

Abstract
Bacillus amyloliquefaciens HB-26, a Gram-positive bacterium was isolated from soil in China. SDS-PAGE analysis showed this strain secreted six major protein bands of 65, 60, 55, 34, 25 and 20 kDa. A bioassay of this strain reveals that it shows specific activity against P. brassicae and nematode. Here we describe the features of this organism, together with the draft genome sequence and annotation. The 3,989,358 bp long genome (39 contigs) contains 4,001 protein-coding genes and 80 RNA genes.
Keywords: Bacillus amyloliquefaciens HB-26, The Next-Generation sequencing, Plasmodiophora brassicae
Introduction
Bacillus amyloliquefaciens (B. amyloliquefaciens) is a species of bacterium in the genus Bacillus with high affinity of Bacillus subtilis. In the growth process, B. amyloliquefaciens can produce numerous antimicrobial or, more generally, bioactive metabolites with well-established activity in vitro such as surfactin, iturin and fengycin [1,2]. The production of all of these antibiotic compounds highlights B. amyloliquefaciens as a good candidate for the development of biocontrol agents [3,4].
Strain HB-26 belongs to the species B. amyloliquefaciens. The type strain of the species produces much bioactive metabolites showing specific activity against Plasmodiophora brassicae which could cause Clubroot, one of the most serious diseases of brassica crops worldwide [5-7]. Heavy infection by this pathogen of Chinese cabbage, cabbage, broccoli, turnip, oilseed rape, and other crucifers can lead to severe economic losses [8-11]. The root systems of infected plants show gall formation, which inhibits nutrient and water transport, stunts plant growth, and increases susceptibility to wilting [12,13]. Otherwise, bioassay results showed strain HB-26 also had some root-knot nematicidal activity.
Here, we present a summary classification and a set of features for B. amyloliquefaciens HB-26, together with the description of the genomic sequencing and annotation in order to improve the understanding of the molecular basis for its ability to inhibit Plasmodiophora brassicae and nematode.
Classification and features
Strain HB-26 colonies were milky white and matte with a wrinkled surface. Microscopy observations indicated that it was a Bacillus species (Figure 1A, Figure 1B and Table 1). SDS-PAGE analysis showed this strain secreted six major protein bands of 65, 60, 55, 34, 25 and 20 kDa (Figure 1C).
Figure 1.

General characteristics of B. amyloliquefaciens HB-26. (A) The colonial morphology pictures of strain HB-26. (B) Phase contrast micrograph of HB-26. (C) SDS-PAGE analysis of proteins of HB-26. Lane M, protein molecular weight marker; Lane 1, proteins of strain HB-26.
Table 1. Classification and general features of B. amyloliquefaciens HB-26.
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Domain Bacteria | TAS [14] | ||
| Phylum Firmicutes | TAS [15-17] | ||
| Class Bacilli | TAS [18,19] | ||
| Current classification | Order Bacillales | TAS [20,21] | |
| Family Bacillaceae | TAS [20,22] | ||
| Genus Bacillus | TAS [20,23,24] | ||
| Species Bacillus amyloliquefaciens | TAS [25-27] | ||
| Gram stain | Gram-positive | NAS | |
| Cell shape | rod-shaped | IDA | |
| Motility | mobile | NAS | |
| Sporulation | Spore-forming | IDA | |
| Temperature range | Room temperature | NAS | |
| Optimum temperature | pH7.0 | IDS | |
| Carbon source | organic carbon source | NAS | |
| Energy source | organic carbon source | NAS | |
| MIGS-6 | Habitat | Soil | IDA |
| MIGS-6.3 | Salinity | salt tolerant | NAS |
| MIGS-22 | Oxygen | Aerobic | NAS |
| MIGS-14 | Pathogenicity | Avirulent | NAS |
| MIGS-4 | Geographic location | Hubei, China | IDA |
| MIGS-4.1 | Latitude | 30.07N | |
| MIGS-4.2 | Longitude | 112.23E | |
| MIGS-4.3 | Depth | 5-10cm | |
| MIGS-4.4 | Altitude | about 35m | |
| MIGS-5 | Sample collection time | 2009 | IDA |
a) Evidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [28].
A representative genomic 16S rDNA sequence of strain HB-26 was searched against GenBank database using BLAST [29]. Sequences showing more than 99% sequence identity to 16S rDNA of HB-26 were selected for phylogentic analysis, and 15 sequences were aligned with ClustalW algorithm. The tree was reconstructed by neighbor-Joining by using Kimura 2-parameter for distance calculation. The phylogenetic tree was assessed by bootstrapped for 1,000 times, and the consensus tree was shown in Figure 2.
Figure 2.
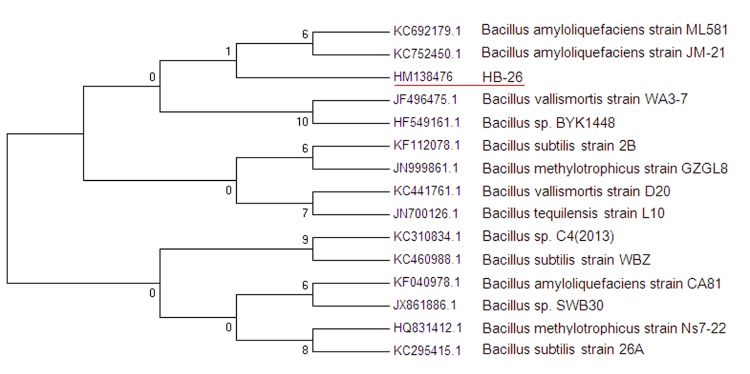
Neighbor-Joining Phylogenetic tree was generated using MEGA 4 based on 16S rRNA sequences. The strains and their corresponding GenBank accession numbers for 16S rDNA sequences are: A: B. amyloliquefaciens ML581 (KC692179.1); B: B. amyloliquefaciens JM-21 (KC752450.1); C: Bacillus strain HB-26 (HM138476); D: B. vallismortis WA3-7 (JF496475.1); E: B. sp.BYK1448 (HF549161.1); F: B. subtilis 2B (KF112078.1); G: B.methylotrophicus GZGL8 (JN999861.1); H: B.vallismortis D20 (KC441761.1); I: B.tequilensis L10 (JN700126.1); J: B. sp. C4(2013) (KC310834.1); K: B. subtilis WBZ (KC460988.1); L: B. Amyloliquefaciens CA81 (KF040978.1) ; M: B. sp. SWB30 (JX861886.1) ; N: B.methylotrophicus Ns7-22 (HQ831412.1); O: B. subtilis 26A (KC295415.1). The phylogenetic tree was constructed by using the neighbor-joining method within the MEGA software [30].
Genome sequencing information
Genome project history
This Bacillus strain was selected for sequencing due to its specific activity against Plasmodiophora brassicae and nematode. The complete high quality draft genome sequence is deposited in GenBank. The Beijing Genomics Institute (BGI) performed the sequencing and the NCBI staffs used the Prokaryotic Genome Annotation Pipeline (PGAAP) to complete the annotation. A summary of the project is given in Table 2.
Table 2. Genome sequencing project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Draft |
| MIGD-28 | Libraries used | One genomic libraries, one Illumina paired-end library (700 bp inserted size) |
| MIGS-29 | Sequencing platform | Illumina Hiseq 2000 |
| MIGS-31.2 | Sequencing coverage | 192 × |
| MIGS-30 | Assemblers | SOAPdenovo 1.05 version |
| MIGS-32 | Gene calling method | Glimmer and GeneMark |
| GenBank Data of Release | August 31, 2016 | |
| NCBI project ID | AUWK00000000 | |
| Project relevance | Agricultural |
Growth conditions and DNA isolation
B. amyloliquefaciens HB-26 was grown in 50 mL Luria-Broth for 6 h at 28°C. DNA was isolated by incubating the cells with lysozyme (10 mg/mL) in 2 mL TE (50 mM Tris base, 10 mM EDTA, 20% sucrose, pH8.0) at 4°C for 6 h. 4 mL of 2% SDS were added and the mixture was incubated at 55°C for 30 min; 2 mL 5M NaCl were added, and the mixture was incubated at 4°C for 10 min. DNA was purified by organic extraction and ethanol precipitation.
Genome sequencing and assembly
The genome of B. amyloliquefaciens HB-26 was sequenced using Illumina Hiseq 2000 platform (with a combination of a 251-bp paired-end reads sequencing from a 700-bp genomic library). Reads with average quality scores below Q30 or more than 3 unidentified nucleotides were eliminated. 2,605,589 paired-end reads (achieving ~192 fold coverage [0.94 Gb]) was de novo assembled using SOAPdenovo 1.05 version [9]. The assembly consists of 39 contigs arranged in 39 scaffolds with a total size of 3,989,358 bp (including chromosome and plasmids).
Genome annotation
Genome annotation was completed using the Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP). Briefly, Protein-coding genes were predicted using a combination of GeneMark and Glimmer [31-33]. Ribosomal RNAs were predicted by sequence similarity searching using BLAST against an RNA sequence database and/or using Infernal and Rfam models [34,35]. Transfer RNAs were predicted using tRNAscan-SE [36]. In order to detect missing genes, a complete six-frame translation of the nucleotide sequence was done and predicted proteins (generated above) were masked. All predictions were then searched using BLAST against all proteins from complete microbial genomes. Annotation was based on comparison to protein clusters and on the BLAST results. Conserved domain Database and Cluster of Orthologous Group information is then added to the annotation.
Genome properties
The draft assembly of the genome consists of 39 contigs in 39 scaffolds, with an overall 47.37% G+C content. Of the 4,114 genes predicted, 4,001 were protein-coding genes, and 80 RNAs were also identified. The majority of the protein-coding genes (54.06%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 3, Table 4 and Figure 3.
Table 3. Genome Statistics.
| Attribute | Value | % of total |
|---|---|---|
| Genome size (bp) | 3,989,358 | 100.00 |
| DNA coding region (bp) | 3,486,615 | 87.39 |
| DNA G+C content (bp) | 1,889,758 | 47.37 |
| Number of scaffolds | 39 | - |
| Extrachromosomal elements | unknown | - |
| Total genes | 4,114 | 100.00 |
| tRNA genes | 76 | 1.85 |
| rRNA genes | 4 | 0.1 |
| rRNA operons | 0** | - |
| Protein-coding genes | 4,001 | 97.25 |
| Pseudo gene (Partial genes) | 0 (36) | 0 (0.87%) |
| Genes with function prediction (proteins) | 2224 | 54.06% |
| Genes assigned to COGs | 2,336 | 56.78% |
| Genes with signal peptides | 328 | 7.97 |
| CRISPR repeats | 0 | 0 |
**: none of the rRNA operons appears to be complete due to unresolved assembly problems.
Table 4. Number of genes associated with the general COG functional categories.
| Code | Value | % age | Description |
|---|---|---|---|
| J | 130 | 3.160 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0.0 | RNA processing and modification |
| K | 262 | 6.368 | Transcription |
| L | 122 | 2.965 | Replication, recombination and repair |
| B | 1 | 0.024 | Chromatin structure and dynamics |
| D | 34 | 0.826 | Cell cycle control, cell division, chromosome partitioning |
| Y | 0 | 0 | Nuclear structure |
| V | 52 | 1.264 | Defense mechanisms |
| T | 153 | 3.719 | Signal transduction mechanisms |
| M | 182 | 4.424 | Cell wall/membrane/envelope biogenesis |
| N | 53 | 1.288 | Cell motility |
| Z | 0 | 0.000 | Cytoskeleton |
| W | 1 | 0.024 | Extracellular structures |
| U | 43 | 1.045 | Intracellular trafficking, secretion, and vesicular transport |
| O | 97 | 2.358 | Posttranslational modification, protein turnover, chaperones |
| C | 177 | 4.302 | Energy production and conversion |
| G | 249 | 6.053 | Carbohydrate transport and metabolism |
| E | 340 | 8.264 | Amino acid transport and metabolism |
| F | 79 | 1.920 | Nucleotide transport and metabolism |
| H | 123 | 2.990 | Coenzyme transport and metabolism |
| I | 117 | 2.844 | Lipid transport and metabolism |
| P | 205 | 4.983 | Inorganic ion transport and metabolism |
| Q | 116 | 2.820 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 435 | 10.574 | General function prediction only |
| S | 287 | 6.976 | Function unknown |
| 856 | 20.81 | Not in COGs |
Figure 3.

Graphical circular map of the Bacillus amyloliquefaciens HB-26 genome. From the outside to the center: genes on forward strand (color by COG categories), genes on reverse strand (color by COG categories), GC content, GC skew. The map was generated with the CGviewer server (Stothard Rearch Group: http://stothard.afns.ualberta.ca/cgview_server/).
Acknowledgments
This work was financially supported by the National Science and Technology Support Program (2008BADA5B03), the National 863 High Technology Research Program of China (2011AA10A201, 2011AA10A203), China 948 Program of Ministry of Agriculture (2011-G25), the National Science and Technology Support Program (2011BAB06B004-02), Hubei Province Development Plan (YJN0077) and the Science and Technology Support Program of Academy of Agricultural Sciences of Hubei Province (2012NKYJJ21).
Reference
- 1.Vilas-Bôas GT, Peruca AP, Arantes OM. Biology and taxonomy of Bacillus cereus, Bacillus anthracis, and Bacillus thuringiensis. Can J Microbiol 2007; 53:673-687 10.1139/W07-029 [DOI] [PubMed] [Google Scholar]
- 2.Chowdhury SP, Dietel K, Rändler M, Schmid M, Junge H, Borriss R, Hartmann A, Grosch R. Effects of Bacillus amyloliquefaciens FZB42 on Lettuce Growth and Health under Pathogen Pressure and Its Impact on the Rhizosphere Bacterial Community. PLoS ONE 2013; 8:e68818 10.1371/journal.pone.0068818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helgason E, Caugant DA, Lecadet MM, Chen Y, Mahillon J, Lovgren A, Hegna I, Kvaloy K, Kolsto AB. Genetic diversity of Bacillus cereus/B. thuringiensis isolates from natural sources. Curr Microbiol 1998; 37:80-87 10.1007/s002849900343 [DOI] [PubMed] [Google Scholar]
- 4.Arguelles-Arias A, Ongena M, Halimi B, Lara Y, Brans A, Joris B, Fickers P. Bacillus amyloliquefaciens GA1 as a source of potent antibiotics and other secondary metabolites for biocontrol of plant pathogens. Microb Cell Fact 2009; 8:63 10.1186/1475-2859-8-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helgason E, Okstad OA, Caugant DA, Johansen HA, Fouet A, Mock M, Hegna I, Kolsto AB. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis--one species on the basis of genetic evidence. Appl Environ Microbiol 2000; 66:2627-2630 10.1128/AEM.66.6.2627-2630.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ticknor LO, Kolsto AB, Hill KK, Keim P, Laker MT, Tonks M, Jackson PJ. Fluorescent Amplified Fragment Length Polymorphism Analysis of Norwegian Bacillus cereus and Bacillus thuringiensis Soil Isolates. Appl Environ Microbiol 2001; 67:4863-4873 10.1128/AEM.67.10.4863-4873.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagaoka T, Doullah MA, Matsumoto S, Kawasaki S, Ishikawa T, Hori H, Okazaki K. Identification of QTLs that control clubroot resistance in Brassica oleracea and comparative analysis of clubroot resistance genes between B. rapa and B. oleracea. Theor Appl Genet 2010; 120:1335-1346 10.1007/s00122-010-1259-z [DOI] [PubMed] [Google Scholar]
- 8.Rocherieux J, Glory P, Giboulot A, Boury S, Barbeyron G, Thomas G, Manzanares-Dauleux MJ. Isolate-specific and broad-spectrum QTLs are involved in the control of clubroot in Brassica oleracea. Theor Appl Genet 2004; 108:1555-1563 10.1007/s00122-003-1580-x [DOI] [PubMed] [Google Scholar]
- 9.Chen XH, Koumoutsi A, Scholz R, Eisenreich A, Schneider K, Heinemeyer I, Morgenstern B, Voss B, Hess WR, Reva O, et al. Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42. Nat Biotechnol 2007; 25:1007-1014 10.1038/nbt1325 [DOI] [PubMed] [Google Scholar]
- 10.Choi K, Yi Y, Lee S, Kang K, Lee E, Hong S, Young J, Park Y, Choi GJ, Kim BJ, Lim Y. Microorganisms against Plasmodiophora brassicae. J Microbiol Biotechnol 2007; 17:873-877 [PubMed] [Google Scholar]
- 11.Helgason E, Tourasse NJ, Meisal R, Caugant DA, Kolsto AB. Multilocus sequence typing scheme for bacteria of the Bacillus cereus group. Appl Environ Microbiol 2004; 70:191-201 10.1128/AEM.70.1.191-201.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schnepf E, Crickmore N, Van Rie J, Lereclus D, Baum J, Feitelson J, Zeigler DR, Dean DH. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol Mol Biol Rev 1998; 62:775-806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576-4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbons NE, Murray RGE. Proposals Concerning the Higher Taxa of Bacteria. Int J Syst Bacteriol 1978; 28:1-6 10.1099/00207713-28-1-1 [DOI] [Google Scholar]
- 16.Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 119-169. [Google Scholar]
- 17.Murray RGE. The Higher Taxa, or, a Place for Everything...? In: Holt JG (ed), Bergey's Manual of Systematic Bacteriology, First Edition, Volume 1, The Williams and Wilkins Co., Baltimore, 1984, p. 31-34. [Google Scholar]
- 18.List of new names and new combinations previously effectively, but not validly, published. List no. 132. Int J Syst Evol Microbiol 2010; 60:469-472 10.1099/ijs.0.022855-0 [DOI] [PubMed] [Google Scholar]
- 19.Ludwig W, Schleifer KH, Whitman WB. Class I. Bacilli class nov. In: De Vos P, Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 3, Springer-Verlag, New York, 2009, p. 19-20. [Google Scholar]
- 20.Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225-420 10.1099/00207713-30-1-225 [DOI] [PubMed] [Google Scholar]
- 21.Prévot AR. In: Hauderoy P, Ehringer G, Guillot G, Magrou. J., Prévot AR, Rosset D, Urbain A (eds), Dictionnaire des Bactéries Pathogènes, Second Edition, Masson et Cie, Paris, 1953, p. 1-692. [Google Scholar]
- 22.Fischer A. Untersuchungen über bakterien. Jahrbücher für Wissenschaftliche Botanik 1895; 27:1-163 [Google Scholar]
- 23.Cohn F. Untersuchungen über Bakterien. Beitr Biol Pflanz 1872; 1:127-224 [Google Scholar]
- 24.Gibson T, Gordon RE. Genus I. Bacillus Cohn 1872, 174; Nom. gen. cons. Nomencl. Comm. Intern. Soc. Microbiol. 1937, 28; Opin. A. Jud. Comm. 1955, 39. In: Buchanan RE, Gibbons NE (eds), Bergey's Manual of Determinative Bacteriology, Eighth Edition, The Williams and Wilkins Co., Baltimore, 1974, p. 529-550. [Google Scholar]
- 25.Priest FG, Goodfellow M, Shute LA, Berkeley RCW. Bacillus amyloliquefaciens sp. nov., nom. rev. Int J Syst Bacteriol 1987; 37:69-71 10.1099/00207713-37-1-69 [DOI] [Google Scholar]
- 26.Wang LT, Lee FL, Tai CJ, Kuo HP. Bacillus velezensis is a later heterotypic synonym of Bacillus amyloliquefaciens. Int J Syst Evol Microbiol 2008; 58:671-675 10.1099/ijs.0.65191-0 [DOI] [PubMed] [Google Scholar]
- 27.Fukomoto J. Studies on the production of bacterial amylase. I. Isolation of bacteria secreting potent amylase and their distribution. Nippon Nogeikagaku Kaishi 1943; 19:487-503 10.1271/nogeikagaku1924.19.7_487 [DOI] [Google Scholar]
- 28.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215:403-410 [DOI] [PubMed] [Google Scholar]
- 29.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 2010; 20:265-272 10.1101/gr.097261.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evo-lutionary distance, and maximum parsimony methods. Mol Biol Evol 2011; 28:2731-2739 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 2001; 29:2607-2618 10.1093/nar/29.12.2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res 1999; 27:4636-4641 10.1093/nar/27.23.4636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lukashin AV, Borodovsky M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res 1998; 26:1107-1115 10.1093/nar/26.4.1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res 2003; 31:439-441 10.1093/nar/gkg006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eddy SR. A memory-efficient dynamic programming algorithm for optimal alignment of a sequence to an RNA secondary structure. BMC Bioinformatics 2002; 3:18 10.1186/1471-2105-3-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955-964 10.1093/nar/25.5.0955 [DOI] [PMC free article] [PubMed] [Google Scholar]