

Abstract
Advances in genome-wide molecular cytogenetics allow identification of novel submicroscopic DNA copy number alterations (aCNAs) and copy-neutral loss of heterozygosity (cnLOH) resulting in homozygosity for known gene mutations in myeloid neoplasms. We describe the use of an oligo-SNP array for genomic profiling of aCNA and cnLOH, together with sequence analysis of recurrently mutated genes, in a patient with myelodysplastic syndrome (MDS) presenting with normal karyotype and FISH results. Oligo-SNP array analysis revealed a hemizygous deletion of 896 kb at chromosome 5q31.2, representing the smallest 5q deletion reported to date. The deletion involved multiple genes, including two tumor suppressor candidate genes (CTNNA1 and HSPA9) that are associated with MDS/AML. The SNP-array study also detected 3 segments of somatic cnLOH: one involved the entire long arm of chromosome 4; the second involved the distal half of the long arm of chromosome 7, and the third encompassed the entire chromosome 22 (UPD 22). Sequence analysis revealed mutations in TET2 (4q), EZH2 (7q), ASXL1 (20q11.21), and RUNX1 (21q22.3). Coincidently, TET2 and EZH2 were located at segments of cnLOH resulting in their homozygosity. Loss of heterozygosity affecting these two chromosomes and mutations in TET2 and EZH2 are indicative of a myelodysplastic syndrome with a poor prognosis. Deletion of the tumor suppressor genes CTNNA1 and HSPA9 is also likely to contribute to a poor prognosis. Furthermore, the original cnLOHs in multiple chromosomes and additional cnLOH 14q in the follow-up study suggest genetic evolution of the disease and poor prognosis. This study attests to the fact that some patients with a myelodysplastic syndrome who exhibit a normal karyotype may have underlying genetic abnormalities detectable by chromosomal microarray and/or targeted mutation analyses.
Keywords: Copy neutral loss of heterozygosity (cnLOH), Uniparental disomy (UPD), MDS, TET2, EZH2, RUNX1, EZH2, ASXL1, CTNNA1, HSPA9
Background
Recent advances in genome-wide molecular cytogenetics allow the identification of novel molecular abnormalities [1-8]. Emerging data demonstrate that myelodysplastic syndrome (MDS) exhibits abundant CNAs and cnLOH, often in the setting of a normal karyotype [9-11]. Loss of heterozygosity (LOH) is an indicator of neoplastic evolution and disease progression [12,13]. Copy-neutral LOH (cnLOH) arises either via a hemizygous deletion in one homolog and duplication of the other, or uniparental disomy (UPD). Both types of somatic LOH have been observed in studies of various cancer types and may explain some of the mechanisms by which tumor suppressor genes (TSGs) are inactivated or activating mutations in oncogenes are duplicated. Acquired UPD (aUPD) is now understood to be common in oncogenesis and appears to be a mechanism to increase the allelic burden of the mutated genes [14-24]. Thus, it is important to know the regions of cnLOH to determine new regions containing potential mutational targets affecting disease pathogenesis and treatment outcome [25,26].
Application of SNP-array technology has led to the identification of recurrent regions of cnLOH in a majority of the chromosomes [4,13,25,27-30] and recurrent pathogenic mutations. These findings have greatly advanced our understanding of the molecular mechanisms of cancer evolution and have led to the development of therapeutics and diagnostic tests. In this respect, mutation analysis helps further stratify neoplasms and their treatment outcome [31-34].
In this study, we used SNP array-based genomic profiling to detect acquired copy number alterations (aCNA) and cnLOHs, together with sequence analysis of genes commonly mutated in MDS, in a patient with normal chromosome and MDS FISH (fluorescence in situ hybridization) panel results.
Clinical presentation
Morphologic and immunophenotypic findings
An 88-year-old woman presented with leukocytosis (16.2×109/L) including minimal monocytosis (1.0×109/L), mild anemia (HGB 105 g/L), and moderate thrombocytopenia (79×109/L). Bone marrow morphologic and flow cytometric examinations revealed hypercellular bone marrow with granulocytic predominance with left-shifted and unusual maturation, and occasional atypical granulocytes and megakaryocytes, but no significant increase in monocytes or myeloblasts (Figure 1). A myeloid neoplasm positive for CD13 and CD33 was diagnosed, best classified as MDS, refractory cytopenia with multilineage dysplasia. Data on therapeutic interventions were not available for this study. At the 5-month follow-up, the overall morphologic and immunophenotypic findings were similar to those at initial presentation.
Figure 1.
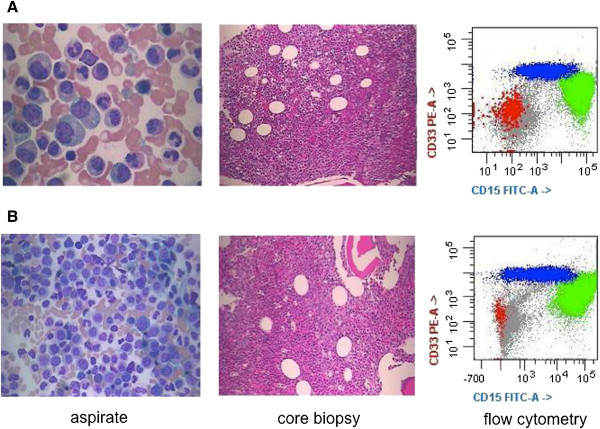
Morphologic and immunophenotypic findings. A) At presentation, there was granulocytic predominance with progressive maturation and no significant increase in blasts or monocytes. The core biopsy shows hypercellular bone marrow with left-shifted granulocytic predominance. Flow cytometry reveals granulocytic predominance (in green) but no significant increase in monocytes (in blue) or myeloblasts (in red). B) At follow-up, the morphologic and immunophenotypic findings are similar to those at presentation.
Chromosome and FISH study
Bone marrow cells were cultivated for 24 and 48 hours in RPMI 1640 or Marrow Max Medium with 10% fetal calf serum (Life Technologies, Grand Island, NY 14072, USA). In total, 20 mitoses were analyzed according to the ISCN (International System for Human Cytogenetic Nomenclature), with a resolution of 300 bands per haploid karyotype. FISH analyses using a panel of MDS probes (-5/5q-, -7/7q-, +8 and 20q-; Vysis, Downers Grove, IL, USA) and BlueGnome probes RP11-114B12 (Illumina, San Diego, CA, USA) for the deleted region 5q31.2 were performed on interphase cells according to the manufacturer’s protocol. Subsequently, 200 cells were examined carefully.
Oligo SNP array
Microdeletion/microduplication screening was performed using an SNP-array platform (CytoScan HD SNP array; Affymetrix, Santa Clara, CA), following the manufacturer’s instructions. The CytoScan HD array has 2.67 million probes, including 1.9 million copy number probes and 0.75 million SNP probes. Array data were analyzed using the Chromosome Analysis Suite (ChAS) software v 2.0 (Affymetrix).
Mutation analysis
Genomic DNA was also tested for mutations in 19 genes that are recurrently mutated in myeloid neoplasms, including ASXL1, EZH2, RUNX1, IDH1, IDH2, KRAS, NRAS and TET2. Sequencing was performed using a TruSeq custom amplicon assay on the MiSeq sequencing platform (Illumina, Hayward, CA). Analysis was performed using SeqPilot software (JSI Medical Systems, Costa Mesa, CA). The assay had sufficient read depth to provide a minimum sensitivity of 5% to 10% for mutation detection.
Results and discussion
During the initial evaluation of the patient, cytogenetic analysis revealed a normal karyotype and FISH studies were negative for aCNAs commonly seen in MDS (MDS panel) (Figure 2A-D). Microarray analysis revealed a microdeletion of approximately 896 kb at the 5q31.2 chromosomal region and three segments of somatic cnLOH for the entire long arm of chromosome 4 (136 Mb), the distal half of the long arm of chromosome 7 (50 Mb), and the entire chromosome 22 (31 Mb). The microdeletion at 5q31.2 extended from 137,821,899 to 138,718,504 bp (UCSC genome Browser; http://genome.ucsc.edu/; hg19 release) and included the ETF1, HSPA9, SNORD63, CTNNA1, LRRTM2, SIL1, SNHG4, MATR3, SNORA74A, PAIP2, and SLC23A1 genes (Figure 3).
Figure 2.
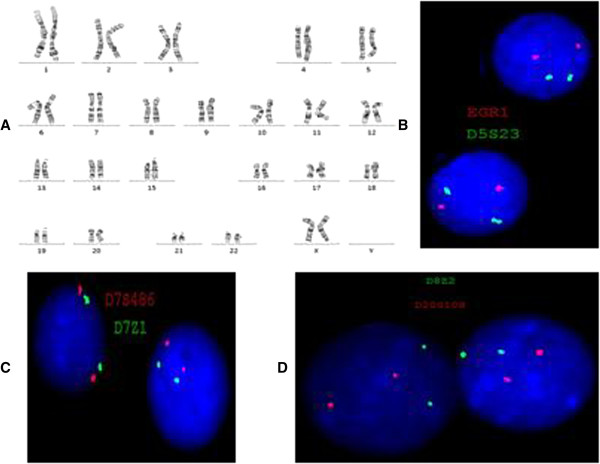
Routine cytogenetic and fluorescence insitu hybridization (FISH) results. A) G-banded chromosome analysis shows a normal female karyotype. B-D) FISH analyses show normal hybridization with an MDS panel using probes for EGR1 (5q31, red signal) and its control probe D5S23 (5p15.2, green signal) in B; D7S486 (7q31, red signal) and its control probe D7Z1 (7centromere, green signal) in C; CEP8 (green signal) for chromosome 8 centromere and D20S108 (20q12, red signal) in D.
Figure 3.
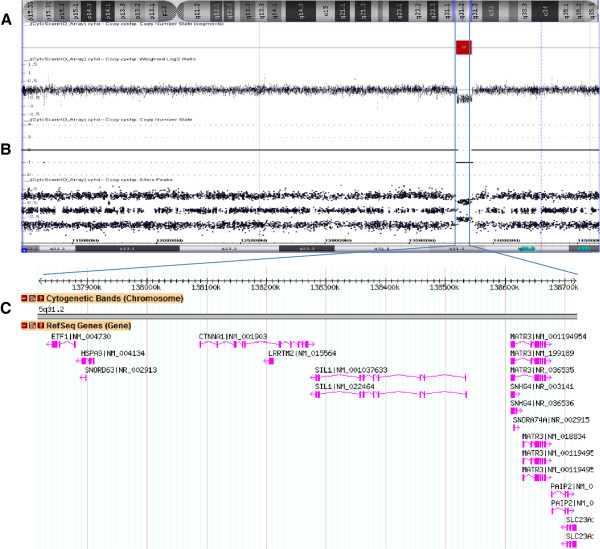
5q31.2 deletion. A) Chromosome 5 with deletion at q31.2. B) SNP-array results, including the weighted log2 ratio, copy number state, and allele peaks at the deleted region. C) Database of genomic variants showing an 896-kb deletion in the short arm of chromosome 5 within band q31.2 (position 137,821,899 to 138,718,504), including the CTNNA1 and HSPA9 genes.
Of the genes deleted due to this microdeletion, two are tumor suppressor candidate genes associated with MDS/AML: CTNNA1, which encodes alpha-1 catenin, and HSPA9, which encodes heat-shock 70-KD protein 9 (mortalin) [35]. Deletion of CTNNA1 was confirmed by applying BlueGnome FISH probes (RP11-114B12) (Figure 4). Alpha-catenins such as that encoded by CTNNA1 are essential for the regulation of cell-cell and cell-matrix interactions in tissues [36]. Loss of expression of the CTNNA1 tumor suppressor gene in hematopoietic stem cells may provide a growth advantage that contributes to human MDS/AML with 5q deletion [37]. Furthermore, loss of the CTNNA1 expression has been associated with leukemia progression or transformation of MDS to AML [38]. The HSPA9 gene is also located at the 5q31.2 region frequently deleted in MDS/AML, making it a candidate tumor suppressor gene; this is consistent with the biological function of its murine homologue. Human mortalin (encoded by HSPA9) was originally identified by its close homology to murine mortalins, which play important roles in cellular senescence [39]. The HSPA9 gene is a novel negative regulator of Raf/MEK/ERK pathway that may be a potential therapeutic target [40].
Figure 4.
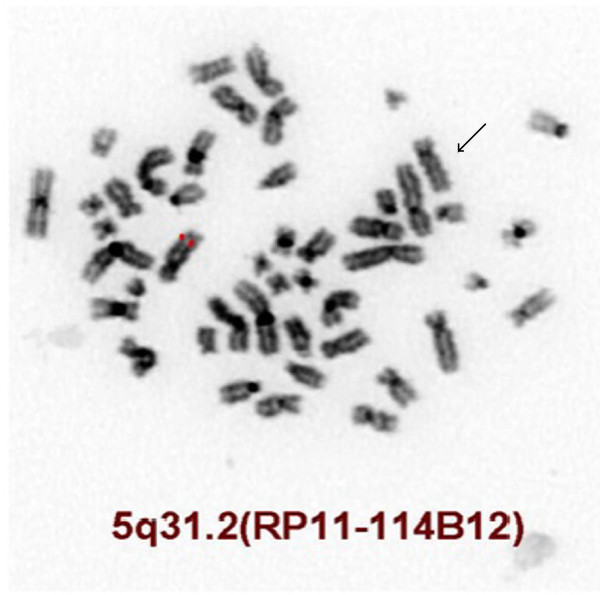
FISH inverted DAPI image showing deletion of the CTNNA1 gene using the BlueGnome FISH probe RP11-114B12 (5q31.2, red signal). The deleted chromosome 5 is indicated by an arrow.
The combined size of cnLOHs spanning at least 10 Mb across the genome was approximately 217.6 Mb (Figure 5). These were detected at the long arm of chromosomes 4, 7, and 22. The cnLOH might result from mitotic recombination or nondisjunction which leads to segmental or whole chromosomal UPD, respectively [13]. Acquired UPD (cnLOH) at diagnosis in our case is indicative of neoplastic evolution [12,13]. An additional UPD was identified at 14q in the follow-up study 5 months later, confirming the genetic progression of disease (Figure 6).
Figure 5.
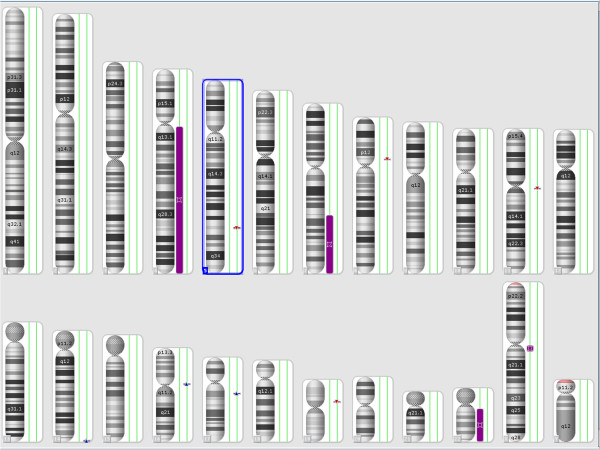
SNP-array results at diagnosis showing loss of heterozygosity (LOH) at the long arm of chromosomes 4, 7, and 22 as purple bars next to the corresponding chromosomes.
Figure 6.
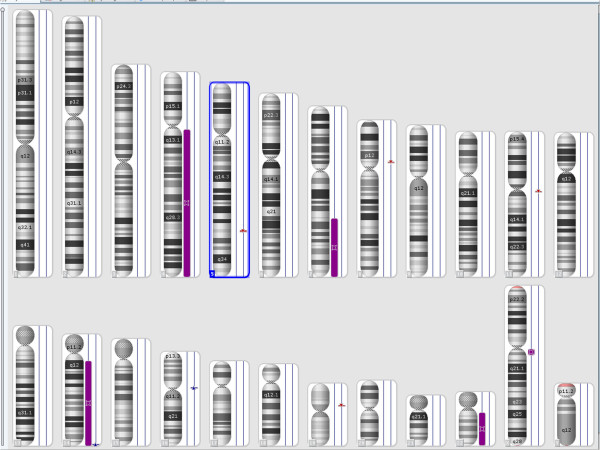
SNP-array results at the follow-up study showing loss of heterozygosity (LOH) of chromosome 14, in addition to the LOH chromosomes 4, 7, and 22 found at initial diagnosis. LOH is shown as purple bar next to the corresponding chromosome.
Recent investigations have indicated that cnLOH can be responsible for homozygosity of mutations in critical genes within the region. Reduction to homozygosity as a consequence of cnLOH was initially thought to be a mechanism for only inactivation of tumor suppressor genes [14,41,42]; however, identification of cnLOH in leukemia has shown that oncogeneic mutations are also targeted [13,16,17].
Mutation analysis of 19 MDS-associated genes revealed an ASXL1 frameshift/stop mutation (Y591*, 41% of reads), an EZH2 point mutation (R690H, 85%), two RUNX1 frameshift/stop mutations (c.474dupT, 19% and c.424_425ins11bp, 7%), and two TET2 frameshift/stop mutations (c.1510_1513delAAAA, 77% and R1465*, 10%). These mutated genes are located on chromosomes 20, 7, 21, and 4, respectively. Recent studies have shown that TET2 mutations are present in up to 30% of MDS/MPN cases, with mutations in ASXL1, EZH2, and RUNX1 also commonly reported [43]. The finding of mutations in all four of these myeloid regulatory genes suggests that they play a critical role in the pathogenesis of this case and demonstrate that mutation analysis is useful in cytogenetically normal myeloid disorders [43].
The cnLOH affecting chromosome 7q and homozygous EZH2 mutation have been reported in 10% of AML and MDS cases. They have been associated with a poor prognosis [9,26,44] and clonal evolution [45,46], supporting the possible role of EZH2 as a tumor suppressor gene for myeloid malignancies.
In contrast to the mutated TET2 and EZH2 genes, no LOH was found for the other two mutated genes (RUNX1 and ASXL1). RUNX1 mutations have been proposed as clinically useful biomarkers to follow disease progression from MDS to AML as well as to monitor minimal residual disease (MRD) [47]. Moreover, RUNX1 mutations were demonstrated to be frequent in de novo AML with non-complex karyotypes and conferred an unfavorable prognosis [48] explained by an association with resistance to chemotherapy [49].
Mutations in ASXL1 have been identified in MDS, AML, chronic myeloid leukemia, chronic myelomonocytic leukemia (CMML), and juvenile myelomonocytic leukemia [50-54], and act as a tumor suppressor in myeloid malignancies [50]. Mutations in ASXL1, TET2, and EZH2 have been found in 41% of MDS cases in a Chinese population [44], similar to the data reported in patients of European decent with MDS [21,23,24,55,56].
Conclusion
In conclusion, our study identified four large cnLOH and a microdeletion at 5q31 harboring two tumor suppressor genes (CTNNA1 and HSPA9) in an MDS case with an apparently normal karyotype. The regions of cnLOH at chromosomes 4, 7, 14, and 22 confirm and extend previous studies, supporting that cnLOHs in myeloid disorders are common and nonrandom. Moreover, the presence of an additional aUPD 14q at follow up, along with persistence of the 5q31 microdeletion and all cnLOHs detected at presentation, indicates genetic progression of the disease. The deletion of tumor suppressor genes CTNNA1 and HSPA9, along with mutation of candidate myeloid regulatory genes TET2 and EZH2, supports the diagnosis of MDS and likely portends a poor prognosis.
This study attests to the fact that some patients with a myelodysplastic syndrome exhibiting a normal karyotype may have underlying genetic abnormalities detectable by chromosomal microarray and/or targeted mutation analyses. Further genomic and molecular studies on a series of patients with MDS may yield information on how to stratify this category of disease to seek further molecular definition.
Ethical approval and consent
These studies were performed on anonymized samples received in the clinical laboratory and thus were exempted from the requirement for consent by an opinion for the Western Institutional Review Board.
Abbreviations
aCNA: Acquired copy number alteration; cnLOH: Loss of heterozygosity; aUPD: Acquired uniparental disomy.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MH, First author; performed chromosome, FISH, and microarray analysis, interpretation of the results, drafting and finalization of the manuscript. WC performed the morphology and immunophenotypic analysis. DJ and YW performed the genomic sequencing. FR performed the comprehensive review of the clinical and laboratory findings. AA reviewed and reported the SNP-array analyses. MEN reviewed the manuscript. FZB reviewed and finalized the manuscript. All authors read and approved the final manuscript.
Contributor Information
Morteza Hemmat, Email: morteza.x.hemmat@questdiagnostics.com.
Weina Chen, Email: Weina.Chen@UTSouthwestern.edu.
Arturo Anguiano, Email: Arturo.L.Anguiano@questdiagnostics.com.
Mohammed El Naggar, Email: Mohamed.M.Elnaggar@questdiagnostics.com.
Frederick K Racke, Email: frederick.k.racke@questdiagnostics.com.
Dan Jones, Email: Dan.Jones@QuestDiagnostics.com.
Yongbao Wang, Email: yongbao.x.wang@questdiagnostics.com.
Charles M Strom, Email: charles.m.strom@questdiagnostics.com.
Karl Chang, Email: karltchang@yahoo.com.
Fatih Z Boyar, Email: fatih.z.boyar@questdiagnostics.com.
Acknowledgments
The authors would like to express their thanks to Maryam Talai and Robert Mcgough (Quest Diagnostics) for their technical support.
References
- Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui CH, Relling MV, Evans WE, Shurtleff SA, Downing JR. Genome-wide analysis of genetic alterations in acute lymphoblastic leukemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- Suela J, Alvarez S, Cifuentes F, Largo C, Ferreira BI, Blesa D, Ardanaz M, Garcia R, Marquez JA, Odero MD, Calasanz MJ, Cigudosa JC. DNA profiling analysis of 100 consecutive de novo acute myeloid leukemia cases reveals patterns of genomic instability that affect all cytogenetic risk groups. Leukemia. 2007;21:1224–1231. doi: 10.1038/sj.leu.2404653. [DOI] [PubMed] [Google Scholar]
- Tyyba kinoja A, Elonen E, Piippo K, Porkka K, Knuutila S. Oligonucleotide array-CGH reveals cryptic gene copy number alterations in karyotypically normal acute myeloid leukemia. Leukemia. 2007;21:571–574. doi: 10.1038/sj.leu.2404543. [DOI] [PubMed] [Google Scholar]
- Gupta M, Raghavan M, Gale RE, Chelala C, Allen C, Molloy G, Chaplin T, Linch DC, Cazier JB, Young BD. Novel regions of acquired uniparental disomy discovered in acute myeloid leukemia. Genes Chromosomes Cancer. 2008;47:729–739. doi: 10.1002/gcc.20573. [DOI] [PubMed] [Google Scholar]
- Kawamata N, Ogawa S, Zimmermann M, Kato M, Sanada M, Hemminki K, Yamatomo G, Nannya Y, Koehler R, Flohr T, Miller CW, Harbott J, Ludwig WD, Stanulla M, Schrappe M, Bartram CR, Koeffler HP. Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray. Blood. 2008;111(2):776–84. doi: 10.1182/blood-2007-05-088310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi T, Ogawa S, Dugas M, Kawamata N, Yamamoto G, Nannya Y, Sanada Miller CW, Yung A, Schnittger S, Haferlach T, Haferlach C, Koeffler HP. Frequent genomic abnormalities in acute myeloid leukemia/myelodysplastic syndrome with normal karyotype. Haematologica. 2009;94:213–223. doi: 10.3324/haematol.13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejewski JP, Tiu RV, O’Keefe C. Application of array based whole genome scanning technologies as a cytogenetic tool in haematological malignancies. Br J Hematol. 2009;146:479–488. doi: 10.1111/j.1365-2141.2009.07757.x. [DOI] [PubMed] [Google Scholar]
- Tiu RV, Gondek LP, O’Keefe CL, Huh J, Sekeres MA, Elson P, McDevitt MA, Wang XF, Levis MJ, Karp JE, Advani AS, Maciejewski JP. New lesions detected by single nucleotide polymorphism array-based chromosomal analysis have important clinical impact in acute myeloid leukemia. J Clin Oncol. 2009;27:5219–5226. doi: 10.1200/JCO.2009.21.9840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrichs S, Kulkarni RV, Bueso-Ramos CE, Levine RL, Loh ML, Li C, Neuberg D, Kornblau SM, Issa JP, Gilliland DG, Garcia-Manero G, Kantarjian HM, Estey EH, Look AT. Accurate detection of uniparental disomy and microdeletions by SNP array analysis in myelodysplastic syndromes with normal cytogenetics. Leukemia. 2009;23(9):1605–13. doi: 10.1038/leu.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel A1, Beier M, Ingenhag D, Servan K, Hein M, Moeller V, Betz B, Hildebrandt B, Evers C, Germing U, Royer-Pokora B. Comprehensive array CGH of normal karyotype myelodysplastic syndromes reveals hidden recurrent and individual genomic copy number alterations with prognostic relevance. Leukemia. 2011;25:387–399. doi: 10.1038/leu.2010.293. [DOI] [PubMed] [Google Scholar]
- Tiu RV1, Gondek LP, O’Keefe CL, Elson P, Huh J, Mohamedali A, Kulasekararaj A, Advani AS, Paquette R, List AF, Sekeres MA, McDevitt MA, Mufti GJ, Maciejewski JP. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood. 2011;117(17):4552–60. doi: 10.1182/blood-2010-07-295857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen CL, Wiuf C, Kruhøffer M, Korsgaard M, Laurberg S, Ørntoft TF. Frequent occurrence of uniparental disomy in colorectal cancer. Carcinogenesis. 2007;28(1):38–48. doi: 10.1093/carcin/bgl086. [DOI] [PubMed] [Google Scholar]
- Raghavan M, Smith LL, Lillington DM, Chaplin T, Kakkas I, Molloy G, Chelala C, Cazier JB, Cavenagh JD, Fitzgibbon J, Lister TA, Young BD. Segmental uniparental disomy is a commonly acquired genetic event in relapsed acute myeloid leukemia. Blood. 2008;112(3):814–21. doi: 10.1182/blood-2008-01-132431. [DOI] [PubMed] [Google Scholar]
- Flotho C, Steinemann D, Mullighan CG, Neale G, Mayer K, Kratz CP, Schlegelberger B, Downing JR, Niemeyer CM. Genome-wide single-nucleotide polymorphism analysis in juvenile myelomonocytic leukemia identifies uniparental disomy surrounding the NF1 locus in cases associated with neurofibromatosis but not in cases with mutant RAS or PTPN11. Oncogene. 2007;26(39):5816–21. doi: 10.1038/sj.onc.1210361. [DOI] [PubMed] [Google Scholar]
- Fitzgibbon J, Smith LL, Raghavan M, Smith ML, Debernardi S, Skoulakis S, Lillington D, Lister TA, Young BD. Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer Res. 2005;65:9152–9154. doi: 10.1158/0008-5472.CAN-05-2017. [DOI] [PubMed] [Google Scholar]
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol. 2002;30(3):229–36. doi: 10.1016/S0301-472X(01)00789-5. [DOI] [PubMed] [Google Scholar]
- Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, Kreil S, Jones A, Score J, Metzgeroth G, Oscier D, Hall A, Brandts C, Serve H, Reiter A, Chase AJ, Cross NC. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood. 2009;113(24):6182–92. doi: 10.1182/blood-2008-12-194548. [DOI] [PubMed] [Google Scholar]
- Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, Tamura A, Honda H, Sakata-Yanagimoto M, Kumano K, Oda H, Yamagata T, Takita J, Gotoh N, Nakazaki K, Kawamata N, Onodera M, Nobuyoshi M, Hayashi Y, Harada H, Kurokawa M, Chiba S, Mori H, Ozawa K, Omine M, Hirai H, Nakauchi H, Koeffler HP, Ogawa S. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009;460(7257):904–8. doi: 10.1038/nature08240. [DOI] [PubMed] [Google Scholar]
- Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, Rataul MS, Szpurka H, Sekeres MA, Wang XF, McDevitt MA, Maciejewski JP. 250 K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008;68(24):10349–57. doi: 10.1158/0008-5472.CAN-08-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, Cervantes F, Oscier D, Boultwood J, Grand FH, Cross NC. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722–6. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, Stevens-Linders E, van Hoogen P, van Kessel AG, Raymakers RA, Kamping EJ, Verhoef GE, Verburgh E, Hagemeijer A, Vandenberghe P, de Witte T, van der Reijden BA, Jansen JH. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41(7):838–42. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tönnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, van der Reijden BA, Jansen JH. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42(8):665–7. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, Lécluse Y, Plo I, Dreyfus FJ, Marzac C, Casadevall N, Lacombe C, Romana SP, Dessen P, Soulier J, Viguié F, Fontenay M, Vainchenker W, Bernard OA. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- Mohamedali A, Gäken J, Twine NA, Ingram W, Westwood N, Lea NC, Hayden J, Donaldson N, Aul C, Gattermann N, Giagounidis A, Germing U, List AF, Mufti GJ. Prevalence and prognostic significance of allelic imbalance by single-nucleotide polymorphism analysis in low-risk myelodysplastic syndromes. Blood. 2007;110(9):3365–73. doi: 10.1182/blood-2007-03-079673. [DOI] [PubMed] [Google Scholar]
- Gondek LP, Tiu R, O'Keefe CL, Sekeres MA, Thiel KS, Maciejewski JP. Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS. Blood. 2008;111:1534–42. doi: 10.1182/blood-2007-05-092304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorletta TA, Gasparini P, D’Elios MM, Trubia M, Pelicci PG, Di Fiore PP. Frequent loss of heterozygosity without loss of genetic material in acute myeloid leukemia with a normal karyotype. Genes Chromosomes Cancer. 2005;44:334–337. doi: 10.1002/gcc.20234. [DOI] [PubMed] [Google Scholar]
- Raghavan M, Lillington DM, Skoulakis S, Debernardi S, Chaplin T, Foot NJ, Lister TA, Young BD. Genome-wide single nucleotide polymorphism analysis reveals frequent partial uniparental disomy due to somatic recombination in acute myeloid leukemias. Cancer Res. 2005;65:375–378. [PubMed] [Google Scholar]
- Fitzgibbon J, Smith LL, Raghavan M, Smith ML, Debernardi S, Skoulakis S, Lillington D, Lister TA, Young BD. Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer Res. 2005;65:9152–9154. doi: 10.1158/0008-5472.CAN-05-2017. [DOI] [PubMed] [Google Scholar]
- Gondek LP, Dunbar AJ, Szpurka H, McDevitt MA, Maciejewski JP. SNP array karyotyping allows for the detection of uniparental disomy and cryptic chromosomal abnormalities in MDS/MPD-U and MPD. PLoS One. 2007;2(11):e122. doi: 10.1371/journal.pone.0001225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett AK, Goldstone AH, Linch DC. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: Analysis of 854 patients from the United Kingdom medical research council. AML 10 and 12 trials. Blood. 2001;98:1752–1759. doi: 10.1182/blood.V98.6.1752. [DOI] [PubMed] [Google Scholar]
- Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, Liso A, Vignetti M, Fazi P, Meani N, Pettirossi V, Saglio G, Mandelli F, Lo-Coco F, Pelicci PG, Martelli MF. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- Baldus CD, Mrozek K, Marcucci G, Bloomfield CD. Clinical outcome of de novo acute myeloid leukaemia patients with normal cytogenetics is affected by molecular genetic alterations: a concise review. Br J Haematol. 2007;137:387–400. doi: 10.1111/j.1365-2141.2007.06566.x. [DOI] [PubMed] [Google Scholar]
- Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood. 2007;110:1262–1270. doi: 10.1182/blood-2006-04-015826. [DOI] [PubMed] [Google Scholar]
- Horrigan SK, Arbieva ZH, Xie HY, Kravarusic J, Fulton NC, Naik H, Le TT, Westbrook CA. Delineation of a minimal interval and identification of 9 candidates for a tumor suppressor gene in malignant myeloid disorders on 5q31. Blood. 2000;95(7):2372–7. [PubMed] [Google Scholar]
- Kask M1, Pruunsild P, Timmusk T. Bidirectional transcription from human LRRTM2/CTNNA1 and LRRTM1/CTNNA2 gene loci leads to expression of N-terminally truncated CTNNA1 and CTNNA2 isoforms. Biochem Biophys Res Commun. 2011;411(1):56–61. doi: 10.1016/j.bbrc.2011.06.085. [DOI] [PubMed] [Google Scholar]
- Liu TX, Becker MW, Jelinek J, Wu WS, Deng M, Mikhalkevich N, Hsu K, Bloomfield CD, Stone RM, DeAngelo DJ, Galinsky IA, Issa JP, Clarke MF, Look AT. Chromosome 5q deletion and epigenetic suppression of the gene encoding alpha-catenin (CTNNA1) in myeloid cell transformation. Nat Med. 2007;13(1):78–83. doi: 10.1038/nm1512. [DOI] [PubMed] [Google Scholar]
- Ye Y, McDevitt MA, Guo M, Zhang W, Galm O, Gore SD, Karp JE, Maciejewski JP, Kowalski J, Tsai HL, Gondek LP, Tsai HC, Wang X, Hooker C, Smith BD, Carraway HE, Herman JG. Progressive chromatin repression and promoter methylation of CTNNA1 associated with advanced myeloid malignancies. Cancer Res. 2009;69(21):8482–90. doi: 10.1158/0008-5472.CAN-09-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Hu Z, Chyna B, Horrigan SK, Westbrook CA. Human mortalin (HSPA9): a candidate for the myeloid leukemia tumor suppressor gene on 5q31. Leukemia. 2000;14(12):2128–34. doi: 10.1038/sj.leu.2401935. [DOI] [PubMed] [Google Scholar]
- Wu PK, Hong SK, Veeranki S, Karkhanis M, Starenki D, Plaza JA, Park JI. A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol Cell Biol. 2013;33(20):4051–67. doi: 10.1128/MCB.00021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavenee WK, Dryja TP, Phillips RA, Benedict WF, Godbout R, Gallie BL, Murphree AL, Strong LC, White RL. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature. 1983;305(5937):779–84. doi: 10.1038/305779a0. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B, Feinberg AP. Somatic deletion and duplication of genes on chromosome 11 in Wilms’ tumours. Nature. 1984;309:176–8. doi: 10.1038/309176a0. [DOI] [PubMed] [Google Scholar]
- Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, O’Keefe CL, Ganetzky R, McDevitt MA, Maciejewski JP. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113(25):6403–10. doi: 10.1182/blood-2009-02-205690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ai X, Gale RP, Xu Z, Qin T, Fang L, Zhang H, Pan L, Hu N, Zhang Y, Xiao Z. TET2, ASXL1 and EZH2 mutations in Chinese with myelodysplastic syndromes. Leuk Res. 2013;37(3):305–11. doi: 10.1016/j.leukres.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Jerez A1, Sugimoto Y, Makishima H, Verma A, Jankowska AM, Przychodzen B, Visconte V, Tiu RV, O’Keefe CL, Mohamedali AM, Kulasekararaj AG, Pellagatti A, McGraw K, Muramatsu H, Moliterno AR, Sekeres MA, McDevitt MA, Kojima S, List A, Boultwood J, Mufti GJ, Maciejewski JP. Loss of heterozygosity in 7q myeloid disorders: clinical associations and genomic pathogenesis. Blood. 2012;119(25):6109–17. doi: 10.1182/blood-2011-12-397620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SN, Jankowska AM, Mahfouz R, Dunbar AJ, Sugimoto Y, Hosono N, Hu Z, Cheriyath V, Vatolin S, Przychodzen B, Reu FJ, Saunthararajah Y, O’Keefe C, Sekeres MA, List AF, Moliterno AR, McDevitt MA, Maciejewski JP, Makishima H. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia. 2013;27(6):1301–9. doi: 10.1038/leu.2013.80. [DOI] [PubMed] [Google Scholar]
- Dicker F, Haferlach C, Sundermann J, Wendland N, Weiss T, Kern W, Haferlach T, Schnittger S. Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia. 2010;24:1528–1532. doi: 10.1038/leu.2010.124. [DOI] [PubMed] [Google Scholar]
- Schnittger S, Dicker F, Kern W, Wendland N, Sundermann J, Alpermann T, Alpermann T, Haferlach C, Haferlach T. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood. 2011;117:2348–2357. doi: 10.1182/blood-2009-11-255976. [DOI] [PubMed] [Google Scholar]
- Gaidzik VI, Bullinger L, Schlenk RF, Zimmermann AS, Rock J, Paschka P, Corbacioglu A, Krauter J, Schlegelberger B, Ganser A, Späth D, Kündgen A, Schmidt-Wolf IG, Götze K, Nachbaur D, Pfreundschuh M, Horst HA, Döhner H, Döhner K. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011;29:1364–1372. doi: 10.1200/JCO.2010.30.7926. [DOI] [PubMed] [Google Scholar]
- Gelsi-Boyer V, Trouplin V, Adélaïde J, Bonansea J, Cervera N, Carbuccia N, Lagarde A, Prebet T, Nezri M, Sainty D, Olschwang S, Xerri L, Chaffanet M, Mozziconacci MJ, Vey N, Birnbaum D. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145(6):788–800. doi: 10.1111/j.1365-2141.2009.07697.x. [DOI] [PubMed] [Google Scholar]
- chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, Ko BS, Wu SJ, Huang SY, Hsu SC, Chen YC, Huang YN, Chang YC, Lee FY, Liu MC, Liu CW, Tseng MH, Huang CF, Tien HF. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood. 2010;115(14):2749–54. doi: 10.1182/blood-2009-11-253070. [DOI] [PubMed] [Google Scholar]
- Boultwood J, Perry J, Zaman R, Fernandez-Santamaria C, Littlewood T, Kusec R, Pellagatti A, Wang L, Clark RE, Wainscoat JS. High-density single nucleotide polymorphism array analysis and ASXL1 gene mutation screening in chronic myeloid leukemia during disease progression. Leukemia. 2010;24(6):1139–45. doi: 10.1038/leu.2010.65. [DOI] [PubMed] [Google Scholar]
- Pérez B, Kosmider O, Cassinat B, Renneville A, Lachenaud J, Kaltenbach S, Bertrand Y, Baruchel A, Chomienne C, Fontenay M, Preudhomme C, Cavé H. Genetic typing of CBL, ASXL1, RUNX1, TET2 and JAK2 in juvenile myelomonocytic leukaemia reveals a genetic profile distinct from chronic myelomonocytic leukaemia. Br J Haematol. 2010;151(5):460–8. doi: 10.1111/j.1365-2141.2010.08393.x. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Muramatsu H, Makishima H, Prince C, Jankowska AM, Yoshida N, Xu Y, Nishio N, Hama A, Yagasaki H, Takahashi Y, Kato K, Manabe A, Kojima S, Maciejewski JP. Spectrum of molecular defects in juvenile myelomonocytic leukaemia includes ASXL1 mutations. Br J Haematol. 2010;150(1):83–7. doi: 10.1111/j.1365-2141.2010.08196.x. [DOI] [PubMed] [Google Scholar]
- Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, Viguié F, Quesnel B, Beyne-Rauzy O, Solary E, Vey N, Hunault-Berger M, Fenaux P, Mansat-De Mas V, Delabesse E, Guardiola P, Lacombe C, Vainchenker W, Preudhomme C, Dreyfus F, Bernard OA, Birnbaum D, Fontenay M. Groupe Francophone des Myélodysplasies. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs) Blood. 2009;114(15):3285–91. doi: 10.1182/blood-2009-04-215814. [DOI] [PubMed] [Google Scholar]
- Thol F, Friesen I, Damm F, Yun H, Weissinger EM, Krauter J, Wagner K, Chaturvedi A, Sharma A, Wichmann M, Göhring G, Schumann C, Bug G, Ottmann O, Hofmann WK, Schlegelberger B, Heuser M, Ganser A. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol. 2011;29(18):2499–506. doi: 10.1200/JCO.2010.33.4938. [DOI] [PubMed] [Google Scholar]