

SUMMARY
The histone modifying complexes PRC2 and TrxG/MLL play pivotal roles in determining the activation state of genes controlling pluripotency, lineage commitment, and cell differentiation. Long non-coding RNAs (lncRNAs) can bind to either complex, and some have been shown to act as modulators of PRC2 or TrxG/MLL activity. Here we show that the lateral mesoderm-specific lncRNA Fendrr is essential for proper heart and body wall development in the mouse. Embryos lacking Fendrr displayed upregulation of several transcription factors controlling lateral plate or cardiac mesoderm differentiation, accompanied by a drastic reduction in PRC2 occupancy along with decreased H3K27 trimethylation and/or an increase in H3K4 trimethylation at their promoters. Fendrr binds to both the PRC2 and TrxG/MLL complexes, suggesting that it acts as modulator of chromatin signatures that define gene activity. Thus, our work identifies a lncRNA that plays an essential role in fine-tuning the regulatory networks which control the fate of lateral mesoderm derivatives.
INTRODUCTION
Embryonic development commences with the formation of a group of pluripotent stem cells which are competent to form all cell types of the body. Development then proceeds through the coordinated action of cellular proliferation, patterning, lineage commitment, and differentiation, which are controlled by transcriptional regulators acting in a cell type-specific manner. The activity of genes encoding such regulatory proteins depends largely on the chromatin structure at their promoters and the associated regulatory elements. The histone modifying Polycomb repressive complexes (PRC1/PRC2) and the Trithorax group/MLL protein complexes (TrxG/MLL) play pivotal roles in the control of chromatin structure and, hence, gene activity of a subset of developmentally important regulators (Schuettengruber et al. 2007). In particular, PRC2 catalyses the methylation of histone H3 at lysine 27 (H3K27me3), which is repressive to gene activity, while the TrxG/MLL complex catalyses the methylation of histone H3 at lysine 4 (H3K4me3), which acts as an activating mark (Margueron et al. 2011; Schuettengruber et al. 2011). Thus, PRC2 and TrxG/MLL have opposing activities, and both are essential for embryonic development (O’Carroll et al. 2001; Yu et al. 1995).
It has been shown that components of both the PRC2 and TrxG/MLL complexes are able to interact with long non-coding RNAs (lncRNAs) (Zhao et al. 2008, 2010; Wang et al. 2011). Recent reports have suggested that lncRNAs may target PRC2 or TrxG/MLL to specific genomic loci, and thus contribute to the histone modification status and activity level of target genes (Rinn et al. 2007; Khalil et al. 2009; Bertani et al. 2011; Guttman et al. 2012). In vitro knock-down experiments have revealed the involvement of lncRNAs in gene regulatory networks controlling ES cell differentiation (Guttman et al. 2011), and functional studies have provided evidence for the roles of several lncRNAs (Wang et al. 2011; Hu et al. 2011; Kretz et al. 2012; Rinn et al. 2007; Gupta et al. 2010; Ulitzky et al. 2011). For instance, the knock-down of HOTTIP by an RCAS-shRNA in the developing chick limb resulted in a shortening of the distal bony elements of the limb, and morpholino-mediated knock-down of megamind and cyrano in zebrafish embryos revealed important roles for these lncRNAs in organogenesis (K. C. Wang et al. 2011; Ulitsky et al. 2011). However, stringent genetic approaches for probing the role of lncRNAs in mouse embryogenesis using loss-of-function analyses have not yet been reported.
Here we have identified a lncRNA, which we have termed Fendrr, that is specifically expressed in nascent lateral plate mesoderm. We inactivated Fendrr by gene targeting in embryonic stem (ES) cells and show that Fendrr is essential for proper development of tissues derived from lateral mesoderm, specifically the heart and the body wall. We illustrate that Fendrr acts by modifying the chromatin signatures of genes involved in the formation and differentiation of the lateral mesoderm lineage through binding to both the PRC2 and TrxG/MLL complexes. Furthermore, we provide evidence that an oligonucleotide corresponding to part of the Fendrr transcript can bind to dsDNA in target promoters.
RESULTS
Fendrr expression is restricted to nascent lateral plate mesoderm
We searched for differentially expressed lncRNAs in six different tissues dissected from early somite-stage mouse embryos (TS12, E8.25, 3–6 somites) using RNA-seq and ChIP-seq analyses. From this data set we identified a gene which is specifically transcribed in the posterior mesoderm. We isolated it by RACE-PCR from embryonic cDNA and determined its 2,397 basepair sequence and gene structure (Figure 1A). The gene consists of seven exons and is transcribed divergently from the transcription factor-coding gene Foxf1. Its transcriptional start site is located 1,250 bp upstream of the 5′-end of Foxf1. We termed this lncRNA Fendrr (Fetal-lethal non-coding developmental regulatory RNA).
Figure 1. Fendrr is transiently expressed in nascent lateral plate mesoderm of developing mouse embryos.

(A) Schematic of the genomic region of Fendrr and Foxf1. (B) Whole-mount in situ hybridization of mouse embryos at E8.25 and E9.5 revealed that Fendrr is transiently expressed and restricted to nascent lateral plate mesoderm (LPM), while Foxf1 transcripts persist in differentiating LPM along the body axis.
Whole-mount in situ hybridization showed that Fendrr is confined to the caudal end of the lateral plate mesoderm (LPM) of mid-gestation embryos, which gives rise to ventral structures such as the heart and body wall (Figure 1B and S1A). We could not detect Fendrr expression in other tissues or in organs of later stage embryos using qPCR analysis (data not shown). In the caudal LPM it is co-expressed with Foxf1, while in more anterior LPM cells which are undergoing differentiation, Fendrr is down-regulated, whereas Foxf1 expression is maintained in the splanchnic mesoderm (Peterson et al. 1997; Mahlapuu, Ormestad, et al. 2001). Quantitative PCR analysis of RNA extracted from the nuclear or cytosolic faction of E9.5 caudal end cells showed that Fendrr RNA is predominantly localized in the nucleus, consistent with its human orthologue in cultured cells (Figure S1B) (Khalil et al. 2009).
Loss of Fendrr causes embryonic lethality
In order to investigate the function of Fendrr in mouse development, we first knocked down Fendrr transcripts using a method for shRNAmir-mediated RNA interference in vivo (Vidigal et al. 2010). A reduction in Fendrr transcripts to 40% of the wild type level caused no phenotype (not shown). Therefore, we consecutively replaced the first exon of Fendrr on both chromosomes in ES cells with a strong transcriptional stop signal (3xpA) by homologous recombination (Friedrich et al. 1991), thereby generating a Fendrr-null mutant (Figure 2A). Successful targeting of both alleles in ES cells was confirmed by Southern blot analysis (Figure S2A). We confirmed loss of Fendrr transcripts in the caudal ends of Fendrr-null embryos generated by tetraploid complementation (Gertsenstein 2011) using whole-mount in situ hybridization (Figure S2B).
Figure 2. Heart and body wall development are impaired in mutants lacking Fendrr transcripts.

(A) Schematic showing the wild type (upper) and targeted (lower) Fendrr-Foxf1 genomic regions; the first exon of Fendrr was replaced with a 3xpA stop cassette by gene targeting in order to prevent transcription of the gene. Arrows indicate the position of primers used for real-time qPCR analysis. (B) E13.75 embryos derived by tetraploid complementation from wild type or homozygous Fendrr mutant ES cells including the neighbouring selection cassettes (Fendrr3xpA[N)/3xpA(H]). In mutant embryos the ventral body wall is perforated by umbilical vessels and parts of the liver; the presence of selection cassettes had no visible effect on the phenotype (see also Figure S3A and S3B). The phenotype was rescued by expression of Fendrr from a modified BAC transgene (Tg(RP23-455G4)Bgh). Note that the tail and limbs have been removed to better observe the ventral malformations. Scale bar: 1 mm. (C) Transverse histological sections of E12.5 wild type and mutant embryos at the mid-trunk level. The body wall of E12.5 mutant embryos is significantly thinner than that of wild type embryos (arrowheads and inlet), leading to perforation by the liver and umbilical vessels at later stages. (D) Transverse histological sections of E12.5 wild type and mutant embryos at the chest level. The thickness of the ventricular wall in the mutant heart is significantly reduced as compared to wild type (inlet). Abbreviations: rv, right ventricle; is, interventricular septum; cu, atrio-ventricular endocardial cushion; la, left atrium; lv, left ventricle. Scale bar: 200 μm. (E) Tissue thickness was measured from Eosin-stained transverse sections of E12.5 wild type and homozygous mutant embryos. Measurements were taken on either side using Zeiss AxioVision software, and values from both sides were combined for paired t-test analysis. Mean +/− s.d. are shown (n=3). (F) Percentage of mitotic cells (H3S10P) in heart ventricles determined on two distinct sections each of three different embryos. Paired t-test analysis and mean +/− s.d. are shown (n=3).
Homozygous mutants (Fendrr3xpA[N]/3xpA[H]) were found to be embryonic lethal around E13.75 (Figure 2B and S3A). At this stage they appeared pale and displayed a prominent omphalocele, wherein parts of the developing liver and all umbilical vessels protruded from the ventral body (88.9%, n=9) (Figures 2B and S3A). Omphalocele and embryonic death persisted after removal of the PGK-Neo and PGK-Hygro selection cassettes (genotype Fendrr3xpA/3xpA; Figures S3A and S3B).
In order to rule out that the phenotype of Fendrr3xpA/3xpA mutant embryos was due to compromised genetic integrity of the ES cells we performed a rescue experiment. We introduced a BAC clone containing a functional Fendrr gene next to an inactivated Foxf1 locus into Fendrr3xpA/3xpA mutant ES cells and generated embryos by tetraploid complementation. These Fendrr3xpA/3xpA;Tg(RP23-455G4)Bgh embryos showed a normal expression pattern of Fendrr at E9.5 (Figure S3C) and expressed approximately half of the wild type level of Fendrr RNA, as expected from a single functional allele (Figure S3D). The Foxf1 expression level was unchanged in rescued embryos in E9.5 caudal ends as compared to wild-type embryos (Figure S3E). They appeared phenotypically normal until E17.5, while at E18.5 rescue was observed in the majority of embryos (Figures S3A and S3F). Thus, the embryonic rescue confirmed the genetic integrity of the mutant ES cells used in the following experiments, and we conclude that the lethal phenotype of Fendrr3xpA[N]/3xpA[H] and Fendrr3xpA/3xpA embryos is entirely due to loss of Fendrr transcripts.
To determine the etiology of the mutant phenotype we examined the morphology of embryos at stage E12.5, when mutants still appeared phenotypically normal. Measurements of the thickness of the ventral body wall, a derivative of the somatic LPM lineage, showed a severe reduction in homozygous mutant as compared to wild-type embryos (Figure 2C and 2E; n=3). This suggests a possible cause for the observed omphalocele – that the weak body wall of the mutant is insufficient to resist the pressure from the growing liver (Figure 2B and S4A).
Besides omphalocele, mutant embryos also displayed blood accumulation in the right heart chamber (Figure S4B). In order to asses heart functioning, which is a derivative of the splanchnic LPM lineage, we incubated E12.5 embryos at 37°C, injected Fluospheres® into the right atrium of the beating heart and followed the blood circulation by time-laps imaging (see Experimental Procedures). In wild type embryos we observed dispersion of the fluorescence through the right ventricle and into the arteries, while in Fendrr mutants the fluorescence accumulated in the right atrium (Figure S4C and Movie S1). This indicated that the heart function of mutant embryos is impaired, providing a plausible explanation for embryonic death observed around E13.5.
To gain further insight into the cellular basis of this heart malfunction, we performed histological analyses of E12.5 mutant hearts. Heart sections revealed hypoplasia of the myocard in the mutants, affecting the ventricular walls and interventricular septum (Figure 2D). Measurements of the thickness of the ventricular walls revealed a severe reduction in mutant as compared to wild-type hearts (Figure 2E). To examine the cellular basis for the hypoplasia we counted the mitotic cells on sections of wild-type and mutant hearts at three developmental stages. No difference was found at E9.5 and E11.5, whereas E12.5 mutant hearts showed a marked decrease of mitotic cells (Figure 2F). We detected no significant apoptosis in E12.5 wild-type or mutant hearts by cleaved caspase-3 (Asp175) staining on sections (data not shown). These data indicate that the cardiac hypoplasia in mutants may be due to impaired proliferation of cardiac myocytes at later stages of heart development.
We examined the expression levels of several cardiac muscle genes by qPCR analysis of heart ventricle tissue. We used background-matched ES cells for all molecular assays and produced the embryos by tetraploid complementation to avoid artifacts caused by variation in the genetic background. This revealed an upregulation of Mef2c, a transcriptional regulator important for controlling a subset of contractile protein genes, along with several other genes involved in cardiac muscle structure and architecture (Figure S4D) (Lin et al. 1997; Olson 2006).
The combined data show that Fendrr mutants exhibit myocardial dysfunction, which most likely is the cause of embryonic death. Moreover, the impaired development of both the heart and the body wall illustrates that loss of Fendrr transcripts in nascent LPM causes impaired development of derivatives of both the somatic and splanchnic LPM lineages.
Loss of Fendrr affects the epigenetic modification and expression of factors controlling lateral mesoderm differentiation
Cardiac mesoderm derives from the first lateral mesoderm formed in the primitive streak at the early to mid-streak stage (E6.5–7). To ensure that Fendrr is expressed in the prospective cardiac mesoderm at this early embryonic stage we isolated EOMES expressing cells from E6.5 mouse embryos by fluorescence activated cell sorting using an EOMES-specific antibody (Figure S4E). Eomes and Myl7 are markers of the early cardiac mesoderm lineage (Costello et al. 2011). We determined the RNA expression level of Eomes, Myl7 and Fendrr in EOMES-positive and EOMES-negative cells in comparison to unlabeled control cells. We found co-expression of Myl7 and Fendrr exclusively in Eomes-positive cells, while neither transcript was detectable in Eomes-negative cells (Figure S4F). Thus, Fendrr is indeed expressed in cardiac mesoderm progenitor cells. Since formation of the heart tube occurs around E8.0, we asked whether the expression of four important regulators of heart development, Gata4, Gata6, Tbx5, and Nkx2-5 were affected at this stage (Olson 2006; Watanabe and Buckingham 2010). We found that Gata6 and Nkx2-5 expression in the heart field of E8.5 mutant embryos was significantly increased in comparison to wildtype tissue, while Gata4 and Tbx5 remain unchanged (Figure 3A).
Figure 3. Loss of Fendrr affects epigenetic promoter modification and expression of transcriptional regulators in E8.5 embryonic hearts and caudal ends.

(A, C) Normalized expression levels of transcriptional regulators in heart fields (A) and caudal ends (C) of Fendrr mutant embryos compared to wild type controls at the developmental stages listed. Several control genes are up-regulated in the mutants. n.d. = not detectable; * p≤0.02 in E8.5, ** p≤0.05 in E9.5. (B, D) Quantitative comparison of the levels of activating H3K4me3 and repressive H3K27me3 methylation marks in the promoters of control genes in E8.5 heart fields (B) and caudal ends (D) from Fendrr mutant and wild type embryos. H3K4 tri-methylation is increased at promoters of genes showing increased expression in the mutants (* p≤0.03). In addition, H3K27 tri-methylation is decreased at the promoters of control genes for LPM differentiation (Foxf1, Pitx2, Irx3) (* p≤0.05). (E) Quantitative comparison of the levels of WDR5 or EZH2 bound to the promoters of control factors in Fendrr mutant or wild type caudal ends. EZH2 occupancy at the Foxf1, Irx3 and Pitx2 promoters is dramatically reduced in the mutant, while other promoters are not affected. WDR5 occupancy is not altered for any of the promoters tested. All measurements were performed by quantitative real-time qPCR. Means +/− s.d. are shown (n=3 for expression and n=2 for ChIP analysis) (* p≤0.05).
Next, we asked whether these changes in gene expression were mirrored by alterations of the histone methylation status at the promoters of these genes. The histone methyltransferase complexes TrxG/MLL and PRC2 deposit activating H3K4me3 and repressive H3K27me3 marks, respectively, and both complexes have been shown to be involved in lineage commitment in vitro (Margueron et al. 2011; Surface et al. 2010; Schuettengruber et al. 2011). We found no significant change in the status of H3K27me3 at the promoters of the heart control genes analyzed, while the H3K4me3 mark of the Gata6 and Nkx2-5, but not Gata4 or Tbx5 promoters was increased (Figure 3B). Thus, loss of Fendrr results in an increase of H3K4 tri-methylation in a subset of heart control gene promoters, mirrored by an increased expression of these genes.
Since Gata6 is widely expressed in the LPM, including nascent LPM and heart progenitor cells, we wanted to determine whether changes in Gata6 expression and promoter histone modifications were already present in nascent LPM. In addition, we analyzed the expression of the LPM control genes Foxf1, Pitx2 and Irx3, which play important roles in determining the splanchnic and somatic LPM lineages, along with Tbx3 (Mahlapuu et al. 2001; Rallis et al. 2005; Kitamura et al. 1999). The presomitic mesoderm (PSM) marker genes Dll1 and Tcf15 were included as controls.
The expression levels of Gata6 and Foxf1 were significantly increased in the caudal ends of E8.5 mutant embryos, while Irx3, Tbx3 and the PSM marker genes were not affected at this stage (Figure 3C). This increase in Gata6 and Foxf1 expression persisted in the nascent LPM of E9.5 mutant embryos. In addition, Irx3 and Pitx2 expression was also increased at this later stage, during which the progenitors of the ventral body wall are generated (Figure 3C). The expression of Foxc2, Foxl1 and Mthfsd, located in close vicinity to Foxf1 and Fendrr, was unchanged, excluding unspecific effects of Fendrr gene locus alterations on neighboring genes n the knockout allele (Figure S5A).
Similarly to what was observed in the heart, changes in gene expression were accompanied by changes in the methylation status of the promoters. The Gata6 and Foxf1 promoters of mutant E8.5 embryos showed a strong increase in H3K4me3, and the Irx3 and Pitx2 promoters a slight increase in H3K4me3 (Figure 3D). In contrast to Gata6, which showed no difference in the repressive mark, the H3K27me3 levels at the Foxf1, Irx3, and Pitx2 promoters of mutant embryos were strongly reduced as compared to wild type (Figure 3D). The methylation status of the control PSM marker gene promoters was not altered for either the H3K4me3 or the H3K27me3 marks.
Next we asked whether the changes in histone modifications observed correlated with altered occupancy of the PRC2 and/or TrxG/MLL complexes at the respective promoters. We analyzed the presence of PRC2 or TrxG/MLL by ChIP with antibodies against EZH2, SUZ12 (PRC2 components) or WDR5 (TrxG/MLL component), followed by qPCR analysis of the promoter regions. Immunoprecipitation of the Foxf1, Irx3 and Pitx2 promoter regions with EZH2 or SUZ12 antibodies from mutant caudal end tissue was drastically reduced compared to wild type tissue, whereas all other promoters were not affected (Figure 3E and S5B). In contrast, promoter occupancy of WDR5 in caudal end tissue was unchanged for all of the promoters tested (Figure 3E). In addition, no changes in EZH2, SUZ12 or WDR5 occupancy were observed in heart tissue for any of the promoters tested (Figure S5C).
These combined data show that Fendrr has differential effects on the histone modification of promoters for transcriptional regulators in the lateral mesoderm and at least one of its derivatives, the cardiac mesoderm. The data suggest that the primary role of Fendrr is to promote occupancy of the PRC2 complex on particular promoters for LPM control genes, resulting in an increase of the repressive H3K27me3 mark, accompanied by a reduction in gene expression. In addition, via a different mechanism, Fendrr is involved in controlling the level of the activating H3K4me3 mark on a subset of promoters, thereby modifying the expression level of those genes.
Fendrr binds in vivo to the PRC2 and TrxG/MLL histone modifying complexes
The ChIP data prompted us to investigate whether Fendrr interacts directly with the PRC2 and/or TrxG/MLL complexes in mouse embryos. We immunoprecipitated the PRC2 complex with specific antibodies directed against EZH2 or SUZ12 from lysates of wild type E9.5 embryonic caudal ends, which express Fendrr, and from forebrains, which are devoid of Fendrr transcripts. Co-precipitated RNA was extracted and quantified by qPCR. The lncRNA HOTAIR served as positive control (Tsai et al. 2010). Fendrr and HOTAIR transcripts were co-precipitated with both PRC2 components from the caudal end tissue where they are expressed, but not from forebrain (Figure 4A). These data demonstrate that Fendrr transcripts bind to PRC2 in the mouse embryo, confirming data previously obtained in cultured human foreskin fibroblasts for the human orthologous lncRNA (Khalil et al. 2009). Next, we tested whether Fendrr transcripts could interact with the TrxG/MLL component WDR5. The lncRNA HOTTIP served as positive control (K. C. Wang et al. 2011). Again, both Fendrr and HOTTIP transcripts were co-immunoprecipitated with WDR5 from the caudal end, but not from forebrain tissue (Figure 4B). In contrast, Fendrr RNA was not co-precipitated with the PRC1 component RING1B, the NuRD complex component LSD1 or SIRT6 (Figure S6A). Thus, Fendrr RNA binds to PRC2 and TrxG/MLL and discriminates between various histone modifying complexes.
Figure 4. Fendrr binds to the PRC2 and TrxG/MLL complexes and to target promoters.
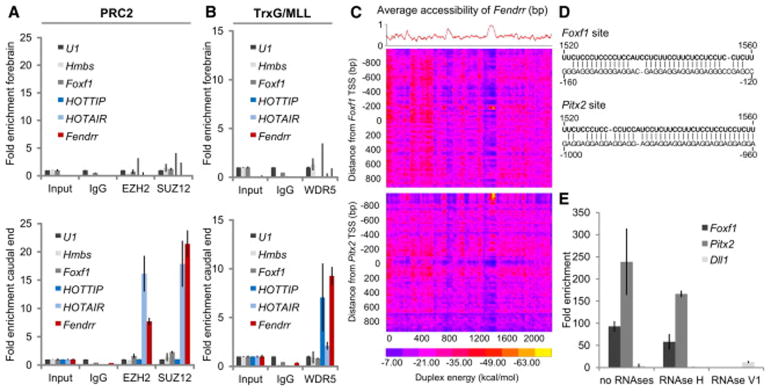
(A, B) RNA co-immunoprecipitation (RIP) from forebrain (upper panels) and caudal end (lower panels) lysates from wild type embryos using antibodies directed against the PRC2 components EZH2 and SUZ12 (A) or the TrxG/MLL component WDR5 (B); normal rabbit IgG was used as a control. Fold enrichment has been normalized to non-enriched input sample and U1 rRNA. Fendrr transcripts co-precipitated with EZH2, SUZ12, and WDR5 from caudal end tissue only, while the control lncRNA Hotair co-precipitated only with EZH2 and SUZ12 and HOTTIP lncRNA co-precipitated only with WDR5 from the caudal end tissue. Foxf1 and Hmbs RNA co-precipitation was used as a negative control. Mean +/− s.d. are shown (n=3). (C) Binding potential between Fendrr and genomic regions. The red curve shows the average probability of single stranded RNA as computed by sfold with a length parameter of 200 and W=1 (Ding et al. 2004). The heat map represents the base-pairing energy for an RNA/RNA duplex model for 40bp regions along the Fendrr transcript and 2,000 bp around the TSS of Foxf1 (top) and Pitx2 (bottom). The duplex energy is computed for each such region, staggered by 20bp. The optimal base pairing occurs in a region of Fendrr that also strongly favors single-stranded RNA, suggesting an open conformation that would enable the binding to the promoter. Probabilities are then averaged for a sliding window of 40bp to give the average RNA accessibility of the region that is binding. (D) Representation of the predicted interaction of the Fendrr RNA region and the promoter DNA region exhibiting the lowest free energy of approximately −70 kcal/mol (see yellow spot in C). (E) In vitro RNA/dsDNA binding assay utilizing biotin tagged RNA oligos as bait. Bars represent normalized enrichment of indicated 2,000 bp promoter fragment over background using a control RNA oligonucleotide (n=3, Mean +/− s.d. is shown).
Fendrr binds to the Foxf1 and Pitx2 promoters in vitro
Since Fendrr binds to the PRC2 complex and loss of Fendrr resulted in a strong reduction of PRC2 occupancy at the Foxf1, Pitx2 and Irx3 promoters, we asked whether Fendrr is able to bind directly to any of these promoters. We calculated the binding potential of Fendrr to fragments covering 1 kb upstream to 1 kb downstream of the transcriptional start site of each of the three genes. The heat map revealed a short stretch in the Fendrr RNA predicted to bind to a complementary region in the Foxf1 and Pitx2 promoters (Figure 4C and 4D). The Irx3 promoter was negative within the region analyzed, just as the promoters of Dll1 and Tcf15, which served as negative controls (Figure S6B). We used a synthetic RNA oligonucleotide coupled to psoralen and biotin at either end and performed binding assays in vitro (Besch et al. 2004). The promoter fragments of both Foxf1 and Pitx2, but not of Dll1, co-precipitated with this RNA oligomer (Figure 4E). Co-precipitation occurred in the presence of RNAseH, but was prevented by RNAseV1 treatment. The former enzyme specifically cuts RNA in DNA/RNA heteroduplexes, while the latter cleaves base-paired nucleotides. The data show that Fendrr can bind to double stranded Foxf1 and Pitx2 promoter fragments, and suggest triplex formation at the complementary region (Buske et al. 2012). The data in combination with findings discussed above confirm that Fendrr acts in cis (at the Foxf1) and in trans (at the Pitx2 and possibly other promoters).
The combined data suggest that Fendrr anchors PRC2 at Fendrr target genes, thereby increasing PRC2 occupancy and H3K27me3 trimethylation at its target promoters, which consequently leads to attenuation of target gene expression. Moreover, the co-expression of the transcriptional regulator Foxf1 and the lncRNA Fendrr in lateral mesoderm links the transcriptional regulatory network with the epigenetic regulatory network acting in this tissue.
DISCUSSION
We have identified a lncRNA, Fendrr, which is specifically transcribed in nascent lateral plate mesoderm of the developing mouse embryo. We analyzed the role of Fendrr in embryogenesis by applying a stringent genetic loss-of-function analysis. We observed malformations of two lateral mesoderm derivatives, the heart and the body wall, in Fendrr-null mutants showing that Fendrr is essential for proper lateral mesoderm differentiation. Similar embryonic phenotypes have been previously observed following gene targeting of transcriptional regulators, which we have identified as targets of Fendrr in this work. The loss of Foxf1, which is located chromosomally adjacent to Fendrr, and is co-expressed with Fendrr in the caudal end, results in improper lateral plate mesoderm differentiation. In particular, Foxf1 is essential for the separation of splanchnic and somatic mesoderm (Mahlapuu, Ormestad, et al. 2001), where it is required to inhibit Irx3 expression in the splanchnic mesoderm and direct it to the somatic mesoderm lineage (Mahlapuu, Ormestad, et al. 2001). Pitx2 is required for heart and ventral body wall development (Kitamura et al. 1999).
Other targets of Fendrr that we identified here are also essential for defining lateral mesoderm derivatives. Gata4 and Gata6 cooperatively regulate cardiovascular development, and control Mef2c expression (Xin et al. 2006). Nkx2-5 is essential for cardiac development and is part of a regulatory network with Gata4, Tbx5, Mef2c, and other transcriptional regulators controlling heart formation (Olson 2006).
Since Fendrr is involved in regulating the expression of transcription factors, it can itself be considered to be a regulatory factor. However, in contrast to the regulatory proteins mentioned above, which primarily function as transcriptional activators, our present data indicate that Fendrr has a repressive role. This is consistent with the finding that Fendrr binds to the PRC2 complex and promotes the repressive histone modification H3K27me3 exerted by this complex, by increasing PRC2 occupancy on target promoters. This follows from the observation that loss of Fendrr causes a dramatic reduction of EZH2 and SUZ12 occupancy and significant decrease of H3K27 trimethylation on the promoters of the LPM control genes Foxf1, Pitx2, and Irx3. Simultaneously, the level of the activating H3K4me3 mark at their promoters was increased, suggesting that the Fendrr/PRC2 complex antagonizes TrxG/MLL at these promoters.
On the other hand, the Gata6 and Nkx2-5 promoters did not show down-regulation of the H3K27me3 level upon loss of Fendrr, but instead solely increased H3K4me3 levels. The latter was not accompanied by increased WDR5 occupancy at their promoters, arguing for an indirect effect resulting from upregulation of factors that are able to induce H3K4 trimethylation at their promoters. In support of this possible explanation, it has been shown in yeast that transcriptional activation can lead to increased levels of H3K4me3 at the 5′-end of the transcription unit (Ng et al. 2003). Nkx2-5 is a target of GATA4 (Olson 2006). Thus, increased H3K4me3 levels at the promoter of Nkx2-5 in Fendrr mutant heart fields might be due to upregulation of Nkx2-5 expression by GATA6, which binds to the same recognition sequence as GATA4, and increased H3K4me3 levels of Gata6 might be due to upregulation of one or a combination of the factors FOXF1, IRX3 or PITX2 in lateral mesoderm. Alternatively, Fendrr binding to the TrxG/MLL complex might interfere with the activity of this complex at the Gata6 and Nkx2-5 promoters. However, at this point of analysis we cannot exclude that Fendrr might also increase TrxG/MLL at some presently unidentified promoters. Whatever the mechanism, the data indicate a differential effect of Fendrr on various promoters in the LPM.
We show that part of the Fendrr RNA is able to bind dsDNA in the promoters of Foxf1 and Pitx2 in vitro. This binding is protected from digestion by RNAseH, a DNA/RNA heteroduplex specific RNAse, but not RNAseV1, which cleaves base-paired nucleotides. This indicates a form of base-pairing between Fendrr RNA and promoter DNA, which is distinct from a heteroduplex, such as a heterotrimeric interaction. In conjunction with the finding that Fendrr is required for increasing PRC2 occupancy at the Foxf1 and Pitx2 promoters, these data strengthen the supposition that Fendrr primarily acts as a scaffold RNA, promoting PRC2 occupancy at its target promoters. This leads to increased H3K27 trimethylation and subsequent reduction of target gene expression. Thus, Fendrr is a regulatory RNA, which mediates the modification of the epigenetic landscape of target promoters thereby causing attenuation of the expression of transcription factors, which are important in lateral mesoderm differentiation.
Our data quite strikingly suggest that quantitative changes in histone modifications within promoters of genes involved in a gene regulatory network (GRN) can cause deleterious effects, which are similar to those seen following the loss of a single crucial transcription factor. In Fendrr mutants, the up-regulation of direct target genes was quantified at an increase of around 30% over the wild type levels. This suggests that GRNs controlling lineage specification and differentiation require considerable fine-tuning to allow for proper tissue and organ development. However, at this point we cannot conclude that the mutant phenotype observed is indeed caused by increased expression of several transcriptional regulators, nor can we exclude that failure of Fendrr binding to TrxG/MLL at presently unidentified promoters leading to down-regulation of target genes in the mutant may contribute to the mutant phenotype. Compound heterozygosity of Gata4 and Gata6 has been shown to result in embryonic lethality demonstrating that a threshold of both genes is required to support cardiovascular development (Xin et al. 2006). Moreover, heterozygosity for Foxf1 or Pitx2 results in haplo-insufficiency phenotypes, indicating that a threshold level of each of these regulators is critical for proper embryonic development (Mahlapuu, Enerbäck, et al. 2001; Gage et al. 1999). While on the basis of current knowledge this is well conceivable, it is harder to envisage how an excess of transcriptional regulators might perturb embryonic processes.
In summary, our data suggest that Fendrr, by interacting with the PRC2 and TrxG/MLL complexes at a specific set of promoters, is required for fine-tuning of the GRN controlling the development of organs and tissues derived from the LPM.
The gene pairing of Fendrr and Foxf1 highlights the intriguing transcriptional and functional coupling of a divergent lncRNA with an adjacent regulatory protein, both of which are essential for development of the same embryonic tissue. Such lncRNA:protein-coding gene neighbours are found throughout the genome (Cabili et al. 2011) and this functional link may reveal a more general mechanism for the control of patterning and lineage commitment (Cabili et al. 2011; Ulitsky et al. 2011; Margueron et al. 2011; Ørom et al. 2010).
Another important feature of Fendrr is its long-term effect. In general, regulatory proteins control expression of genes in a cell-autonomous manner. In contrast, the chromatin signatures of promoters set early in a differentiation process can persist through several stages of differentiation until the gene is finally activated by a transcription factor. The increased H3K4me3 level that we observed at the Nkx2-5 promoter in the cardiac mesoderm of Fendrr mutant embryos indicates that Fendrr is involved in the histone modification of genes activated in descendants of the cells in which Fendrr was, but no longer is, active. Thus, disturbances in the epigenetic prepattern within the early precursor cells of organs and tissues may have far-reaching consequences on subsequent cell proliferation, patterning or differentiation processes in the descendants of those cells. It is particularly intriguing that cardiac myocyte proliferation in Fendrr mutant hearts is affected not before E12.5, six days after Fendrr expression in the cardiac progenitor cells of the lateral mesoderm has been lacking. Future work has to address how this late effect in cardiac tissue is triggered by Fendrr loss in cardiac progenitor cells.
EXPERIMENTAL PROCEDURES
Isolation of Fendrr and generation of transgene constructs
Full length Fendrr was isolated by standard rapid amplification of cDNA-ends with polymerase chain reaction (RACE-PCR) on RNA derived from the caudal ends of E9.5 C57BL/6J embryos. In order to generate a genetic null of Fendrr, we replaced the genomic region coding for the first exon of Fendrr with a stop cassette containing a strong transcriptional stop signal (3xpA) (Friedrich et al. 1991). For rescue a neomycin resistance cassette was inserted into the coding region of Foxf1 to generate a Foxf1 mutant BAC that contains a wild-type Fendrr locus. The BAC was linearized with PI-SceI (NEB) and stably integrated into Fendrr3xpA/3xpA ES cells.
Whole mount in situ hybridization
Whole-mount in situ hybridization was carried out using standard procedures described on the MAMEP website (http://mamep.molgen.mpg.de/index.php). Probes were generated by PCR from E8.5 whole embryo cDNA and subcloned into pCRII-TOPO (Life Technologies) or pBluescript II SK(+) (Agilent Technologies). After verification of the probe templates by sequencing, antisense in situ probes were generated as described on the MAMEP website using T7 polymerase (Promega). The in situ probe against Fendrr covers 698 bases at the 3′ end of the Fendrr transcript and the probe against Foxf1 is complementary to nucleotides 80–885 (805 bases) of the Foxf1 protein coding sequence.
Isolation of EOMES-positive cells by FACS
30 wild type embryos of stage E6.5 were isolated, washed once with PBS and trypsinized on ice for 5 minutes to obtain a single cell suspension. The cells were washed three times with 1% BSA/PBS and fixed in 4% paraformaldehyde/PBS for 10 minutes on ice. Cells were washed again three times with 1% BSA/PBS, followed by a 30 minute incubation with 1% BSA/1% FCS/PBST (PBS+0.1% Tween-20). Cells were split in two equal fractions and incubated 30 minutes with or without the primary (anti-EOMES, Abcam, 1:250) antibody, washed 3 times with PBST and incubated another 30 minutes with anti-rabbit-Cy3 (GE Healthcare, 1:250) antibody. Cells were washed three times with PBST and sorted on a FACS Aria II SORP (BD Bioscience) directly into RLT buffer (Qiagen). RNA was isolated (Qiagen), reverse transcribed with random hexamers and subjected to real-time PCR analysis.
Genetic manipulation of ES cells and generation of embryos
The genetic background of all ES cells and embryos generated in this work is identical (129S6/C57BL6 (G4)) (George et al. 2007). ES cells were cultured and modified according to standard procedures. Briefly, 10×106 ES cells were electroporated with 25μg of linearized targeting construct and cultivated with selection media containing 250 μg/ml G418 (Life Technologies) or 125 μg/ml Hygromycin B (Life Technologies) for the first and second targeting, respectively. Resistant clones were isolated, and successful gene targeting was confirmed by Southern blot analysis of the Fendrr/Foxf1 locus (Figure S2A). Embryos and live animals were generated by tetraploid complementation (Gertsenstein 2011). Heterozygous Fendrr+/3xpA[N] ES cells generated 21 mice from four foster mothers, confirming their integrity and usability in subsequent developmental assays. The selection cassettes consisting of PGK::Neo-SV40pA (abbreviated “N”) or PGK::Hygro-SV40pA (abbreviated “H”) were flanked by FRT sites. Absence of Fendrr expression in embryos was confirmed by in situ hybridization (Figure S2B).
Transient transfection of pCAGGS-flpE-puro using Lipofectamine 2000 (Life Technologies) into Fendrr3xpA[N]/3xpA[H] ES cells removed the selection cassettes (Beard et al. 2006), resulting in Fendrr3xpA/3xpA ES cells. Rescue of the Fendrr3xpA/3xpA embryos was achieved by stable random integration of the rescue BAC construct. Briefly, 6×106 ES cells (Fendrr3xpA/3xpA) were electroporated with 5 μg linearized BAC (RP23-455G4) DNA and cultivated in 250 μg/ml G418 (Life Technologies) selection media. Resistant clones were isolated and integration was confirmed by Southern blot analysis. The expression level of Fendrr was analysed by qPCR in embryos derived by tetraploid complementation (Figure S2D).
Embryo preparation and histology
Staged embryos were dissected from uteri into PBS and fixed in fresh 4% paraformaldehyde/PBS overnight at 4°C. For histology, embryos were embedded in paraffin. Sections (4–6 μm thickness) were mounted onto Superfrost® Plus microscope slides (Thermo scientific) and Eosin (Sigma) stained according to standard procedures.
Immunohistochemistry on sections were carried out using standard procedures. Antibodies used for the detection of mitotic cells was anti-H3S10P (Millipore) and cleaved caspase 3 (Asp175) (Cell Signaling) and slides were mounted in Vectashield with DAPI (Vector Laboratories). For quantification of mitotic cells, DAPI positive cells were counted on two sections each of three different embryos, and subsequently the number of H3S10P positive cells in the counted area was determined.
All image documentation was carried out on a Zeiss SteREO Discovery. V12 microscope and captured with Zeiss AxioVision 4.8.2 software.
Fluorescent imaging
Embryos were dissected at E12.5 into warm M2 medium, and kept at 37°C. Embryos were fixed onto temperature adjusted SYLGARD® 184 Silicone Elastomer mounting plates using needles in order to expose their thoraxes. Sonicated FluoSpheres® (20nm, Life Technologies) were diluted 1:10 with PBS and injected into the right atrium using a glass microcapillary for 10 seconds with mild pressure. Hearts were imaged under a Leica MZ 16FA microscope and videos recorded for 140s using the Leica LAS AF software.
Real-time quantitative PCR analysis
RNA was isolated using the Qiagen RNAeasy mini kit according to the manufacturer’s protocol. Quantitative PCR (qPCR) analysis was carried out on a StepOnePlus™ Real-Time PCR System (Life Technologies) using Power SYBR® Green PCR Master Mix (Promega). RNA levels were normalized to housekeeping genes and chromatin immunoprecipitation (ChIP) levels were normalized to intergenic genomic control regions. Quantification was calculated using the ΔΔCt method (Muller et al. 2002). Pmm2 served as housekeeping control gene for qPCR, and an intergenic region between Msgn1 and Kcns3, devoid of any specific histone methylation signatures, served as a reference for ChIP. Error bars indicate the standard error from biological replicates, each consisting of technical triplicates. A list of oligonucleotides can be found in Table S1. Oligonucleotide primers used in Figure S4D were a gift from S. Sperling (Charité, Berlin) and sequences are available upon request.
Chromatin immunoprecipitation (ChIP)
E8.5 embryos (8–12 somites) were dissected, and tissue from eight embryos were pooled for ChIP analysis. Samples were treated with 0.75% formaldehyde at room temperature (RT) for 10 minutes, and crosslinking was stopped by adding 1/10 volume of 1.25 M Glycin/PBS solution for five minutes at RT. Tissues were washed twice with ice-cold PBS and 200 μl of ChIP lysis buffer was added (50 mM TrisHCl pH8.1, 10 mM EDTA, 1% SDS, Roche Complete Protease Inhibitor Cocktail). In case of ChIP with antibodies against EZH2 (Active Motif), SUZ12 (Abcam) and WDR5 (Bethyl Laboratories, Inc.) the nuclei were isolated from cells in cell lysis buffer (5 mM PIPES-KOH, pH8.0, 85 mM KCl, 0.5% IGEPAL). Chromatin was fragmented with a Branson Digital Sonifier II W-450 (3 mm tip) using 6×10 second pulses with 50 second breaks, and fragmentation was verified on an agarose gel (mean size ~250–350bp). ChIP lysate was diluted in dilution buffer (16.7 mM TrisHCl pH8.1, 155 mM NaCl, 1.1% Triton X-100, 1.2 mM EDTA, 0.01% SDS) containing 1:1 Dynabeads® Protein A/Dynabeads® Protein G (40 μl each per ChIP), pre-adsorbed with 1 μg (5 μg in case of anti-EZH2 and anti-WDR5) of antibody. The following antibodies were used: anti-H3K4me3 (Abcam), anti-H3K27me3 (Upstate). ChIP lysates were incubated for 4h at 4°C and washed 8 minutes at 4°C in the following order: 2x Low salt wash buffer (20 mM Tris-HCl, pH 8.1, 140 mM NaCl, 0.1% SDS, 1% Triton X-100, 1 % Na-Deoxycholate, 2 mM EDTA), 1x High salt wash buffer (as Low salt wash buffer, but with 500 mM NaCl) and 1x LiCl wash buffer (20 mM TrisHCl pH8.1, 250 mM LiCl, 0.5% IGEPAL, 0.5% Na-Deoxycholate, 2 mM EDTA) and 1x TE. Bound DNA/histone complexes were eluted by incubating the beads twice for 15 minutes at 37°C with 10 mM TrisHCl pH 8.1, 1 mM EDTA, 140 mM NaCl, 5 mM DTT, 2% SDS. DNA was purified using the Qiagen PCR Cleanup Kit.
RNA co-immunoprecipitation (RIP)
RNA co-immunoprecipitation was carried out as previously described (Galgano et al. 2008). Magnetic Protein A and G beads (Life Technologies) were used for isolation of antibody-bound protein/RNA complexes. Co-precipitated RNA was reverse transcribed using random hexamers, and cDNA content quantified by qPCR. Antibodies used were: anti-EZH2 (Active Motif), anti-SUZ12 (Abcam), and anti-WDR5 (Bethyl Laboratories, Inc.). Oligonucleotides used for cDNA quantification can be found in Table S1 or in (Zhao et al. 2008).
RNA/dsDNA interaction assay
RNA/DNA interaction was essentially carried out as described (Besch et al. 2004). Briefly, 1 pmol of promoter fragment (−1kb to +1kb from the transcriptional start site) were incubated with 100 pmol of PsoralenC6-UCCCCUCCAUCCUCUUCCUUCUCCUCCUCCUCUUCUUU-BiotinTEG (Fendrr) or unspecific PsoralenC6-UCCCCUGUGGGUGGGGGUGGGGGGUCUUU-BiotinTEG RNA oligonucleotides (Biomers) (Schmitz et al. 2010) and UV (265nm) crosslinked as described. Pre-blocked M270 Streptavidin beads (Life Technologies) were used to precipitate bound DNA in the presence or absence of RNAse H or V1. Fold enrichment was determined by the ratio of specific to unspecific DNA precipitate obtained from three replicates.
Supplementary Material
Acknowledgments
We thank Silke Sperling and Cornelia Dorn for discussion on the heart phenotype and qPCR primers for heart marker analysis, members of the animal facility for breeding of mice and strain maintenance, and Tracie Pennimpede for comments on the manuscript. We are grateful to Mikhail Sukchev for providing the modified RP23-455G4 BAC.
References
- Beard C, Hochedlinger K, Plath K, Wutz A, Jaenisch R. Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis. 2006;44:23–8. doi: 10.1002/gene.20180. [DOI] [PubMed] [Google Scholar]
- Bertani S, Sauer S, Bolotin E, Sauer F. The Noncoding RNA Mistral Activates Hoxa6 and Hoxa7 Expression and Stem Cell Differentiation by Recruiting MLL1 to Chromatin. Molecular Cell. 2011;43:1040–1046. doi: 10.1016/j.molcel.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Besch R, Giovannangeli C, Schuh T, Kammerbauer C, Degitz K. Characterization and quantification of triple helix formation in chromosomal DNA. Journal of molecular biology. 2004;341:979–89. doi: 10.1016/j.jmb.2004.05.079. [DOI] [PubMed] [Google Scholar]
- Buske Fa, Bauer DC, Mattick JS, Bailey TL. Triplexator: detecting nucleic acid triple helices in genomic and transcriptomic data. Genome research. 2012;22:1372–81. doi: 10.1101/gr.130237.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-vega B, Regev Aviv, Rinn John L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes & Development. 2011;25:1–13. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Chan CY, Lawrence CE. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic acids research. 2004;32:W135–41. doi: 10.1093/nar/gkh449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich G, Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes & Development. 1991;5:1513–23. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Suh H, Camper Sa. Dosage requirement of Pitx2 for development of multiple organs. Development (Cambridge, England) 1999;126:4643–51. doi: 10.1242/dev.126.20.4643. [DOI] [PubMed] [Google Scholar]
- Galgano A, Forrer M, Jaskiewicz L, Kanitz A, Zavolan M, Gerber AP. Comparative analysis of mRNA targets for human PUF-family proteins suggests extensive interaction with the miRNA regulatory system. PloS one. 2008;3:e3164. doi: 10.1371/journal.pone.0003164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George SHL, Gertsenstein M, Vintersten K, Korets-Smith E, Murphy J, Stevens ME, Haigh JJ, Nagy A. Developmental and adult phenotyping directly from mutant embryonic stem cells. Proc Natl Acad Sci USA. 2007;104:4455–60. doi: 10.1073/pnas.0609277104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertsenstein M. In: Tetraploid Complementation Assay. 1. Pease S, Saunders TL, editors. Berlin, Heidelberg: Springer Berlin Heidelberg; 2011. [Google Scholar]
- Gupta Ra, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–6. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295–300. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Rinn John L. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–346. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Yuan B, Flygare J, Lodish HF. Long noncoding RNA-mediated anti-apoptotic activity in murine erythroid terminal differentiation. Genes & Development. 2011;25:2573–2578. doi: 10.1101/gad.178780.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K, et al. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development. 1999;126:5749–58. doi: 10.1242/dev.126.24.5749. [DOI] [PubMed] [Google Scholar]
- Kretz M, et al. Suppression of progenitor differentiation requires the long noncoding RNA ANCR. Genes & Development. 2012;26:338–343. doi: 10.1101/gad.182121.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997;276:1404–7. doi: 10.1126/science.276.5317.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlapuu M, Enerbäck S, Carlsson P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development. 2001;128:2397–406. doi: 10.1242/dev.128.12.2397. [DOI] [PubMed] [Google Scholar]
- Mahlapuu M, Ormestad M, Enerbäck S, Carlsson P. The forkhead transcription factor Foxf1 is required for differentiation of extra-embryonic and lateral plate mesoderm. Development. 2001;128:155–66. doi: 10.1242/dev.128.2.155. [DOI] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P, Janovjak H, Miserez A, Dobbie Z. Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques. 2002;32:1372–1378. [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- Olson Eric N. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–7. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Carroll D, Erhardt S, Pagani M, Barton SC, Surani AM, Jenuwein T. The Polycomb-Group Gene Ezh2 Is Required for Early Mouse Development. Mol Cell Biol. 2001;21:4330–4336. doi: 10.1128/MCB.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson RS, Lim L, Ye H, Zhou H, Overdier DG, Costa RH. The winged helix transcriptional activator HFH-8 is expressed in the mesoderm of the primitive streak stage of mouse embryos and its cellular derivatives. Mechanisms of Development. 1997;69:53–69. doi: 10.1016/s0925-4773(97)00153-6. [DOI] [PubMed] [Google Scholar]
- Rallis C, Del Buono J, Logan MPO. Tbx3 can alter limb position along the rostrocaudal axis of the developing embryo. Development. 2005;132:1961–70. doi: 10.1242/dev.01787. [DOI] [PubMed] [Google Scholar]
- Rinn JL, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz K, Mayer C, Postepska A, Grummt I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes & Development. 2010;24:2264–2269. doi: 10.1101/gad.590910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–45. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Schuettengruber B, Martinez AM, Iovino N, Cavalli G. Trithorax group proteins: switching genes on and keeping them active. Nature Reviews Molecular Cell Biology. 2011;12:799–814. doi: 10.1038/nrm3230. [DOI] [PubMed] [Google Scholar]
- Surface LE, Thornton SR, Boyer La. Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell. 2010;7:288–98. doi: 10.1016/j.stem.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang Jordon K, Lan F, Shi Y, Segal Eran, Chang Howard Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science (New York, NY) 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved Function of lincRNAs in Vertebrate Embryonic Development despite Rapid Sequence Evolution. Cell. 2011;147:1537–1550. doi: 10.1016/j.cell.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidigal Ja, Morkel M, Wittler L, Brouwer-Lehmitz a, Grote P, Macura K, Herrmann BG. An inducible RNA interference system for the functional dissection of mouse embryogenesis. Nucleic Acids Research. 2010:1–7. doi: 10.1093/nar/gkq199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–4. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Buckingham M. The formation of the embryonic mouse heart: heart fields and myocardial cell lineages. Annals of the New York Academy of Sciences. 2010;1188:15–24. doi: 10.1111/j.1749-6632.2009.05078.x. [DOI] [PubMed] [Google Scholar]
- Xin M, Davis Ca, Molkentin JD, Lien CL, Duncan Sa, Richardson Ja, Olson Eric N. A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11189–94. doi: 10.1073/pnas.0604604103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378:505–8. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- Zhao J, et al. Genome-wide Identification of Polycomb-Associated RNAs by RIP-seq. Molecular Cell. 2010;40:939–53. doi: 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin Ja, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–6. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ørom UA, et al. Long Noncoding RNAs with Enhancer-like Function in Human Cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.