

Abstract
Cancer of the stomach is the fourth most common cancer worldwide. The single strongest risk factor for gastric cancer is Helicobacter pylori-associated chronic gastric inflammation. Among persons with H. pylori infection, strain-specific components, host immune responses, and environmental factors influence the risk for gastric disease, including adenocarcinoma of the stomach, although only a small proportion of infected persons develop the malignancy. Recent advances in DNA sequencing technology have uncovered a complex community of non-cultivatable inhabitants of the human stomach. The interaction between these inhabitants, collectively referred to as the gastric microbiota, and H. pylori likely impacts gastric immunobiology and possibly the sequelae of H. pylori infection. Thus, characterization of the gastric microbiota in subjects with and without H. pylori infection could provide new insight into gastric homeostasis and the pathogenesis of H. pylori-associated disease, including gastric cancer.
Keywords: Cancer, stomach, H. pylori, microbiota
Introduction
Gastric adenocarcinoma is the second leading cause of cancer-related death worldwide, with approximately 700,000 new cases diagnosed each year. An extremely aggressive malignancy, gastric cancer has a 5-year survival rate in the United States of less than 15%1. Gastric cancer is classified into the less common diffuse type and the more common intestinal type. Diffuse type gastric adenocarcinoma tends to affect younger people and does not progress through distinct histological stages, whereas intestinal type adenocarcinoma is characterized by histological progression from H. pylori-associated inflammatory cell invasion to atrophic gastritis, intestinal metaplasia, dysplasia and ultimately adenocarcinoma.
The strongest risk factor for gastric cancer is chronic H. pylori infection. Consequently, gastric cancer is more common in developing countries, where H. pylori is endemic, than in developed countries, where H. pylori is substantially less common. However, only 1–2% of persons infected with H. pylori develop stomach cancer2. Factors that contribute to susceptibility to gastric cancer include virulence among H. pylori strains3,4, duration of infection5, host genetic polymorphisms6,7 and environmental factors such as diet8,9. Similar to the influence of the microbiota in the lower gastrointestinal tract on human health and disease10, the microbiota of the stomach likely influences gastric immunobiology and possibly gastric disease.
In this review, we first discuss the co-evolutionary history of H. pylori and humans and the possible implications of the eradication of this ancient member of the human gastric microbiota. We next summarize the role of H. pylori in gastric cancer pathogenesis. We then discuss how H. pylori may influence the overall composition of the gastric microbiota and the potential role of the stomach microbiota in the progression to stomach cancer.
The Ancient Relationship between Humans and H. pylori
Helicobacter species are present in the gastrointestinal tracts of many mammals and birds, often exhibiting a host-specific relationship11. These associations suggest a co-evolutionary relationship between the bacteria and its host. Evidence for H. pylori-host co-evolution is provided by molecular epidemiology studies that compared nucleotide sequences of different H. pylori strains and in vivo mutation rates to calculate the minimum amount of time since the appearance of the last common ancestor of H. pylori12. These studies indicate that the last common bacterial ancestor existed at least 2,500 to 11,000 years ago. Furthermore, the genetic diversity among H. pylori strains, like genetic diversity among humans, decreases with distance from East Africa, suggesting H. pylori co-evolved with humans at least since their migration out of East Africa nearly 60,000 year ago12,13. Thus, H. pylori can be regarded as a component of the normal gastric microbiota in humans. Importantly, the phylogenetic origin of H. pylori is a strong predictor of the risk for gastric cancer, as H. pylori Europe strains are more strongly associated with advanced histological lesions and increased DNA damage than H. pylori Africa strains14.
The co-evolution of H. pylori with anatomically modern humans is the basis for speculation that the relatively sudden eradication of the bacterium in the modern era through improved hygiene and the widespread use of antibiotics, at least in the developed world, have affected stomach physiology and immunology15. Indeed, the rise of disorders, such as acid-related esophageal diseases, metabolic diseases, and allergic disorders, correlates with the decreasing prevalence of H. pylori, particularly in industrialized countries during the past century. Conversely, in regions of the world where H. pylori is still relatively common, human populations appear protected from these same diseases and disorders.
H. pylori most commonly colonizes the corpus, rather than the antrum, of the stomach, leading to chronic pan-gastric inflammation. Corpus-dominant infection leads to gastric mucosal atrophy, with an increase in gastric pH due to loss of acid-producing parietal cells and more closely mimics the microenvironment of the stomach of our early ancestors. Thus, from an evolutionary perspective, the reduced prevalence of H. pylori colonization and the resultant high stomach acid content could be considered a physiological deviation from the historically achlorhydric stomach11. Accordingly, the reduced prevalence of H. pylori beginning in the 20th century correlates with an increase in the rate of gastroesophageal reflux disease, Barrett’s esophagus, and esophageal adenocarcinoma, at least in North America and Europe16.
The gastric corpus produces most of the body’s ghrelin, a hormone that signals immediate hunger and appetite17. H. pylori-infected persons have lower ghrelin levels due to the decrease in ghrelin-producing cells caused by gastric atrophy, and H. pylori eradication increases ghrelin levels18,19. Consistent with these observations, cross-sectional studies show that H. pylori-infected children tend to weigh less than uninfected children20. Similarly, H. pylori-infected adults in East Asia tend to have a lower body mass index than non-infected adults, and eradication of H. pylori is associated with weight gain21,22. Although direct evidence is lacking, the available data provide a plausible link between the absence of H. pylori and metabolic diseases such as obesity and type 2 diabetes.
According to the hygiene hypothesis, childhood exposure to infections stimulate the developing immune system and protects against allergic and autoimmune diseases through the induction of regulatory T cell (Treg) responses. In recent years, the disappearing microbiota hypothesis has been proposed as an alternative hypothesis15. This hypothesis contends that the organisms that protect against allergy and autoimmunity are members of the human microbiota, such as H. pylori, that have long been present in the species but are gradually disappearing. In support of this hypothesis, the declining prevalence of H. pylori bearing the virulence factor CagA correlates, in part, with an increasing prevalence of certain atopic and allergic diseases23. Mechanistically, such an association can be explained, in part, by the ability of H. pylori to induce a strong gastric and systemic Treg response24–26.
The Role of H. pylori in Gastric Cancer Pathogenesis
The two best studied H. pylori virulence factors, VacA and CagA, have been linked to the bacterium’s carcinogenic potential. The protein VacA is present in all strains of H. pylori, although considerable DNA sequence variation in the vacA gene results in variable degrees of cellular toxicity and disease severity27. VacA induces pore formation in target host cells and promotes apoptosis of gastric epithelial cells, possibly by interfering with mitochondrial function28. VacA can also bind to CD4+ T cells and, after internalization, prevents dephosphorylation of the transcription factor nuclear factor of activated T cells (NFAT). Phosphorylated NFAT remains in the cytosol and therefore cannot activate its target genes such as interleukin (IL)-2 and the IL-2 receptor, resulting in inhibition of antigen-dependent T cell proliferation29. In addition, VacA exerts immunosuppressive effects by inducing dendritic cells to express and release the anti-inflammatory cytokines IL-10 and IL-18, which promote Treg differentiation30,31. These immunosuppressive activities of VacA likely promote H. pylori evasion of the host immune system and a dampened immune response that may enhance gastric tumor survival. Indeed, H. pylori strains that express more active forms of VacA in vitro are associated with higher rates of stomach cancer than strains that possess less active forms of the virulence factor27,32,33.
The cag pathogenicity island, a collection of genes that, unlike the vacA gene, is not present in every strain of H. pylori. Genes within the cag pathogenicity island encode proteins that form a type IV bacterial secretion system (T4SS) that injects bacterial components into host cells. One such component, the protein CagA, can undergo phosphorylation upon entering gastric epithelial cells, leading to morphological aberrations, including cell scattering and elongation. In addition, unmodified CagA binds to and inactivates Par1b, resulting in the loss of cell polarity34. Transgenic expression of CagA in mice results in gastric epithelial cell proliferation (and a corresponding resistance to apoptosis) and the appearance of carcinomas, leading to the classification of CagA as a bacterial oncoprotein35. In addition to CagA itself, the T4SS also injects H. pylori peptidoglycan into gastric epithelial cells, activating the PI3K pathway and stimulating cell migration, which also may contribute to carcinogenesis36.
A host protein that has received strong attention for its role in the development of carcinogenesis in association with H. pylori is β-catenin, a downstream component of the Wnt signal transduction pathway. In the absence of Wnt ligand, cytoplasmic β-catenin is bound within an inhibitory complex consisting of axin, adenomatous polyposis coli (APC), and glycogen synthase kinase-3β (GSK3β). GSK3β constitutively phosphorylates β-catenin, leading to its degradation. Upon Wnt binding to the receptor Frizzled, Dishevelled is activated, preventing GSK3β from phosphorylating β-catenin. Non-phosphorylated β-catenin then is able to translocate into the nucleus and direct the transcription of target genes involved in carcinogenesis. β-catenin is over-expressed or APC is mutated in 50% of gastric adenocarcinoma samples, and its localization in the nucleus is increased in gastric adenomas and areas of dysplasia37, suggesting aberrant activation of β-catenin is a prerequisite step in the development of stomach cancer. H. pylori induces expression of β-catenin target genes in stomach mucosal tissue in vitro and in co-cultures with gastric epithelial cells, providing support for the possibility that activation of β-catenin is an important event in which H. pylori initiates carcinogenesis.
H. pylori induces β-catenin activity through injection of CagA into gastric epithelial cells. In addition to its cytoplasmic form, β-catenin also exists in a membrane-bound form that links the E-cadherin receptor to the actin cytoskeleton. Intracellular CagA interacts with E-cadherin, disrupting its association with β-catenin and allowing its translocation into the nucleus. The result of this translocation is the activation of genes involved in gastric cancer, such as caudal type homeobox 1 (CDX1)38, which encodes an intestine-specific transcription factor that is required for the intestinal metaplasia observed in intestinal type gastric adenocarcinoma. CagA also induces β-catenin activity through the activation of the PI3K-AKT pathway. AKT is phosphorylated by PI3K. Phosphorylated AKT then phosphorylates and inactivates GSK3β, releasing β-catenin for nuclear translocation. Sustained activity of the PI3K-AKT pathway is made possible by CagA binding to the hepatocyte growth factor receptor MET39. In addition to CagA, other H. pylori constituents such as VacA40 and the adhesion molecule OipA41 have been shown to mediate β-catenin nuclear localization.
Activation of the PI3K-AKT axis in gastric epithelial cells is also made possible by transactivation of the epidermal growth factor receptor (EGFR) by H. pylori. Transactivation is dependent on the cleavage of EGFR membrane-bound ligands by the disintegrin and metalloproteinase (ADAM) family of proteinases42. Upon ligand stimulation, EGFR dimerizes and phosphorylates PI3K, leading not only to β-catenin nuclear localization but also to increased migration43 and resistance to apoptosis44.
The Relationship between H. pylori and Other Members of the Gastric Microbiota
Culture-based studies previously suggested that the stomach was essentially sterile in normal subjects due to its hostile gastric environment. However, recent advances in both DNA sequencing of the conserved rRNA genes for phylogenetic analysis and computational methods have uncovered an exceedingly rich and complex microbiota in the human gastrointestinal tract10, including the stomach. The majority of the gastric microbiota in humans belong to five phyla, including Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria, and Fusobacteria. Among these phyla, more than 100 phylotypes have been identified45.
Chronic H. pylori infection causes reduced gastric acidity and perturbs nutrient availability and local innate responses. Together, these H. pylori-associated changes in gastric physiology and immunology likely predispose to alterations in the composition of the gastric microbiota (Figure 1). The effect of H. pylori on the composition of the gastric microbiota, however, is incompletely understood. In specific pathogen free (SPF) BALB/c mice, the colonization by H. pylori results in the reduced abundance of Lactobacillus spp.46 In transgenic insulin-gastrin (INS-GAS) mice, H. pylori elevated abundance of Firmicutes and a reduction in Bacteroidetes47. In contrast, H. pylori infection had no effect on the gastric microbiota composition in conventional C57BL/6 mice49. However, the administration of antibiotics to C57BL/6 mice altered the composition of the gastric microbiota, and antibiotic-treated mice responded to H. pylori infection with less severe inflammation than untreated mice50. The antibiotic-treated mice contained increased numbers of cluster IV and XIVa Clostridium spp., bacteria known to promote Treg responses in colonic mucosa51. The ability of H. pylori to alter the gastric microbiota of mice is likely determined by several factors, including the genetic background of the mouse, the strain of H. pylori, and the length of infection.
Figure 1.
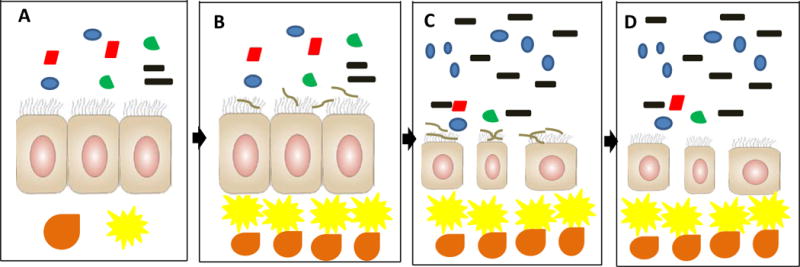
Model for cross-talk between the microbiota and H. pylori in predisposing the gastric mucosa for the development of cancer. A) In the H. pylori-free stomach, the microbiota coexists peacefully with the gastric mucosa. B) Initial H. pylori colonization of the stomach, initiating cross-talk with the microbiota. C) After years of co-colonization, the gastric mucosa atrophies, leading to an increase in pH and changes in nutrient availability and innate immune responses that together promote outgrowth of bacteria that otherwise would not tolerate the gastric environment. H. pylori and possibly certain members of the expanded microbiota elicit a strong innate and adaptive inflammatory response that damages the epithelial cell layer, establishing conditions that may promote carcinogenesis. D) After decades of colonization with loss of its ecological niche, H. pylori may clear, but the atrophy and non-indigenous microbiota persist, along with their permissive carcinogenic potential. Symbol code: Black, non-H. pylori Proteobacteria; Blue, Acidobacteria; Red, Actinobacteria; Green, Firmicutes; Brown, H. pylori. Yellow cell, dendritic cell; Orange cell, pro-inflammatory T cell. Relative abundances of bacterial phyla based on Maldonado-Contreras, A, et al., ISME J, 2011.
The effect of H. pylori on the microbiota of the human stomach has been examined in a limited number of studies. Bik et al.45 reported that H. pylori had no effect on the composition of the adult human gastric microbiota, whereas Maldonado-Contreras et al.52 showed that H. pylori-infected adults tended to have a higher abundance of Spirochetes, Acidobacteria and non-Helicobacter Proteobacteria and a relative decrease in members of the Actinobacteria, Bacteroidetes and Firmicutes compared with uninfected adult subjects. The difference between the results of these two studies could be related to differences in the method used to detect bacterial DNA. Bik et al. used a cloning approach, while Maldonado-Contreras et al. performed DNA microarrays. Another group has reported that the stomach mucosa of children is rarely colonized by non-Helicobacter bacteria, regardless of H. pylori status53. However, the later group used a culture-based approach that would have excluded non-cultivatable bacteria from detection.
The Role of the Gastric Microbiota in Gastric Cancer
Intestinal-type gastric adenocarcinoma is the final stage in the progression from H. pylori-induced gastritis to gastric atrophy, metaplasia, dysplasia, and finally carcinoma. Despite the effects of H. pylori on carcinogenesis summarized above, mucosal atrophy seems to be the most important step in the pathogenesis of gastric cancer. Although treatment for presumed H. pylori infection in patients that have developed gastric atrophy may sometimes limit the development of cancer, the lesion nevertheless frequently progresses to cancer54. In contrast, eradication of H. pylori before the onset of atrophy appears to protect against gastric cancer55. Thus, a key, albeit unresolved, issue is how the gastric microbiota interacts with H. pylori, namely, does the gastric microbiota facilitate a more virulent H. pylori or, vice versa, does H. pylori impact the microbiota to promote carcinogenesis, (Figure 2).
Figure 2.
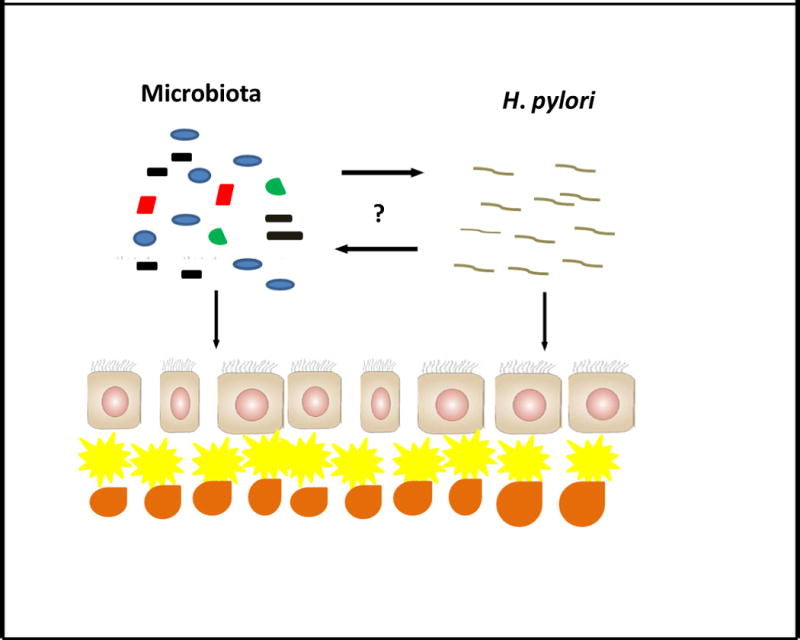
Cross-talk between the microbiota and H. pylori. Prolonged interaction between the microbiota and H. pylori may facilitate the outgrowth of communities of bacteria with a more carcinogenic profile, such as nitrosamine-producing species. Alternatively, the microbiota may promote a more virulent H. pylori with an enhanced carcinogenic potential. Yellow cell, dendritic cell; Orange cell, pro-inflammatory T cell.
In the INS-GAS mouse model of spontaneous gastric cancer, H. pylori accelerated the development of gastric intraepithelial neoplasia, and mice given H. pylori-eradicating antibiotics at 8 weeks post-infection were significantly more protected from neoplasia than mice that received antibiotics at 12 and 22 weeks post-infection56. Importantly, similar eradication therapy in age-matched Helicobacter-free INS-GAS mice also significantly delayed the onset of neoplasia. Gastric atrophy due to autoimmune gastritis also is a risk factor for gastric adenocarcinoma. Taken together, these findings suggest that gastric atrophy induced by H. pylori or other factors creates favorable conditions, such as overgrowth of non-H. pylori bacteria from the intestine, for gastric carcinogenesis. The observation that earlier antibiotic therapy was more effective in protecting against gastric cancer in both H. pylori-infected and non-infected mice may be related to earlier eradication of other cancer-potentiating gastric microbiota.
Further evidence that the gastric microbiota contributes to stomach cancer was provided by Lofgren et al.47. These investigators demonstrated that germ-free INS-GAS mice were slower to develop atrophic gastritis and intra-epithelial neoplasia than specific pathogen free (SPF) INS-GAS mice. H. pylori-mono-association accelerated development of the atrophy and neoplasia compared with mice that remained germ-free, but the gastritis was less severe and the onset of the neoplasia was delayed compared with H. pylori-infected INS-GAS mice that possessed a complex microbiota. Interestingly, male INS-GAS mice infected with H. pylori displayed earlier and more severe gastric pathology than their female counterparts, similar to the higher proportion of stomach cancer in men than women48.
The mechanism(s) by which H. pylori and its associated microbiota promotes the atrophy that lead to cancer is unclear. One hypothesis is that DNA-damaging reactive oxygen species (ROS) and reactive nitrogen species (RNS) produced by the bacterial communities contributes to carcinogenesis. Mowat et al.57 compared the effects of the proton pump inhibitor omeprazole, a common component in H. pylori-eradication therapy, on H. pylori-positive and negative patients. During treatment, H. pylori-infected persons had a higher gastric pH than non-infected subjects and harbored a significantly greater proportion of non-H. pylori bacteria in their stomachs. Some of the non-H. pylori bacteria included nitrosating species, bacteria capable of converting nitrite and other nitrogen compounds in gastric fluid to potentially carcinogenic N-nitroso compounds. Nitrite in a healthy stomach is virtually undetectable due to the action of stomach acid, but nitrite levels are increased during hypochlorhydria, providing a mechanistic link between atrophy and stomach cancer.
A major goal of microbiota studies is to identify the bacterial species responsible for promoting pathogenic changes that might contribute to carcinogenesis in the stomach. Lertpiriyapong et al.58 addressed this issue using gnotobiotic INS-GAS mice that were colonized with only three species of Altered Schaedler’s flora (ASF), including ASF356 Clostridium species, ASF361 Lactobacillus murinus and ASF519 Bacteroides species. Compared with germ-free INS-GAS mice, mice that were colonized with the restricted ASF (rASF) had more pronounced gastric pathology, including gastric corpus inflammation, epithelial hyperplasia, and dysplasia. Interestingly, rASF mice had similar pathology to H. pylori-free SPF mice, although neither group developed gastric neoplasia at the age equivalent of seven months post-infection with H. pylori. However, 46% of rASF-colonized mice and 53% of mice colonized by intestinal flora and H. pylori developed gastric neoplasia seven months post-infection, in contrast to none of H. pylori mono-associated mice. Male mice colonized by H. pylori and either ASF or intestinal flora displayed the highest levels of gastric and systemic pro-inflammatory cytokines and cancer-related gene expression compared with the other groups of mice. Taken together, these results suggest that H. pylori can act synergistically with even a limited gastric microbiota to promote gastric neoplasia and that a community of bacteria contribute to cancer risk.
In contrast to the above study, Dicksved et al.59 reported that the composition of the gastric microbiota was not significantly different between stomach cancer patients and dyspeptic subjects. However, the study had considerable limitations, including small sample size and the lack of the most current high-throughput sequencing technology. Indeed, the results of this study underscore the importance of using sequence technology together with computational bioinformatics to define the gastric bacterial composition.
Conclusion
Our understanding of the biology of H. pylori and the host response to the bacterium has advanced remarkably during the past three decades through a large body of elegant scientific and clinical investigation. An important consequence of this work is the recognition that H. pylori-induced chronic gastritis is the major risk factor for the development of gastric cancer. Recently, high-throughput sequencing technology has uncovered an extensive and complex microbiota in the lower gastrointestinal tract that impacts both health and an array of diseases, but characterization of the microbiota in the upper gastrointestinal tract, especially the stomach, has lagged behind advances in defining the lower tract microbiota. Consequently, information regarding the role of the commensal microbiota in gastric disease, particularly H. pylori infection, is in its infancy, especially when compared with rapidly emerging information on the microbiota at other sites. Thus, we predict that elucidating the role of the gastric microbiota in H. pylori infection will uncover new insights into the pathogenesis of gastric cancer.
Acknowledgments
This work was supported by National Institutes of Health grants AI083127, RR-20136 and DK064400 (Mucosal HIV and Immunobiology Center); Immunology, Autoimmunity and Transplantation Strategic Planning (IAT); the Crohn’s and Colitis Foundation of America; and the Research Service of the Veterans Administration. K.M.B. was supported by Basic Mechanism of AIDS Pathogenesis training grant (T32-AI007493). The UAB Microbiome Resource is supported by the School of Medicine, Comprehensive Cancer Center (P30AR050948), Center for AIDS Research (5P30AI027767), Center for Clinical Translational Science (UL1TR000165) and the Heflin Center.
References
- 1.Polk DB, Peek RM., Jr Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 2010;10:403–414. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herrera V, Parsonnet J. Helicobacter pylori and gastric adenocarcinoma. Clin Microbiol Infect. 2009;15:971–976. doi: 10.1111/j.1469-0691.2009.03031.x. [DOI] [PubMed] [Google Scholar]
- 3.Atherton Jc, Cao P, Peek JRM, et al. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. J Biol Chem. 1995;270:17771–17777. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]
- 4.Ziel KA, Campbell CC, Wilson GL, et al. Ref-1/Ape is critical for formation of the hypoxia-inducible transcriptional complex on the hypoxic response element of the rat pulmonary artery endothelial cell VEGF gene. FASEB J. 2004;18:986–988. doi: 10.1096/fj.03-1160fje. [DOI] [PubMed] [Google Scholar]
- 5.Harris PR, Smythies LE, Smith PD, et al. Role of childhood infection in the sequelae of H. pylori disease. Gut Microbes. 2013;4:1–13. doi: 10.4161/gmic.26943. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El-Omar EM, Carrington M, Chow W-H, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 7.Lee WP, Tai DI, Lan KH, et al. The −251T allele of the interleukin-8 promoter is associated with increased risk of gastric carcinoma featuring diffuse-type histopathology in Chinese population. Clin Cancer Res. 2005;11:6431–6441. doi: 10.1158/1078-0432.CCR-05-0942. [DOI] [PubMed] [Google Scholar]
- 8.Correa P, Fontham ET, Bravo JC, et al. Chemoprevention of gastric dysplasia: fandomized trial of antioxidant supplements and anti-Helicobacter pylori therapy. J Natl Cancer Inst. 2000;92:1881–1888. doi: 10.1093/jnci/92.23.1881. [DOI] [PubMed] [Google Scholar]
- 9.Shikata K, Kiyohara Y, Kubo M, et al. A prospective study of dietary salt intake and gastric cancer incidence in a defined Japanese population: the Hisayama study. Int J Cancer. 2006;119:196–201. doi: 10.1002/ijc.21822. [DOI] [PubMed] [Google Scholar]
- 10.Knight ZA, Tan K, Birsoy K, et al. Molecular profiling of activated neurons by phosphorylated ribosome capture. Cell. 2012;151:1126–1137. doi: 10.1016/j.cell.2012.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009;119:2475–87. doi: 10.1172/JCI38605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linz B, Balloux F, Moodley Y, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–918. doi: 10.1038/nature05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falush D, Wirth T, Linz B, et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299:1582–1585. doi: 10.1126/science.1080857. [DOI] [PubMed] [Google Scholar]
- 14.de Sablet T, Piazuelo MB, Shaffer CL, et al. Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut Microbes. 2011;60:1189–1195. doi: 10.1136/gut.2010.234468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.el-Serag HB, Sonnenberg A. Opposing time trends of peptic ulcer and reflux disease. Gut. 1998;43:327–333. doi: 10.1136/gut.43.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kojima M, Kangawa K. Ghrelin: structure and function. Physiol Rev. 2005;85:495–522. doi: 10.1152/physrev.00012.2004. [DOI] [PubMed] [Google Scholar]
- 18.Isomoto H, Nakazato M, Ueno H, et al. Low plasma ghrelin levels in patients with Helicobacter pylori-associated gastritis. Am J Med. 2004;117:429–432. doi: 10.1016/j.amjmed.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 19.Nwokolo CU, Freshwater DA, O’Hare P, et al. Plasma ghrelin following cure of Helicobacter pylori. Gut. 2003;52:637–640. doi: 10.1136/gut.52.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richter T, List S, Muller DM, et al. Five- to 7-year-old children with Helicobacter pylori infection are smaller than Helicobacter-negative children: a cross-sectional population-based study of 3,315 children. J Pediatr Gastroenterol Nutr. 2001;33:472–475. doi: 10.1097/00005176-200110000-00010. [DOI] [PubMed] [Google Scholar]
- 21.Azuma T, Suto H, Ito Y, et al. Gastric leptin and Helicobacter pylori infection. Gut. 2001;49:324–329. doi: 10.1136/gut.49.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furuta T, Shirai N, Xiao F, et al. Effect of Helicobacter pylori infection and its eradication on nutrition. Aliment Pharmacol Ther. 2002;16:799–806. doi: 10.1046/j.1365-2036.2002.01222.x. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Blaser MJ. Inverse associations of Helicobacter pylori with asthma and allergy. Arch Intern Med. 2007;167:821–827. doi: 10.1001/archinte.167.8.821. [DOI] [PubMed] [Google Scholar]
- 24.Lundgren A, Stromberg E, Sjoling A, et al. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect Immun. 2005;73:523–531. doi: 10.1128/IAI.73.1.523-531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harris PR, Wright SW, Serrano C, et al. Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology. 2008;134:491–499. doi: 10.1053/j.gastro.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 26.Serrano C, Wright SW, Bimczok D, et al. Downregulated Th17 responses are associated with reduced gastritis in Helicobacter pylori-infected children. Muc Immunol. 2013 doi: 10.1038/mi.2012.133. ePub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhead JL, Letley DP, Mohammadi M, et al. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology. 2007;133:926–936. doi: 10.1053/j.gastro.2007.06.056. [DOI] [PubMed] [Google Scholar]
- 28.Cover TL, Krishna US, Israel DA, et al. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003;63:951–957. [PubMed] [Google Scholar]
- 29.Gebert B, Fischer W, Weiss E, et al. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science. 2003;301:1099–1102. doi: 10.1126/science.1086871. [DOI] [PubMed] [Google Scholar]
- 30.Oertli M, Sundquist M, Hitzler I, et al. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest. 2012;122:1082–1096. doi: 10.1172/JCI61029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kao JY, Zhang M, Miller MJ, et al. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2010;138:1046–1054. doi: 10.1053/j.gastro.2009.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerhard M, Lehn N, Neumayer N, et al. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci USA. 1999;96:12778–12783. doi: 10.1073/pnas.96.22.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miehlke S, Kirsch C, Agha-Amiri K, et al. The Helicobacter pylori vacA s1, m1 genotype and cagA is associated with gastric carcinoma in Germany. Int J Cancer. 2000;87:322–327. [PubMed] [Google Scholar]
- 34.Murata-Kamiya N. Pathophysiological functions of the CagA oncoprotein during infection by Helicobacter pylori. Microbes Infect. 2011;13:799–807. doi: 10.1016/j.micinf.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 35.Ohnishi N, Yuasa H, Tanaka S, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA. 2008;105:1003–1008. doi: 10.1073/pnas.0711183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaparakis M, Turnbull L, Carneiro L, et al. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol. 2010;12:372–385. doi: 10.1111/j.1462-5822.2009.01404.x. [DOI] [PubMed] [Google Scholar]
- 37.Tsukashita S, Kushima R, Bamba M, et al. Beta-catenin expression in intramucosal neoplastic lesions of the stomach. Comparative analysis of adenoma/dysplasia, adenocarcinoma and signet-ring cell carcinoma Oncology. 2003;64:251–258. doi: 10.1159/000069310. [DOI] [PubMed] [Google Scholar]
- 38.Murata-Kamiya N, Kurashima Y, Teishikata Y, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26:4617–4626. doi: 10.1038/sj.onc.1210251. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki M, Mimuro H, Kiga K, et al. Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe. 2009;5:23–34. doi: 10.1016/j.chom.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 40.Nakayama M, Hisatsune J, Yamasaki E, et al. Helicobacter pylori VacA-induced inhibition of GSK3 through the PI3K/Akt signaling pathway. J Biol Chem. 2009;284:1612–1619. doi: 10.1074/jbc.M806981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Franco AT, Johnston E, Krishna U, et al. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 2008;68:379–387. doi: 10.1158/0008-5472.CAN-07-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keates S, Sougioultzis S, Keates AC, et al. cag+ Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J Biol Chem. 2001;276:48127–48134. doi: 10.1074/jbc.M107630200. [DOI] [PubMed] [Google Scholar]
- 43.Nagy TA, Frey MR, Yan F, et al. Helicobacter pylori regulates cellular migration and apoptosis by activation of phosphatidylinositol 3-kinase signaling. J Infect Dis. 2009;199:641–651. doi: 10.1086/596660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maeda S, Yoshida H, Mitsuno Y, et al. Analysis of apoptotic and antiapoptotic signalling pathways induced by Helicobacter pylori. Gut. 2002;50:771–778. doi: 10.1136/gut.50.6.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bik EM, Eckburg PB, Gill SR, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci USA. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aebischer T, Fischer A, Walduck A, et al. Vaccination prevents Helicobacter pylori-induced alterations of the gastric flora in mice. FEMS Immunol Med Microbiol. 2006;46:221–229. doi: 10.1111/j.1574-695X.2005.00024.x. [DOI] [PubMed] [Google Scholar]
- 47.Lofgren JL, Whary MT, Ge Z, et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology. 2011;140:210–220. doi: 10.1053/j.gastro.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Devesa SS, Blot WJ, Fraumeni JF., Jr Changing patterns in the incidence of esophageal and gastric carcincoma in the United States. Cancer. 1998;83:2049–2053. [PubMed] [Google Scholar]
- 49.Tan MP, Kaparakis M, Galic M, et al. Chronic Helicobacter pylori infection does not significantly alter the microbiota of the murine stomach. Appl Environ Microbiol. 2007;73:1010–1013. doi: 10.1128/AEM.01675-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rolig AS, Cech C, Ahler E, et al. The degree of Helicobacter pylori-triggered inflammation is manipulated by preinfection host microbiota. Infect Immun. 2013;81:1382–1389. doi: 10.1128/IAI.00044-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atarashi K, Tanoue T, Oshima K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 52.Maldonado-Contreras A, Goldfarb KC, Godoy-Vitorino F, et al. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 2011;5:574–579. doi: 10.1038/ismej.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kato S, Fujimura S, Kimura K, et al. Non-Helicobacter bacterial flora rarely develops in the gastric mucosal layer of children. Dig Dis Sci. 2006;51:641–646. doi: 10.1007/s10620-006-3185-0. [DOI] [PubMed] [Google Scholar]
- 54.Levine SM, Lin EA, Emara W, et al. Plastic cells and populations: DNA substrate characteristics in Helicobacter pylori transformation define a flexible but conservative system for genomic variation. FASEB J. 2007;21:3458–3467. doi: 10.1096/fj.07-8501com. [DOI] [PubMed] [Google Scholar]
- 55.Wong BC, Lam SK, Wong WM, et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA. 2004;291:187–194. doi: 10.1001/jama.291.2.187. [DOI] [PubMed] [Google Scholar]
- 56.Lee CW, Rickman B, Rogers AB, et al. Helicobacter pylori eradication prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2008;68:3540–3548. doi: 10.1158/0008-5472.CAN-07-6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mowat C, Williams C, Gillen D, et al. Omeprazole, Helicobacter pylori status, and alterations in the intragastric milieu facilitating bacterial N-nitrosation. Gastroenterology. 2000;119:339–347. doi: 10.1053/gast.2000.9367. [DOI] [PubMed] [Google Scholar]
- 58.Lertpiriyapong K, Whary MT, Muthupalani S, et al. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut. 2014;63:54–63. doi: 10.1136/gutjnl-2013-305178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dicksved J, Lindberg M, Rosenquist M, et al. Molecular characterization of the stomach microbiota in patients with gastric cancer and in controls. J Med Microbiol. 2009;58:509–516. doi: 10.1099/jmm.0.007302-0. [DOI] [PubMed] [Google Scholar]