

Abstract
Characterizing the genomic sequences of influenza A viruses is important for pathophysiological and evolutionary studies. Noncoding regions (NCR) of influenza A virus have been shown to play critical roles in replication and transcription but their sequences are infrequently determined. In this study, a method employing poly(A) addition and SMART (switching mechanism at 5′ end of RNA transcript) technology is described for directly determining and discriminating both NCR ends of viral RNA (vRNA), complementary RNA (cRNA), or NCR and cap sequences from viral mRNA. This modified method may also be used to characterize the NCRs of influenza A virus samples in which the RNA has been degraded.
Keywords: Influenza A virus, Noncoding regions, RACE (random amplification of cDNA ends)
1. Introduction
Influenza A virus (IAV), of the family Orthomyxoviridae, is a single-stranded, negative-sense, enveloped RNA virus. The RNA genome of IAV comprises eight distinct gene segments, which encode 10–14 proteins (Hutchinson et al., 2010; Jagger et al., 2012; Palese and Shaw, 2007; Wise et al., 2012). The central coding regions of each segment are flanked at both ends by short noncoding regions (NCRs) that range from 19 to 58 bases long (Liang et al., 2008). The NCRs are conserved with variations among different strains and segments. Compared to the more well-investigated coding regions of IAV, the biological significance of NCR variation between IAV strains is less well understood. The first 12-to-13 non-coding bases at the 3′ and 5′ termini of the viral NCRs are highly conserved and are partially complementary to each other and form the unique panhandle structure. This double-strand panhandle structure was demonstrated to be critical for IAV polymerase binding, cap-snatching, transcription initiation and replication (Fodor et al., 1995; Neumann et al., 2004). Internal to these ends, the NCRs in IAV contain segment-specific noncoding regions (averaging about 25 nucleotides). Together with the adjacent coding regions of the IAV genome, the segment-specific NCRs can serve as components of the signals required for efficient vRNA packaging (Hutchinson et al., 2010). Phylogenetic studies have also suggested that the NCRs are more conserved than the viral open reading frames and might be associated with host adaptation or functional viral changes (Furuse and Oshitani, 2011). It is therefore important to develop accurate and rapid methods to determine the NCRs sequences of IAV.
During IAV replication, which occurs in the host cell nucleus, the negative-sense viral RNA segments (vRNA), encapsidated with viral nucleoprotein and already associated with the heterotrimeric viral RNA-dependent RNA polymerase complex, serve as templates for the transcription of positive-sense viral mRNAs and cRNA. Viral mRNA is transcribed to make viral proteins, and the viral genome replicates via positive-stranded cRNA intermediates (Neumann et al., 2004). Viral mRNA synthesis requires cellular RNA polymerase II activity (Engelhardt et al., 2005) and viral mRNA transcription is primed using a 5′ fragment of 7-methyl guanosine-capped host cell pre-mRNA caps ‘snatched’ from host cell pre-mRNAs (Krug, 1981; Plotch et al., 1981). Cap-snatching initiates transcription of influenza A virus mRNA in which the host pre-mRNA cap, along with 10–13 nucleotides of the pre-mRNA itself, is cleaved from the rest of the pre-mRNA by the endonuclease domain of the viral polymerase PA subunit (Dias et al., 2009; Plotch et al., 1981; Yuan et al., 2009). It is thought that stuttering of the viral polymerase on a short stretch of polyuridines near the 5′ vRNA end (complementary to the 3′ end of the mRNA transcript) results in the addition of a polyadenosine tail mimicking host cell mRNAs (Fodor et al., 1994; Palese and Shaw, 2007). Consequently, viral mRNA lack full-length NCRs on their 3′ ends. Full-length positive sense replication intermediates, cRNA, have full-length NCRs on both ends, and are needed for the synthesis of new vRNAs. In contrast to mRNAs, cRNA and vRNA synthesis terminates without a poly(A) tail (Neumann et al., 2004; Scull and Rice, 2010).
Various methods have been used to clone the IAV NCRs. A commonly applied method for cloning the full-length IAV genomic segments is performing RT-PCR using the ‘universal’ primers (Hoffmann et al., 2001). These primers are based on the highly conserved terminal sequences in the NCRs. Another pair of primers used to amplify the eight genomic RNA segments simultaneously is MBTuni-12 and MBTuni-13, which were also designed from the conserved vRNA termini (Zhou et al., 2009). Such primer-based amplification methods have facilitated IAV gene segment cloning for reverse genetics and other assays, but primer sequences used for reverse transcription and/or amplification may not always match the native sequences of the initial viral RNA template, for example at the known fourth base polymorphism at the 3′-end of the vRNA (Lee and Seong, 1998b). Other methods for cloning and sequencing of the ends of IAV gene segments can be time consuming and labor intensive (Desselberger et al., 1980; Robertson, 1979). In two recent papers, a T4 RNA ligase-based viral RNA ligation was used for circularizing or concatenating the viral gene segments to determine viral NCRs sequences (de Wit et al., 2007; Szymkowiak et al., 2003). Although such methods can be used to accurately sequence the NCRs of IAV, they requiring manipulating RNA samples as well as requiring high quality viral RNA for better results.
In order to overcome the difficulties for cloning and determining the sequences of IAV NCRs, the SMART (switching mechanism at 5′ end of RNA template) technology was employed to synthesize 5′-RACE (rapid amplification of cDNA ends) cDNA (Wellenreuther et al., 2004) and employed a poly (A) tailing tool for making 3′ RACE cDNA. Using only one-ended RACE cDNA with either 5′-end or 3′-end specific primers, both 5′ and 3′ NCRs of IAV gene segments can be cloned. Furthermore, the cloned NCRs can be discriminated by RNA template as to vRNA, cRNA and mRNA, and the sequences of capped primers that were snatched from the host mRNAs can also be elucidated.
2. Materials and method
2.1. Viruses
The representative influenza A viruses (IAVs) used for the study included wild type strains A/New York/470/2004 (H3N2) (Memoli et al., 2009) [NY470], A/New York/312/2001 (H1N1) (Qi et al., 2009) [NY312] and A/WSN/1933(H1N1). The 2009 pandemic H1N1 strain A/California/04/2009 (H1N1) was acquired by reverse genetic rescue (Jagger et al., 2010) [CA04]. Viruses were propagated in Madin-Darby canine kidney (MDCK) cells at 37 °C, maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and then were washed with PBS and infected with the representative viruses at an MOI of 0.01 in Opti-MEM® I Reduced Serum Media for 1 h at 37 °C. The inoculum was removed and cells were incubated with Opti-MEM® I Reduced Serum Media with 1 μg/mL TPCK-Trypsin (Sigma–Aldrich). Viruses were then harvested at 48 h post infection or once full cytopathic effect (CPE) was achieved.
2.2. RNA isolation
Viral RNA from MDCK-propagated viruses was extracted using the QIAamp Viral RNA extraction kit by following the manufacturer’s protocol (Qiagen Inc., Valencia, CA). RNA was eluted from the columns using 60 μl AVE buffer, and stored at −70 °C. Total RNA from viral infected cells and tissues was extracted with Trizol reagent (Invitrogen, Carlsbad, CA) followed by the manufacturer’s instructions. The amount of total RNA and viral RNA were quantified by NanoDrop ND-1000 Spectrophotometer. Samples from formalin-fixed, paraffin-embedded lung blocks were first treated with CitriSolv (Fisher HealthCare, Houston, TX) and protease K (Krafft et al., 1997), and then RNA was isolated using Trizol reagents (Invitrogen, Carlsbad, CA).
2.3. Poly(A) addition
Poly(A) addition was performed using Poly(A) polymerase (Ambion Austin, TX). In brief, the viral RNA or total RNA was used in this study in a 10 μl reaction with 2 μl of 5 × E. coli Polymerase buffer, 1 μl of 10 mM ATP solution, 1 μl of 25 mM MnCL2 and 0.5 μl of2 unit/μl of E. coli polymerase. Reaction was incubated at 37 °C for 1 h. The minimum amount used for viral RNA or total RNA ranged was 10 ng.
2.4. First-strand cDNA synthesis
Influenza A virus RNA with poly(A) addition as described above was used for the cDNA synthesis by using the SMART™ RACE cDNA amplification kit (Clontech, Mountain View, CA). Based on the manufacturer’s instructions, 3 μl poly(A) addition RNA with 1 μl of 10 μM 5′-RACE CDS primer (Clontech) and 1 μl of 10 μM SMART II A oligonucleotide (Clontech) were used to synthesize the 5′-RACE cDNA. For the 3′-RACE cDNA, the same amount of sample RNA used in 5′ end RACE and 1 μl of 10 μM 3′- RACE CDS primer (Clontech) were added in the reaction. After 2 min incubation at 70 °C, 2 μl of 5× first-strand buffer, 1 μl 20 mM DTT, 1 μl of 10 mM dNTP mix, and 1 μl MMLV reverse transcriptase were added to the 5′-RACE and 3′-RACE reactions individually. After 90 min incubation at 42 °C, Tricine-EDTA was added to stop the reactions at 72 °C for 7 min.
2.5. Primer design
Specific primers for NCRs cloning of the influenza A viruses used in this experiment were designed based on the sequences from the NCBI Influenza Virus Resource [http://www.ncbi.nlm.nih.gov/genomes/FLU/Database] (Tables 1, 2A and 2B).
Table 1.
Sequence-specific RACE primers for influenza A/New York/470/2004 (H3N2) and A/WSN/1933(H1N1) viruses.
| Gene | A/NEW YORK/470/2004 (H3N2) | A/WSN/1933(H1N1) |
|---|---|---|
| PB2 | 5′-RACE: 5′-GAGTTTGTCCTTGTTCATTTCTCTCCGGA-3′ | 5′-RACE: 5′-GTACTTGTCACTGGTCCATTCCTATTCC-3′ |
| 3′-RACE: 5′-AGATACGGACCAGCATTAAGCATCA-3′ | 3′-RACE: 5′-TCTAGCATACTTACTGACAGCCAG-3′ | |
| PB1 | 5′-RACE: 5′-GTGTTCTGTTGACTGTGTCCATGGTGTA-3′ | 5′-RACE: 5′-AGGGAAAGTTGTGCTTATAGCATT-3′ |
| 3′-RACE: 5′-GAGGAACCGCTCTATTCTCAACACA-3′ | 3′-RACE: 5′-GAAGATCTGTTCCACCATTGAAGA-3′ | |
| PA | 5′-RACE: 5′-CAGAAAAGGTTCAATTTGGGC-3′ | 5′-RACE: 5′-GATCGCCAAGTTCTACGACTATTGACTC-3′ |
| 3′-RACE: 5′-GGGGCTATATGAAGCAATTGAGGAG-3′ | 3′-RACE: 5′-CAACTCCTTCCTCACACATGCATT-3′ | |
| HA | 5′-RACE: 5′-ATCAAGGATCTGATGAGGACTGTCG-3′ | 5′-RACE: 5′-TGTTCGCATGGTAGCCTATACATA-3′ |
| 3′-RACE: 5′-CATTAAACAACCGGTTCCAGATCAA-3′ | 3′-RACE: 5′-TCAGTTTCTGGATGTGTTCTAATG-3′ | |
| NP | 5′-RACE: 5′-GCACCATTTTCTCTATTGTCAAGCTGTTC-3′ | 5′-RACE: 5′-GCATTCTGGCGTTCTCCATCAGTC-3′ |
| 3′-RACE: 5′-ACCTCCCATTTGAAAAGTCAACCAT-3′ | 3′-RACE: 5′-CCTTTGACATGAGTAATGAAGGAT-3′ | |
| NA | 5′-RACE: 5′-TTCACACAGCATCACTTGGTTGTTT-3′ | 5′-RACE: 5′-AGGCTCTCTTATGACAAAAACGTCTCCT-3′ |
| 3′-RACE: 5′-TTTTCTCTGTTGAAGGCAAAAGCTG-3′ | 3′-RACE: 5′-TGAATAGTGATACTGTAGATTGGT-3′ | |
| M | 5′-RACE: 5′-CCATGAGAGCCTCAAGATCTGTGTT-3′ | 5′-RACE: 5′-CTTAGTCAGAGGTGACAGGATTGGTCTT-3′ |
| 3′-RACE: 5′-TATCGACTCTTCAAACACGGCCTTA-3′ | 3′-RACE: 5′-AGGAACAGCAGAATGCTGTGGATG-3′ | |
| NS | 5′-RACE: 5′-TTCCTCTTAGGGACCTCTGATCTCG-3′ | 5′-RACE: 5′-AGAAAGCAATCTACCTGAAAGCTT-3′ |
| 3′-RACE: 5′-GATTCGCTTGGAGAAGCAGTAATGA-3′ | 3′-RACE: 5′-GCTTGAAGTGGAGCAAGAGATAAG-3′ |
Table 2.
A Sequence-specific RACE primers for influenza A/New York/312/2001 (H1N1) virus.
| Gene | A/New York/312/2001 (H1N1) |
|---|---|
| PB2 | 5′-RACE: 5′-GCGAGATTGTGACATCAGATTCCTT-3′ |
| 3′-RACE: 5′-GACTCTAGCATACTTACTGACAGCCA-3′ | |
| PB1 | 5′-RACE: 5′-CTTATAGCATTTTGTGCTGGCACTTTT-3′ |
| 3′-RACE: 5′-ATAAAGACCTGCTCCACCATTGAA-3′ | |
| PA | 5′-RACE: 5′-CCTCTCCATACTCTTTCATTGCCTTTT-3′ |
| 3′-RACE: 5′-CTAACACATGCATTGAGATAGTTA-3′ | |
| HA | 5′-RACE: 5′-TTGAGTTGTTGGCATGGTAGCCTATAC-3′ |
| 3′-RACE: 5′-CAGCTTCTGGATGTGTTCCAATGG-3′ | |
| NP | 5′-RACE: 5′-AGTCTCCATCTGTTCGTAAGACCGTTT-3′ |
| 3′-RACE: 5′-GCTCTCGGACGAAAGGGCAACGAA-3′ | |
| NA | 5′-RACE: 5′-GGATTGAGTGACTAGCCCATATTGAAA-3′ |
| 3′-RACE: 5′-CTGGTCTTGGCCAGACGGTGCTGA-3′ | |
| M | 5′-RACE: 5′-AGACATCTTCAAGTCTCTGTGCGATCT-3′ |
| 3′-RACE: 5′-GTACCAGAGTCTATGAGGGAAGAA-3′ | |
| NS | 5′-RACE: 5′-AGGAAGCAATCTACCTGAAAGCTTGAC-3′ |
| 3′-RACE: 5′-CATTTATGCAAGCATTACAGCTAT-3′ | |
|
| |
| B Sequence-specific RACE primers for influenza A/California/04/2009 (H1N1) virus. | |
|
| |
| Gene | A/California/04/2009(H1N1) |
|
| |
| PB2 | 5′-RACE: 5′-TCGCGAGTGCGGGACTGCGACATT-3′ |
| 3′-RACE: 5′-TCTAGCATAC1TACTGACAGCCAGA-3′ | |
| PB1 | 5′-RACE: 5′-GCTTATGGCATiTTGCGCTGGAA-3′ |
| 3′-RACE: 5′-CATGAAGATCTG1TCCACCA1TGA-3′ | |
| HA | 5′-RACE: 5′-GTGTCTGCAT1TGCGG1TGCAA-3′ |
| 3′-RACE: 5′-GGATGTGCTCTAATGGGTCTCTA-3′ | |
| NP | 5′-RACE: 5′-ACCAGTCTCCAT1TGTTCATATGATCG1T-3′ |
| 3′-RACE: 5′-GACATGAGTAATGAAGGGTC1TA-3′ | |
2.6. PCR and cloning
Conventional polymerase chain reaction (PCR) was applied to amplify the NCRs using platinum PCR supermix high fidelity reagents (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. Typically, 42 μl supermix reagent, 3 μl of 10 mM specific primer, 5 μl of 10× universal primer A mix and 1 μl of either 5′-RACE or 3′-RACE cDNA reactions. Samples were amplified by PCR as follows: 94 °C 2 min and 40 cycles of 94 °C 30s, 62 °C 30s, and 68 °C 1 min. PCR products were evaluated on 1% agarose gels in 1× modified TAE buffer (Millpore, Billerica, Massachusetts), and were purified using ultrafree-DA centrifugal units (Millpore, Bil-lerica, Massachusetts) if need. PCR products were then cloned into the TOPO TA cloning kit (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. Colony PCR was performed using M13 Forward and M13 Reverse primers at 94 °C 2 min for 40 cycles of 94 °C 30s, 55 °C 30 s, and 68 °C 30 s with the platinum PCR supermix system (Invitrogen, Carlsbad, CA).
2.7. DNA sequencing
All PCR products were subjected to Sanger sequencing using either M13 forward and reverse primers. The sequences were then analyzed by BLAST (http://www.ncbi.nlm.nih.gov/genomes/FLU/Database).
3. Results
3.1. Cloning both 3′ and 5′ NCRs from 3′ RACE cDNA
Both IAV vRNA and cRNA are present in virus-infected samples although the ratio of vRNA to cRNA varies in different infectious phases. The 3′ end of cRNA is complementary to the 5′ end of vRNA, and vice versa. Thus, a poly(A) tail can be added at both 3′-end of vRNA and cRNA in vitro by using Poly(A) poly-merases, and then 3′-RACE cDNA can be performed using a poly(T) oligonucleotide primer (Fig. 1). With genome specific primers, the synthesized 3′-RACE cDNA products therefore can yield the 3′ NCR of vRNA from the vRNA-generated 3′-RACE cDNA and yield the 5′ NCR of vRNA from cRNA-generated strands (Fig. 2A; Table 3).
Fig. 1.
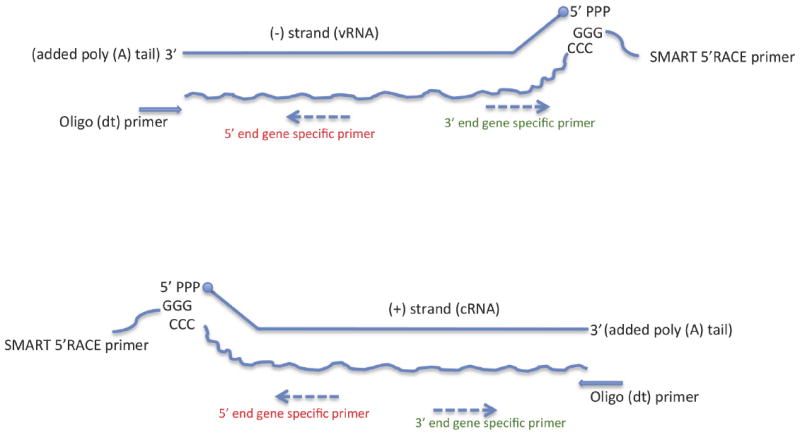
Diagram of influenza A virus RACE methods to determine both 5′ and 3′ noncoding region (NCR) sequences. The technique employees SMART technology to perform 5′-RACE (rapid amplification of cDNA ends) cDNA and employees poly(A) tailing to perform 3′ RACE cDNA from different influenza viral RNA templates (see Table 1).
Fig. 2.
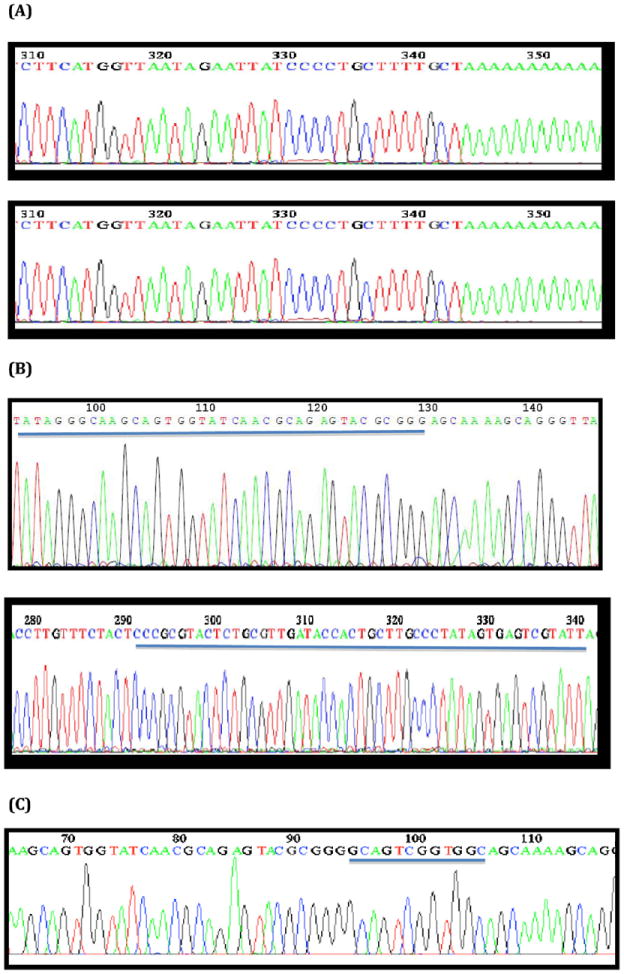
Representative DNA sequence chromatograms of cloned influenza A virus noncoding region (NCR) sequences as determined by RACE methods. (A) Poly(A) addition at the 3′ end of the vRNA strand (in sense orientation) of the NY470 hemagglutinin (HA) segment; and Poly(A) addition at the 3′ end of the cRNA strand of the NY470 HA segment. (B) SMART oligo ligated to the 5′ end of the cRNA strand of the NY470 nucleoprotein (NP) segment (underlined); and SMART oligo ligated to the 5′ end of the vRNA strand (in sense orientation) of the NY470 NP segment (underlined). (C) Chromatogram showing cloned CAP sequences at the 5′ end of mRNA of NY312 polymerase A (PA) (underlined).
Table 3.
Identification of both 5′- and 3′-NCRs from influenza A virus vRNA, cRNA, and mRNA templates.
| Influenza A virus template source | 5′ RACE cDNA [SMART tag + 5′ primer]a | 5′ RACE cDNA [SMART tag + 3′ primer]a | 3′ RACE cDNA [Poly(A) + 5′ primer]a | 3′ RACE cDNA [Poly(A) + 3′ primer]a |
|---|---|---|---|---|
| vRNA | 5′ NCRb | 3′ NCRb | ||
| cRNA | 3′ NCRb | 5′ NCRb | ||
| mRNA | mRNA/CAP |
Based on cDNA sequences and orientation.
Based on vRNA genome sequence orientation.
3.2. Cloning both 3′ and 5′ NCRs from 5′ RACE cDNA
SMART utilizes the specific features of some MMLV reverse transcriptases to add a few non-template-dependent cytosine residues at the 3′ end of the newly synthesized first strand cDNA, and then the SMART oligonucleotide, containing a terminal stretch of dG residues, can anneal to the dC-rich cDNA tail to serve as an extended template for reverse transcription (Fig. 1). With IAV genome specific primers, the 5′-RACE cDNA products therefore can yield the 5′ NCR of vRNA from the vRNA-templated 5′-RACE cDNA and yield the 3′ NCR of vRNA from cRNA-templated 5′-RACE cDNA (Fig. 2B). Thus, either 5′-RACE or 3′-RACE cDNA reactions can be used to clone both NCRs of the IAV genome using both vRNA and cRNA as templates (Table 3).
3.3. CAP sequences of mRNA
SMART-generated 5′-RACE cDNA can also be used to clone the capped 5′-NCR sequences of IAV mRNAs (Fig. 2C; Table 3). Example CAP sequences from representative IAV are shown in Fig. 3. Cloned 5′-NCRs with CAP sequences confirmed their template origin as viral mRNA in 5′ RACE cDNA reactions.
Fig. 3.
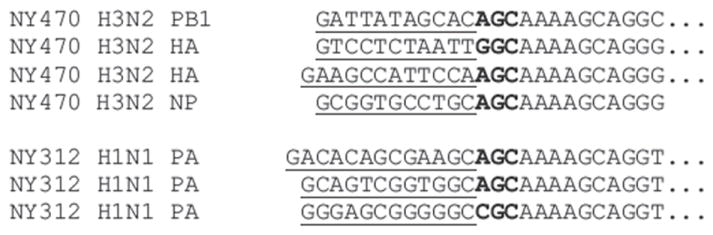
Influenza A virus CAP and 5′ mRNA sequences. CAP sequences (underlined) isolated from the SMART oligo 5′-RACE cDNAs with 5′-end specific primers from NY470 polymerase B1 (PB1) hemagglutinin (HA), nucleoprotein (NP) genes and NY312 polymerase A (PA) genes.
3.4. Variations at the first base of 3′-end vRNA
To test the RACE cloning methods, the IAV NCRs were cloned from both ends of the representative wild type strains, including A/New York/470/2004 (H3N2) [NY470], A/New York/312/2001 (H1N1) [NY312],A/WSN/1933 (H1N1) [WSN], and the rescued 2009 pandemic strain A/California/04/2009 (H1N1) [CA04]. Both NCRs of the viral genome from the representative viruses were sequenced and confirmed in two ways by using both 5′-end and 3′-end specific primers with both 5′-RACE and 3′-RACE cDNAs. The NCR sequences from all the representative viral genomes are shown in Figs. 4–7 in the sense orientation. Variations of the first nucleotide of the IAV genome were identified at the 3′ vRNA NCRs of these representative viruses. The first nucleotide of the 3′ NCR of vRNA segments has been reported to be a uridine (Palese and Shaw, 2007), but using this method, the first position was found to be variable, and while presdominantly uridine, also included clones with a first nucleotide of cytidine, guanosine or adenine. Two hundred forty RACE derived clones were picked for identifying the segment terminal sequences from the representative influenza A viruses in this study, and in this set 160 (66.7%) clones reached to the first base sequence of the 3′-end of vRNA. Among these clones, 74.4% were uridine, 12.5% were guanosine, 11.9% were cytidine and 1.2% were adenine. These first nucleotide variations were identified by using poly(A)-based 3′-RACE PCR with 5′-end specific primers, which can directly yield vRNA 3′ NCR sequences, as well as by using SMART-generated 5′-RACE RCR with 5′-end specific primers that can yield vRNA 3′ NCR sequences through cRNA. Similarly, polymorphisms in the first nucleotide of the 3′-end vRNA were also identified from rescued influenza A viruses that were derived from plasmids that contain the NCRs of A/New York/312/2001 (H1N1) and A/WSN/1933 (H1N1) with a 5′ adenine at the first base of the NCR in the plasmid (representing a 3′ vRNA uridine as the first nucleotide in all segments).
Fig. 4.

Influenza A virus noncoding region (NCR) sequences as determined using RACE methods as shown in Fig. 1. Representative RACE results from rescued 2009 pandemic H1N1 and from the degraded viral RNA extracted from a formalin-fixed, paraffin-embedded (FFPE) postmortem lung sample from a fatal 2009 pandemic H1N1 influenza sample (Gill et al., 2010). 5′ noncoding region sequences (5′-NCR) from four viral gene segments shown.
Fig. 7.

Influenza A/WSN/1933 (H1N1) virus noncoding region (NCR) sequences as determined using 5′ and 3′ RACE methods as shown in Fig. 1. Sequences are shown in sense orientation. Start and stop codons are boxed and frame the viral segment open reading frames (not shown). Quasispecies are identified with the degenerate code: M = A/C, R = A/G, W = A/T, S = C/G, Y = C/T and K = G/T.
3.5. Cloning the NCRS from the degraded IAV RNA samples
Because Poly(A) can be added at any RNA fragment with a 3′-hydroxyl, and the SMART technology allows an oligonucleotide to be added to the 5′ end of either capped or uncapped RNA, the RACE PCR techniques described here do not require full length template viral RNA. Therefore, samples containing degraded IAV RNA can also be used for NCR sequence determination. As examples, this method was successfully used to clone 1918 pandemic H1N1 NCRs from 1918 human lung tissue (data not shown), and also applied to degraded samples from formalin-fixed paraffin-embedded lung tissue sections from a 2009 H1N1 influenza pandemic autopsy case (Gill et al., 2010), and the sequences of 5′ NCRs of viral segments of pandemic 2009 influenza A virus cloned matching the NCR sequences obtained from CA04 intact RNA samples (Fig. 4).
4. Discussion
Characterization of the NCR sequences of IAV is important for understanding viral evolution and pathogenesis. In this paper, a method is described for a rapid and efficient way to clone both 3′ and 5′ NCRs by combining Poly(A) addition and SMART technologies. This method neither requires extensive handling of RNA nor is not dependent on full-length RNA gene segments or transcripts for sample preparation. Using specific primers designed from the IAV genome, the method can be used not only to clone the sequences of NCRs but also to discriminate whether the cloned NCRs were derived from vRNA, cRNA or mRNA templates, which should aid the study of IAV evolution and replication. Another advantage of this method is that it can be applied to samples with degraded viral RNA templates. RNA from formalin-fixed, paraffin-embedded tissue blocks was also used to determined NCR sequences with good results (Fig. 4A).
Variation in the fourth nucleotide of the 3′ end vRNA (either a cytidine or a uridine) has been reported (Lee and Seong, 1998a) and previous studies have demonstrated that nucleotide differences at this position played a critical role for viral polymerase binding, transcriptional regulation, and replication (Jiang et al., 2010; Lee et al., 2003). Using the method described here, the clones demonstrated variations in the reference IAV genomes at this position (Figs. 5–7), without a clear segment specific pattern between reference genomes analyzed.
Fig. 5.

Influenza A/New York/312/2001 (H1N1) virus noncoding region (NCR) sequences as determined using 5′ and 3′ RACE methods as shown in Fig. 1. Sequences are shown in sense orientation. Start and stop codons are boxed and frame the viral segment open reading frames (not shown). Quasispecies are identified with the degenerate code: M = A/C, R = A/G, W = A/T, S = C/G, Y = C/T and K = G/T.
An unexpected finding in this study was the identification of sequence variation in the first nucleotide at the 3′ end of the vRNA. Based on the NCRs of the references sequences identified in this study, the first nucleotide of the 3′-end of vRNA was variable, with a cytidine, adenine or guanosine, in addition to the expected uridine (Palese and Shaw, 2007). These polymorphisms were found in clones derived from the same viral isolation, likely indicating the co-existence viral quasispecies at this site. To rule out the possibility that the variations might being attributed solely to cap snatching during the maturation of mRNA transcripts, the first nucleotide polymorphisms at the 3′ end of vRNA in our reference samples were individually cloned and identified from both cRNA-derived 5′-RACE PCR and vRNA-generated 3′-RACE PCR using 5′-end specific primers. Although the variations were not found using T4 RNA ligase method (Szymkowiak et al., 2003; Yuan et al., 2009) as the comparison study in this paper, other data from our laboratory have provided support for these findings. After re-mining the high throughput sequencing data generated from randomprimed cDNA sequencing libraries prepared from 1918 and 2009 influenza pandemic autopsy samples (Xiao et al., 2013), variations of the first nucleotides of UTRs from different segments from both 1918 and 2009 pandemic virus were also identified (Table 4). However, more work is needed to understand the significance of these first nucleotide polymorphisms. It will be useful to design detailed experiments to quantify the occurrence of the polymorphisms in each segment, and in different IAV strains and subtypes. Although statistical analyses of the ratios of the variable nucleotides at the first base were not performed, differences in the polymorphisms among the segments and viral strains examined were noticed.
Table 4.
Deep-sequencing identification of variations of the first nucleotides of the NCRs from different segments from 1918 and 2009 pandemic influenza viruses.
| Segment | 1918 pandemic
|
2009 pandemic
|
||
|---|---|---|---|---|
| The 1st nucleotide | Number of identified reads | The 1st nucleotide | Number of identified reads | |
| PB2 | Agcgaaa… | 26 | Agcgaaa… | 5 |
| Cgcgaaa… | 65 | |||
| Tgcgaaa… | 10 | |||
| PB1 | Agcgaaa… | 57 | Agcgaaa… | 384 |
| PA | Agcgaaa… | 106 | Agcaaa… | 93 |
| Tgcgaaa… | 7 | |||
| HA | Cgcaaaa… | 29 | ||
| NP | Cgcaaaa… | 11 | Agcaaaa… | 644 |
| Cgcaaaa… | 17 | |||
| NA | ???????… | Agcaaaa… | 1 | |
| MP | Agcaaaa… | 47 | Agcaaaa… | 92 |
| Cgcaaaa… | 3 | |||
| NS | Agcaaaa… | 36 | ?gcaaaa… | |
The upper case letters are the 1st bases of 5′ UTRs from recovered cDNA sequences. ? indicates that sequences recovered did not reach the 5′ end.
IAV transcription and replication takes place in the nucleus of the infected cells. The viral polymerase synthesizes viral mRNAs using short-capped primers derived from cellular transcripts by a unique ‘cap-snatching’ mechanism (Dias et al., 2009). With the method described in this paper, viral mRNA CAP sequences can be cloned using SMART and specific primers. Previous studies have shown that CA-terminated capped fragments were preferentially utilized as primers for viral mRNA synthesis in vitro (Rao et al., 2003). However, not all the CAP sequences identified here contained CA-terminated sequences (Figs. 2C and 3). Furthermore it was also demonstrated here that the CAP sequences vary among mRNA transcripts from different gene segments and even from the same segment during viral infection.
In summary, the NCRs of IAV need to be studied more extensively to evaluate the biological significance of the observed polymorphisms. Further sequence analysis of host-derived capped primers of viral mRNA will also shed light on viral transcription and host response. The simple methods described here provided an efficient way for cloning IAV NCR sequences.
Fig. 6.

Influenza A/New York/470/2004 (H3N2) virus noncoding region (NCR) sequences as determined using 5′ and 3′ RACE methods as shown in Fig. 1. Sequences are shown in sense orientation. Start and stop codons are boxed and frame the viral segment open reading frames (not shown). Quasispecies are identified with the degenerate code: M = A/C, R = A/G, W = A/T, S = C/G, Y = C/T and K = G/T.
Acknowledgments
This work was supported by the Intramural Research Program of NIAID.
References
- de Wit E, Bestebroer TM, Spronken MI, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. Rapid sequencing of the non-coding regions of influenza A virus. J Virol Methods. 2007;139:85–89. doi: 10.1016/j.jviromet.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Desselberger U, Racaniello VR, Zazra JJ, Palese P. 3′-Terminal and 5′-terminal sequences of influenza-A, influenza-B and influenza-C virus-RNA segments are highly conserved and show partial inverted complementarity. Gene. 1980;8:315–328. doi: 10.1016/0378-1119(80)90007-4. [DOI] [PubMed] [Google Scholar]
- Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW. The cap-snatching endonuclease of influenza virus poly-merase resides in the PA subunit. Nature. 2009;458:914–918. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- Engelhardt OG, Smith M, Fodor E. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J Virol. 2005;79:5812–5818. doi: 10.1128/JVI.79.9.5812-5818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor E, Pritlove DC, Brownlee GG. The influenza virus panhandle is involved in the initiation of transcription. J Virol. 1994;68:4092–4096. doi: 10.1128/jvi.68.6.4092-4096.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor E, Pritlove DC, Brownlee GG. Characterization of the RNA-fork model of virion RNA in the initiation of transcription in influenza A virus. J Virol. 1995;69:4012–4019. doi: 10.1128/jvi.69.7.4012-4019.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse Y, Oshitani H. Evolution of the influenza A virus untranslated regions. Infect Genet Evol : J Mol Epidemiol Evol Genet Infect Dis. 2011;11:1150–1154. doi: 10.1016/j.meegid.2011.04.006. [DOI] [PubMed] [Google Scholar]
- Gill JR, Sheng ZM, Ely SF, Guinee DG, Beasley MB, Suh J, Deshpande C, Mollura DJ, Morens DM, Bray M, Travis WD, Taubenberger JK. Pulmonary pathologic findings of fatal 2009 pandemic influenza A/H1N1 viral infections. Arch Pathol Lab Med. 2010;134:235–243. doi: 10.5858/134.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson EC, von Kirchbach JC, Gog JR, Digard P. Genome packaging in influenza A virus. J Gen Virol. 2010;91:313–328. doi: 10.1099/vir.0.017608-0. [DOI] [PubMed] [Google Scholar]
- Jagger BW, Memoli MJ, Sheng ZM, Qi L, Hrabal RJ, Allen GL, Dugan VG, Wang R, Digard P, Kash JC, Taubenberger JK. The PB2-E627K mutation attenuates viruses containing the 2009 H1N1 influenza pandemic polymerase. MBio. 2010;1 doi: 10.1128/mBio.00067-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, Dalton RM, Lo A, Efstathiou S, Atkins JF, Firth AE, Taubenberger JK, Digard P. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science. 2012;337:199–204. doi: 10.1126/science.1222213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Zhang S, Wang Q, Wang J, Geng L, Toyoda T. Influenza virus genome C4 promoter/origin attenuates its transcription and replication activity by the low polymerase recognition activity. Virology. 2010;408:190–196. doi: 10.1016/j.virol.2010.09.022. [DOI] [PubMed] [Google Scholar]
- Krafft AE, Duncan BW, Bijwaard KE, Taubenberger JK, Lichy JH. Optimization of the isolation and amplification of RNA from formalin-fixed, paraffin-embedded tissue: The Armed Forces Institute of Pathology Experience and Literature Review. Mol Diagn. 1997;2:217–230. doi: 10.1054/MODI00200217. [DOI] [PubMed] [Google Scholar]
- Krug RM. Priming of influenza viral RNA transcription by capped heterologous RNAs. Curr Top Microbiol Immunol. 1981;93:125–149. doi: 10.1007/978-3-642-68123-3_6. [DOI] [PubMed] [Google Scholar]
- Lee KH, Seong BL. The position 4 nucleotide at the 3′ end of the influenza virus neuraminidase vRNA is involved in temporal regulation of transcription and replication of neuraminidase RNAs and affects the repertoire of influenza virus surface antigens. J Gen Virol. 1998a;79:1923–1934. doi: 10.1099/0022-1317-79-8-1923. [DOI] [PubMed] [Google Scholar]
- Lee KH, Seong BL. The position 4 nucleotide at the 3′ end of the influenza virus neuraminidase vRNA is involved in temporal regulation of transcription and replication of neuraminidase RNAs and affects the repertoire of influenza virus surface antigens. J Gen Virol. 1998b;79(Pt 8):1923–1934. doi: 10.1099/0022-1317-79-8-1923. [DOI] [PubMed] [Google Scholar]
- Lee MK, Bae SH, Park CJ, Cheong HK, Cheong C, Choi BS. A single-nucleotide natural variation (U4 to C4) in an influenza A virus promoter exhibits a large structural change: implications for differential viral RNA synthesis by RNA-dependent RNA polymerase. Nucleic Acids Res. 2003;31:1216–1223. doi: 10.1093/nar/gkg214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Huang T, Ly H, Parslow TG. Mutational analyses of packaging signals in influenza virus PA, PB1, and PB2 genomic RNA segments. J Virol. 2008;82:229–236. doi: 10.1128/JVI.01541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memoli MJ, Jagger BW, Dugan VG, Qi L, Jackson JP, Taubenberger JK. Recent human influenza A/H3N2 virus evolution driven by novel selection factors in addition to antigenic drift. J Infect Dis. 2009;200:1232–1241. doi: 10.1086/605893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann G, Brownlee GG, Fodor E, Kawaoka Y. Orthomyxovirus replication, transcription, and polyadenylation. Curr. Top. Microbiol. Immunol. 2004;283:121–143. doi: 10.1007/978-3-662-06099-5_4. [DOI] [PubMed] [Google Scholar]
- Palese P, Shaw ML. Orthomyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott, Williams & Wilkins; Philadelphia: 2007. pp. 1647–1690. [Google Scholar]
- Plotch SJ, Bouloy M, Ulmanen I, Krug RM. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell. 1981;23:847–858. doi: 10.1016/0092-8674(81)90449-9. [DOI] [PubMed] [Google Scholar]
- Qi L, Kash JC, Dugan VG, Wang R, Jin G, Cunningham RE, Taubenberger JK. Role of sialic acid binding specificity of the 1918 influenza virus hemagglutinin protein in virulence and pathogenesis for mice. J Virol. 2009;83:3754–3761. doi: 10.1128/JVI.02596-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P, Yuan W, Krug RM. Crucial role of CA cleavage sites in the cap-snatching mechanism for initiating viral mRNA synthesis. Embo J. 2003;22:1188–1198. doi: 10.1093/emboj/cdg109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JS. 5′ and 3′ terminal nucleotide sequences of the RNA genome segments of influenza virus. Nucleic Acids Res. 1979;6:3745–3757. doi: 10.1093/nar/6.12.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scull MA, Rice CM. A big role for small RNAs in influenza virus replication. Proc Natl Acad Sci U S A. 2010;107:11153–11154. doi: 10.1073/pnas.1006673107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymkowiak C, Kwan WS, Su Q, Toner TJ, Shaw AR, Youil R. Rapid method for the characterization of 3′ and 5′ UTRs of influenza viruses. J Virol Methods. 2003;107:15–20. doi: 10.1016/s0166-0934(02)00184-2. [DOI] [PubMed] [Google Scholar]
- Wellenreuther R, Schupp I, Poustka A, Wiemann S. SMART amplification combined with cDNA size fractionation in order to obtain large full-length clones. BMC Genomics. 2004;5:36. doi: 10.1186/1471-2164-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise HM, Hutchinson EC, Jagger BW, Stuart AD, Kang ZH, Robb N, Schwartz-man LM, Kash JC, Fodor E, Firth AE, Gog JR, Taubenberger JK, Digard P. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog. 2012;8:e1002998. doi: 10.1371/journal.ppat.1002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao YL, Kash JC, Beres SB, Sheng ZM, Musser JM, Taubenberger JK. High-throughput RNA sequencing of a formalin-fixed, paraffin-embedded autopsy lung tissue sample from the 1918 influenza pandemic. J Pathol. 2013;229:535–545. doi: 10.1002/path.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T, Fodor E, Rao Z, Liu Y. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature. 2009;458:909–913. doi: 10.1038/nature07720. [DOI] [PubMed] [Google Scholar]
- Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M, Kawaoka Y, Went-worth DE. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J Virol. 2009;83:10309–10313. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]