoacetogenic growth
The genome of D. gibsoniae contains putative genes that code for the enzymes of the complete tricarboxylic acid (TCA) cycle: Citrate synthase, Desgi_1296, 2412; aconitase, Desgi_1576; isocitrate dehydrogenase, Desgi_4665; 2-oxoacid:ferredoxin oxidoreductase, Desgi_0085-0088, 2095, 2585-2588, 3041-3044; succinyl-CoA synthetase, Desgi_1954-1955; succinate dehydrogenase, Desgi_0077-0080, 3996-3998; fumarase, Desgi_0075, 1952-1953; malate dehydrogenase, Desgi_1960. These genes could be involved in the complete oxidation to CO2 by D. gibsoniae. Moreover, the complete acetyl-CoA pathway is also present in the genome of D. gibsoniae (Figure 7). However, D. gibsoniae is not able to grow on acetate with or without sulfate. The acetyl-CoA pathway in D. gibsoniae does not perform acetate oxidation, as described in D. kuznetsovii [48], but facilitates complete oxidation of substrates leading to acetyl-CoA, autotrophic growth on H2 + CO2 (or formate) in the presence of sulfate as electron acceptor, and slow homoacetogenic growth on pyruvate, crotonate, formate, hydrogen plus carbon dioxide, and methoxylated aromatic compounds [1].
Figure 7.

Acetyl-CoA pathway in D. gibsoniae based on genomic data. Enzymes are depicted in bold italic. Next to these enzymes are the possible encoding genes, and their locus tags. Genes with the locus tags Desgi_2048 and Desgi_2050 putatively code for the small subunit and the large subunit of the iron-sulfur protein, respectively. This protein is involved in transferring the methyl from tetrahydrofolate to acetyl-CoA. Abbreviations: A-CoA S, acetyl-CoA synthetase; AcsA, carbon monoxide dehydrogenase; AcsB, acetyl-CoA synthase; CFeSP, iron-sulfur protein; CH3, methyl; THF, tetrahydrofolate; MeTr, methyltransferase.
Three putative acetyl-CoA synthase encoding genes can be found in the D. gibsoniae genome (Figure 8). All three genes have a putative carbon monoxide dehydrogenase catalytic subunit encoding gene (cooS) downstream. However, only Desgi_2051 is part of an operon structure containing other genes coding for enzymes involved in the acetyl-CoA pathway.
Figure 8.
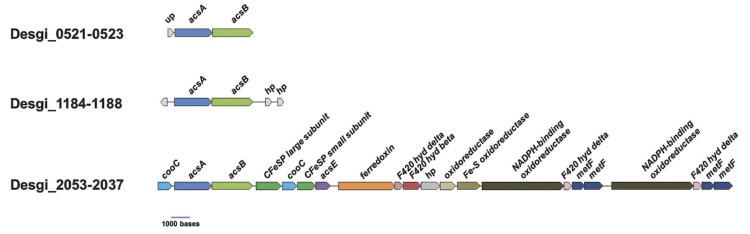
Gene orientation of putative coding acetyl-CoA synthase and neighboring genes in the genome of D. gibsoniae. Abbreviations: acsA, carbon monoxide dehydrogenase; acsB, acetyl-CoA synthase; acsE, methyl-tetrahydrofolate methyltransferase; CFeSP, iron-sulfur protein; cooC, carbon monoxide dehydrogenase maturation factor; Fe-S, Iron sulfur; hp, hypothetical protein; hyd, hydrogenase; metF, methylene-tetrahydrofolate reductase; up, uncharacterized protein.
C1 compound degradation
In addition to the three cooS genes downstream of the genes coding for the acetyl-CoA synthase, D. gibsoniae has two other cooS genes in its genome, Desgi_2753, and Desgi_3080. The latter has a transcriptional regulator (Desgi_3081) downstream and a ferredoxin (Desgi_3079) and a nitrite reductase (Desgi_3078) upstream. Growth tests on CO have not yet been performed. However, the presence of multiple cooS genes with neighbor genes like ferredoxin and nitrate reductase, or genes coding for the acetyl-CoA pathway indicates that D. gibsoniae may grow on CO.
D. gibsoniae can grow on formate coupled to sulfate reduction. In the genome, two putative formate dehydrogenases (FDHs) were found. One FDH (Desgi_1522-23) is translocated over the membrane and bound to a polysulfide reductase (NrfD)-like protein containing 10 trans-membrane helixes (Desgi_1524). The alpha subunit contains a twin-arginine translocation (tat) motif and genes encoding proteins of the Tat system; TatA (Desgi_1521) and TatC (Desgi_1526) were found near the alpha subunit coding gene. The second FDH (Desgi_2136-2139) might be a confurcating FDH. Desgi_2138 shows similarity with the NADH binding 51kD subunit of NADH:ubiquinone oxidoreductase and Fe-S cluster binding motifs, which were found in all subunits.
No methanol methyltransferase genes can be found in the genome of D. gibsoniae, which correlates with the absence of growth on methanol [1]. Other methyltransferase genes that might point to growth with methylated amines were not found, except for a possible dimethylamine methyltransferase beta subunit (Desgi_3904) and a cobalamin binding protein (Desgi_3903). However, another methyltransferase gene, mtbA, which is absent from the genome, is necessary for growth with dimethylamine.
Propionate and butyrate oxidation
The genome of D. gibsoniae contains at least one copy of genes putatively encoding enzymes involved in propionate oxidation via the methylmalonyl-CoA pathway (Figure 9A). This includes genes in a methylmalonyl-CoA (mmc) cluster (Desgi_1951-1961), which have a genetic organization similar to those seen D. kuznetzovii (Desku_1358-1369) and Pelotomaculum thermopropionicum (Pth_1355-1368) [48-50]. However, a few differences were found. The genome of D. gibsoniae lacks genes coding for methylmalonyl-CoA decarboxylase epsilon and gamma subunits. Moreover, the mmc cluster of D. gibsoniae contains a single gene encoding the alpha subunit of succinyl-CoA synthase (Desgi_1955), whereas the mmc clusters of D. kuznetzovii and P. thermopropionicum contain two encoding genes. Bifurcating hydrogenases may be used to re-oxidize ferredoxin, which is generated by pyruvate:ferredoxin oxidoreductase and NADH, which in turn is generated from malate dehydrogenation for the formation of hydrogen. The membrane-anchored extracellular formate dehydrogenases and hydrogenases may be involved in generating a proton motive force for succinate reduction.
Figure 9.

(A) Propionate and (B) butyrate degradation pathways in D. gibsoniae based on genomic data. The two main gene clusters for propionate and butyrate degradation are indicated in red and blue colors. For butyrate degradation, additional gene clusters were found, shown in different colors. For the other steps in the propionate degradation pathway, genes were found to be located in different places in the genome. Abbreviations: PCT, propionate CoA transferase; MCE, methylmalonyl-CoA epimerase; MCM, methylmalonyl-CoA mutase; SCS, succinyl-CoA synthase; SDH, succinate dehydrogenase; FHT, fumarase (fumarate hydratase); MDH, malate dehydrogenase; ODC, oxaloacetate decarboxylase; PFO, pyruvate:ferredoxin oxidoreductase; PFL, pyruvate formate lyase; POT, propionyl-CoA:oxaloacetate transcarboxylase; BAT, butyryl-CoA:acetate-CoA transferase; ACD, acyl-CoA dehydrogenase; ECH, enoyl-CoA hydratase; 3-HCD, 3-hydroxybutyryl-CoA dehydrogenase; ACA, acetyl-CoA acetyltransferase.
Genes putatively coding for butyrate β-oxidation enzymes are also present in the genome of D. gibsoniae. One complete cluster of genes putatively encoding all the enzymes required to convert butyrate is present (Desgi_4671-4675, Figure 9B). Gene organization in this cluster is similar to that found in D. reducens (Dred_1493-1489), which can also utilize butyrate (Figure 10A). In D. gibsoniae another gene cluster (Desgi_1916-1925) is present which only lacks one gene coding for butyryl-CoA:acetate CoA-transferase (Figure 11). Desgi_1918 and Desgi_1920-1925 have a similar organization to genes found in D. acetoxidans (Dtox_1697-1703) [51]. In addition to the genes encoding enzymes involved in butyrate β-oxidation, these clusters contain genes for electron transfer flavoproteins (Desgi_1920-1921 and Dtox_1698-1699) and for Fe-S oxidoreductases (Desgi_1922 and Dtox_1700). Although Dtox_1700 is annotated as a cysteine-rich unknown protein, a protein blast of these ORFs against the D. gibsoniae genome revealed 53.65% identity (Evalue = 0.0) with the putative Fe-S oxidoreductase encoded by Desgi_1922. Two genes encoding acyl-CoA synthetases (Desgi_1916-1917) are present upstream of the acetyl-CoA dehydrogenase gene in D. gibsoniae (Desgi_1918), but these are not found near this cluster in D. acetoxidans. However, these genes are present in the same gene cluster location in other butyrate-degrading sulfate-reducing bacteria (SRB), namely D. alcoholivorax (H569DRAFT_00537-00530), D. kuznetsovii (Desku_1226-1234) and Desulfurispora thermophila (B064DRAFT_00829-00837). Acyl-CoA synthetases are most likely involved in the biosynthesis of coenzyme A [52]. Several other clusters of genes containing at least three genes encoding enzymes involved in butyrate conversion can be found in the genome of D. gibsoniae.
Figure 10.
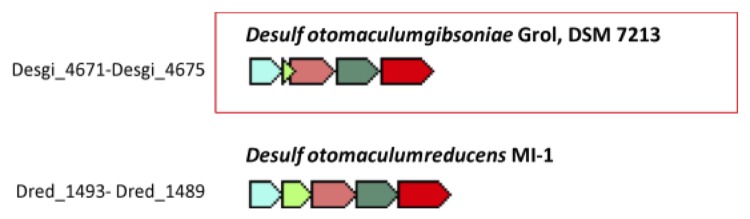
Orthologous neighborhood genes for gene cluster Desgi_4671- 4675, encoding enzymes necessary for butyrate degradation. Light blue – 3-hydroxybutyryl-CoA dehydrogenase (3-HCD); green – enoyl-CoA hydratase (ECH); old pink – acetyl-CoA acetyltransferase (ACA); dark green – Acyl-CoA dehydrogenase (ACD); red – butyryl-CoA:acetate CoA-transferase (BAT).
Figure 11.

Orthologous neighborhood genes for gene cluster Desgi_1916- 1925, encoding enzymes necessary for butyrate degradation. Purple/pink – acyl-CoA synthetase; red – acyl-CoA dehydrogenase (ACD); orange/dark purple – electron transfer flavoprotein; yellow – Fe-S oxidoreductase; old pink – acetyl-CoA acetyltransferase (ACA); light blue – 3-hydroxybutyryl-CoA dehydrogenase (3-HCD); green – enoyl-CoA hydratase (ECH).
Sulfate reduction
The genome contains single copies of the sulfate adenyltransferase (Desgi_3703), adenosine-5´-phosphosulfate (APS) reductase (Desgi_3701–3702) and dissimilatory sulfite reductase (Desgi 4661-4662) as are found in most of the other members of the genus [18-20]. A membrane-bound pyrophosphatase (Desgi_4294) is used for energy regeneration as in other Desulfotomaculum spp. The QmoABC complex contains only the A and B subunit, the C subunit is lacking (Desgi_3699–3700). In all members of the genus Desulfotomaculum the QmoAB is followed by HdrCB (Desgi_3697–3698). This arrangement is identical to that seen in the closely related species “Desulforhudis audaxivator”, Desulfurispora thermophila and the Gram-negative Desulfarculus baarsii and strain NaphS2, which possess a Gram-positive AprBA [53]. Interestingly, the same organization is also found in some phototrophic sulfur-oxidizing bacteria, such as Thiobacillus dentrificans, Thiothrix nivea and Sedimentibacter selenatireducens [54]. Other closely related Gram-positive SRB like Desulfovirgula thermoconiculi and Ammonifex degensii have a complete QmoABC system like all other SRB and the Green Sulfur Bacteria, or have QmoAB linked to a Fe-S oxidoreductase/HdrD as seen in Desulfosporosinus spp. This latter modification is also seen in other Gram-negative SRB, which have a Gram-positive AprBA-like Desulfomonile tiedjei and Syntrophomonas fumaroxidans [55]. It seems that both Desulfotomaculum sp. and Desulfosporosinus have been the source of the entire aps reductase/ QmoA complex for members of the Gram-negative Syntrophobacterales [55]. The genomes of Syntrophobacter fumaroxidans and of Desulfovirgula thermoconiculi have two different systems that can be linked to the aps reductase.
In D. gibsoniae the dsrAB (Desgi_4661–4662) is linked to the same truncated dsr operon coding only for dsrC and dsrMK (Desgi 4648–4649) as in other Desulfotomaculum spp [48,51,56].
Hydrogenases
D. gibsoniae has six [FeFe] and three [NiFe] hydrogenases, suggesting a lower redundancy in the case of [FeFe] enzymes than other members of the genus. The [FeFe] hydrogenases include one membrane-associated protein (Desgi_0926-0928) that contains a tat motif in the alpha subunit (Desgi_0926), suggesting an extracellular localization; one monomeric hydrogenase (Desgi_0935) encoded close to the membrane-bound enzyme, which suggests the possibility of co-regulation; two copies of trimeric NAD(P)-dependent bifurcating hydrogenases (Desgi_4669-4667 and Desgi_3197-3195); one enzyme (Desgi_0771) that is part of a multi-gene cluster encoding two flavin-dependent oxidoreductases that is also present in other Desulfotomaculum spp., and one HsfB-type hydrogenase (Desgi_3194) encoding a PAS-sensing domain that is likely involved in sensing and regulation, and possibly with the bifurcating Desgi_3195 hydrogenase.
The [NiFe] hydrogenases include one enzyme (Desgi_1398 – 1397) that may also be bound to the membrane by a cytochrome b (Desgi_1402); one simple dimeric enzyme (Desgi_1231-1230); and one trimeric group 3 hydrogenase (Desgi_1166-1164), similar to methyl-viologen reducing hydrogenases from methanogens, and which is encoded next to a HdrA-like protein (Desgi_1163).
Nitrogenases
A cluster of nitrogenase genes, specifically genes encoding nitrogenase iron protein, nitrogen regulatory protein PII, nitrogenase molybdenum-iron protein alpha chain, nitrogenase molybdenum-iron protein beta chain, nitrogenase molybdenum-iron cofactor biosynthesis protein NifE, nitrogenase molybdenum-iron protein, alpha and beta chains, nitrogenase cofactor biosynthesis protein NifB; ferredoxin, iron only nitrogenase protein AnfO (AnfO_nitrog) (Desgi_2428-2419) were detected within the annotated genome sequence. Thus, D. gibsoniae probably has the capacity for nitrogen fixation. However, the fixation of molecular nitrogen has not been analyzed in this species so far.
Acknowledgements
The work conducted by the U.S. Department of Energy Joint Genome Institute was supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, and was also supported by grants CW-TOP 700.55.343, ALW 819.02.014 of the Netherlands Science Foundation (NWO), ERC (project 323009), and BE-Basic (project F07.002.03).
References
- 1.Kuever J, Rainey FA, Hippe H. Description of Desulfotomaculum sp. Groll as Desulfotomaculum gibsoniae sp. nov. Int J Syst Bacteriol 1999; 49:1801-1808 10.1099/00207713-49-4-1801 [DOI] [PubMed] [Google Scholar]
- 2.Kuever J, Kulmer J, Jannsen S, Fischer U, Blotevogel KH. Isolation and characterization of a new spore-forming sulfate-reducing bacterium growing by complete oxidation of catechol. Arch Microbiol 1993; 159:282-288 10.1007/BF00248485 [DOI] [PubMed] [Google Scholar]
- 3.Widdel F. The Genus Desulfotomaculum In: Rosenberg E., Stackebrandt E., Thompson F., Lory S., DeLong E. (Ed.) The Prokaryotes (3rd edition) – 4. Bacteria: Firmicutes, Cyanobacteria: SpringerReference (www.springerreference.com). Springer-Verlag Berlin Heidelberg, 2006. DOI: 10.1007/SpringerReference_3746 2011-01-31 23:00:00 UTC. [DOI]
- 4.Liu Y, Karnauchow TM, Jarrell KF, Balkwill DL, Drake GR, Ringelberg D, Clarno R, Boone DR. Description of two new thermophilic Desulfotomaculum spp., Desulfotomaculum putei sp. nov., from a deep terrestrial subsurface, and Desulfotomaculum luciae sp. nov., from a hot spring. Int J Syst Bacteriol 1997; 47:615-621 10.1099/00207713-47-3-615 [DOI] [Google Scholar]
- 5.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576-4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gibbons NE, Murray RGE. Proposals Concerning the Higher Taxa of Bacteria. Int J Syst Bacteriol 1978; 28:1-6 10.1099/00207713-28-1-1 [DOI] [Google Scholar]
- 8.Murray RGE. The Higher Taxa, or, a Place for Everything...? In: Holt JG (ed), Bergey's Manual of Systematic Bacteriology, First Edition, Volume 1, The Williams and Wilkins Co., Baltimore, 1984, p. 31-34. [Google Scholar]
- 9.List of new names and new combinations previously effectively, but not validly, published. List no. 132. Int J Syst Evol Microbiol 2010; 60:469-472 10.1099/ijs.0.022855-0 [DOI] [PubMed] [Google Scholar]
- 10.Rainey FA. Class II. Clostridia class nov. In: De Vos P, Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 3, Springer-Verlag, New York, 2009, p. 736. [Google Scholar]
- 11.Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225-420 10.1099/00207713-30-1-225 [DOI] [PubMed] [Google Scholar]
- 12.Prévot AR. In: Hauderoy P, Ehringer G, Guillot G, Magrou. J., Prévot AR, Rosset D, Urbain A (eds), Dictionnaire des Bactéries Pathogènes, Second Edition, Masson et Cie, Paris, 1953, p. 1-692. [Google Scholar]
- 13.Rogosa M. Peptococcaceae, a new family to include the Gram-positive, anaerobic cocci of the genera Peptococcus, Peptostreptococcus and Ruminococcus. Int J Syst Bacteriol 1971; 21:234-237 10.1099/00207713-21-3-234 [DOI] [Google Scholar]
- 14.Campbell LL, Postgate JR. Classification of the spore-forming sulfate-reducing bacteria. Bacteriol Rev 1965; 29:359-363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell LL. Genus IV. Desulfotomaculum Campbell and Postgate 1965, 361. In: Buchanan RE, Gibbons NE (eds), Bergey's Manual of Determinative Bacteriology, Eighth Edition, The Williams and Wilkins Co., Baltimore, 1974, p. 572-573. [Google Scholar]
- 16.BAuA. 2010, Classification of bacteria and archaea in risk groups. http://www.baua.de TRBA 466, p. 74.
- 17.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2012; 40:D571-D579 10.1093/nar/gkr1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mavromatis K, Land ML, Brettin TS, Quest DJ, Copeland A, Clum A, Goodwin L, Woyke T, Lapidus A, Klenk HP, et al. The fast changing landscape of sequencing technologies and their impact on microbial genome assemblies and annotation. PLoS ONE 2012; 7:e48837 10.1371/journal.pone.0048837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.List of growth media used at DSMZ: http://www.dsmz.de/microorganisms/media_list.php
- 21.JGI website. http://www.jgi.doe.gov/
- 22.The Phred/Phrap/Consed software package. http://www.phrap.com
- 23.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821-829 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han C, Chain P. Finishing repeat regions automatically with Dupfinisher. In: Proceeding of the 2006 international conference on bioinformatics & computational biology. Arabnia HR, Valafar H (eds), CSREA Press. June 26-29, 2006: 141-146. [Google Scholar]
- 25.Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008. [Google Scholar]
- 26.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal Prokaryotic Dynamic Programming Genefinding Algorithm. BMC Bioinformatics 2010; 11:119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC. The DOE-JGI Standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci 2009; 1:63-67 10.4056/sigs.632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pati A, Ivanova N, Mikhailova N, Ovchinikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A Gene Prediction Improvement Pipeline for microbial genomes. Nat Methods 2010; 7:455-457 10.1038/nmeth.1457 [DOI] [PubMed] [Google Scholar]
- 29.Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271-2278 10.1093/bioinformatics/btp393 [DOI] [PubMed] [Google Scholar]
- 30.Evans WC, Fuchs G. Anaerobic degradation of aromatic compounds. Annu Rev Microbiol 1988; 42:289-317 10.1146/annurev.mi.42.100188.001445 [DOI] [PubMed] [Google Scholar]
- 31.Heider J, Fuchs G. Anaerobic Metabolism of Aromatic Compounds. Eur J Biochem 1997; 243:577-596 10.1111/j.1432-1033.1997.00577.x [DOI] [PubMed] [Google Scholar]
- 32.Müller JA, Galushko AS, Kappler A, Schink B. Anaerobic degradation of m-cresol by Desulfobacterium cetonicum is initiated by formation of 3-hydroxybenzylsuccinate. Arch Microbiol 1999; 172:287-294 10.1007/s002030050782 [DOI] [PubMed] [Google Scholar]
- 33.Müller JA, Galushko AS, Kappler A, Schink B. Initiation of anaerobic degradation of p-cresol by formation of 4-hydroxybenzylsuccinate in Desulfobacterium cetonicum. J Bacteriol 2001; 183:752-757 10.1128/JB.183.2.752-757.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wöhlbrand L, Jacob JH, Kube M, Mussmann M, Jarling R, Beck A, Amann R, Wilkes H, Reinhardt R, Rabus R. Complete genome, catabolic sub-proteomes and key-metabolites of Desulfobacula toluolica Tol2, a marine, aromatic compound-degrading, sulfate-reducing bacterium. Environ Microbiol 2013; 15:1334-1355 10.1111/j.1462-2920.2012.02885.x [DOI] [PubMed] [Google Scholar]
- 35.Rabus R, Nordhaus R, Ludwig W, Widdel F. Complete oxidation of toluene under strictly anoxic conditions by a new sulfate-reducing bacterium. Appl Environ Microbiol 1993; 59:1444-1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schleinitz KM, Schmeling S, Jehmlich N, von Bergen M, Harms H, Kleinsteuber S, Vogt C, Fuchs G. Phenol degradation in strictly anaerobic iron-reducing bacterium Geobacter metallireducens GS-15. Appl Environ Microbiol 2009; 75:3912-3919 10.1128/AEM.01525-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wischgoll S, Heintz D, Peters F, Erxleben A, Sarnighausen E, Reski R, Van Dorsselaer A, Boll M. Gene clusters involved in anaerobic benzoate degradation of Geobacter metallireducens. Mol Microbiol 2005; 58:1238-1252 10.1111/j.1365-2958.2005.04909.x [DOI] [PubMed] [Google Scholar]
- 38.Oberender J, Kung JW, Seifert J, von Bergen M, Boll M. Identification and characterization of a succinyl-coenzyme A (CoA):benzoate CoA transferase in Geobacter metallireducens. J Bacteriol 2012; 194:2501-2508 10.1128/JB.00306-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuchs G, Boll M, Heider J. Microbial degradation of aromatic compounds – from one strategy to four. Nat Rev Microbiol 2011; 9:803-816 10.1038/nrmicro2652 [DOI] [PubMed] [Google Scholar]
- 40.Boll M, Fuchs G. Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K172. Eur J Biochem 1995; 234:921-933 10.1111/j.1432-1033.1995.921_a.x [DOI] [PubMed] [Google Scholar]
- 41.Kung JW, Löffler C, Dörner K, Heintz D, Gallien S, Van Dorsselaer A, Friedrich T, Boll M. Identification and characterization of the tungsten-containing class of benzoyl-coenzyme A reductases. Proc Natl Acad Sci USA 2009; 106:17687-17692 10.1073/pnas.0905073106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Löffler C, Kuntze K, Vazquez Ramos J, Rugor A, Kung J, Böttcher A, Boll M. Occurrence, genes and expression of the W/Se-containing class II benzoyl-coenzyme A reductases in anaerobic bacteria. Environ Microbiol 2011; 13:696-709 10.1111/j.1462-2920.2010.02374.x [DOI] [PubMed] [Google Scholar]
- 43.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, et al Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 2008, Jul 1;36(Web Server issue)W465-9. 10.1093/nar/gkn180 Epub 2008 Apr 19. [DOI] [PMC free article] [PubMed]
- 44.Kuever J, Könneke M, Galushko A, Drzyzga O. Reclassification of Desulfobacterium phenolicum as Desulfobacula phenolica comb. nov. and description of strain SaxT as Desulfotignum balticum gen. nov., sp. nov. Int J Syst Evol Microbiol 2001; 51:171-177 [DOI] [PubMed] [Google Scholar]
- 45.Schink B, Thiemann V, Laue H, Friedrich MW. Desulfotignum phosphitoxidans sp. nov., a new marine sulfate-reducer that oxidizes phosphate to phosphate. Arch Microbiol 2002; 177:381-391 10.1007/s00203-002-0402-x [DOI] [PubMed] [Google Scholar]
- 46.Ommedal H, Torsvik T. Desulfotignum toluenicum sp. nov., a novel toluene-degrading, sulfate-reducing bacterium isolated from an oil-reservoir model column. Int J Syst Evol Microbiol 2007; 57:2865-2869 10.1099/ijs.0.65067-0 [DOI] [PubMed] [Google Scholar]
- 47.Finster K, Liesack W, Tindall BJ. Desulfospira joergensenii, gen. nov., sp. nov., a new sulfate-reducing bacterium isolated from marine surface sediment. Syst Appl Microbiol 1997; 20:201-208 10.1016/S0723-2020(97)80066-5 [DOI] [Google Scholar]
- 48.Vischer M, Worm P, Muyzer G, Pereira IAC, Schaap PJ, Plugge CM, Kuever J, Parshina SN, Nazina TN, Ivanova AE, et al Genome analysis of Desulfotomaculum kuznetsovii strain 17T reveals a physiological similarity twith Pelotomculum thermopropionicum strain SIT Stand Genom Sci, 2013 8: 69-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kato S, Kosaka T, Watanabe K. Substrate-dependent transcriptomic shifts in Pelotomaculum thermopropionicum grown in syntrophic co-culture with Methanothermobacter thermoautotrophicus. Microb Biotechnol 2009; 2:575-584 10.1111/j.1751-7915.2009.00102.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kosaka T, Uchiyama T, Ishii S, Enoki M, Imachi H, Kamagata Y, Ohashi A, Harada H, Ikenaga H, Watanabe K. Reconstruction and Regulation of the Central Catabolic Pathway in the Thermophilic Propionate-Oxidizing Syntroph Pelotomaculum thermopropionicum. J Bacteriol 2006; 188:202-210 10.1128/JB.188.1.202-210.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spring S, Lapidus A, Schröder M, Gleim D, Sims D, Meincke L, Del Rio TG, Tice H, Copeland A, Cheng JF, et al. Complete genome sequence of Desulfotomaculum acetoxidans type strain (5575). Stand Genomic Sci 2009; 1:242-253 10.4056/sigs.39508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sieber JR, Sims DR, Han C, Kim E, Lykidis A, Lapidus AL, McDonnald E, Rohlin L, Culley DE, Gunsalus R, McInerney MJ. The genome of Syntrophomonas wolfei: new insights into syntrophic metabolism and biohydrogen production. Environ Microbiol 2010; 12:2289-2301 [DOI] [PubMed] [Google Scholar]
- 53.Meyer B, Kuever J. Phylogeny of the alpha and beta subunits of the dissimilatory adenosine-5'-phosphosulfate (APS) reductase from sulfate-reducing prokaryotes – origin and evolution of the dissimilatory sulfate-reduction pathway. Microbiology UK 2007a; 153:2026-2044 10.1099/mic.0.2006/003152-0 [DOI] [PubMed] [Google Scholar]
- 54.Meyer B. Kuever, J Molecular analysis of the distribution and phylogeny of dissimilatory adenosine-5'-phosphosulfate reductase-encoding genes (aprBA) among sulfur-oxidizing prokaryotes. Microbiology UK 2007b; 153:3478-3498 10.1099/mic.0.2007/008250-0 [DOI] [PubMed] [Google Scholar]
- 55.Meyer B, Kuever J. Homology Modeling of Dissimilatory APS Reductases (AprBA) of Sulfur-Oxidizing and Sulfate-ReducingProkaryotes. PLoS ONE 2008; 3: e1514. [DOI] [PMC free article] [PubMed]
- 56.Spring S, Visser M, Lu M, Copeland A, Lapidus A, Lucuas S, Cheng JF, Han C, Tapia R, Goodwin LA, et al. Complete genome sequence of the sulfate-reducing firmicute Desulfotomaculum ruminis type strain (DLT). Stand Genomic Sci 2012; 7:304-319 10.4056/sigs.3226659 [DOI] [PMC free article] [PubMed] [Google Scholar]