

Summary
Germline mutations of BRCA1 confer hereditary susceptibility to breast and ovarian cancer. However, somatic mutation of BRCA1 is infrequent in sporadic breast cancers. The BRCA1 protein C-terminus BRCT domains interact with multiple proteins and are required for BRCA1's tumor suppressor function. In this study, we demonstrated that Abraxas, a BRCA1 BRCT domain-interacting protein, plays a role in tumor suppression. Abraxas exerts its function through binding to BRCA1 to regulate DNA repair and maintain genome stability. Both homozygous and heterozygous Abraxas knockout mice exhibited decreased survival and increased tumor incidence. The gene encoding Abraxas suffers from gene copy loss and somatic mutations in multiple human cancers including breast, ovarian, and endometrial cancers, suggesting that mutation and loss of function of Abraxas may contribute to tumor development in human patients.
Introduction
The hereditary breast and ovarian cancer tumor suppressor protein BRCA1 plays critical roles in DNA repair, cell cycle checkpoint control, and maintenance of genomic stability. The C-terminal BRCT domains of BRCA1 constitute a phospho-peptide recognition domain that binds specifically to phospho-peptides containing a pSPxF (phosphor-serine-proline-x-phenylalanine) motif (Manke et al., 2003; Rodriguez et al., 2003; Yu et al., 2003). BRCT domains are frequently targeted by many clinically important mutations and most of these mutations disrupt the binding surface of the BRCT domains to phosphorylated peptides (Bouwman et al., 2013; Glover, 2006). Mice carrying a BRCT mutant of BRCA1 defective in recognition of phosphorylated proteins are prone to tumor development indicating that BRCT phosphoprotein recognition is required for BRCA1 tumor suppression (Shakya et al., 2011). Three proteins, Abraxas (also known as Abra1, FAM175A, CCDC98), Bach1 (also known as Brip1, FancJ) and CtIP (also known as RBBP8), directly interact with the BRCA1 BRCT domains through the pSPxF motif in a phosphorylation-dependent manner forming mutually exclusive complexes, which were thus named as the A, B and C complexes of BRCA1 (Greenberg et al., 2006; Kim et al., 2007; Liu et al., 2007; Wang et al., 2007a; Yu et al., 2003).
The BRCA1 A complex, comprised of Abraxas, and other components, NBA1, BRE, BRCC36, and Rap80, is found to play an important role in mediating the recruitment of BRCA1 to DNA double strand breaks for DNA damage checkpoint regulation and efficient DNA repair. In the BRCA1 A complex, Abraxas appears to be a central adaptor protein, as it harbors several domains necessary for the interactions with BRCA1 and other components in the complex (Wang, 2012). Abraxas interacts with the BRCT domains of BRCA1 through the pSPxF motif at its C-terminus. The N-terminus of Abraxas including a MPN domain is important for interactions with NBA1, BRE and Rap80. In addition, Abraxas dimerizes with BRCC36 through a coiled-coil domain present on both proteins. As a vital component of the BRCA1-A complex, Abraxas appears to be a key player in the DNA damage response. However it's role in DNA repair, BRCA1 signaling and tumor suppression is not clear.
In this study, by generating an Abraxas-deficient mouse model and further analysis, we demonstrated that Abraxas is a tumor suppressor gene and the interaction of Abraxas and BRCA1 is critical for Abraxas' function in repair of DNA and maintenance of genome stability. We also showed evidences that loss of Abraxas function by copy number loss, reduced expression and mutations occur in multiple human cancers.
Results
Abraxas-deficient mice are viable and display increased sensitivity to ionizing radiation
To explore the function of Abraxas in vivo, we generated knockout mice of the Abraxas/Fam175A gene encoding the Abraxas protein (Figure 1 and S1). We first generated a gene-targeting construct containing exon 5 of the Abraxas genomic sequence flanked by two loxP sites and a selection marker NEO gene flanked by two FRT sites inserted in the intron region between exon 4 and 5 of the Abraxas gene (Figure 1A). Upon deletion of exon 5, due to a frame-shift in the targeted allele, a premature stop codon will be encountered immediately after exon 4. Any processed exon 5-deleted transcript would generate a truncated product of 97 amino acids that lacks all functional domains and should be inactive (Figure S1). The construct was introduced into murine ES cells for homologous recombination replacing one copy of the Abraxas gene. Properly targeted ES cells were identified by Southern blot and used for the generation of chimeric mice (Figure S1). The chimeras were then crossed with C56BL/6 mice to identify germline transmission and generation of AbraxasNeo/+ mice. Abraxas+/Δ animals were generated after Cre recombinase mediated deletion of exon 5. Intercrossing of Abraxas+/Δ mice indicated that AbraxasΔ/Δ mice were viable and born at expected Mendelian ratios. RT-PCR using primers located in exon 5 and exon 6 revealed disruption of the full length Abraxas transcript in AbraxasΔ/Δ MEFs (Figure S1). Sequencing of the RT-PCR products spanning exon 2 to exon 6 confirmed that the exon 5-deleted transcript contains a premature STOP codon and has likely undergone nonsense mediated decay indicated by decreased transcript levels (Figure S1). The loss of Abraxas full-length protein was also confirmed in AbraxasΔ/Δ MEFs by immunoblotting (Figure 1B and S1) with an antibody recognizing the C-terminus region of the mouse Abraxas protein. Abraxas-/- (AbraxasΔ/Δ) mice did not exhibit any gross developmental defects and were indistinguishable from their wild type littermates after birth. Thus, unlike BRCA1 and the BRCA1-BRCT interacting CtIP, Abraxas is not required for embryonic and postnatal development.
Figure 1. Abraxas plays critical roles in cellular responses to DNA damage in vivo.
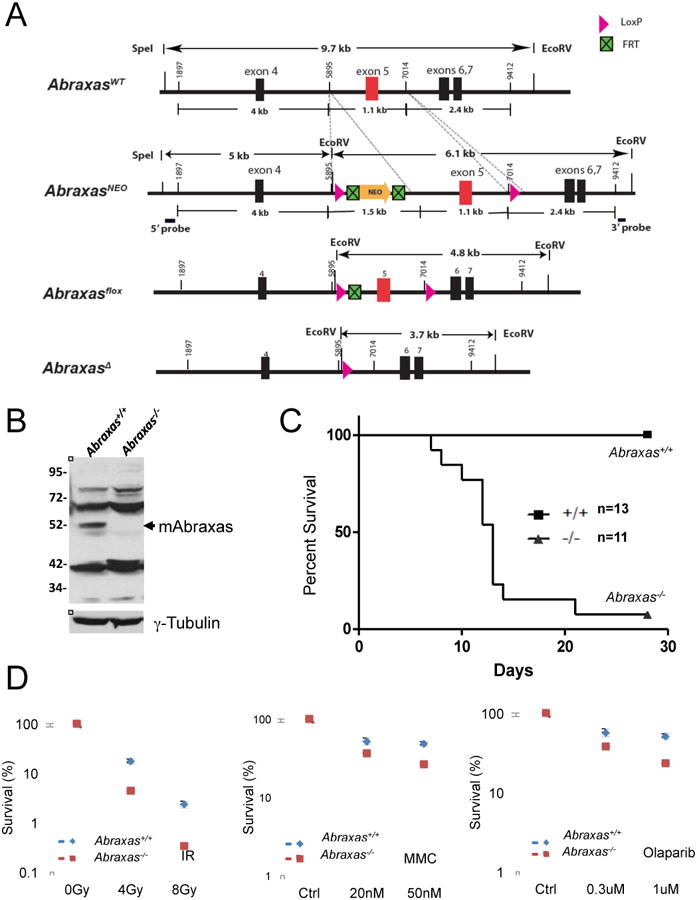
(A) Generation of Abraxas-deficient mice. A diagram of mouse Abraxas alleles generated for deletion of exon 5. NEO, neomycin resistance gene. (B) Abraxas protein is absent in Abraxas-/- MEFs. Cell lysates from Abraxas+/+ and Abraxas-/- MEFs were separated in SDS-PAGE gel. Western blots were carried out with antibodies to mouse Abraxas and tubulin. (C) Abraxas-deficient mice show increased sensitivity to ionizing radiation. Five-week old wild type and Abraxas-/- littermates were subjected to 7.5 Gy IR. Mice were monitored daily and survival was assessed by Kaplan-Meir curve and log-rank test. (D) Abraxas-/- MEF cells are sensitive to IR, MMC and PARP1 inhibitor (Olaparib) as measured by clonogenic survival assay. Cells were incubated with indicated doses of MMC or PARP1 inhibitor during the course of incubation. For colony survival studies, cells were seeded at low density and treated with respective doses of DNA damaging agents. Colonies were counted and normalized to untreated samples to calculate percent survival. Error bars represent standard deviation across triplicates. Three independent experiments were performed.
To determine whether Abraxas plays a role in the DNA damage response in vivo, we treated wild type and Abraxas-/- mice with 7.5 Gy IR. After treatment, the majority of Abraxas-/- mice died between 7-28 days post irradiation, while none of the wild type mice died during this same time frame (Figure 1C). Thus, Abraxas null mice are more sensitive to IR. Similarly, MEF cells prepared from Abraxas-/- mice displayed increased sensitivity to IR, the DNA crosslinking agent mytomycin C (MMC) and poly ADP ribose polymerase (PARP) inhibitor Olaparib (Figure 1D). Abraxas forms IR induced DNA damage foci (IRIF) (Kim et al., 2007; Liu et al., 2007; Wang et al., 2007b). The IRIF formation of Abraxas was undetectable in Abraxas-/- MEF cells as a result of loss of the Abraxas protein (Figure S1). Consistent with the previous notion that Abraxas plays a critical role in the accumulation of BRCA1 to DNA damage sites and formation of the BRCA1-A complex, the IRIF formation of BRCA1 and Rap80 in Abraxas-/- MEF cells were diminished (Figure S1) (Wang and Elledge, 2007; Wang et al., 2009). Together, these data indicate Abraxas plays an important role in the DNA damage response in vivo in mice.
Abraxas-BRCA1 interaction is crucial for double strand break (DSB) repair in response to IR
Abraxas knockout mice and MEF cells are sensitive to IR indicating a deficiency in DNA repair. In order to investigate whether Abraxas-BRCA1 interaction is needed for repair of DSBs, we first examined whether Abraxas-/- MEF cells are defective in DNA repair. Using staining of γH2AX as a marker for damaged DNA, we found that while the WT cells continued to show a decline in the percentage of cells containing γH2AX foci over time and by 7 h post treatment returned to pre-IR levels, the Abraxas-/- cells, however, consistently displayed a high percentage of cells with γH2AX foci through the duration of the experiment indicating a deficiency in DNA repair (Figure 2A). This is confirmed in a comet assay to measure DNA breaks and monitor cells' ability to repair induced DNA breaks. Compared to wild type cells, Abraxas-/- cells appeared to have much higher percentage of cells having unrepaired DNA (increased tail moments) at 6 hour post IR treatment (Figure 2B and S2). As a comparison, BRCA1-deficient Brca1Δ11/Δ11 MEF cells were also defective in DNA repair in the comet assay (Figure S2). With previous implications of Abraxas in homologous recombination (Coleman and Greenberg, 2011; Hu et al., 2011; Wang et al., 2007b), we investigated the role of Abraxas in homologous recombination by analysis of sister chromatid exchange. Compared to wild type cells Abraxas-/- cells showed reduced sister chromatid exchanges per metaphase (Figure 2C) indicating that Abraxas is required for homologous recombination. In consistent, accumulation of Rad51 (Rad51 foci), a central participant in homologous recombination, to the DNA damage sites was also decreased in Abraxas-/- cells (Figure S2).
Figure 2. Abraxas-BRCA1 interaction is crucial for double strand breaks repair in response to IR.
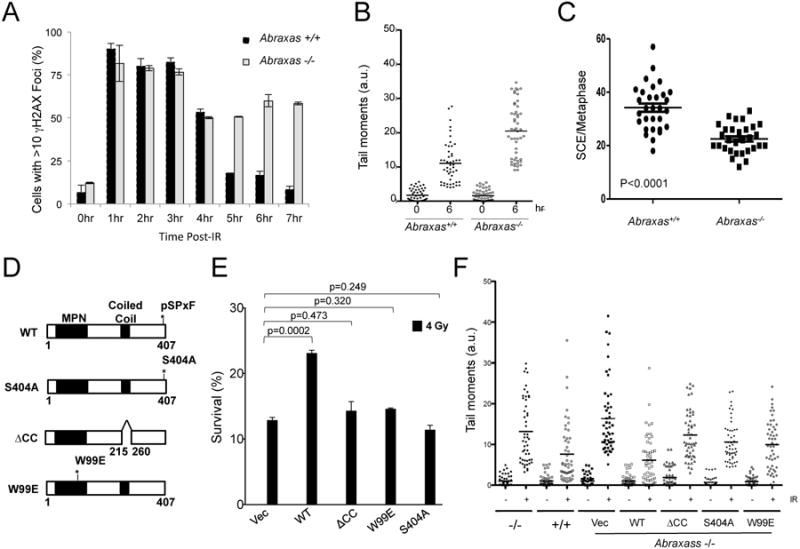
(A) Abraxas-/- cells were defective in repairing damaged DNA after 2 Gy IR treatment. Wild type and Abraxas-/- MEF cells were treated with 2 Gy IR, incubated for indicated times, fixed, and stained with γH2AX antibodies followed by appropriated secondary antibodies. At least 300 cells were counted for each time point for quantification. Cells containing >10 foci were counted as positive. (B) Abraxas-/- cells were defective in DNA repair using comet assay. DNA damage was assessed by comet assay in either untreated or 5 Gy treated primary MEF cells at indicated time points. Tail moments were quantified from 50 cells and the mean value is calculated. The experiment has been repeated three times. (C) Loss of Abraxas resulted in decreased sister chromatid exchange (SCE). After labeling with BrdU for 20hrs, cells were treated with 250nM mitomycin C (MMC) and analyzed for SCE 12hrs post MMC treatment. Data are representative of 30 metaphases per sample. (D) Generation of Abraxas mutants. (E) Wild type but not mutants of Abraxas rescued the increased sensitivity of Abraxas-/- MEF cells to IR as measured by clonogenic survival assay. Error bars represent standard deviation across triplicates. Three independent experiments were performed. (F) Mutants of Abraxas failed to rescue DNA repair deficiency of Abraxas-/- MEF cells. DNA damage was assessed by comet assay in either untreated or 5 Gy treated cells 6 hr post-IR. Tail moments were quantified from 50 cells and damage percentages were calculated based on 150 cells per sample.
We then examined whether interaction with BRCA1 is needed for Abraxas' function in DNA repair. We generated a S404A mutant of mouse Abraxas protein with the serine residue (S404) present in the pSPxF motif mutated to alanine. Mutation of a corresponding serine mutant S406A of the human Abraxas protein abolished the binding of Abraxas to BRCA1 (Wang et al., 2007b). Since the formation of the BRCA1-A complex is important for recruitment of BRCA1 to DNA damage sites, we also generated mutants of Abraxas which fail to form an intact BRCA1-A complex, a coiled-coil deletion (ΔCC) mutant and a W99E mutant (Figure 2D). Previously it was shown deletion of the coiled-coil domain in human Abraxas (ΔCC) disrupts the binding of Abraxas to BRCC36 (Wang et al., 2009) and mutation of W99E in the MPN domain of Abraxas disrupts interaction with other members of the BRCA1-A complex including Rap80, BRE, NBA1 and BRCC36 (Patterson-Fortin et al., 2010). These mutants, when expressed, behaved as expected for interactions with BRCA1 and other A-complex proteins in immunoprecipitation analysis (Figure S2). In the analysis of Abraxas-/- MEFs infected with retroviral constructs stably expressing HA-tagged mouse Abraxas wild type or mutants, the Abraxas mutants failed to increase the survival of Abraxas-/- MEFs in response to IR (Figure 2E). The mutants also failed to rescue the DNA repair deficiency of Abraxas-/- cells using a comet assay to assess DNA repair efficiency (Figure 2F). Together, these results indicate that the interaction of Abraxas with BRCA1, as well as the formation of the BRCA1-A complex, is critical for efficient DNA repair. It suggests that Abraxas is part of the BRCA1 signaling in cellular resistance to DNA damaging agents and DNA repair.
Abraxas is not required for protecting stalled replication forks
BRCA1 is implicated in a repair-independent mechanism in protecting stalled replication forks from degradation to maintain genome stability (Schlacher et al., 2012). We tested whether Abraxas is required for such a role. As previously shown, BRCA1-deficient cells show significant shortening of fork length following hydroxyurea (HU) treatment indicating degradation of stalled replication forks. However, unlike BRCA1, loss of Abraxas resulted in subtle changes in the length of stalled replication forks (Figure 3 and S3). In addition, Abraxas did not appear to be required for efficient replication restart after HU treatment (data not shown), consistent with published observation that BRCA1 is not required for replication recovery (Schlacher et al., 2012). It suggests that the role of BRCA1 in protecting stalled replication fork is independent of Abraxas.
Figure 3. Abraxas is not required for protecting HU-stalled replication fork.

(A) Schematic of experimental design. (B) BRCA1 deficiency results in shortening of stalled replication fork. Fiber assay was performed with UWB1+BRCA1 (BRCA1+/+) or UWB1 (BRCA1-/-) isogenic cell lines following 4 mM hydroxyurea (HU) treatment. UWB1 is a BRCA1-null ovarian cancer cell line. The mean fiber lengths were calculated and indicated (BRCA1-/-, p<0.0001). (C) Abraxas deficiency does not result in shortening of stalled replication fork. Fiber assay was performed with two pairs of Abraxas+/+ or Abraxas-/- MEFs. The mean fiber lengths are calculated and indicated (Abraxas-/-, p=0.073). The experiments were repeated three times with two pairs of Abraxas MEF cell lines (also see Figure S7) and with reversing the analogs and the HU pulse with the same result. More than fifty fibers were measured for each condition.
Abraxas interacts with FancD2 and is recruited to cross-linked DNA damage sites for repair
Since Abraxas-/- cells are sensitive to the DNA crosslinking agent MMC (Figure 1D), we examined whether Abraxas is involved in DNA crosslink repair. Abraxas accumulated to DNA damage sites in cells treated with MMC (Figure 4A). In addition, Abraxas interacted with FancD2 in co-immunoprocipitation assays and the interaction appeared not to be induced by DNA damaging agents such as IR or MMC treatment (Figure 4B). Rap80, another component in the BRCA1-A complex also interacted with FancD2, and similarly, the interaction was independent of DNA damage (Figure S4). We then tested whether Abraxas is directly recruited to DNA crosslink sites. Using an episomal replication-based chromatin IP (eCHIP) assay which allows the identification of DNA damage response proteins at cross-linked DNA lesions in the context of Epstein-Barr nuclear antigen-1 (EBNA)-dependent episomal replication (Shen et al., 2009), we found that Abraxas and Rap80 were significantly enriched on the crosslink-bearing substrate compared with the unmodified substrate (Figure 4C). In addition, the enrichment was further enhanced by DNA replication of the crosslinked substrate, as indicated by the dramatic increase of its enrichment in cells expressing EBNA. It thus suggests that the further recruitment of Abraxas and Rap80 depends on the presence of stalled replication forks. Since BRCA1 interacts with FancD2 and mediates FancD2 foci formation after treatment with MMC (Bunting et al., 2012; Garcia-Higuera et al., 2001; Vandenberg et al., 2003), we wished to examine whether Abraxas affects FancD2 recruitment or ubiquitination in response to crosslink agent. Due to lack of antibodies recognizing mouse FancD2 protein, we tested whether Abraxas is required for FancD2 foci formation or FancD2 ubiquitination after MMC treatment in human cells treated with shRNAs against Abraxas. We found that Abraxas is not required for FancD2 foci formation or ubiquitination in response to MMC treatment (Figure S4). Thus, Abraxas does not appear to play a role in BRCA1 mediated recruitment of FancD2 suggesting Abraxas might have an additional role independent of BRCA1 in the crosslink repair. If this is true, Abraxas mutant not interacting with BRCA1 might be able to rescue the defects of Abraxas-/- cells in crosslink repair. We thus tested whether the S404A mutant could rescue the sensitivity of Abraxas-/- cells to MMC. We noticed that indeed, the S404A mutant appeared to, at least partially, rescue the MMC sensitivity of Abraxas-/- cells (Figure 4D and S4), suggesting that Abraxas possesses a role independent of BRCA1 in the crosslink repair.
Figure 4. Abraxas is involved in cross-linked DNA damage repair.

(A) Abraxas accumulated to DNA damage sites in response to cross-linking agent MMC. Abraxas+/+ and Abraxas-/- MEFs were treated with 1.5uM MMC and collected at 6 hr post-treatment for immunofluorescence analysis. (B) Abraxas interacts with FancD2. Co-immunoprecipitation of Abraxas and FancD2 were carried out using lysates of 293T human cells untreated or treated with 10 Gy IR or 1.5 uM MMC and antibodies against FancD2 or Abraxas. “*” represents a band of IgG heavy chain. (C) Abraxas and Rap80 accumulated to crosslinked DNA damage sites. The eChIP analysis of Abraxas and Rap80 enrichment to a defined psoralen lesion in the absence (293 XL) or presence (EBNA XL) of DNA replication in 293 cells. Error bars for chromatin fraction and eChIP analyses represent SDs derived from three independent experiments. Recruitment of ERCC1, a critical component of the nucleotide excision repair (NER)-lesion bypass-based DNA interstrand cross link repair, was used as a control. (D) MEF cells were treated with MMC with indicated doses for 1 hr followed by two washes of PBS and addition of fresh medium for incubation at 37°C for two weeks. Rescue of the increased sensitivity to MMC of the Abraxas-/- MEF cells as measured by clonogenic survival assay. Error bars represent standard deviation across triplicates. Experiments were repeated three times. Another set of data was presented in supplemental Figure S8.
Abraxas-BRCA1 interaction is crucial for maintaining genome stability
To monitor the effects of Abraxas deficiency on genome stability, we prepared metaphase chromosome spreads from MEF cell lines. Untreated Abraxas-/- MEFs showed a nearly 3-fold increase of spontaneous single chromatid breaks compared to wild type cell lines (Figure 5A and 5B), suggesting an intrinsic defect in genome stability in Abraxas-/- cells. When treated with 2 Gy IR, Abraxas-/- cells displayed a more severe deficiency in maintaining genome stability. IR treated Abraxas-/- MEFs exhibited a significant increase in the percentage of cells containing chromosomal abnormalities compared to wild type cells. In addition, while chromosomal aberrations in wild type cells are mostly single breaks and very rarely fusions and radial structures, IR treated Abraxas-/- MEFs manifested multiple breaks per metaphase as well as significant increases in chromosomal fusion events and radial structures (Figure 5C and 5D). Thus, loss of Abraxas results in the accumulation of both spontaneous and IR-induced chromosomal defects. In addition, DAPI staining of Abraxas-/- MEFs displayed an increased incidence of abnormal nuclear morphology in Abraxas-/- cells compared to wild type. Nuclear fragmentation and micronuclei were more frequently observed in Abraxas-deficient cells, which also suggest elevated levels of chromosomal breaks and defects (Figure S5).
Figure 5. Abraxas-BRCA1 interaction is critical for maintaining genomic stability.

(A) and (B) Abraxas-/- cells displayed increased spontaneous DNA breaks. Primary Abraxas+/+ MEF cells were used in the metaphase spread analysis for spontaneous DNA breaks. Representative image and the percentage of cells containing at least one DNA break are shown. Quantified data are based on >300 metaphases per sample from 3 replicate analyses. (C) and (D) Abraxas-/- cells displayed increased severity of chromosomal aberrations, consisting of multiple DNA breaks, fusions, and radial structures, in response to IR. Metaphase spread analysis was carried out with primary early passage Abraxas+/+ and Abraxas-/- MEF cells treated with 2 Gy IR. Percentage of normal and abnormal metaphase was quantitated. Normal metaphase contains no breaks. Abnormal metaphase contains a single break, more than two breaks, a fusion, a radial structure, or a combination of breaks and fusion/radial structures (complex). The percentage of each type of abnormal metaphase was also calculated. Quantified data are based on >40 metaphases per sample. Error bars represent S.D. (E) Mutants of Abraxas failed to rescue genome instability of the Abraxas-/- MEF cells. Metaphase spread analysis was carried out in immortalized Abraxas-/- MEFs complemented with empty vector, wild type or mutants of Abraxas 4 hr after treated with 2Gy IR. Metaphases consisting “single break”, “multiple breaks (>1)”, or “other” (chromosomal aberrations of fusion/radial, or a combination of breaks and fusion/radial structure) were quantified. Quantified data are based on >40 metaphases per sample.
We then tested whether the interaction of Abraxas and BRCA1 is important for Abraxas to maintain genome stability (Figure 5E). The IR-induced chromosome aberrations in Abraxas null cell were greatly reduced when the null cells were complemented with expression of wild type Abraxas but not with the S404A mutant that is defective in interacting with BRCA1. In addition, mutants that disrupt the formation of the BRCA1-A complex also failed to reduce the chromosome aberrations as much as the wild type Abraxas. Together, these results indicate that the interaction of Abraxas with BRCA1, as well as the formation of the BRCA1-A complex, is critical for Abraxas' function in maintaining genome stability.
Abraxas suppresses tumor development in mice
Since Abraxas interacts with BRCA1 and is critical for maintaining genome stability, we examined whether Abraxas plays a tumor suppressor role in mice. We monitored survival and tumor development in a cohort of Abraxas+/+, Abraxas+/- and Abraxas-/- mice during organismal ageing. Compared to wild-type mice, Abraxas+/- and Abraxas-/- mice had a significantly reduced life span exhibiting decreased disease-free survival (Figure 6A). Pathologic analysis of the tissue slides from the end stage mice revealed a clear increase in the tumor incidence in Abraxas+/- and Abraxas-/- mice. From the mice we analyzed, over 60% of Abraxas-/- nullizygous and Abraxas+/- heterozygous mice developed cancer including lymphoma and other types of tumors (Figure 6B). Although tumors were detected in some Abraxas+/- or Abraxas-/- mice as early as 8 months of age, the mean age of Abraxas-/- and Abraxas+/- mice that developed tumor was 17 months and 24 months respectively. Tumors developed in Abraxas+/- and Abraxas-/- mice were primarily lymphoma and a few cases of solid tumors in lung and liver (Figure 6C). Lymphoma prominently involved the spleen and lymph nodes including the mesenteric and cervical lymph nodes (Figure S6). The majority of the lymphomas developed in Abraxas+/- and Abraxas-/- mice were found to infiltrate various non-lymphoid organs, including liver, lung, kidney and intestine (Figure 6D). Majority of the lymphomas appeared to be B cell lymphoma indicated by B cell marker B220 positive staining in immunohistochemistry (IHC) and B220 and IgM double positive staining in flow cytometry analysis of tumor cells (Figure S6). To investigate whether tumor formation in Abraxas+/- mice is attributed to haploid insufficiency, we performed microdisscetion to isolate tumor cells and analyzed the Abraxas genotype by PCR and Abraxas expression by western blot. The tumor samples analyzed appeared to retain the wild type allele and expression of Abraxas indicating the remaining wild-type allele is expressed (Figure S7) suggesting that haploinsufficiency of Abraxas leads to tumorigenesis in mice. Together, these data indicate that Abraxas is a tumor suppressor in mice.
Figure 6. Abraxas+/- and Abraxas-/- mice exhibit an increased susceptibility to tumor formation.
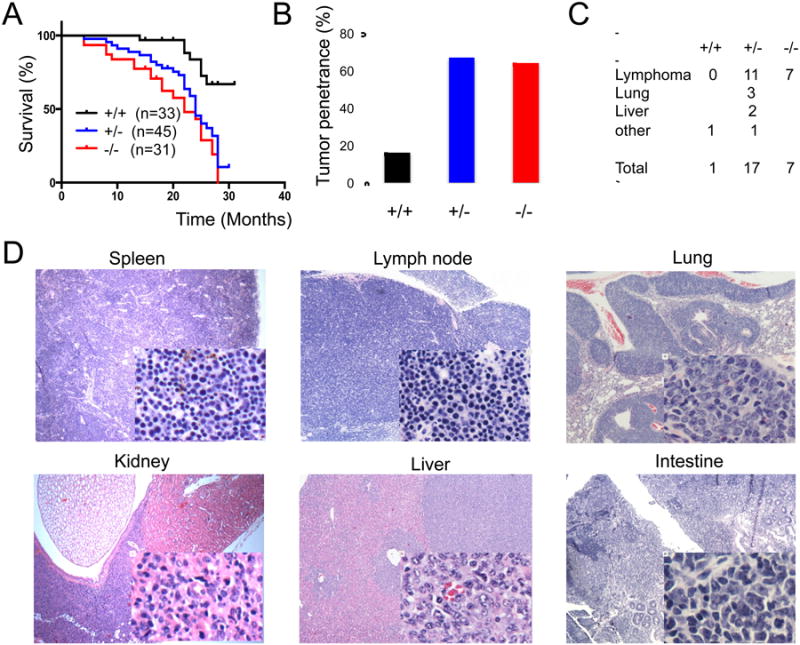
(A) Disease free survival analyzed by the Kaplan-Meier method (p<0,0001). A matched cohort of 33 Abra1+/+, 45 Abra1+/- and 31 Abra1-/- mice were monitored over 30 months for spontaneous tumor development. (B) Spontaneous tumor incidence in Abraxas+/+, Abraxas+/- and Abraxas-/- mice. From the mice we analyzed, 1 out of 6 Abraxas+/+ mice (17%), 14 out of 21 Abraxas+/- mice (67%) and 7 out of 11 Abraxas-/- mice (64%) developed tumor. (C) Summary of the spontaneous tumor spectrum. Pathologic analysis revealed that 1 Abraxas+/+ mouse developed tumor that is most likely histocytic sarcoma; Among 14 Abraxas+/- mice which developed tumor, 2 mice developed both lymphoma and liver tumor, one mouse developed lung adenocarcinoma, liver tumor and lymphoma, one mouse developed histocytic sarcoma; 7 Abraxas-/- mice developed lymphoma. (D) Lymphoma in spleen, lymph node and lymphoid infiltrates to non-lymphoid organs. Representative histological images (H&E staining) of detected tumors were shown.
Indication of loss of Abraxas/FAM175A function in human tumors
Since knockout of Abraxas leads to spontaneous tumor development in mice, we investigated whether Abraxas is also critical for tumor suppression in human. To this end, we analyzed the Abraxas expression level, gene copy number alteration and somatic mutation status in various human tumors in the TCGA (http://cancergenome.noih.gov/) and COSMIC (Forbes et al., 2010) database. Reduced Abraxas gene expression is observed in multiple types of cancers (bladder, breast, cervical, head and neck, renal papillary, endometrial, and thyroid) compared to those from autologous normal tissues (Figure 7A). Gene copy number loss of the Abraxas locus at chromosome 4q21 is frequently found in ovarian, breast (especially basal subtype), lung, and colon cancers, which involves the loss of whole chromosome 4q arm (Figure 7B and Supplemental Table S1). In addition, copy loss of Abraxas correlated well with reduced Abraxas expression in ovarian and breast cancer (Figure 7C), suggesting loss of gene copy number is one of the major mechanisms to down-regulate Abraxas expression in these tumors. Furthermore, somatic mutations of Abraxas are found in endometrial, colon, lung, liver, kidney cancers, and in leukemia (Supplemental Table S1 and S2) with the highest mutation rate found in endometrial cancer (2.5%). Despite the low mutation frequency, the distribution of mutation sites in the Abraxas gene displays a remarkable pattern indicative of targeted inactivation by human tumors. Among all 26 non-synonymous mutations found in Abraxas, five nonsense mutations and one frame-shift insertion were found to generate truncated or abnormal protein products that lack the pSPxF motif and incapable of binding to the BRCA1 protein. Structural and biochemical analysis of BRCA1 BRCT domains bound to optimized phosphopeptides has revealed that the phenylalanine residue in the pSPxF motif is essential for the recognition of this motif by the BRCT domains (Rodriguez et al., 2003; Shiozaki et al., 2004; Williams et al., 2004). The finding of an amino acid mutation of phenylalanine (F409C) in the pSPxF motif located at the very C-terminus of Abraxas in one endometrial tumor also highlights the importance of Abraxas binding to BRCA1. Notably, the same R252* nonsense mutation within the coiled-coil domain was found twice in two endometrial tumor samples. Another seven mutations were found inside the MPN- domain, which is a domain critical for the formation of the BRCA1-A complex. In addition, seven missense mutations were gathered around a small region including a bipartite nuclear localization signal (NLS) (Table S2). A germline mutation of Abraxas (R361Q) in familiar breast cancer patients in this region was previously reported to disrupt nuclear localization of Abraxas (Solyom et al., 2012). Taken together, our cancer genomics data analyses strongly argue that Abraxas is a bona fide tumor suppressor gene in human and Abraxas-BRCA1 interaction is likely to be important for the role of Abraxas in tumor suppression.
Figure 7. Compromise of Abraxas function in human cancers by reduced gene expression, copy number loss, and somatic mutation.
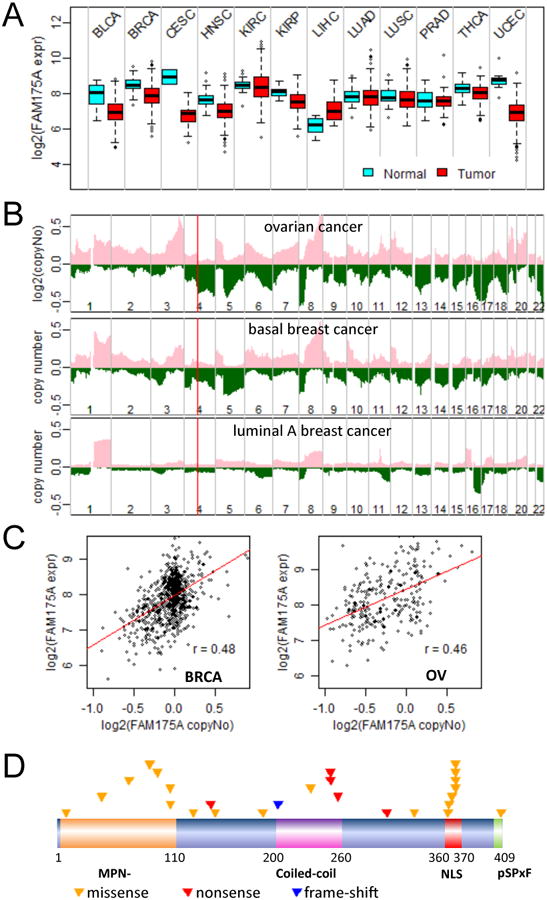
(A) Down-regulation of Abraxas mRNA expression in multiple tumors. Boxplots of logged RNAseq RSEM were made for multiple cancers. BLCA, bladder; BRCA, breast; CESC, cervical; HNSC, head and neck; KIRC, renal clear cell; KIRP, renal papillary; LIHC, liver; LUAD, lung adenocarcinoma; LUSC, lung squamous carcinoma; PRAD, prostate; THCA, thyroid; UCEC, endometrial cancer. (B) Copy number loss of Abraxas/FAM175A locus (4q21) in ovarian cancer and basal breast cancer, but not in luminal A breast cancer. Cumulative gene copy number alterations were drawn from SNP6 data to show frequency of gene copy number changes on each chromosome. Pink/green, copy number gains/losses; the red line marks the position of Abraxas gene on chromosome 4 (chr4). (C) Correlation of reduced Abraxas expression level with copy number loss in breast and ovarian cancer. (D) A graphical summary of non-silent somatic mutations of Abraxas gene identified in human tumors. Each triangle represents a mutation identified from an individual tumor.
Discussion
Abraxas interacts with BRCA1 and is required for accumulation of BRCA1 to DNA damage sites playing important roles in the DNA damage response (Kim et al., 2007; Liu et al., 2007; Wang et al., 2007b). In vivo study of this gene is necessary to further understand its role in DNA repair and tumor suppression. In addition, assessing the clinical relevance of this gene in human cancer patients will provide invaluable insights to the function of this gene in tumor suppression. In this study, we demonstrate Abraxas is essential for DNA repair and tumor suppression in vivo in mouse. In addition, reduced expression, gene copy loss and mutation of Abraxas are also observed in multiple types of human tumor including breast and ovarian cancer.
Analysis from a number of mouse models for BRCA1 inactivation has indicated that BRCA1 is essential for embryonic development and conditional inactivation of BRCA1 in mammary and ovarian tissues predisposes to tumor (Dine and Deng, 2013). Abraxas null mice are viable and born at expected Mendelian ratios indicating that Abraxas gene, unlike BRCA1, is not essential for embryonic development and thus is not likely to play a major role in BRCA1's function in embryonic development. Importantly, Abraxas is essential for tumor suppression. Both Abraxas null and heterozygous mice are tumor prone with over 60% tumor incidence developing lymphomas and other tumors. The tumor suppressing function of Abraxas is likely due to its role in DNA repair. Ability to efficiently repair damaged DNA is crucial for cells to maintain chromosomal stability and prevention of cancer. Our study showed that Abraxas is not only involved in the IR induced double strand break repair, it also plays a role in the crosslink repair. As a result, compared to wild type cells, Abraxas-/- cells displayed increased spontaneous breaks and IR induced chromosome aberrations including multiple breaks, fusions and radial structures. In addition, lack of Abraxas in mice results in hypersensitivity of mice to IR and MEF cells to DNA damaging agents such as IR, MMC and PARPi. Repair of DNA crosslinking lesions require Fanconi anemia (FA) proteins. A FA core complex consisting of FANCA-C, E-G, L and M is formed for damage induced monoubiquitination of FancD2/FancI (Kee and D'Andrea, 2010). Both Abraxas and its interacting protein Rap80 interact with FancD2 and are recruited to DNA crosslink damage sites and the recruitment appears to depend on the presence of stalled replication forks suggesting that Abraxas is involved in replication mediated crosslink repair. Since Abraxas does not appear to be required for FancD2 ubiquitination and recruitment to crosslink damage sites, it is more likely the function of Abraxas and the BRCA1-A complex in DNA crosslink repair is downstream or independent of FancD2 ubiquitination. A group of breast cancer susceptibility (BRCA) genes including BRCA2/FancD1, BACH1/FancJ, PALB2/FancN (Wang, 2007) have been shown to be enriched on crosslink DNA damage sites, and involved in homologous recombination, but not required for monoubiquitination of the FancD2/FancI complex (Shen et al., 2009). The exact mechanism of how Abraxas is involved in DNA repair is still not clear. Nevertheless, despite undefined mechanism, Abraxas is required for efficient DNA repair and plays a critical role in maintaining genome stability and tumor suppression.
Although it has been shown that the BRCT domains of BRCA1 are essential for BRCA1's tumor suppressor function (Shakya et al., 2011), it remains largely unknown how the BRCT associated complexes transmit the BRCA1 signaling in mechanisms of the DNA damage response and whether the BRCA1 interaction is important for tumor suppression. Our study evaluated the importance of Abraxas-BRCA1 interaction in DNA repair and genomic stability. Mutant of Abraxas lacking the BRCA1 interaction motif (S404A mutant) fails to rescue the deficiency of Abraxas-/- cells in DNA damage DSB repair and chromosome stability. Although it is not yet clear the role of Abraxas-BRCA1 interaction in tumor suppression in mice, given its importance in DNA repair and maintenance of genomic stability, the Abraxas-BRCA1 interaction is likely to play a critical role in tumor suppression as well. The importance of Abraxas and BRCA1 interaction in tumor suppression is also suggested by identification of loss of function mutation in Abraxas resulting in truncated products lacking BRCA1-binding motif and a F409C mutation in the pSPxF BRCA1 BRCT binding motif predicted to abolish the binding to BRCA1 (Shiozaki et al., 2004; Williams et al., 2004) in human cancer patient. Thus, Abraxas is part of the BRCA1 signaling in DNA repair, maintenance of genome stability and likely tumor suppression as well. However, it is also not surprising that Abraxas does not participate in all the function of BRCA1. For example, Abraxas, unlike BRCA1, is not required for embryonic development or stalled replication fork protection. On the other hand, Abraxas might have additional roles than BRCA1 signaling in DNA repair and tumor suppression, such as a BRCA1-independent role in the crosslink repair.
Abraxas appears to be a tumor suppressor gene in human from analysis of the human cancer genomics. Among the 26 non-synonymous mutations identified in tumors in the Abraxas gene, at least 6 (23%) are inactivating mutants which result in truncated product of the protein that lose the binding to BRCA1. According to the “20/20” rule proposed by Vogelstein et al. for predicting driver oncogene/tumor suppressor genes (Vogelstein et al., 2013), Abraxas could be classified as a tumor suppressor gene with more than 20% of the mutations are inactivating. Abraxas also fits the criteria as a tumor suppressor gene using the Tumor Suppressor and Oncogene (TUSON) Explorer, a recently proposed computational method for predicting oncogene/tumor suppressor genes (Davoli et al., 2013). The fact that many missense mutations of Abraxas occur at functional domains of Abraxas critical for binding to BRCA1 or interactions with other components of the BRCA1-A complex suggests the functional importance of these domains in prevention of cancer. This is consistent with our experimental findings that the MPN, coiled-coil and pSPxF motif are important for Abraxas' role in DNA repair and genomic stability. We also identified a mutational “hot spot” in Abraxas which includes a previously identified bipartite nuclear localization signal (NLS). A germ-line Abraxas mutation (R361Q) from familiar breast cancer patients was reported to disrupt nuclear localization of Abraxas in this region (Solyom et al., 2012). It is important to note that further analysis is needed to determine whether these mutations are truly pathogenic.
Hereditary germline mutations or genetic/epigenetic inactivation of BRCA1 predispose women to breast and ovarian cancer. Although tumors developed in Abraxas-/- mice are mostly lymphoma, it is known that genetic background plays a role in both the type of tumor that develops and tumor latency in mice (Donehower and Lozano, 2009). Indeed, bioinformatics analysis of the TCGA database indicates that loss/reduced function of Abraxas may contribute to multiple types of human cancers including breast and ovarian cancer. The chromosome locus of Abraxas/FAM175A (4q21) is frequently lost in ovarian and breast tumors. Interestingly, in breast cancer, loss of 4q21 is most frequently found in basal but less common in luminal tumors. Although it is possible multiple tumor suppressor gene including Abraxas exist at 4q21 are lost during chromosome 4 (Chr4) copy loss in tumors, loss of copy correlates well with reduced expression of Abraxas in these tumors suggesting loss of gene copy number is one of the major mechanisms to down-regulate Abraxas expression in these tumors and reduced Abraxas expression may play a role in the development of ovarian and breast tumor. In breast cancer and ovarian cancer, somatic BRCA1 mutation rate is low. Thus, loss of ABRA1 expression may be a surrogate of BRCA1 functional loss. This is consistent with Abraxas plays an important role in BRCA1 signaling, mediating at least partly of BRCA1's role in double strand break repair and maintaining genome stability.
Taken together, our study implicates Abraxas is a bona fide tumor suppressor playing a role in DNA repair and maintaining genome stability. It is also likely to be part of the BRCA1 signaling that contributes to tumor suppression.
Experimental Procedures
Generation of Abraxas null mice
Details are described in the Supplemental experimental procedures. All experiments with mice followed protocols approved by the MD Anderson Cancer Center Institutional Animal Care and Use Committee, protocol 110812132, and conformed to the guidelines of the United States Animal Welfare Act and the National Institutes of Health.
Mouse radiosensitivity assay
Five-week-old WT and Abraxas-/- littermates were subjected to 7.5 Gy ionizing radiation. Mice were monitored daily for survival and Kaplan-Meier survival cures were generated. A log-rank test was used for statistical anslysis. Thirteen Abraxas+/+ mice and 11 Abraxas-/- mice were included for the study.
Plasmids and antibodies
Retroviral expression constructs for expression of wild type and Abraxas mutants were made using MSCV vectors containing HA-Flag tag at the N terminus as described (Schlabach et al., 2008). Deletion mutants of Abraxas were generated by cloning the corresponding cDNA fragments into the aforementioned retroviral vector. Site-directed mutagenesis was performed with the QuickChange II site-directed mutagenesis kit (Stratagene, CA) to generate Abraxas point mutations. Purified Gst-tagged C-terminal fragment of Abraxas (281-407 aa) was used for Abraxas antibody production. Other antibodies used were BRCA1 (M20, Santa Cruz), γH2AX (Upstate), HA (cell signaling), FancD2 (Santa Cruz, Genetex).
Cell culture and generation of MEFs
MEFs were generated from 13.5 day embryos using standard procedures. Immortalization was carried out by standard 3T3 assay. About 3×105 cells were seeded every three days. 293T cells and MEFs were cultured in DMEM supplemented with 10% FBS and 100ug/ml penicillin/streptomycin. Stable cells were generated by infecting 293T cells or MEFs with retrovirus containing HA-tagged proteins, followed by puromycin selection.
Immunofluorescence
Cells grown on coverslips were fixed in 3% paraformaldehyde/2% sucrose fixative solution for 10 minutes, permeablized with 0.5% Triton X-100, blocked with 5% bovine serum albumin, and incubated with primary antibodies at 37°C for 1hr. Appropriate secondary antibodies conjugated to Alexa -488, -555, -647 (Invitrogen) and Cy3 (Amersham Biosciences) fluorophores were used. All images were captured on a Nikon 2000U inverted microscope using a Photometric CoolSnap HQ camera.
Clonogenic survival assay
MEF cells were seeded at low density and treated with various DNA damaging agents, i.e. ionizing radiation, MMC, etc. Cells were then cultured at 37°C for 10-14 days to allow colonies to form. Colonies were stained with 2% crystal violet/50% methanol. Colonies containing 50 or more cells were counted and statistical data were analyzed by t-test analysis.
Analysis of Metaphase Chromosomes
Cells were seeded to obtain 50-70% confluency following overnight culture. For IR induced chromosome breaks, cells were irradiated with 2Gy IR and cultured for 2h. Cells were then incubated in 25ng/ml colcemid (Invitrogen) for 1-3h. Cells were harvested by mitotic shake off, washed, incubated in hypotonic solution (0.06M KCl) and fixed in 10ml of Carnoy's fixative. Metaphase spreads were prepared on glass slides and dried. Dried slide preparations were stained with Giemsa or DAPI and examined for the presence of chromatid breaks, fusions, and structural defects.
Comet Assay
Trypsinized 2 × 105 cells were pelleted and suspended in 1ml ice-cold 1 × PBS buffer followed by standard protocol. Details are described in the Supplemental experimental procedures. Nuclear DNA on slides was visualized under a fluorescent microscope (Leica DM4000 B) and images were captured at 400 × magnifications using Leica DFC 300 FX color camera. To measure levels of DNA damage, 50 cells of each sample were analyzed for tail moment (a.u.) by the CometScore software (TriTek Corp.). Quantitative results of tail moment were graphed by Prism 5 software.
DNA Fiber Assays
Cells were labeled with CldU (100 uM) for 30min, washed, labeled with IdU (250 uM) for 30min, and then treated with HU (4mM) for 4hrs. Alternatively, cells were labeled with CldU, followed by HU, and then IdU for the same times indicated. DNA fibers were spread as described (Jackson and Pombo, 1998). Briefly, cells were diluted 1:10 with unlabeled cells, and 2.5ul of the cells suspended in PBS (∼106 cell/ml) were spotted on to a glass slide. After briefly drying, 7.5ul of spreading buffer (0.5% SDS, 200mM Tris-HCl pH7.4, 50mM EDTA) was dropped on the cells and incubated for 10min. Slides were tilted (∼15°) to spread lysed cells across the slide. Slides were air dried, fixed in methanol:acetic acid (3:1) for 3min, and stored at 4°C overnight before staining. CldU and IdU tracts were detected using Mouse anti-BrdU (BD Biosciences) (1:50) and Rat-anti-BrdU (OBT0030, AbDSerotec) (1:100) 1hr at 37°C, followed by Alexa-488 anti-mouse (1:100) and Cy3-Anti Rat (Jackson ImmunoResearch) (1:100) 30min at 37°C, and mounted with VectaShield (Vector labs). Fibers were imaged at 40× with a Zeiss Axio Observer inverted microscope and Metamorph software for acquisition. Statistics were performed using a two-tailed Mann-Whitney test with Prism 6 (GraphPad) software.
eChIP Assay
Preparation of lesion-defined substrates and the eChIP assay were carried out according to protocols described earlier (Shen et al., 2009)
Histology
Tissues were fixed in 10% buffered Formalin (Fisher Scientific). Paraffin blocks were cut at 5 um and were stained with hematoxylin and eosin (H&E) stain. For immunohistochemistry, B220 (550286; BD PharMingen), Ki67 (VP-RM04; Vector), CD3 (ab5690; Abcam) antibodies were used. Images were obtained using a Nikon Eclipse90i microscope.
Cancer Genomics Data Analysis
Level 3 RNAseqV2 data for gene expression and level 3 SNP array data for gene copy numbers were downloaded from The Cancer Genome Atlas (TCGA) data portal (https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm). Somatic mutation data including mutation rate and gene copy loss rate were downloaded from the COSMIC website (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/).
Supplementary Material
Highlights.
Abraxas is a tumor suppressor gene.
Abraxas deficiency causes defective DNA repair and genomic instability.
Abraxas-BRCA1 interaction is crucial for DNA repair and genomic stability.
Somatic mutation and gene copy loss result in loss of Abraxas function in human cancers.
Acknowledgments
We thank Michael Huang, Drs. Jianxin Liu and Ling Wu for providing technical support, Henry P. Adams for assistance with the use of the microscope, Dr. Xiaochun Yu for providing mouse BRCA1 antibody used in this work. We also thank Drs. Dong Zhang, Randy Legerski, Guillermina Lozano for helpful discussions. A.C. was a recipient of the Training Grant in Molecular Genetics of Cancer (CA009299). A.P. is a recipient of the Schissler Foundation Fellowship for Translational Studies in Cancer Research. L.L. is supported by NIH CA127945 and CA179441 grant. M.J.Y. is supported by NIH CA164346 grant and funds from the MD Anderson Cancer Center (Leukemia SPORE CA100632, Center for Inflammation and Cancer, Center for Genetics and Genomics, IRG, SINF, and Physician Scientist Award). This work was supported by the National Institutes of Health grant (CA155025 to B.W.), the Mel Klein Family Fund and funds from the University of Texas MD Anderson Cancer Center (IRG, Center for Genetics and Genomic Pilot Award).
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
Author contribution: A.C. and B.W. designed the study. A.C., A.P., B.S., T.H., Y.W., and S.A.Y. performed the experiments. M.J.Y. carried out pathological analysis of mice tumors. J.Y. performed computational analysis. J.T., L.L, L.Z., J.Y. and B.W. analyzed the data. A.C. and B.W. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bouwman P, van der Gulden H, van der Heijden I, Drost R, Klijn CN, Prasetyanti P, Pieterse M, Wientjens E, Seibler J, Hogervorst FB, et al. A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov. 2013;3:1142–1155. doi: 10.1158/2159-8290.CD-13-0094. [DOI] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Kozak ML, Kim JM, Wong N, Lopez-Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A, et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell. 2012;46:125–135. doi: 10.1016/j.molcel.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman KA, Greenberg RA. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J Biol Chem. 2011;286:13669–13680. doi: 10.1074/jbc.M110.213728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dine J, Deng CX. Mouse models of BRCA1 and their application to breast cancer research. Cancer Metastasis Rev. 2013;32:25–37. doi: 10.1007/s10555-012-9403-7. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Lozano G. 20 years studying p53 functions in genetically engineered mice. Nature reviews Cancer. 2009;9:831–841. doi: 10.1038/nrc2731. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, Kok CY, Jia M, Ewing R, Menzies A, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Research. 2010;38:D652–657. doi: 10.1093/nar/gkp995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Glover JN. Insights into the molecular basis of human hereditary breast cancer from studies of the BRCA1 BRCT domain. Fam Cancer. 2006;5:89–93. doi: 10.1007/s10689-005-2579-z. [DOI] [PubMed] [Google Scholar]
- Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011;25:685–700. doi: 10.1101/gad.2011011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee Y, D'Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24:1680–1694. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Huang J, Chen J. CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nat Struct Mol Biol. 2007;14:710–715. doi: 10.1038/nsmb1277. [DOI] [PubMed] [Google Scholar]
- Liu Z, Wu J, Yu X. CCDC98 targets BRCA1 to DNA damage sites. Nat Struct Mol Biol. 2007;14:716–720. doi: 10.1038/nsmb1279. [DOI] [PubMed] [Google Scholar]
- Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302:636–639. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- Patterson-Fortin J, Shao G, Bretscher H, Messick TE, Greenberg RA. Differential regulation of JAMM domain deubiquitinating enzyme activity within the RAP80 complex. J Biol Chem. 2010;285:30971–30981. doi: 10.1074/jbc.M110.135319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M, Yu X, Chen J, Songyang Z. Phosphopeptide binding specificities of BRCA1 COOH-terminal (BRCT) domains. J Biol Chem. 2003;278:52914–52918. doi: 10.1074/jbc.C300407200. [DOI] [PubMed] [Google Scholar]
- Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, Zhao Z, Smogorzewska A, Sowa ME, Ang XL, et al. Cancer Proliferation Gene Discovery Through Functional Genomics. Science. 2008;319:620–624. doi: 10.1126/science.1149200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin CS, deRooij DG, Hirsch S, Ravi K, Hicks JB, et al. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science. 2011;334:525–528. doi: 10.1126/science.1209909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Do H, Li Y, Chung WH, Tomasz M, de Winter JP, Xia B, Elledge SJ, Wang W, Li L. Recruitment of fanconi anemia and breast cancer proteins to DNA damage sites is differentially governed by replication. Mol Cell. 2009;35:716–723. doi: 10.1016/j.molcel.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozaki EN, Gu L, Yan N, Shi Y. Structure of the BRCT repeats of BRCA1 bound to a BACH1 phosphopeptide: implications for signaling. Mol Cell. 2004;14:405–412. doi: 10.1016/s1097-2765(04)00238-2. [DOI] [PubMed] [Google Scholar]
- Solyom S, Aressy B, Pylkas K, Patterson-Fortin J, Hartikainen JM, Kallioniemi A, Kauppila S, Nikkila J, Kosma VM, Mannermaa A, et al. Breast cancer-associated Abraxas mutation disrupts nuclear localization and DNA damage response functions. Sci Transl Med. 2012;4:122ra123. doi: 10.1126/scitranslmed.3003223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg CJ, Gergely F, Ong CY, Pace P, Mallery DL, Hiom K, Patel KJ. BRCA1-independent ubiquitination of FANCD2. Mol Cell. 2003;12:247–254. doi: 10.1016/s1097-2765(03)00281-8. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B. BRCA1 tumor suppressor network: focusing on its tail. Cell & Bioscience. 2012;2:6. doi: 10.1186/2045-3701-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:20759–20763. doi: 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Hurov K, Hofmann K, Elledge SJ. NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev. 2009;23:729–739. doi: 10.1101/gad.1770309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007a;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 Form a BRCA1 Protein Complex Required for the DNA Damage Response. Science. 2007b;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nature reviews Genetics. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- Williams RS, Lee MS, Hau DD, Glover JN. Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nat Struct Mol Biol. 2004;11:519–525. doi: 10.1038/nsmb776. [DOI] [PubMed] [Google Scholar]
- Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.