

Introduction
Physiologic processes including sleep are regulated in part by humoral substances. Cytokines as pleiotropic signaling molecules are involved in the regulation of many such processes. The study of the humoral regulation of sleep began almost 100 years ago with the publication of Ishimori's1 and Legendre and Pieron's2 work showing that the transfer of cerebrospinal fluid from sleep-deprived dogs into normal dogs enhanced sleep in the recipients. These findings led to several searches dedicated to the identification of the responsible substances. These searches were conceptually based on the hypotheses that; (1) sleep is regulated, in part, by humoral agents within the nervous system, and (2) those agents undergo concentration or conformational changes that track, and thereby potentially provide an index of, cumulative sleep/wake history. Although many have replicated those original findings in similar experiments (eg, Ref.3 and reviewed in Ref.4), this search continues. Many substances are implicated in sleep regulation. These sleep regulatory substances (SRSs) range from low-molecular-weight substances with short half-lives (eg, adenosine, nitric oxide [NO]), to longer-lived peptides, such as growth hormone-releasing hormone and orexin, and proteins including the cytokines. Until recently, the brain mechanisms that index sleep/wake history to SRS activity were unknown. Two cytokines, interleukin 1 β (IL-1) and tumor necrosis factor α (TNF), are well characterized for their roles in sleep regulation and are used to show these newer ideas in this review.
Many experimental approaches have been used to discover and characterize SRSs (reviewed in Refs.4–6). All of these approaches, including methods such as the use of transgenic animals, epigenetic and posttranslational modifications, and proteomic and genome-wide searches, are limited because sleep cannot be isolated as an independent variable. Virtually every physiologic parameter (eg, body temperature, hormonal levels, respiration rate, urinary output, brain metabolism, feeding and reproductive behaviors) changes with sleep. As a consequence, it is not possible to definitively know, for example, whether the change in expression of a particular molecule that correlates with sleep or sleep loss does so as a direct consequence of sleep or of some other concurrent physiologic process. Sleep researchers have developed lists of criteria that candidate SRSs need to meet before they can be reasonably proposed as being involved in sleep regulation (Box 1).6–9 The usefulness of a multiple criteria approach to identify SRSs is that it limits false detection. Adherence to these criteria is especially important because many substances are capable of altering sleep (eg, alcohol). To date, only a few substances have met all of these criteria; IL-1 and TNF are among them.
Box 1 Criteria for SRSs.
The SRS should enhance a sleep phenotype (eg, duration of non–rapid eye movement sleep (NREMS) or electroencephalographic (EEG) δ wave power).
Inhibition of the SRS should reduce spontaneous sleep.
The SRS levels in the brain should correlate with sleep propensity.
The SRS should act on putative sleep regulatory circuits
The SRS levels in disease should correlate with sleepiness.
Our knowledge of SRS involvement in processes believed to be unrelated to sleep has led to unexpected developments in our understanding of sleep mechanisms and how the brain organizes sleep. For example, our view of what exactly it is that sleeps has shifted from whole organisms to neural networks such as cortical columns (also called neuronal assemblies or neuronal groups). Our departure from the canonical view that sleep is a global process distributed across the brain was deduced from the fact that all SRSs identified play a role in activity-dependent neural plasticity. This finding suggests that 1 important function of sleep is to facilitate neural connectivity. The roles that cytokines play in these developments are discussed later.
TNF and IL-1 Meet all the Criteria for SRSs
Systemic or central injection of either TNF or IL-1 enhances duration of NREMS and EEG δ wave power during NREMS in every species thus far tested including, mice, rats, rabbits, cats, sheep, monkeys, and humans (criterion 1; see Box 1) (reviewed in Refs.4,10,11). After intracerebroventricular (ICV) or intraperitoneal (IP) injections of either IL-1 or TNF, increases in NREMS manifest within the first hour and, depending on dose, last up to 8 to 12 hours. The effects on NREMS can be large (eg, after 3 μg of TNF IP, mice spent an extra 90 minutes of NREMS over the first 9 postinjection hours12 and after 600 fmol ICV IL-1 rabbits spent an extra 2 hours in NREMS over the first 12 post-injection hours).10 The effects on rapid eye movement sleep (REMS) are dependent on route of administration, time of day, and dose. For instance, low somnogenic doses usually do not alter duration of REMS, whereas high somnogenic doses inhibit REMS. High doses of either IL-1 or TNF inhibit sleep; the sleep responses after these high doses resemble the sleep that occurs during severe infectious disease (eg, sleep episode duration is shortened). Somnogenic doses of IL-1 or TNF also increase δ wave power during NREMS,12,13 a measure of sleep intensity.
The brain is susceptible to sleep disruptions, often over periods of days or more. Prolonged bouts of wakefulness are followed by sleep rebound, sometimes over multiple subsequent sleep periods. Sleep rebound is characterized by increased time spent in sleep and increased sleep intensity as defined by larger amplitude of EEG δ waves. Sleep homeostasis is a defining characteristic of sleep and its mechanisms likely involve the production and release of SRSs, including IL-1 and TNF. Thus, injection of exogenous IL-1 or TNF induces an NREMS that resembles sleep after sleep loss in that its duration and intensity are greater. Further, if either IL-1 or TNF is inhibited during sleep loss, the expected subsequent sleep rebound is greatly attenuated (reviewed in Ref.14). These latter findings coupled with the evidence presented in the previous paragraph strongly implicate IL-1 and TNF in sleep homeostasis.
Inhibition of either IL-1 or TNF using several different approaches reduces spontaneous NREMS (criterion 2; see Box 1). For example, the IL-1 receptor antagonist (an endogenous gene product), IL-1 and TNF soluble receptors (also endogenous substances), and anti-IL-1 or anti-TNF antibodies inhibit NREMS if given to experimental animals [reviewed in Ref.4). In humans, the TNF soluble receptor is a normal constituent of cerebrospinal fluid and inhibits sleep15 and fatigue.16 Substances that inhibit the production, release, or actions of IL-1 or TNF also inhibit duration of NREMS. For example, glucocorticoids, IL-4, IL-10, and IL-13, and corticotrophin-releasing hormone all inhibit IL-1 and TNF and reduce spontaneous NREMS (reviewed in Ref.4).
Another approach to inhibit SRSs is to remove 1 or more of the genes in its signaling pathway. Knockout mice that lack either the IL-1 type I receptor (IL1R1),13 the brain-specific IL1R1 accessory protein,17 the TNF 55-kD receptor12 or both IL1R1 and TNF receptor (TNFR)18 have less spontaneous sleep than control strains of mice. The NREMS deficits in the TNF 55-kD receptor knockout mice occur mostly during the first hours of daylight, whereas the NREMS deficits in the IL1R1 and double TNF/IL-1 receptor knockout mice occur mostly during the nighttime. The results from those studies suggest some independence of the somnogenic actions of IL-1 and TNF, although these cytokines induce each other in brain in vivo.13 Further, the TNF receptor knockout mice show NREMS responses if given IL-113 and the IL-1 receptor knockout mice do likewise if given TNF.12
Brain levels of either IL-1 or TNF or their respective mRNAs vary with sleep propensity (criterion 3; see Box 1). For example, IL-1 cerebrospinal fluid levels in cats vary with the sleep/wake cycle.19 Spontaneous brain levels of IL-1 mRNA and TNF mRNA vary with sleep propensity in rats, with highest levels occurring at the onset of daylight hours (reviewed in Ref.4). Rat hypothalamic (an NREMS regulatory network) levels of both IL-120 and TNF21 are highest at the time when spontaneous NREMS duration is greatest. Cerebral cortical levels of IL-1 and TNF also vary with the time of day. If sleep propensity is enhanced by sleep deprivation, both IL-1 mRNA and TNF mRNA levels increase in brain (reviewed in Ref.4). Further, if rats are fed a cafeteria diet their NREMS is enhanced, as are their hypothalamic IL-1 mRNA levels.22 During infectious disease states when sleep in enhanced, brain levels of IL-1 and TNF mRNAs are enhanced (eg, during influenza virus infections in mice).23
If either IL-1 or TNF is microinjected into sleep regulatory circuits, NREMS is enhanced (criterion 4; see Box 1). Thus, microinjection of TNF into the anterior hypothalamus is associated with dose-dependent increases in NREMS.24 In contrast, injection of the TNF soluble receptor into the anterior hypothalamus inhibits spontaneous NREMS. Similarly, an extensive study of IL-1-responsive sites indicated that sites near the ventricles and subarachnoid sites near the hypothalamus are associated with enhanced NREMS.25 Further, IL-1 receptive hypothalamic neurons also are receptive to growth hormone-releasing hormone, another well-characterized SRS (reviewed in Ref.4), and those neurons are γ-aminobutyric acid-releasing (GABA)-ergic.26 If growth-hormone-releasing hormone (GHRH) is microinjected into the hypothalamus, it induces sleep responses and activity in sleep-active neurons.27,28 Sleep-active hypothalamic neuron firing rates are enhanced by IL-1, whereas wake-active hypothalamic neurons are inhibited.29 Classic brain stem sleep/wake circuits are also modulated by SRSs. For example, microinjection of either IL-1 or TNF into the locus coeruleus30 or IL-1 into the dorsal raphe31 enhances NREMS. IL-1 suppresses wake-active serotonergic neurons in the dorsal raphe32 by affecting GABA33 and possibly its receptor availability.34 Collectively, these data indicate that IL-1 and TNF act on sleep regulatory circuits to enhance NREMS. Both cytokines also have the capacity to act directly on the cerebral cortex to enhance sleep intensity regionally, and that suggests that these substances can act throughout the neuraxis to alter state within neuronal assemblies. This view of brain organization of sleep is discussed later.
Many diseases with associated changes in sleep propensity also alter cytokines (criterion 5; see Box 1). The changes in hypothalamic cytokines associated with influenza virus in mice have already been mentioned. Human studies have greatly enriched the literature relating circulating cytokines to disease-associated sleepiness. TNF plasma levels are increased in multiple diseases associated with enhanced sleepiness, including patients with AIDS, chronic fatigue, insomnia, myocardial infarct, excessive daytime sleepiness, postdialysis fatigue, preeclampsia, alcoholism, obesity, sleep apnea (reviewed in Ref.4), and Alzheimer disease.35 The TNF polymorphic variant, G-308A, is linked to metabolic syndrome36 insulin resistance,37 sleep apnea,38 and heart disease.39 Systemic endotoxin, a gram-negative bacterial cell wall product, enhances sleep and plasma TNF levels in humans.40 Clinically approved inhibitors of TNF (eg, etanercept) reverse the sleepiness and fatigue associated with sleep apnea,15 rheumatoid arthritis,16 ankylosing spondylitis,41 and alcoholism.42 Surgical treatment of, or CPAP treatment of, obstructive sleep apnea reduces TNF/TNFR plasma levels.43–45
Blood levels of IL-1 in humans may also vary with sleep propensity, but this literature is not as clear as that for TNF. IL-1 plasma levels peak at the onset of sleep46 and are enhanced during sleep deprivation.47,48 Circulating levels of either TNF or IL-1 affect sleep via the vagus nerve because vagotomy blocks IP TNF-enhanced49 or IL-1-enhanced50 NREMS. Systemic injections of either IL-1 or TNF enhance brain levels of IL-1 and TNF mRNAs.51 Vagotomy also blocks the IP IL-1-enhanced hypothalamic IL-1 mRNA.52 Collectively, it seems that the sleep disturbances associated with disease are mediated in part via IL-1 and TNF (reviewed in Ref.53). Given the strong relationship of IL-1 and TNF with sleep in both health and disease states, their importance in sleep medicine will continue to grow.
Associated Mechanisms of IL-1-Enhanced and TNF-Enhanced Sleep
The regulation of the brain cytokine network is not fully understood. Regardless, a variety of cytokines and cytokine-associated substances have been shown to alter sleep. Several of these such as the IL-1 and TNF soluble receptors have already been mentioned. Cytokine-associated substances such as the IL-1 receptor antagonist and several antisomnogenic substances, such as IL4, IL-10, IL-13, and transforming growth factor b, inhibit spontaneous NREMS. In contrast, other cytokines such as IL-6, IL-18, acidic fibroblast growth factor, interferon γ, nerve growth factor, brain-derived neurotrophic factor, and glia-derived neurotrophic factor, promote NREMS (reviewed in Ref.4). There are some cytokines that apparently do not affect sleep, at least under the conditions tested; these include interferon β54 and basic fibroblast growth factor.55 Nevertheless, IL-1 and TNF affect many other molecules that in turn affect sleep. Nuclear factor κ B (NFκB) and c-Fos (AP-1) are transcription factors that are activated by IL-1 and TNF (reviewed in Refs.4,53). These transcription factors promote production of IL-1 and TNF and many other substances implicated in sleep regulation including multiple cytokines, the purine type 1 receptor adenosine A1 receptor (A1AR), cyclooxygenase 2, and GHRH receptor. NFκB is activated within the hypothalamus and cortex by sleep deprivation.56,57 Adenosine also elicits NFκB nuclear translocation in basal forebrain slices via the A1AR.58 An inhibitor of NFκB inhibits NREMS.59 IL-1 and TNF also affect many small molecules with short half-lives that are involved in sleep regulation. including NO, adenosine and prostaglandins (reviewed in Ref.,4 eg, Ref.60). For example, inhibition of NO synthase blocks IL-1-induced increased NREMS responses.61 Cytokines also interact with multiple neurotransmitters involved in sleep regulation including GABA, norepinephrine, serotonin, and acetylcholine (reviewed in Ref.4). The cytokine network is characterized by redundancy, positive feedback loops, extensive cross-talk, autoregulation, and many other complexities; most of it remains to be studied within the context of sleep. The exact somnogenic biochemical pathways affected by cytokines likely depend on circumstances such as time of day, waking activity, and disease, although it seems clear that known SRSs work in concert with each other to affect sleep.
Brain Organization of Sleep: Cytokine Involvement in Cortical Column State
Sleep researchers have yet to reach consensus as to exactly what it is that sleeps. This problem has the potential to confuse discussions of sleep regulation. For instance, traditionally sleep was considered a whole-animal phenomenon: either the subject was asleep or awake. However, it is now clear that some marine mammals show unihemispheric sleep (reviewed in Ref.62). Further, some characteristics of sleep such as EEG δ wave activity, metabolism, and blood flow manifest regionally depending on previous waking activity in those regions. In addition, a fundamental meta-finding within sleep research is that regardless of where a lesion in the brain may occur, if the subjects survive, they sleep. This finding strongly indicates that sleep is an intrinsic property of any viable neuronal network and, contrary to the prevailing sleep regulatory paradigm, that sleep regulatory circuits do not impose sleep on the brain, because if they are lesioned, the animal sleeps (reviewed in Ref.63).
These considerations led us and others to propose that sleep is a fundamental property of neural networks.64–67 It is possible that individual cells may sleep, but if one entertains this hypothesis, definitional problems are confronted (eg, is a silent neuron, or a bursting neuron, asleep? Most likely not, because such firing patterns can be found in conditions not associated with sleep). There also seems to be little chance of causally connecting activity of a single neuron to a state beyond correlation of firing rates. The positing of a brain organization level at which sleep emerges allows falsifiable hypotheses to be made at the appropriate level of organization. By way of analogy, to study the heat capacity, osmotic properties, vapor pressure, or taste of water, one does not study H or O; these emergent properties are the result of combining H and O and are fundamentally not predictable from our current knowledge of H or O. To relate our hypothesis to our past work with cytokines, we framed it within a biochemical mechanistic causal proposal (Box 2). There is now considerable evidence for the hypothesis and it is discussed in this section.
Box 2 Sleep mechanisms.
There is activity-dependent production of SRSs.
Activity-dependent SRSs act locally on nearby neurons/glia to change their electrical/receptive properties and thereby alter the input-output relationships of the networks within which they are found.
Altered input-output relationships within neuronal assemblies indicate functional state changes of the assemblies.
Synchrony of state between semiautonomous neural assemblies occurs because they are loosely connected via neurons and humoral substances.
Sleep regulatory circuits coordinate neuronal assembly functional state changes into organism sleep.
There is cell activity-dependent expression of cytokines in brain (step 1; see Box 2). This is well known for cytokines such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) (reviewed in Ref.68), but is less studied for IL-1 and TNF. Conditions such as kindling, sleep deprivation, or extracellular glutamate enhance brain IL-1 or TNF, suggesting that excessive activity or excitatory stimuli are responsible.69–72 Data from our laboratory indicate that within cerebral cortical neurons or glia, TNF and IL-1 are enhanced if afferent neuronal activity into the specific column is enhanced.73,74 Collectively, such data strongly suggest that cytokine expression in neurons/glia is activity dependent.
The activity-dependent cytokines act on neurons to change their electrical and responsive properties (step 2; see Box 2). For some cytokines such as NGF and BDNF, this process is well established. For IL-1 and TNF, it has also been studied, but within the context of the fever literature (reviewed in Ref.75). For instance, IL-1 or TNF alter hypothalamic neuron sensitivity to temperature. TNF upregulates, whereas IL-1 downregulates glutamatergic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor expression in neurons (see later discussion), and changed populations of AMPA receptors alter neuronal response patterns. IL-1 receptors on hypothalamic neurons colocalize with GHRH receptors on GABAergic cells.26 IL-1 enhances presynaptic release of GABA in hypothalamic cells.76 IL-1 enhances hypothalamic sleep-active neurons and inhibits wake-active neurons.29 There is thus ample evidence indicating that cytokines act on neurons to change their electrical properties.
These cytokine-induced altered neuronal properties affect sleep phenotype. Thus if either IL-177 or TNF78 is applied to the surface of the cortex unilaterally, there is a dose-dependent and state-dependent increase in EEG δ power on the side receiving the cytokine. The increases occur during NREMS but not during REMS or waking and are confined to the 0.5-Hz to 4-Hz frequency band. Further, if TNF expression is unilaterally inhibited using a small-interfering RNA (siRNA) within the cortex, there is a reduction of EEG δ power unilaterally.79 If rats are deprived of sleep and pre-treated with a TNF soluble receptor or an IL-1 soluble receptor, the enhanced EEG δ wave power that occurs during subsequent NREMS is attenuated.77,78 Together, these data suggest that TNF and IL-1 are produced in response to activity and act locally on networks to change input-output properties, resulting in a regionally more intense sleep or if inhibited a regionally less intense NREMS.
There is direct evidence that neuronal assemblies oscillate between 2 or more functional states and one of these states is induced by TNF and is sleeplike in character (step 3; see Box 2). If cortical columns are probed with afferent stimulation and subsequent amplitudes of evoked potentials are measured, different functional states can be determined.80 One of those states correlates with whole-animal sleep and the probability of entering that state is dependent on previous afferent input to the column and past state status. Excessive afferent input to a cortical column increases the likelihood that the column enters the sleeplike state. Similarly, the longer the column is in a wakelike state, the higher the probability that later it enters the sleeplike state. These properties of cortical column sleeplike states are also properties of whole-animal sleep. Further, cortical column state affects behavior. If rats are trained to lick in response to stimulation of a whisker, the error rate is higher if the cortical column of the stimulated whisker is in the sleeplike state than if it is in the wakelike state.81,82 Localized injection of TNF onto cortical columns induces the sleeplike state in the affected columns.78 These data suggest that sleep is a fundamental property of neuronal assemblies.
During organism sleep and wake, most of the columns are in their respective sleeplike and wakelike states, suggesting synchrony of state between columns (step 4; see Box 2). Columns are topographically organized, and in general, the closer a column is to another, the more tightly the 2 are linked by neural and humoral connections. Because they are linked, it is likely from a theoretic view that they functionally synchronize with each other.83,84
Cortical columns are also connected to subcortical sleep regulatory circuits (step 5; see Box 2). Unilateral injection of either TNF or IL-1 onto the cerebral cortex activates reticular thalamic neurons as determined by fos expression.85,86 Further, prefrontal cortical neurons, ventral lateral preoptic neurons, and medial preoptic neurons are also activated by IL-1.86 These data suggest that the status of cortical column state could be relayed to these NREMS regulatory networks. It is also possible that these regulatory networks are thus involved in coordinating whole-animal sleep using cortical column state status information. Thus in this view sleep is (1) dependent on previous cellular activity, (2) initiated at the cortical column level, (3) a self-organized state being coordinated between columns and being a statistical property of the number of columns is the sleeplike state, and (4) it is refined and timed into whole-animal sleep by sleep regulatory networks. Each of these is a falsifiable hypothesis.
A Neuroconnectivity Function for Sleep: Cytokine Involvement
Sleep as a subject of neurobiology is unusual because its function has not been experimentally defined. Its importance is shown by the facts that during sleep one does not reproduce, eat, drink, or socialize and one is subject to predation. These are high evolutionary costs to overcome by whatever the beneficial effects of sleep are. So what could be so important to the brain to allow such a disadvantaged state to persist? There are many theories of sleep function positing that the answer is neural connectivity.66,87–91 In this review, we focus on just 2 (our own89 and that of Kavanau66) because the logic of the 2 is similar and both are derived in part from the earlier proposal of Roffwarg.92 The central idea of both theories was the recognition that use-dependent changes in synaptic efficacy and connectivity would lead to dysfunction unless there were processes to stabilize synaptic networks that are constantly being modified by activity. This process is now termed synaptic scaling. Synaptic scaling serves to regulate Hebbian plasticity; thus, an increase in network activity causes a slow compensatory decrease in excitatory synaptic efficacy, whereas a decrease in network activity enhances excitatory synaptic strength.93 The stabilization mechanism proposed by us was SRS-induced changes in local electrical properties, whereas the mechanism proposed by Kavanau was intrinsic spontaneous electrical activity. These mechanisms are not mutually exclusive and both are scaling mechanisms. More recent sleep-connectivity theories have also invoked synaptic scaling (reviewed in Refs.94,95).
Of importance to this review is that TNF is involved in synaptic scaling. Thus, TNF promotes AMPAreceptor expression and enhances cytosolic Ca++ levels.96 This TNF action is physiologic because an inhibitor of TNF inhibits AMPA-induced postsynaptic potentials97 and AMPA-induced changes in cytosolic Ca++.96 A TNF siRNA applied to the cortex inhibits gluR1 mRNA levels79; gluR1 is a subunit of the AMPA receptor. AMPA receptors are involved in EEG synchronization98 and synaptic plasticity.99 Direct evidence for the involvement of TNF in synaptic scaling has been described.100 IL-1 may also affect AMPA receptor expression.101 AMPA receptors in layer V are involved in downscaling during NREMS.102 Overall, these data suggest a cytokine-dependent mechanism for the reconfiguration of synaptic weights during NREMS.
Connecting Activity to Cytokines: The Adenosine Triphosphate-Cytokine-Adenosine Hypothesis
A major tenant of the neuroconnectivity theories is their dependence on activity. Indeed, within the brain, a major stimulus for IL-1 and TNF production and release is neuronal activity. Adenosine triphosphate (ATP) is coreleased with neurotransmitters.103 ATP in turn induces IL-1104 and TNF105 release from glia via P2X receptors (reviewed in Refs.106,107). ATP is present in neuronal synaptic vesicles. The concentration of ATP in the vesicles is 10 to 50 times higher than in the cytosol. In the brain, ATP is coreleased in GABAergic, cholinergic, noradrenergic, and glutamatergic synapses. ATP is also considered a gliotransmitter. Once released, some extracellular ATP is converted to adenosine. In turn, adenosine binds to the other major purine receptor types, P1 receptors (Fig. 1). The action of adenosine is fast, occurring within milliseconds to seconds, and it results in increased K+ permeability and hyperpolarization.
Fig. 1.
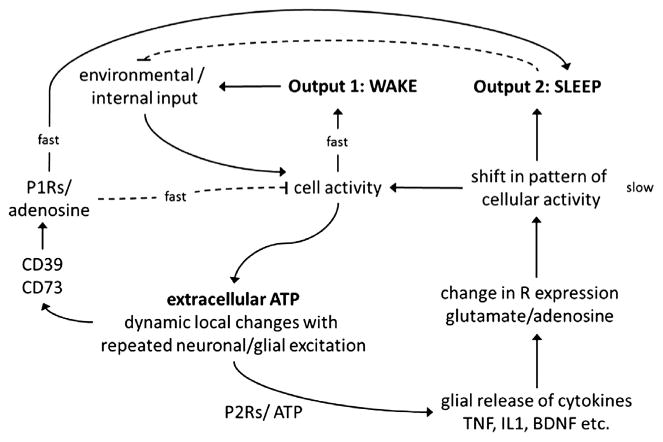
The sleep homeostat. Molecular networks operating on different timescales (fast vs slow) comprise the sleep homeostat. Cellular activity is induced and sustained by environmental stimuli. Cellular activity increases arousal (output 1) and causes ATP to be coreleased with glutamate and other neurotransmitters. This extracellular ATP provides a way for the brain to track previous activity; some ATP is enzymatically converted to adenosine (fast) and some binds with glial P2 receptors to facilitate SRS secretion. SRSs induce fast-acting labile substances like adenosine and NO. Alternatively, SRSs and their effectors can lead to alterations (slow) in receptor content and receptor-mediated ion gating, causing a shift in overall cellular activity. Resulting changes in receptor populations predispose affected networks (within the diffusible range of extracellular ATP) to sleep (output 2).
Some of the released extracellular ATP acts on glial P2Rs and causes the release of IL-1, TNF, and BDNF as well as additional ATP (see Fig. 1). IL-1 precursor is processed by caspase 1, which is triggered via ATP activation of P2X7 receptors.108,109 TNF and IL-1 released from glia act more slowly (minutes to hours) on adjacent neurons, leading to the activation of NFκB. NFκB promotes transcription of receptor mRNAs such as the adenosine A1R and the glutamate AMPA receptor-gluR1 mRNAs. Translation of those mRNAs into their respective proteins and their subsequent expression on the cell membrane would change sensitivity of the postsynaptic neuron over longer periods. This is a prototypical scaling effect, because the expression of postsynaptic receptors is modulated by the activity of the presynaptic neuron (ie, the amount of released ATP). Thus, the sensitivity of the postsynaptic neuron is scaled to the previous use of the synapse. As mentioned, TNF and BDNF are the 2 molecules most firmly linked to synaptic scaling. Thus, ATP levels are affected by metabolism and neural activity and in turn affect extracellular levels of adenosine and cytokines, thereby providing direct links between neural activity, metabolism, and sleep regulation. This idea fostered our ATP-adenosine-cytokine hypothesis.89 Mechanistically, our hypothesis is summarized as follows: (1) neuronal activity is associated with gliotransmission and neurotransmission corelease of ATP; (2) the consequent increase in extracellular ATP thus provides an index of previous local neuronal activity; (3) the ATP is detected by nearby purine type 2 receptors, causing the release of sleep regulatory cytokines such as TNF, IL-1, and BDNF, and this provides for the translation of previous neuronal activity into local levels of SRSs; (4a) these substances in turn, by a slow process (gene transcription/translation), alter electrical properties of nearby neurons by altering their own production and that of receptor populations, such as glutamate and adenosine receptors; (4b) the SRSs also, by a fast process (diffusion for short distances), directly interact with their receptors on neurons and alter electrical properties; (4c) further, ATP itself breaks down, releasing extracellular adenosine, which in turn acts on adenosine receptors, again altering electrical potentials on the nearby neurons. These events are happening locally and the collective electrical changes result in a shift in input-output relationships within the local neuronal assemblies that originally showed the increase in activity (ie, a state shift). In a mathematical model, the local states of neuronal assemblies rapidly synchronize, or phase lock, with each other because they are loosely connected to each other via neurons and humoral substances.84 Well-characterized sleep regulatory circuits and associated activation networks play a critical role in both sleep and waking by ensuring the synchronization of neuronal assembly state for niche-adaptation purposes. Consistent with this model, ATP agonists promote NREMS, whereas ATP antagonists inhibit sleep.110 Further, after sleep deprivation, mice lacking the P2X7 receptor have attenuated duration of NREMS and EEG d wave power during NREMS compared with control mice.110
Summary
IL-1 and TNF are well-characterized SRSs that form part of the sleep homeostat (see Fig. 1). Our knowledge of cytokine sleep mechanisms has led to a view of brain organization of sleep positing that sleep is a local property of neural networks being initiated, for example, within cortical columns. Cortical columns oscillate between functional states; the sleeplike state of cortical columns is promoted by TNF. Further, TNF is involved in glutamatergic AMPA receptor expression and in synaptic scaling mechanisms. Cytokine-mediated sleep mechanisms provide support for the hypothesis that sleep serves a synaptic-connectivity function and is tightly coupled to cerebral metabolism. IL-1 and TNF release from glia is enhanced by neuronal activity via ATP and, in turn, IL-1 and TNF activate NFkB, adenosine, and other downstream mechanisms.
Key Points.
Interleukin 1 (IL-1) and tumor necrosis factor (TNF) are well-characterized sleep regulatory substances that form part of the sleep homeostat.
A large literature showing of cytokine involvement in the brain organization of sleep corroborates the view that sleep is an emergent property of neural networks such as cortical columns.
Cortical columns oscillate between functional states; the sleeplike state of cortical columns is promoted by cytokines. Further, cytokines are involved in synaptic scaling mechanisms and are therefore capable of modulating network activity.
The function(s) of sleep is still debated; however cytokine-mediated sleep mechanisms support the hypothesis that sleep serves a synaptic-connectivity function.
How the brain tracks activity and mediates sleep homeostasis is posited in our ATP-cytokine-adenosine hypothesis, wherein IL-1 and TNF release from glia is enhanced by neuronal activity via adenosine triphosphate and, in turn, IL-1 and TNF activate nuclear factor κ B, adenosine, and other downstream effectors to locally influence state.
Acknowledgments
This work was supported by the National Institutes of Health grant numbers NS031453, NS025378 and HD036520.
References
- 1.Ishimori K. True cause of sleep–a hypnogenic substance as evidenced in the brain of sleep-deprived animals. Tokyo Igakkai Zasshi. 1909;23:429–57. [Google Scholar]
- 2.Legendre R, Pieron H. Recherches sur le besoin de sommeil consécutif à une veille prolongée. Z Allg Physiol. 1913;14:235–62. [Google Scholar]
- 3.Pappenheimer JR, Miller TB, Goodrich CA. Sleep-promoting effects of cerebrospinal fluid from sleep-deprived goats. Proc Natl Acad Sci U S A. 1967;58:513–7. doi: 10.1073/pnas.58.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Obal F, Jr, Krueger JM. Biochemical regulation of non-rapid-eye-movement sleep. Front Biosci. 2003;8:d520–50. doi: 10.2741/1033. [DOI] [PubMed] [Google Scholar]
- 5.Borbely AA, Tobler I. Endogenous sleep-promoting substances and sleep regulation. Physiol Rev. 1989;69:605–70. doi: 10.1152/physrev.1989.69.2.605. [DOI] [PubMed] [Google Scholar]
- 6.Inoue S. Biology of sleep substances Boca Raton (FL) CRC Press; 1989. [Google Scholar]
- 7.Jouvet M. Neuromédiateurs et facteurs hypnogénes. Rev Neurol (Paris) 1984;140:389–400. [PubMed] [Google Scholar]
- 8.Borbely AA, Tobler I. The search for an endogenous “sleep substance”. Trends Pharmacol Sci. 1980;1:356–8. [Google Scholar]
- 9.Krueger JM, Obal F. Sleep factors. In: Saunders NA, Sullivan CE, editors. Sleep and breathing. New York: Marcel Dekker; 1994. pp. 79–112. [Google Scholar]
- 10.Krueger JM, Walter J, Dinarello CA, et al. Sleep-promoting effects of endogenous pyrogen (interleukin-1) Am J Physiol. 1984;246:R994–9. doi: 10.1152/ajpregu.1984.246.6.R994. [DOI] [PubMed] [Google Scholar]
- 11.Shoham S, Davenne D, Cady AB, et al. Recombinant tumor necrosis factor and interleukin 1 enhance slow-wave sleep. Am J Physiol. 1987;253:R142–9. doi: 10.1152/ajpregu.1987.253.1.R142. [DOI] [PubMed] [Google Scholar]
- 12.Fang J, Wang Y, Krueger JM. Mice lacking the TNF 55 kDa receptor fail to sleep more after TNFalpha treatment. J Neurosci. 1997;17:5949–55. doi: 10.1523/JNEUROSCI.17-15-05949.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang J, Wang Y, Krueger JM. Effects of interleukin-1 beta on sleep are mediated by the type I receptor. Am J Physiol. 1998;274:R655–60. doi: 10.1152/ajpregu.1998.274.3.R655. [DOI] [PubMed] [Google Scholar]
- 14.Krueger JM, Clinton JM, Winters BD, et al. Involvement of cytokines in slow wave sleep. Prog Brain Res. 2011;193:39–47. doi: 10.1016/B978-0-444-53839-0.00003-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vgontzas AN, Zoumakis E, Lin HM, et al. Marked decrease in sleepiness in patients with sleep apnea by etanercept, a tumor necrosis factor-alpha antagonist. J Clin Endocrinol Metab. 2004;89:4409–13. doi: 10.1210/jc.2003-031929. [DOI] [PubMed] [Google Scholar]
- 16.Franklin CM. Clinical experience with soluble TNF p75 receptor in rheumatoid arthritis. Semin Arthritis Rheum. 1999;29:172–81. doi: 10.1016/s0049-0172(99)80028-6. [DOI] [PubMed] [Google Scholar]
- 17.Taishi P, Davis CJ, Bayomy O, et al. Brain-specific interleukin-1 receptor accessory protein in sleep regulation. J Appl Physiol. 2012;112:1015–22. doi: 10.1152/japplphysiol.01307.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baracchi F, Opp MR. Sleep-wake behavior and responses to sleep deprivation of mice lacking both interleukin-1 beta receptor 1 and tumor necrosis factor-alpha receptor 1. Brain Behav Immun. 2008;22:982–93. doi: 10.1016/j.bbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lue FA, Bail M, Jephthah-Ochola J, et al. Sleep and cerebrospinal fluid interleukin-1-like activity in the cat. Int J Neurosci. 1988;42:179–83. doi: 10.3109/00207458808991595. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen KT, Deak T, Owens SM, et al. Exposure to acute stress induces brain interleukin-1beta protein in the rat. J Neurosci. 1998;18:2239–46. doi: 10.1523/JNEUROSCI.18-06-02239.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Floyd RA, Krueger JM. Diurnal variation of TNF alpha in the rat brain. Neuroreport. 1997;8:915–8. doi: 10.1097/00001756-199703030-00020. [DOI] [PubMed] [Google Scholar]
- 22.Hansen MK, Taishi P, Chen Z, et al. Cafeteria feeding induces interleukin-1beta mRNA expression in rat liver and brain. Am J Physiol. 1998;274:R1734–9. doi: 10.1152/ajpregu.1998.274.6.R1734. [DOI] [PubMed] [Google Scholar]
- 23.Alt JA, Bohnet S, Taishi P, et al. Influenza virus-induced glucocorticoid and hypothalamic and lung cytokine mRNA responses in dwarf lit/lit mice. Brain Behav Immun. 2007;21:60–7. doi: 10.1016/j.bbi.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 24.Kubota T, Li N, Guan Z, et al. Intrapreoptic microinjection of TNF-alpha enhances non-REM sleep in rats. Brain Res. 2002;932:37–44. doi: 10.1016/s0006-8993(02)02262-x. [DOI] [PubMed] [Google Scholar]
- 25.Terao A, Matsumura H, Saito M. Interleukin-1 induces slow-wave sleep at the prostaglandin D2-sensitive sleep-promoting zone in the rat brain. J Neurosci. 1998;18:6599–607. doi: 10.1523/JNEUROSCI.18-16-06599.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De A, Churchill L, Obal F, Jr, et al. GHRH and IL1-beta increase cytoplasmic Ca(2+) levels in cultured hypothalamic GABAergic neurons. Brain Res. 2002;949:209–12. doi: 10.1016/s0006-8993(02)03157-8. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Obal F, Jr, Zheng T, et al. Intrapreoptic microinjection of GHRH or its antagonist alters sleep in rats. J Neurosci. 1999;19:2187–94. doi: 10.1523/JNEUROSCI.19-06-02187.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peterfi Z, McGinty D, Sarai E, et al. Growth hormone-releasing hormone activates sleep regulatory neurons of the rat preoptic hypothalamus. Am J Physiol Regul Integr Comp Physiol. 2010;298:R147–56. doi: 10.1152/ajpregu.00494.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alam MN, McGinty D, Bashir T, et al. Interleukin-1beta modulates state-dependent discharge activity of pre-optic area and basal forebrain neurons: role in sleep regulation. Eur J Neurosci. 2004;20:207–16. doi: 10.1111/j.1460-9568.2004.03469.x. [DOI] [PubMed] [Google Scholar]
- 30.De Sarro G, Gareri P, Sinopoli VA, et al. Comparative, behavioural and electrocortical effects of tumor necrosis factor-alpha and interleukin-1 microinjected into the locus coeruleus of rat. Life Sci. 1997;60:555–64. doi: 10.1016/s0024-3205(96)00692-3. [DOI] [PubMed] [Google Scholar]
- 31.Nistico G, De Sarro G, Rotiroti D. Behavioral and electrocortical spectrum power changes of interleukins and tumor necrosis factor after microinjection into different areas of the brain. In: Smirne S, Francesch M, Ferini-Stambi L, editors. Sleep, hormones, and immunological system Milan (Italy): Mason. 1992. pp. 11–22. [Google Scholar]
- 32.Manfridi A, Brambilla D, Bianchi S, et al. Interleukin-1beta enhances non-rapid eye movement sleep when microinjected into the dorsal raphe nucleus and inhibits serotonergic neurons in vitro. Eur J Neurosci. 2003;18:1041–9. doi: 10.1046/j.1460-9568.2003.02836.x. [DOI] [PubMed] [Google Scholar]
- 33.Brambilla D, Franciosi S, Opp MR, et al. Interleukin-1 inhibits firing of serotonergic neurons in the dorsal raphe nucleus and enhances GABAergic inhibitory post-synaptic potentials. Eur J Neurosci. 2007;26:1862–9. doi: 10.1111/j.1460-9568.2007.05796.x. [DOI] [PubMed] [Google Scholar]
- 34.Serantes R, Arnalich F, Figueroa M, et al. Interleukin-1beta enhances GABAA receptor cell-surface expression by a phosphatidylinositol 3-kinase/Akt pathway: relevance to sepsis-associated encephalopathy. J Biol Chem. 2006;281:14632–43. doi: 10.1074/jbc.M512489200. [DOI] [PubMed] [Google Scholar]
- 35.Chen R, Yin Y, Zhao Z, et al. Elevation of serum TNF-α levels in mild and moderate Alzheimer patients with daytime sleepiness. J Neuroimmunol. 2012;244:97–102. doi: 10.1016/j.jneuroim.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 36.Sookoian SC, Gonzalez C, Pirola CJ. Meta-analysis on the G-308A tumor necrosis factor alpha gene variant and phenotypes associated with the metabolic syndrome. Obes Res. 2005;13:2122–31. doi: 10.1038/oby.2005.263. [DOI] [PubMed] [Google Scholar]
- 37.Gupta V, Gupta A, Jafar T, et al. Association of TNF-alpha promoter gene G-308A polymorphism with metabolic syndrome, insulin resistance, serum TNF-alpha and leptin levels in Indian adult women. Cytokine. 2012;57:32–6. doi: 10.1016/j.cyto.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 38.Riha RL, Brander P, Vennelle M, et al. Tumour necrosis factor-alpha-308 gene polymorphism in obstructive sleep apnoea-hypopnoea syndrome. Eur Respir J. 2005;26:673–8. doi: 10.1183/09031936.05.00130804. [DOI] [PubMed] [Google Scholar]
- 39.Zhang HF, Xie SL, Wang JF, et al. Tumor necrosis factor-alpha G-308A gene polymorphism and coronary heart disease susceptibility: an updated meta-analysis. Thromb Res. 2011;127:400–5. doi: 10.1016/j.thromres.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 40.Mullington J, Korth C, Hermann DM, et al. Dose-dependent effects of endotoxin on human sleep. Am J Physiol Regul Integr Comp Physiol. 2000;278:R947–55. doi: 10.1152/ajpregu.2000.278.4.R947. [DOI] [PubMed] [Google Scholar]
- 41.Karadag O, Nakas D, Kalyoncu U, et al. Effect of anti-TNF treatment on sleep problems in ankylosing spondylitis. Rheumatol Int. 2011;17:358–62. doi: 10.1007/s00296-011-1907-x. [DOI] [PubMed] [Google Scholar]
- 42.Irwin MR, Olmstead R, Valladares EM, et al. Tumor necrosis factor antagonism normalizes rapid eye movement sleep in alcohol dependence. Biol Psychiatry. 2009;66:191–5. doi: 10.1016/j.biopsych.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arias MA, Garcia-Rio F, Alonso-Fernandez A, et al. CPAP decreases plasma levels of soluble tumour necrosis factor-alpha receptor 1 in obstructive sleep apnoea. Eur Respir J. 2008;32:1009–15. doi: 10.1183/09031936.00007008. [DOI] [PubMed] [Google Scholar]
- 44.Steiropoulos P, Kotsianidis I, Nena E, et al. Long-term effect of continuous positive airway pressure therapy on inflammation markers of patients with obstructive sleep apnea syndrome. Sleep. 2009;32:537–43. doi: 10.1093/sleep/32.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eun YG, Kim MG, Kwon KH, et al. Short-term effect of multilevel surgery on adipokines and pro-inflammatory cytokines in patients with obstructive sleep apnea. Acta Otolaryngol. 2010;130:1394–8. doi: 10.3109/00016489.2010.495134. [DOI] [PubMed] [Google Scholar]
- 46.Moldofsky H, Lue FA, Eisen J, et al. The relationship of interleukin-1 and immune functions to sleep in humans. Psychosom Med. 1986;48:309–18. doi: 10.1097/00006842-198605000-00001. [DOI] [PubMed] [Google Scholar]
- 47.Hohagen F, Timmer J, Weyerbrock A, et al. Cytokine production during sleep and wakefulness and its relationship to cortisol in healthy humans. Neuropsychobiology. 1993;28:9–16. doi: 10.1159/000118993. [DOI] [PubMed] [Google Scholar]
- 48.Uthgenannt D, Schoolmann D, Pietrowsky R, et al. Effects of sleep on the production of cytokines in humans. Psychosom Med. 1995;57:97–104. doi: 10.1097/00006842-199503000-00001. [DOI] [PubMed] [Google Scholar]
- 49.Kubota T, Fang J, Guan Z, et al. Vagotomy attenuates tumor necrosis factor-alpha-induced sleep and EEG delta-activity in rats. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1213–20. doi: 10.1152/ajpregu.2001.280.4.R1213. [DOI] [PubMed] [Google Scholar]
- 50.Hansen MK, Krueger JM. Subdiaphragmatic vagotomy blocks the sleep- and fever-promoting effects of interleukin-1beta. Am J Physiol. 1997;273:R1246–53. doi: 10.1152/ajpregu.1997.273.4.R1246. [DOI] [PubMed] [Google Scholar]
- 51.Churchill L, Taishi P, Wang M, et al. Brain distribution of cytokine mRNA induced by systemic administration of interleukin-1beta or tumor necrosis factor alpha. Brain Res. 2006;1120:64–73. doi: 10.1016/j.brainres.2006.08.083. [DOI] [PubMed] [Google Scholar]
- 52.Hansen MK, Taishi P, Chen Z, et al. Vagotomy blocks the induction of interleukin-1beta (IL-1beta) mRNA in the brain of rats in response to systemic IL-1beta. J Neurosci. 1998;18:2247–53. doi: 10.1523/JNEUROSCI.18-06-02247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Majde JA, Krueger JM. Links between the innate immune system and sleep. J Allergy Clin Immunol. 2005;116:1188–98. doi: 10.1016/j.jaci.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 54.Kimura M, Majde JA, Toth LA, et al. Somnogenic effects of rabbit and recombinant human interferons in rabbits. Am J Physiol. 1994;267:R53–61. doi: 10.1152/ajpregu.1994.267.1.R53. [DOI] [PubMed] [Google Scholar]
- 55.Knefati M, Somogyi C, Kapas L, et al. Acidic fibroblast growth factor (FGF) but not basic FGF induces sleep and fever in rabbits. Am J Physiol. 1995;269:R87–91. doi: 10.1152/ajpregu.1995.269.1.R87. [DOI] [PubMed] [Google Scholar]
- 56.Brandt JA, Churchill L, Rehman A, et al. Sleep deprivation increases the activation of nuclear factor kappa B in lateral hypothalamic cells. Brain Res. 2004;1004:91–7. doi: 10.1016/j.brainres.2003.11.079. [DOI] [PubMed] [Google Scholar]
- 57.Chen Z, Gardi J, Kushikata T, et al. Nuclear factor-kappaB-like activity increases in murine cerebral cortex after sleep deprivation. Am J Physiol. 1999;276:R1812–8. doi: 10.1152/ajpregu.1999.276.6.R1812. [DOI] [PubMed] [Google Scholar]
- 58.Basheer R, Rainnie DG, Porkka-Heiskanen T, et al. Adenosine, prolonged wakefulness, and A1-activated NF-kappaB DNA binding in the basal forebrain of the rat. Neuroscience. 2001;104:731–9. doi: 10.1016/s0306-4522(01)00111-7. [DOI] [PubMed] [Google Scholar]
- 59.Kubota T, Kushikata T, Fang J, et al. Nuclear factor-kappaB inhibitor peptide inhibits spontaneous and interleukin-1beta-induced sleep. Am J Physiol Regul Integr Comp Physiol. 2000;279:R404–13. doi: 10.1152/ajpregu.2000.279.2.R404. [DOI] [PubMed] [Google Scholar]
- 60.Luk WP, Zhang Y, White TD, et al. Adenosine: a mediator of interleukin-1beta-induced hippocampal synaptic inhibition. J Neurosci. 1999;19:4238–44. doi: 10.1523/JNEUROSCI.19-11-04238.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kapas L, Shibata M, Kimura M, et al. Inhibition of nitric oxide synthesis suppresses sleep in rabbits. Am J Physiol. 1994;266:R151–7. doi: 10.1152/ajpregu.1994.266.1.R151. [DOI] [PubMed] [Google Scholar]
- 62.Rattenborg NC, Amlaner CJ, Lima SL. Behavioral, neurophysiological and evolutionary perspectives on unihemispheric sleep. Neurosci Biobehav Rev. 2000;24:817–42. doi: 10.1016/s0149-7634(00)00039-7. [DOI] [PubMed] [Google Scholar]
- 63.Krueger JM, Obal F., Jr Sleep function. Front Biosci. 2003;8:d511–9. doi: 10.2741/1031. [DOI] [PubMed] [Google Scholar]
- 64.Krueger JM, Obal F. A neuronal group theory of sleep function. J Sleep Res. 1993;2:63–9. doi: 10.1111/j.1365-2869.1993.tb00064.x. [DOI] [PubMed] [Google Scholar]
- 65.Tononi G, Cirelli C. Sleep and synaptic homeostasis: a hypothesis. Brain Res Bull. 2003;62:143–50. doi: 10.1016/j.brainresbull.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 66.Kavanau JL. Sleep and dynamic stabilization of neural circuitry: a review and synthesis. Behav Brain Res. 1994;63:111–26. doi: 10.1016/0166-4328(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 67.Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol. 1995;45:347–60. doi: 10.1016/0301-0082(94)00057-o. [DOI] [PubMed] [Google Scholar]
- 68.Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23:639–45. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- 69.de Bock F, Dornand J, Rondouin G. Release of TNF alpha in the rat hippocampus following epileptic seizures and excitotoxic neuronal damage. Neuroreport. 1996;7:1125–9. doi: 10.1097/00001756-199604260-00004. [DOI] [PubMed] [Google Scholar]
- 70.De A, Krueger JM, Simasko SM. Glutamate induces the expression and release of tumor necrosis factor-alpha in cultured hypothalamic cells. Brain Res. 2005;1053:54–61. doi: 10.1016/j.brainres.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 71.Schneider H, Pitossi F, Balschun D, et al. A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci U S A. 1998;95:7778–83. doi: 10.1073/pnas.95.13.7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yi PL, Tsai CH, Lin JG, et al. Kindling stimuli delivered at different times in the sleep-wake cycle. Sleep. 2004;27:203–12. doi: 10.1093/sleep/27.2.203. [DOI] [PubMed] [Google Scholar]
- 73.Churchill L, Rector DM, Yasuda K, et al. Tumor necrosis factor alpha: activity dependent expression and promotion of cortical column sleep in rats. Neuroscience. 2008;156:71–80. doi: 10.1016/j.neuroscience.2008.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fix C, Churchill L, Hall S, et al. The number of tumor necrosis factor alpha-immunoreactive cells increases in layer IV of the barrel field in response to whisker deflection in the rats. Sleep. 2006;29:A11. [Google Scholar]
- 75.Shibata M. Hypothalamic neuronal responses to cytokines. Yale J Biol Med. 1990;63:147–56. [PMC free article] [PubMed] [Google Scholar]
- 76.Tabarean IV, Korn H, Bartfai T. Interleukin-1beta induces hyperpolarization and modulates synaptic inhibition in preoptic and anterior hypothalamic neurons. Neuroscience. 2006;141:1685–95. doi: 10.1016/j.neuroscience.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 77.Yasuda T, Yoshida H, Garcia-Garcia F, et al. Interleukin-1beta has a role in cerebral cortical state-dependent electroencephalographic slow-wave activity. Sleep. 2005;28:177–84. doi: 10.1093/sleep/28.2.177. [DOI] [PubMed] [Google Scholar]
- 78.Yoshida H, Peterfi Z, Garcia-Garcia F, et al. State-specific asymmetries in EEG slow wave activity induced by local application of TNFalpha. Brain Res. 2004;1009:129–36. doi: 10.1016/j.brainres.2004.02.055. [DOI] [PubMed] [Google Scholar]
- 79.Taishi P, Churchill L, Wang M, et al. TNFalpha siRNA reduces brain TNF and EEG delta wave activity in rats. Brain Res. 2007;1156:125–32. doi: 10.1016/j.brainres.2007.04.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rector DM, Topchiy IA, Carter KM, et al. Local functional state differences between rat cortical columns. Brain Res. 2005;1047:45–55. doi: 10.1016/j.brainres.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 81.Phillips DJ, Schei JL, Meighan PC, et al. Cortical evoked responses associated with arousal from sleep. Sleep. 2011;34:65–72. doi: 10.1093/sleep/34.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Walker JL, Walker BM, Fuentes FM, et al. Rat psychomotor vigilance task with fast response times using a conditioned lick behavior. Behav Brain Res. 2011;216:229–37. doi: 10.1016/j.bbr.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strogatz SH, Stewart I. Coupled oscillators and biological synchronization. Sci Am. 1993;269:102–9. doi: 10.1038/scientificamerican1293-102. [DOI] [PubMed] [Google Scholar]
- 84.Roy S, Krueger JM, Rector DM, et al. A network model for activity-dependent sleep regulation. J Theor Biol. 2008;253:462–8. doi: 10.1016/j.jtbi.2008.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Churchill L, Yasuda K, Yasuda T, et al. Unilateral cortical application of tumor necrosis factor alpha induces asymmetry in Fos- and interleukin-1beta-immunoreactive cells within the corticothalamic projection. Brain Res. 2005;1055:15–24. doi: 10.1016/j.brainres.2005.06.052. [DOI] [PubMed] [Google Scholar]
- 86.Yasuda K, Churchill L, Yasuda T, et al. Unilateral cortical application of interleukin-1beta (IL1beta) induces asymmetry in fos, IL1beta and nerve growth factor immunoreactivity: implications for sleep regulation. Brain Res. 2007;1131:44–59. doi: 10.1016/j.brainres.2006.11.051. [DOI] [PubMed] [Google Scholar]
- 87.Benington JH, Frank MG. Cellular and molecular connections between sleep and synaptic plasticity. Prog Neurobiol. 2003;69:71–101. doi: 10.1016/s0301-0082(03)00018-2. [DOI] [PubMed] [Google Scholar]
- 88.Best J, Diniz Behn C, Poe GR, et al. Neuronal models for sleep-wake regulation and synaptic reorganization in the sleeping hippocampus. J Biol Rhythms. 2007;22:220–32. doi: 10.1177/0748730407301239. [DOI] [PubMed] [Google Scholar]
- 89.Krueger JM, Rector DM, Roy S, et al. Sleep as a fundamental property of neuronal assemblies. Nat Rev Neurosci. 2008;9:910–9. doi: 10.1038/nrn2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62. doi: 10.1016/j.smrv.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 91.Aton SJ, Seibt J, Dumoulin M, et al. Mechanisms of sleep-dependent consolidation of cortical plasticity. Neuron. 2009;61:454–66. doi: 10.1016/j.neuron.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roffwarg HP, Muzio JN, Dement WC. Ontogenetic development of the human sleep-dream cycle. Science. 1966;152:604–19. doi: 10.1126/science.152.3722.604. [DOI] [PubMed] [Google Scholar]
- 93.Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3(Suppl):1178–83. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- 94.Krueger JM, Tononi G. Local use-dependent sleep; synthesis of the new paradigm. Curr Top Med Chem. 2011;11:2490–2. doi: 10.2174/156802611797470330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krueger JM, Wisor JP. Local use-dependent sleep. Curr Top Med Chem. 2011;11:2390–1. doi: 10.2174/156802611797470295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.De A, Krueger JM, Simasko SM. Tumor necrosis factor alpha increases cytosolic calcium responses to AMPA and KCl in primary cultures of rat hippocampal neurons. Brain Res. 2003;981:133–42. doi: 10.1016/s0006-8993(03)02997-4. [DOI] [PubMed] [Google Scholar]
- 97.Beattie EC, Stellwagen D, Morishita W, et al. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–5. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 98.Bazhenov M, Timofeev I, Steriade M, et al. Model of thalamocortical slow-wave sleep oscillations and transitions to activated states. J Neurosci. 2002;22:8691–704. doi: 10.1523/JNEUROSCI.22-19-08691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 100.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–9. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 101.Lai AY, Swayze RD, El-Husseini A, et al. Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J Neuroimmunol. 2006;175:97–106. doi: 10.1016/j.jneuroim.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 102.Czarnecki A, Birtoli B, Ulrich D. Cellular mechanisms of burst firing-mediated long-term depression in rat neocortical pyramidal cells. J Physiol. 2007;578:471–9. doi: 10.1113/jphysiol.2006.123588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Farber K, Kettenmann H. Purinergic signaling and microglia. Pflugers Arch. 2006;452:615–21. doi: 10.1007/s00424-006-0064-7. [DOI] [PubMed] [Google Scholar]
- 104.Bianco F, Pravettoni E, Colombo A, et al. Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J Immunol. 2005;174:7268–77. doi: 10.4049/jimmunol.174.11.7268. [DOI] [PubMed] [Google Scholar]
- 105.Hide I, Tanaka M, Inoue A, et al. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem. 2000;75:965–72. doi: 10.1046/j.1471-4159.2000.0750965.x. [DOI] [PubMed] [Google Scholar]
- 106.Suzuki T, Hide I, Ido K, et al. Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci. 2004;24:1–7. doi: 10.1523/JNEUROSCI.3792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev. 2007;87:659–797. doi: 10.1152/physrev.00043.2006. [DOI] [PubMed] [Google Scholar]
- 108.Ferrari D, Pizzirani C, Adinolfi E, et al. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–83. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 109.Duan S, Neary JT. P2X(7) receptors: properties and relevance to CNS function. Glia. 2006;54:738–46. doi: 10.1002/glia.20397. [DOI] [PubMed] [Google Scholar]
- 110.Krueger JM, Taishi P, De A, et al. ATP and the purine type 2 X7 receptor affect sleep. J Appl Physiol. 2010;109:1318–27. doi: 10.1152/japplphysiol.00586.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]