

Abstract
Methanoplanus limicola Wildgruber et al. 1984 is a mesophilic methanogen that was isolated from a swamp composed of drilling waste near Naples, Italy, shortly after the Archaea were recognized as a separate domain of life. Methanoplanus is the type genus in the family Methanoplanaceae, a taxon that felt into disuse since modern 16S rRNA gene sequences-based taxonomy was established. Methanoplanus is now placed within the Methanomicrobiaceae, a family that is so far poorly characterized at the genome level. The only other type strain of the genus with a sequenced genome, Methanoplanus petrolearius SEBR 4847T, turned out to be misclassified and required reclassification to Methanolacinia. Both, Methanoplanus and Methanolacinia, needed taxonomic emendations due to a significant deviation of the G+C content of their genomes from previously published (pre-genome-sequence era) values. Until now genome sequences were published for only four of the 33 species with validly published names in the Methanomicrobiaceae. Here we describe the features of M. limicola, together with the improved-high-quality draft genome sequence and annotation of the type strain, M3T. The 3,200,946 bp long chromosome (permanent draft sequence) with its 3,064 protein-coding and 65 RNA genes is a part of the Genomic Encyclopedia of Bacteria and Archaea project.
Keywords: anaerobic, motile, mesophilic, methanogen, swamp, improved-high-quality draft, Methanomicrobiaceae, GEBA
Introduction
Strain M3T (= DSM 2279 = ATCC 35062 = OCM 101) is the type strain of the species Methanoplanus limicola [1,2], one out of currently three species in the genus Methanoplanus [1,2]. Strain M3T was originally isolated from the mud of a drilling swamp near Baia, Naples Area, Italy [1]. The genus name was derived from the Neo-Latin therm “methanum”, pertaining to methane, and the Latin adjective “planus”, meaning a flat plate, which refers to its flat cell morphology [1]. The species epithet was derived from the Latin word limicola, a dweller in the mud, inhabitant of a swamp [1]. When Wildgruber et al. described the type strain of the novel species in 1982 [1] they not only realized the striking similarity to the square-shaped flat bacterium that was reported two years earlier by Walsby [3], but also classified it as the type strain of the type species in the type genus of Methanomicrobiales Family III, ‘Methanoplanaceae’ [1]. However, when years later 16S rRNA sequences became available for phylogenetic analyses it became clear that the strains which represent the species Methanoplanus are closely related to Methanomicrobiaceae (including the genera Methanomicrobium, Methanogenium, and Methanoculleus). Since that time, the genus Methanoplanus is generally placed within the Methanomicrobiaceae, and Methanoplanaceae Wildgruber et al. 1984 has fallen into disuse [4], although the genus Methanoplanus was never formally reclassified. In the 31 years since strain M3T was first characterized, only two follow-up projects have reported the use of M. limicola in comparative analyses; Ivanov and Stabnikova [5] used M. limicola for a study on the molecular phylogeny of methanogenic archaea based on the G+C content, and Liu et al. used the species in a study on air tolerance and water stress [6].
Here we present a summary classification and a set of features for M. limicola M3T, together with the description of the genomic sequencing and annotation.
Classification and features
The single genomic 16S rRNA sequence of M. limicola M3T was compared with the Greengenes database for determining the weighted relative frequencies of taxa and (truncated) keywords as previously described [7]. The most frequently occurring genera were Methanoculleus (51.9%), Methanoplanus (18.5%), Methanogenium (16.8%), Methanosphaerula (5.3%) and Methanomicrobium (3.7%) (52 hits in total). Regarding the two hits to sequences from members of the species, the average identity within HSPs was 99.9%, whereas the average coverage by HSPs was 92.8%. Regarding the five hits to sequences from other members of the genus, the average identity within HSPs was 96.6%, whereas the average coverage by HSPs was 95.0%. Among all other species, the one yielding the highest score was M. endosymbiosus (FR733674), which corresponded to an identity of 99.5% and an HSP coverage of 99.7%. (Note that the Greengenes database uses the INSDC (= EMBL/NCBI/DDBJ) annotation, which is not an authoritative source for nomenclature or classification.) The highest-scoring environmental sequence was EU420694 ('Archaeal and Kao-Mei Wetland clone KM07-Da-3'), which showed an identity of 95.7% and an HSP coverage of 98.0%. The most frequently occurring keywords within the labels of all environmental samples which yielded hits were 'temperatur' (4.7%), 'bioreactor' (4.4%), 'anaerob' (4.0%), 'methanogen' (3.3%) and 'archaeal' (2.9%) (198 hits in total) fit to the features known from the habitat of strain M3T. Environmental samples which yielded hits of a higher score than the highest scoring species were not found.
Figure 1 shows the phylogenetic neighborhood of M. limicola in a 16S rRNA based tree. The sequence of the single 16S rRNA gene copy in the genome does not differ from the previously published 16S rRNA sequence (M59143), which contains 23 ambiguous base calls.
Figure 1.

Phylogenetic tree highlighting the position of M. limicola relative to the type strains of the other species within the family Methanomicrobiaceae. The tree was inferred from 1,271 aligned characters of the 16S rRNA gene sequence under the maximum likelihood (ML) criterion and rooted as previously described [7]. The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 250 ML bootstrap replicates [8] (left) and from 1,000 maximum-parsimony bootstrap replicates [9] (right) if larger than 60%. Lineages with type-strain genome sequencing projects registered in GOLD [10] are labeled with one asterisk, those also listed as 'Complete and Published' with two asterisks [11-14] (for Methanoregula boonei and Methanosphaerula palustris see CP000780 and CP001338, respectively).
The tree depicted in Figure 1 reveals discrepancies between the current classification of the group and 16S rRNA phylogenetic analysis, as the genus Methanoplanus appeared polyphyletic, with M. petrolearius appearing as sister group of Methanolacinia payntneri with maximum support. We conducted a constraint analysis as previously described [15], enforcing the monophyly of all genera (which only affects Methanoplanus in this dataset, see Figure 1). The best-known ML tree had a log likelihood of -7,097.90, whereas the best tree found under the constraint had a log likelihood of -7,144.12. The constrained tree was significantly worse than the globally best one in the Shimodaira-Hasegawa test as implemented in RAxML [8] (α = 0.01). The best-known MP trees had a score of 1,090, whereas the best constrained trees found had a score of 1,115 and were significantly worse in the Kishino-Hasegawa test as implemented in PAUP* [9] (α = 0.01).
M. limicola M3T cells stain Gram negative [1] and are plate-shaped with sharp crystal-like edges 1−3 µm long and 1−2 µm wide (Figure 2 and [1]). Weak motility was observed and motility genes were identified in the genome (see below). Polar tufts of flagella were also reported [1], but not visible in Figure 2. Granules with putative reserve material were observed in thin section EM images, as were curious ‘bone-shaped’ cells [1]. Cell envelopes consist of an S-layer glycoprotein with a hexagonal surface pattern [1]. Cultures grow with H2 or formate as sole substrates supplemented with ≥ 0.1% acetate essentially required [1]. Growth temperatures span from 17−41°C (optimum 40°C) in the presence of 0.4−5.4% NaCl (optimum 1%) [1]. A summary of the classification and features is presented in Table 1.
Figure 2.

Scanning electron micrograph of M. limicola M3T
Table 1. Classification and general features of M. limicola M3T according to the MIGS recommendations [16] published by the Genomic Standards Consortium [17].
| MIGS ID | Property | Term | Evidence code |
|---|---|---|---|
| Current classification | Domain Bacteria | TAS [18] | |
| Phylum Euryarchaeota | TAS [19] | ||
| Class Methanomicrobia | TAS [20] | ||
| Order Methanomicrobiales | TAS [21-25] | ||
| Family Methanomicrobiaceae | TAS [21,22] | ||
| Genus Methanoplanus | TAS [1,2] | ||
| Species Methanoplanus limicola | TAS [1,2] | ||
| Type strain M3 | TAS [1] | ||
| Gram stain | negative | TAS [1] | |
| Cell shape | plate-like | TAS [1] | |
| Motility | weakly motile | TAS [1] | |
| Sporulation | not reported | ||
| Temperature range | mesophile, 17-41°C | TAS [1] | |
| Optimum temperature | 40°C | TAS [1] | |
| Salinity | 0.4 - 5.4% NaCl (w/v), optimum 1.0% | TAS [5] | |
| MIGS-22 | Oxygen requirement | anaerobe | TAS [1] |
| Carbon source | CO2, formate | TAS [1] | |
| Energy metabolism | methanogen, chemoorganotrophic | TAS [1] | |
| MIGS-6 | Habitat | swamps of fresh water and seawater | TAS [1] |
| MIGS-15 | Biotic relationship | free living | TAS [1] |
| MIGS-14 | Pathogenicity | none | NAS |
| Biosafety level | 1 | TAS [26] | |
| MIGS-23.1 | Isolation | mud of drilling swamp | TAS [1] |
| MIGS-4 | Geographic location | near Baia, Naples Area, Italy | TAS [1] |
| MIGS-5 | Sample collection time | 1981 or earlier | NAS |
| MIGS-4.1 | Latitude | 40.629 | NAS |
| MIGS-4.2 | Longitude | 14.362 | NAS |
| MIGS-4.3 | Depth | not reported | |
| MIGS-4.4 | Altitude | not reported |
Evidence codes - TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). Evidence codes are from of the Gene Ontology project [27].
Chemotaxonomy
No chemotaxonomical results were reported for strain M3T, except for an estimation of 47.5% for the G+C content of the genome determined by a melting point in 0.1 × SSC [1].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [28], and is part of the Genomic Encyclopedia of Bacteria and Archaea project [29]. The genome project is deposited in the Genomes On Line Database [10] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI) using state of the art sequencing technology [30]. A summary of the project information is shown in Table 2.
Table 2. Genome sequencing project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Improved-high-quality-draft |
| MIGS-28 | Libraries used | Three genomic libraries: one 454 pyrosequence standard library, one 454 PE library (5 kb insert size), one Illumina library |
| MIGS-29 | Sequencing platforms | Illumina GAii, 454 GS FLX Titanium |
| MIGS-31.2 | Sequencing coverage | 834.5 × Illumina; 33.4 × pyrosequence |
| MIGS-30 | Assemblers | Newbler version 2.3, Velvet 1.0.13, phrap version SPS - 4.24 |
| MIGS-32 | Gene calling method | Prodigal |
| INSDC ID | CM001436, AHKP00000000 | |
| GenBank Date of Release | January 24, 2012 | |
| GOLD ID | Gi02923 | |
| NCBI project ID | 61291 | |
| Database: IMG | 2506381025 | |
| MIGS-13 | Source material identifier | DSM 2279 |
| Project relevance | Tree of Life, GEBA |
Growth conditions and DNA isolation
M. limicola strain M3T, DSM 2279, was grown anaerobically under H2/CO2 gas phase in DSMZ medium 141 (Methanogenium medium; MMG medium + 0.1% acetate; substrate: H2 or formate; stimulated by YE or peptone, + vitamins) [31] at 35−40°C. DNA was isolated from 0.5-1 g of cell paste using MasterPure Gram-positive DNA purification kit (Epicentre MGP04100) following the standard protocol as recommended by the manufacturer with modification st/LALM for cell lysis as described in Wu et al. 2009 [29]. DNA is available through the DNA Bank Network [32].
Genome sequencing and assembly
The genome was sequenced using a combination of Illumina and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website [33]. Pyrosequencing reads were assembled using the Newbler assembler (Roche). The initial Newbler assembly consisting of 760 contigs in ten scaffolds was converted into a phrap [34] assembly by making fake reads from the consensus, to collect the read pairs in the 454 paired end library. Illumina GAii sequencing data (3,470.2 Mb) was assembled with Velvet [35] and the consensus sequences were shredded into 1.5 kb overlapped fake reads and assembled together with the 454 data. The 454 draft assembly was based on 332.3 Mb 454 draft data and all of the 454 paired end data. Newbler parameters are -consed -a 50 -l 350 -g -m -ml 20. The Phred/Phrap/Consed software package [34] was used for sequence assembly and quality assessment in the subsequent finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution [33], Dupfinisher [36], or sequencing cloned bridging PCR fragments with subcloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J.-F. Chang, unpublished). A total of 159 additional reactions were necessary to close some gaps and to raise the quality of the final sequence. Illumina reads were also used to correct potential base errors and increase consensus quality using a software Polisher developed at JGI [37]. The error rate of the final genome sequence is less than 1 in 100,000. Together, the combination of the Illumina and 454 sequencing platforms provided 867.9 x coverage of the genome. The final assembly contained 421,665 pyrosequence and 44,481,858 Illumina reads.
Genome annotation
Genes were identified using Prodigal [38] as part of the DOE-JGI [39] genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [40]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. These data sources were combined to assert a product description for each predicted protein. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [41].
Genome properties
The genome consists of one scaffold (circularity not experimentally proven) of 3,200,946 bp length with a 42.2% G+C content (Table 3 and Figure 3). Of the 3,128 genes predicted, 3,064 were protein-coding genes, and 65 RNAs; 122 pseudogenes were also identified. The majority of the protein-coding genes (60.8%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.
Table 3. Genome Statistics.
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 3,200,946 | 100.00% |
| DNA coding region (bp) | 2,799,644 | 87.46% |
| DNA G+C content (bp) | 1,350,606 | 42.20% |
| Number of replicons | 1 | |
| Extrachromosomal elements | 0 | |
| Total genes | 3,129 | 100.00% |
| RNA genes | 65 | 2.08% |
| rRNA operons | 1* | |
| tRNA genes | 56 | 1.79% |
| Protein-coding genes | 3,064 | 97.92% |
| Pseudo genes | 122 | 3.90% |
| Genes with function prediction (proteins) | 1,901 | 60.75% |
| Genes in paralog clusters | 1,568 | 50.11% |
| Genes assigned to COGs | 2,204 | 70.44% |
| Genes assigned Pfam domains | 2,149 | 68.68% |
| Genes with signal peptides | 129 | 4.12% |
| Genes with transmembrane helices | 748 | 23.91% |
| CRISPR repeats | 0 |
*but five genes for 5S rRNA
Figure 3.
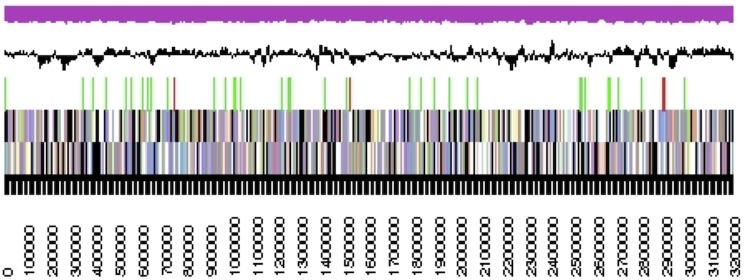
Graphical map of the chromosome. From bottom to the top: Genes on forward strand (colored by COG categories), Genes on reverse strand (colored by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content (black), GC skew (purple/olive).
Table 4. Number of genes associated with the general COG functional categories.
| Code | value | %age | Description |
|---|---|---|---|
| J | 155 | 6.5 | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.0 | RNA processing and modification |
| K | 133 | 5.6 | Transcription |
| L | 129 | 5.4 | Replication, recombination and repair |
| B | 3 | 0.1 | Chromatin structure and dynamics |
| D | 17 | 0.7 | Cell cycle control, cell division, chromosome partitioning |
| Y | 0 | 0.0 | Nuclear structure |
| V | 33 | 1.4 | Defense mechanisms |
| T | 191 | 8.0 | Signal transduction mechanisms |
| M | 90 | 4.8 | Cell wall/membrane biogenesis |
| N | 79 | 3.3 | Cell motility |
| Z | 1 | 0.0 | Cytoskeleton |
| W | 0 | 0.0 | Extracellular structures |
| U | 27 | 1.1 | Intracellular trafficking and secretion, and vesicular transport |
| O | 82 | 3.4 | Posttranslational modification, protein turnover, chaperones |
| C | 173 | 7.2 | Energy production and conversion |
| G | 75 | 3.1 | Carbohydrate transport and metabolism |
| E | 147 | 6.1 | Amino acid transport and metabolism |
| F | 61 | 2.6 | Nucleotide transport and metabolism |
| H | 157 | 6.6 | Coenzyme transport and metabolism |
| I | 28 | 1.2 | Lipid transport and metabolism |
| P | 115 | 4.8 | Inorganic ion transport and metabolism |
| Q | 8 | 0.3 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 355 | 14.8 | General function prediction only |
| S | 332 | 13.8 | Function unknown |
| - | 925 | 29.6 | Not in COGs |
Insights into the genome sequence
The G+C content in the genus Methanoplanus
When calculated from the genome sequences, the G+C content of M. limicola DSM 2279 amounts to 42.2%, whereas the previously published value, determined using traditional (“wet-lab”) techniques, is 47.5% [1]. Similarly, the G+C content of M. petrolearius was given as 50% [42], whereas the analysis of the genome sequence of the type strain SEBR 4837T (DSM 11571) yielded 47.4% [11]. It was frequently stated in the literature that “organisms that differ by more than 10 mol% do not belong to the same genus and that 5 mol% is the common range found within a species” [43]. A recent study [44] has shown that when calculated from genome sequences the G+C content varies at most 1% within species and that larger variances are caused by the limitations of the traditional techniques for analyses. It has thus been recommended to conduct emendations of species descriptions in the case of discrepancies larger than 1%, and to also conduct emendations of genus descriptions if the species emendations yield values that do not fit into the range of the G+C content given in the literature for the respective genus [44].
Considerations about the polyphyletic genus Methanoplanus
The phylogenetic tree presented in Figure 1 shows Methanoplanus as a polyphyletic taxon with the members of Methanomicrobium and Methanolacinia interspersed between the members of Methanoplanus. Given the high bootstrap support for the branches in that section of the phylogenetic tree, this situation calls for some attention, mainly due to the location of M. petrolearius [42]. The conflict between 16S rRNA gene data and the classification is significant, as revealed by the bootstrap values and the paired-site tests described above.
The problematic local structure of the phylogenetic tree might be caused by the fact that most of the five species located in the respective part of the tree were already decribed in the early days of Archaea research when only a limited number of reference sequences were available: M. limicola dates from 1982 [1], M. endosymbiosus from 1986 [45], M. petrolearius from 1997 [42], M. paynteri from 1983 [46] (renamed in 1989 [47]), and Methanomicrobium mobilis even from 1968 [48]. State-of-the-art techniques for the initial taxonomic characterization of the then novel bacteria were much less advanced than today, e.g. Sanger sequencing had just been invented (in 1977) when M. limicola was characterized with DNA-RNA hybridizations as decisive technique [49], and still not yet generally used for taxonomic work when M. endosymbiosus was characterized four years later. When the latest of the three Methanoplanus species with a validly published name, M. petrolearius, was added in 1997 16S rRNA sequences were used, but the ones from M. paynteri (closest neighbor in the phylogenetic tree in Figure 1) and M. mobilis were not yet available or at least not used for comparative analyses [42].
The completion of the Sequencing Orphan Species (SOS) initiative early last year [50], closed the last gaps in the availability of high-quality 16S rRNA reference sequences for phylogenetic trees. However, a decade after the first genome-based investigations into the history of the domain Archaea [51] and the systematic overview of their evolution, physiology, and molecular biology [52], a significant fraction of draft genome sequences as such generated in the genomic Encyclopedia of Bacteria and Archaea [29] are still very much needed to cover all of the diversity of the Archaea, especially from difficult-to-grow organisms and from type strains of remote clades such as the Methanomicrobiaceae.
With all these limitations, a closer inspection of the positions of the members of Methanoplanus in Figure 1 might still be worthwhile. M. petrolearius appears to be clearly separated from the other two members of the genus, M. limicola and M. endosymbiosus, but closely linked to M. paynteri with a 99.8% 16S rRNA gene sequence identity. Table 5 shows a summary of the features of all members of the genera Methanoplanus and Methanolacinia, indicating that based on the higher optimal growth temperature, the lack of observed flagella and observed motility (although the flagellin genes are encoded in the genome), the usage of CO2+2-propanol as a substrate, and the higher G+C content of the genome [42], M. petrolearius clusters rather with M. paynteri than with the other two members of Methanoplanus.
Table 5. Features of the type strains within the genera Methanoplanus and Methanolacinia.
| M. limicola [1] DSM 2279 | M. endosymbiosus [45] DSM 3599 | M. petrolearius [42] DSM 11571 | M. paynteri [46,47] DSM 2545 | |
|---|---|---|---|---|
| Source | swamp | marine ciliate | oil well | marine sediment |
| Temperature range (Topt) °C | 17−41 (32) | 16−36 (32) | 28−43 (37) | unknown (40) |
| motility | motile, flagella | flagella reported | non-motile | non-motile |
| pH range (pHopt) | ND (6.5−7.5) | 6.1−8.0 (6.8−7.3) | 5.3−8.2 (7.0) | ND |
| NaCl conc. % (opt.) | 0.4−5.4 (1) | 0−4.5 (1.5) | 0−5 (1−3) | |
| Substrates used | H2+CO2, formate | H2+CO2, formate | H2+CO2, formate, CO2+2-propanol | H2+CO2, CO2+2-propanol, CO2+2-butanol, CO2+2-cyclopentanol |
| G+C content | 42.2% genome (was 47.5% pre-genome) |
38.7% melting curve | 47.4% genome (was 50% pre-genome) |
44.8% buoyant density |
Although the genome sequence of M. petrolearius SEBR 4847T (DSM 11571) was recently published [11], the one for Ml. paynteri was still lacking, as well as information about a wet lab DNA-DNA hybridization (DDH) between the type strains of the two species. Given the high degree of 16S rRNA sequence identity between the two strains (99.8%), established thresholds of species delimitations, 97% [53], even under recently published relaxed recommendations, 98.2−99% [54], definitely demands such an analysis for the purpose of species discrimination. Whereas the rather large difference of 2.6% in the G+C content of the two genomes (Table 5, based on currently available mixed data from genome sequence and buoyant density measurement) predicts a rather low DDH value as the outcome of such an experiment, the recently observed significant deviations between previously published G+C values and G+C values inferred from genome sequences [44] do not, however, allow for definitive conclusions from the difference in G+C values.
For this reason, we have obtained a draft genome sequence for M. paynteri DSM 2546T using Illumina-MiSeq as a sequencing platform in order to obtain paired-end reads of 250 bp and Velvet [35] for the assembly. The draft genome comprised 54 contigs and is available from NCBI under the accession number AXDV00000000 and from IMG under the object ID pending. Digital DDH similarities between Ml. paynteri DSM 2546T (AXDV00000000) and M. petrolearius SEBR 4847T (DSM 11571, CP002117) were calculated with the GGDC web server [55,56] version 2.0 [57] under the recommended settings. The inter-genomic distance (formula 2) was 0.0753, corresponding to a DDH estimate of 48.50% ± 2.61%. The probability of a DDH value > 70% was accordingly only 0.1514.
In conclusion, from the topology of 16S rRNA gene sequence-based phylogenetic tree supported by the distribution of the characteristic features listed in Table 4 we can conclude that strain SEBR 4847T should rather be classified as a member of the genus Methanolacinia than as M. petrolearius, whereas the digital DDH results clearly indicate that Ml. paynteri (represented by the type strain G-2000, DSM 2545) and M. petrolearius (represented by the type strain SEBR 4847, DSM 11571) are distinct species. Thus, we propose Methanolacinia petrolearia comb. nov. to accommodate M. petrolearius, with SEBR 4847 being the type strain.
The situation between M. limicola (type species of Methanoplanus) and M. endosymbiosus is only slightly better than the relationship between M. paynteri and M. petrolearius discussed above. Based on the above reported Greengenes analysis the 16S rRNA gene sequences of the two type strains show 99.5% sequence identity and an HSP coverage of 99.7%. Again, by all accepted standards of species discrimination [53,54] such a close similarity would call for a DDH experiment to resolve the close relationship, but such data are not available. Also a digital DDH cannot be performed because only the genome sequence of M. limicola presented here is available, but not that of DSM 3599, the type strain of M. endosymbiosus. Table 5 indicates that the two strains share almost all of the listed features (except habitat), except for a 3.5% difference in the G+C content, which, in case it would be confirmed and not biased by a technical artifact in the melting curve measurement done for M. endosymbiosus, indicated a sufficiently low level of DDH to distinguish the two species [44]. Nevertheless, the probability that the digital DDH value between the two type strains might surpass the 70% species discrimination (once the genome sequence of M. endosymbiosus is resolved) threshold cannot be neglected. It might be too early to draft the obituary for M. endosymbiosus, but it is better to be prepared in case the once trispecific polyphyletic genus Methanoplanus becomes monospecific, an event that may occur once the drafts of all needed type strain genomes (the core objective of GEBA) are deciphered. Depending on the availability of enough cell material, M. endosymbiosus should now be scheduled as a sequencing target for the upcoming phases the GEBA, e.g. the Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes (KMG-I) projects [58], to resolve the question about the exact relationship between M. limicola and M. endosymbiosus.
Taxonomic consequences
As explained in detail above, the differences in the reported G+C contents of M. limicola and M. petrolearius to the ones calculated from their genome sequences justifies an emendation of the species descriptions. Moreover, M. petrolearius should be placed within the genus Methanolacinia. The descriptions of the two genera should be emended accordingly.
Emended description of the species Methanoplanus limicola Wildgruber et al. 1982
The description of the species Methanoplanus limicola is the one given by Wildgruber et al. 1982 [1], with the following modification.
The G+C content is 42%.
Emended description of the species Methanoplanus petrolearius Ollivier et al. 1997
The description of the species Methanoplanus petrolearius is the one given by Ollivier et al. 1997 [42], with the following modification.
The G+C content is 47%.
Description of Methanolacinia petrolearia, comb. nov.
Basonym: Methanoplanus petrolearius Ollivier et al. 1997
The description of the species is the same as given for Methanoplanus petrolearius Ollivier et al. 1997 with the emendation given above.
Emended description of the genus Methanoplanus
The description is the one given by Wildgruber et al. [1] with the following modifications:
The G+C content is 39-42%.
Emended description of the genus Methanolacinia
The description is the one given by Zellner et al. [47] with the following modifications:
The G+C content is 45-47%.
Acknowledgements
We would like to gratefully acknowledge the help of Evelyne-Marie Brambilla (DSMZ) for DNA extractions and quality control. This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, UT-Battelle and Oak Ridge National Laboratory under contract DE-AC05-00OR22725, as well as German Research Foundation (DFG) INST 599/1-2. AL was in part supported by the Russian Ministry of Science Mega-grant no.11.G34.31.0068; SJ O'Brien Principal Investigator. The Council of Scientific and Industrial Research (CSIR, India) and DAAD, Germany, provided a Fellowship to Shanmugam Mayilraj.
References
- 1.Wildgruber G, Thomm M, König H, Ober K, Rucchiuto T, Stetter KO. Methanoplanus limicola, a plate-shaped methanogen representing a novel family, the Methanoplanaceae. Arch Microbiol 1982; 132:31-36 10.1007/BF00690813 [DOI] [Google Scholar]
- 2.List Editor Validation List no. 14. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol 1984; 34:270-271 10.1099/00207713-34-2-270 [DOI] [Google Scholar]
- 3.Walsby EA. A square bacterium. Nature 1980; 283:69-71 10.1038/283069a0 [DOI] [Google Scholar]
- 4.Huber H, Huber G, Stetter KO. Genus VI. Methanoplanus Wildgruber, Thomm and Stetter. 1984, 270VP (Effective publication: Wildgruber, Thomm and Stetter in Wildgruber, Thomm, König, Ober, Ricchiuto and Stetter 1982, 36). In: Boone DR, Castenholz RW, Garrity (eds): Bergey's Manual of Systematic Bacteriology, second edition, vol. 1 (The Archaea and the deeply branching and phototrophic Bacteria), Springer-Verlag, New York, 2001, pp. 259-261..
- 5.Ivanov VN, Stabnikova EV. Use of the level of G+C n DNA for studying the molecular phylogeny of methanogenic archaebacteria. [in Russian] Mikrobiologiia 1999; 68:710-715 [PubMed] [Google Scholar]
- 6.Liu CT, Miyaki T, Aono T, Oyaizu H. Evaluation of methanogenic strains and their ability to endure aeration and water stress. Curr Microbiol 2008; 56:214-218 10.1007/s00284-007-9059-7 [DOI] [PubMed] [Google Scholar]
- 7.Göker M, Lapidus A, Spring S, Yasawong M, Lucas S, Glavina del Rio T, Chen F, Tice H, Cheng JF, Goodwin LA, et al. Complete genome sequence of Ignisphaera aggregans type strain (AQ1.S1T). Stand Genomic Sci 2010; 3:66-75 10.4056/sigs.1072907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 2008; 57:758-771 10.1080/10635150802429642 [DOI] [PubMed] [Google Scholar]
- 9.Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4.0 b10. Sinauer Associates, Sunderland, 2002. [Google Scholar]
- 10.Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2012; 40:D571-D579 10.1093/nar/gkr1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brambilla E, Ngatchou-Djao OD, Daligault H, Lapidus A, Lucas S, Hammon N, Nolan M, Tice H, Cheng JF, Han C, et al. Complete genome sequence of Methanoplanus petrolearius type strain (SEBR 4847T). Stand Genome Sci 2010; 2:203-211 10.4056/sigs.791633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson IJ, Sieprawska-Lupa M, Lapidus A, Nolan M, Copeland A, Glavina del Rio T, Tice H, Dalin E, Barry K, Saunders E, et al. Complete genome sequence of Methanoculleus marisnigri Romesser et al 1981 type strain JR1. Stand Genomic Sci 2009; 1:189-196 10.4056/sigs.32535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson IJ, Sieprawska-Lupa M, Goltsman E, Lapidus A, Copeland A, Glavina del Rio T, Tice H, Dalin E, Barry K, Pitluck S, et al. Complete genome sequence of Methanocorpusculum labreanum type strain Z. Stand Genomic Sci 2009; 1:197-203 10.4056/sigs.35575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson I, Ulrich LE, Lupa B, Susanti D, Porat I, Hooper SD, Lykidid A, Sieprawska-Lupa M, Dharmarajan L, Goltsman E, et al. Genomic characterization of methanomicrobiales reveals three classes of methanogens. PLoS ONE 2009; 4:e5797 10.1371/journal.pone.0005797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abt B, Han C, Scheuner C, Lu M, Lapidus A, Nolan M, Lucas S, Hammon N, Deshpande S, Cheng JF, et al. Complete genome sequence of the termite hindgut bacterium Spirochaeta coccoides type strain (SPN1T), reclassification in the genus Sphaerochaeta as Sphaerochaeta coccoides comb. nov. and emendations of the family Spirochaetaceae and the genus Sphaerochaeta. Stand Genomic Sci 2012; 6:194-209 10.4056/sigs.2796069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Field D, Amaral-Zettler L, Cochrane G, Cole JR, Dawyndt P, Garrity GM, Gilbert J, Glöckner FO, Hirschman L, Karsch-Mzrachi I, et al. The Genomic Standards Consortiu.m PLoS Biol 2011; 9:e1001088 10.1371/journal.pbio.1001088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms. Proposal for the domains Archaea and Bacteria. Proc Natl Acad Sci USA 1990; 87:4576-4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrity GM, Holt JG. Phylum AII. Euryarchaeota phy. nov. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 211-355. [Google Scholar]
- 20.Garrity GM, Bell JA, Lilburn T. Taxonomic outline of the Procaryotes. In: Garrity GM, Bell JA, Lilburn TG (eds), Taxonomic Outline of the Procaryotes, Bergey's Manual of Systematic Bacteriology, Second Edition. Release 4.0, Fourth Edition, Springer-Verlag, New York, 2003, p. 1-39. [Google Scholar]
- 21.Balch WE, Fox GE, Magrum LJ, Woese CR, Wolfe RS. Methanogens: reevaluation of a unique biological group. Microbiol Rev 1979; 43:260-296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.List Editor Validation List no. 6. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol 1981; 31:215-218 10.1099/00207713-31-2-215 [DOI] [Google Scholar]
- 23.Garrity GM, Labeda DP, Oren A. Judicial Commission of the International Committeeon Systematics of Prokaryotes; XIIth International (IUMS) Congress of Bacteriology and Applied Microbiology. Minutes of the meetings, 3, 4 and 6 August 2008, Istanbul, Turkey. Int J Syst Evol Microbiol 2011; 61:2775-2780 . 10.1099/ijs.0.037366-0 [DOI] [Google Scholar]
- 24.Judicial Commission of the International Committee on Systematics of Prokaryotes The nomenclatural types of the orders Acholeplasmatales, Halanaerobiales, Halobacteriales, Methanobacteriales, Methanococcales, Methanomicrobiales, Planctomycetales, Prochlorales, Sulfolobales, Thermococcales, Thermoproteales and Verrucomicrobiales are the genera Acholeplasma, Halanaerobium, Halobacterium, Methanobacterium, Methanococcus, Methanomicrobium, Planctomyces, Prochloron, Sulfolobus, Thermococcus, Thermoproteus and Verrucomicrobium, respectively. Opinion 79. Int J Syst Evol Microbiol 2005; 55:517-518 . 10.1099/ijs.0.63548-0 [DOI] [PubMed] [Google Scholar]
- 25.Euzéby JP, Tindall BJ. Nomenclatural type of orders: corrections necessary according to Rules 15 and 21a of the Bacteriological Code (1990 Revision), and designation of appropriate nomenclatural types of classes and subclasses. Request for an opinion. Int J Syst Evol Microbiol 2001; 51:725-727 [DOI] [PubMed] [Google Scholar]
- 26.BAuA. 2010, Classification of bacteria and archaea in risk groups. http://www.baua.de TRBA 466, p. 131.
- 27.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175-182 10.1016/j.syapm.2010.03.003 [DOI] [PubMed] [Google Scholar]
- 29.Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven Genomic Encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056-1060 10.1038/nature08656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mavromatis K, Land ML, Brettin TS, Quest DJ, Copeland A, Clum A, Goodwin L, Woyke T, Lapidus A, Klenk HP, et al. The fast changing landscape of sequencing technologies and their impact on microbial genome assemblies and annotation. PLoS ONE 2012; 7:e48837 10.1371/journal.pone.0048837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.List of growth media used at DSMZ: http://www.dsmz.de/catalogues/catalogue-microorganisms/culture-technology/list-of-media-for-microorganisms.html
- 32.Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, Berendsohn WG, Wägele JW. The DNA Bank Network: the start from a German initiative. Biopreserv Biobank 2011; 9:51-55 10.1089/bio.2010.0029 [DOI] [PubMed] [Google Scholar]
- 33.The DOE Joint Genome Institute www.jgi.doe.gov
- 34.Phrap and Phred for Windows. MacOS, Linux, and Unix. www.phrap.com
- 35.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821-829 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han C, Chain P. Finishing repeat regions automatically with Dupfinisher. In: Proceedings of the 2006 international conference on bioinformatics & computational biology. Arabnia HR, Valafar H (eds), CSREA Press. June 26-29, 2006: 141-146. [Google Scholar]
- 37.Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008. [Google Scholar]
- 38.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal Prokaryotic Dynamic Programming Genefinding Algorithm. BMC Bioinformatics 2010; 11:119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC. The DOE-JGI Standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci 2009; 1:63-67 10.4056/sigs.632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pati A, Ivanova N, Mikhailova N, Ovchinikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A Gene Prediction Improvement Pipeline for microbial genomes. Nat Methods 2010; 7:455-457 10.1038/nmeth.1457 [DOI] [PubMed] [Google Scholar]
- 41.Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271-2278 10.1093/bioinformatics/btp393 [DOI] [PubMed] [Google Scholar]
- 42.Ollivier B, Cayol JL, Patel BKC, Magot M, Fardeau ML, Garcia JL. Methanoplanus petrolearius sp. nov., a novel methanogenic bacterium from an oil-producing well. FEMS Microbiol Lett 1997; 147:51-56 10.1111/j.1574-6968.1997.tb10219.x [DOI] [PubMed] [Google Scholar]
- 43.Rossello-Mora R, Amann R. The species concept for prokaryotes. FEMS Microbiol Rev 2001; 25:39-67 10.1016/S0168-6445(00)00040-1 [DOI] [PubMed] [Google Scholar]
- 44.Meier-Kolthoff JP, Klenk HP, Göker M. Taxonomic use of the G+C content and DNA:DNA hybridization in the genomic age. Int J Syst Evol Microbiol 2014; 64:352-256.. 10.1099/ijs.0.056994-0 [DOI] [PubMed] [Google Scholar]
- 45.van Bruggen JJA, Zwart KB, Hermans JGF, van Hove EM, Stumm CK, Vogels GD. Isolation and characterization of Methanoplanus endodymbiosus sp. nov., an endosymbiont of the marine sapropelic ciliate Metopus contortus Quennerstedt. Arch Microbiol 1986; 144:367-374 10.1007/BF00409886 [DOI] [Google Scholar]
- 46.Rivard CJ, Henseon JM, Thomas MV, Smith PH. Isolation and characterization of Methanobacterium paynteri sp. nov., a mesophilic methanogen isolated from marine sediments. Appl Environ Microbiol 1983; 46:484-490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zellner G, Messner P, Kneifel H, Tindall BJ, Winter J, Stackebrandt E. Methanolacinia gen nov., incorporating Methanomicrobium paynteri as Methanolacinia paynteri comb. nov. J Gen Appl Microbiol 1989; 35:185-202 10.2323/jgam.35.185 [DOI] [Google Scholar]
- 48.Paynter MJB, Hungate RE. Characterization of Methanobacterium mobilis, sp. n., isolated the bovine rumen. J Bacteriol 1968; 95:1943-1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tu JK, Prangishvili D, Huber H, Wildgruber G, Zillig W, Stetter KO. Taxonomic relations between archaebacteria including 6 novel genera examined by cross hybridizations of DNA and 16s rRNA. J Mol Evol 1982; 18:109-114 10.1007/BF01810829 [DOI] [PubMed] [Google Scholar]
- 50.Yarza P, Spröer C, Swiderski J, Mrotzek N, Spring S, Tindall BJ, Gronow S, Pukall R, Klenk HP, Lang E, et al. Sequencing orphan species initiative (SOS): Filling the gaps in the 16S rRNA gene sequence database for all species with validly published names. Syst Appl Microbiol 2013; 36:69-73 10.1016/j.syapm.2012.12.006 [DOI] [PubMed] [Google Scholar]
- 51.Forterre P, Gribaldo S, Brochier-Armanet C. Natural history of the archaeal domain. In: RA Garrett & HP Klenk (eds) Archaea: evolution, physiology, and molecular biology. Wiley-Blackwell 2007, pp 17-28. [Google Scholar]
- 52.Garrett RA. HP Klenk HP (eds) Archaea: evolution, physiology, and molecular biology. Wiley-Blackwell 2007. [Google Scholar]
- 53.Stackebrandt E, Goebel BM. Taxonomic note: a place for DNA-DNA reassiciations and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol 1994; 44:846-849 10.1099/00207713-44-4-846 [DOI] [Google Scholar]
- 54.Meier-Kolthoff JP, Göker M, Spröer C, Klenk HP. When should a DDH experiment be mandatory in microbial taxonomy? Arch Microbiol 2013; 195:413-418 10.1007/s00203-013-0888-4 [DOI] [PubMed] [Google Scholar]
- 55.Auch AF, Klenk HP, Göker M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci 2010; 2:142-148 10.4056/sigs.541628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Auch AF, Von Jan M, Klenk HP, Göker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci 2010; 2:117-134 10.4056/sigs.531120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 2013; 14:60 10.1186/1471-2105-14-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kyrpides NC, Wojke T, Eisen JA, Garrity GM, Whitman WB, Hugenholtz P, Klenk HP. Genomic encyclobedia of type strains, phase I: the one thousand microbial genomes (KMG-I) project. Stand Genomic Sci. 2013; 9:628-634 [DOI] [PMC free article] [PubMed] [Google Scholar]