

Abstract
Naphtoquinones have been used as promising scaffolds for drug design studies against protozoan parasites. Considering the highly toxic and limited therapeutic arsenal, the global negligence with tropical diseases and the elevated prevalence of co-morbidities especially in developing countries, the parasitic diseases caused by various Leishmania species (leishmaniasis) became a significant public health threat in 98 countries. The aim of this work was the evaluation of antileishmanial in vitro potential of thirty-six 2-hydroxy-3-phenylsulfanylmethyl-[1,4]-naphthoquinones obtained by a three component reaction of lawsone, the appropriate aldehyde and thiols adequately substituted, exploiting the in situ generation of o-quinonemethides (o-QM) via the Knoevenagel condensation. The antileishmanial activity of the naphthoquinone derivatives was evaluated against promastigotes and intracellular amastigotes of Leishmania (Leishmania) infantum and their cytotoxicity was verified in mammalian cells. Among the thirty-six compounds, twenty-seven were effective against promastigotes, with IC50 values ranging from 8 to 189 µM; fourteen compounds eliminated the intracellular amastigotes, with IC50 values ranging from 12 to 65 µM. The compounds containing the phenyl groups at R1 and R2 and with the fluorine substituent at the phenyl ring at R2, rendered the most promising activity, demonstrating a selectivity index higher than 15 against amastigotes. A QSAR (quantitative structure activity relationship) analysis yielded insights into general structural requirements for activity of most compounds in the series. Considering the in vitro antileishmanial potential of 2-hydroxy-3-phenylsulfanylmethyl-[1,4]-naphthoquinones and their structure-activity relationships, novel lead candidates could be exploited in future drug design studies for leishmaniasis.
Introduction
Leishmaniasis is a complex of diseases caused by protozoan parasites of the genus Leishmania. Leishmaniasis is still one of the most neglected diseases, affecting the poorest population in developing countries [1]. The disease is endemic in 98 countries, with a global incidence estimated at approximately 0.9–1.6 million cases occurring each year and Brazil is among the 10 countries with the highest estimated case counts [2]. Currently, the leishmaniasis treatment is based on the use of few drugs such as pentavalent antimonials, miltefosine and amphotericin B, with medium to severe toxic side effects and long-term administration [3]. Over the past years, the lack of new medicines targeting parasitic diseases affecting people in developing countries has become a global concern [4]. Therefore, an urgent need remains for safer and more effective drug candidates.
Naphthoquinones are a class of chemical compounds exhibiting a variety of anticancer [5], antiviral, trypanocidal, immunomodulatory and antimicrobial activities [6]. Among the 1,4-naphthoquinone derivatives in literature, some promising antimalarials have been described [7]–[9]. Some other series of 1,4-naphthoquinone derivatives have also been widely evaluated against Mycobacterium tuberculosis [10], Plasmodium falciparum [9], and as molluscicidal candidates against Biomphalaria glabrata [11]. A number of reports have also shown the antiprotozoal potential of 1,4-naphthoquinone derivatives against Trypanosoma cruzi and Leishmania (L.) donovani [12]–[13]. In the present study, a series of thirty-six 2-hydroxy-3-phenylsulfanylmethyl-[1,4]-naphthoquinones (1–36) was synthesized and evaluated against the extracellular and the clinically relevant form of the parasite, the intracellular amastigotes of L. (L.) infantum, which is the etiologic agent of visceral leishmaniasis in Brazil and the Mediterranean region. The in vitro toxicity of these compounds against mammalian cells was also studied to provide the selectivity index. Finally, a QSAR analysis was conducted in order to obtain insights into the structural requirements for activity and selectivity within this series of naphthoquinone derivatives.
Materials and Methods
General Procedures
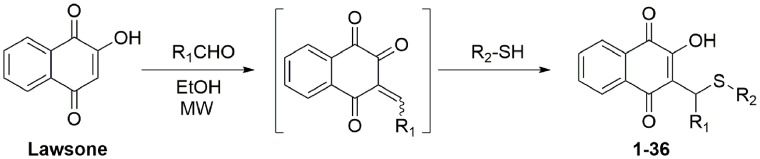
The compounds 1–36 were obtained by reaction of lawsone with formaldehyde in situ generating the intermediate ortho-quinonemethide, followed by nucleophilic addition of thiols adequately substituted (Figure 1). This reaction explores in situ generation of o-quinonemethides (o-QM) via the Knoevenagel condensation reaction between lawsone and formaldehyde [9].
Figure 1. General scheme for preparing the 2-hydroxy-3-alkyl[1,4]naphthoquinones or 3-arylsulfanylmethyl[1,4]naphthoquinones under microwave irradiation.
Ethics Statement: Bioassay Procedures
BALB/c mice and Golden hamsters (Mesocricetus auratus) were supplied by the Animal Breeding Facility at the Adolfo Lutz Institute of São Paulo. They were maintained in sterilized cages, receiving water and food ad libitum, under climate-controlled (22°C±2 and relative humidity- 60%) and photoperiod-controlled (12 h light-dark cycles) environment. To avoid pain, animals were euthanized using carbon dioxide (purity 99.99%) in a gas chamber, in a flow rate of 20% of the chamber volume per minute, according to the Newcastle Consensus Meeting on Carbon Dioxide Euthanasia of Laboratory Animals (http://www.nc3rs.org.uk/downloaddoc.asp?id=416&page=292&skin=0). Animal procedures were performed with the approval of the Research Ethics Commission of Instituto Adolfo Lutz/Instituto Pasteur (project CEUA-IAL/Pasteur 04/2011) in agreement with the Guide for the Care and Use of Laboratory Animals from the National Academy of Sciences (http://www.nas.edu) and all efforts were made to minimize suffering.
Parasites and Macrophages Maintenance
L. (L.) infantum (MHOM/BR/1972/LD) promastigotes were maintained in M-199 medium supplemented with 10% fetal bovine serum (FBS) and 0.25% hemin at 24°C. L. (L.) infantum was maintained in golden hamsters for up to approximately 60–70 days post-infection. The amastigotes were obtained from the spleens of previously infected hamsters by differential centrifugation. The macrophages were collected from the peritoneal cavity of BALB/c mice by washing with RPMI-1640 (without phenol red and supplemented with 10% FBS). NCTC (clone 929) murine conjunctive cells were maintained in RPMI-1640 (without phenol red and supplemented with 10% FBS) at 37°C in a humidified containing 5% CO2.
Determination of the Antileishmanial Activity
To determine the 50% inhibitory concentration (IC50) against L. (L.) infantum, drugs were dissolved previously in dimethyl sulphoxide (DMSO) and diluted with M-199 medium. Promastigotes were counted in a Neubauer hemocytometer and seeded at 1×106 cells per well in 96-well microplates using miltefosine as standard drug. The tested compounds were incubated at the highest concentration of 200 µM for 48 h at 24°C. Parasite viability was determined using the MTT assay [14]. Briefly, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) was dissolved in phosphate-buffered saline (PBS) at 5 mg/mL, sterilized through 0.22 µm membranes and incubated with cells (20 µL/well) for 4 h at 24°C. The extraction of the mitochondrial formazan was done with 80 µL of 10% SDS for 18 h at 24°C. The optical density was determined in FilterMax F5 (Molecular Devices) at 570 nm. Promastigotes incubated without compounds were used as the viability control (100% viability) and without cells (blank).
The activity against intracellular L. (L.) infantum amastigotes was determined in infected macrophages. Macrophages were obtained as previously described and seeded for 24 h at 1×105 cells/well in 16-well slide chambers (Nunc). Amastigotes were isolated from the spleens of previously infected hamsters, separated by differential centrifugation and added to the macrophages at a ratio of 1∶10 (macrophage/amastigotes) for 24 h at 37°C. Non-internalized parasites were removed by washing once with medium and the cells were then incubated with the test compounds for 120 h at 37°C in 5% CO2, using miltefosine as standard drug. At the end of the assay, the cells were fixed in methanol, stained with Giemsa and observed under a light microscope to determine the number of intracellular parasites. The number of amastigotes was determined in 400 macrophages from the drug-treated and control wells [15].
Cytotoxicity against Mammalian Cells
The 50% cytotoxic concentration (CC50) was determined in NCTC clone 929 murine conjunctive cells. NCTC cells were seeded at 6×104 cells/well in 96-well microplates at 37°C in a 5% CO2. The mammalian cells were incubated with tested compounds to the highest concentration of 200 µM for 48 h at 37°C, using miltefosine as standard drug. The viability of the cells was determined by MTT assay at 570 nm [15]. The selectivity index (SI) was determined considering the following equation: CC50 NCTC cells/IC50 amastigotes.
Statistical Analysis
The obtained data represent the mean and standard deviation of two independent experiments performed in duplicate. The IC50 and CC50 values were calculated using sigmoid dose-response curves performed using GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA), and the 95% confidence intervals (95% CI) were included. The ANOVA test was performed to evaluate the significance (p<0.05) of data.
QSAR Analysis
The biological activity data (IC50 values against L. (L.) infantum promastigotes available for twenty-seven compounds) were transformed to a molar dimension and used for the QSAR analysis in form of their negative decadic logarithms (pIC50) [16]. These values were in a range from 3.72 to 5.09. 3D molecular models of these twenty-seven compounds were generated with the modeling package MOE rel. 2011.10 (CCG, Montreal). For each structure, a conformational search was performed using a low-mode molecular dynamics method (default parameters, MMFF94X force field) as implemented in MOE. The lowest energy conformer was then energy minimized using the AM1 Hamiltonian as included in MOE's MOPAC module. The optimized geometries thus obtained were used to calculate molecular descriptors. In this study, a total of 308 descriptors were taken into consideration, i.e. analyzed for linear correlation with the target value, pIC50. The full descriptor table is available from the authors (TJS) on request. Descriptor selection was performed with a genetic algorithm – multiple linear regression method (GA-MLR) as available in the MOE program GA.svl [available for MOE users on the CCG SVL exchange site, http://www.chemcomp.com/Support-SVL_Exchange.htm]. This algorithm was applied to the data set using three different descriptor blocks such that (a) only 2D descriptors (n = 186) (b) only 3D descriptors (n = 122) and (c) all 308 descriptors were taken into account. For each setting, three GA runs (population size = 100) were performed, in which the allowed number of descriptors was 3, 4 and 5. The optimization criterion for the GA was a minimization of the LOF (lack of fit), the predefined maximum number of generations was 50.000 and the termination criterion was a failure to achieve an improvement (i.e. further decrease) of the LOF within 1000 generations. After termination, the final model population of each run was validated using leave-one-out cross validation and the model with the highest squared correlation coefficient (Q2) between the cross validation predictions and the experimental biological data was chosen for further evaluation. This was achieved by applying partial least squares (PLS) regression to the best descriptor combinations found by the GA-MLR calculations. The best model found in this way is described by equation (1).
Results
Antileishmanial Activity
The antileishmanial activity of the thirty-six naphthoquinone derivatives was evaluated against promastigotes of L. (L.) infantum by MTT reduction assay, as shown in Table 1. After 48 h of incubation, twenty-seven compounds were active with IC50 values in the range of 8.09 to 189.91 µM; compound 32 was the most active. According to the colorimetric assay of MTT and light microscopy, the active compounds killed 100% of parasites at the highest tested concentration. When tested against the intracellular amastigotes, fourteen compounds resulted in IC50 values in the range of 12.98 to 65.52 µM. Miltefosine was used as a standard drug and resulted in IC50 values of 16.85 and 17.80 µM, respectively. Among the tested compounds, compound 11 was the most effective, with an IC50 value of 12.98 µM against intracellular amastigotes. Compounds 11, 20 and 21 showed a similar effectiveness to the standard drug miltefosine. Other tested compounds (2, 3, 4, 5, 6, 7, 12, 19, 25, 26, 27, 28 and 34) were effective against promastigotes, but exhibited no activity against intracellular amastigotes at the highest tested concentration. In contrast, the naphthoquinone derivatives (1, 9, 13, 14, 15, 24, 31, 35 and 36) showed no activity (>200 µM) against both forms of L. (L.) infantum.
Table 1. Antileishmanial and cytotoxicity effects of 2-hydroxy-3-phenylsulfanylmethyl-[1,4]-naphthoquinones.
| Compounds | R1 | R2 | Promastigotes | Amastigotes | Cytotoxicity | S.I. |
| IC50 (µM) | IC50 (µM) | CC50 (µM) | ||||
| (C.I.95%) | (C.I.95%) | (C.I.95%) | ||||
| 1 | H | 4-CH3C6H4 | na | na | 89.09 (82.77–95.88) | nd |
| 2 | H | -C6H5 | 124.3 (115.8–133.3) | na | 84.66 (77.02–93.05) | nd |
| 3 | H | 4-OCH3C6H4 | 75.0 (71.14–79.08) | na | 92.57 (90.14–95.05) | nd |
| 4 | H | 4-ClC6H4 | 85.38 (78.54–92.82) | na | 82.20 (74.47–90.72) | nd |
| 5 | H | 4-FC6H4 | 129.0 (115.6–143.90) | na | 97.94 (85.80–111.80) | nd |
| 6 | H | 4-CH3SC6H4 | 188.5 (177.2–200.50) | na | 100.50 (86.81–116.30) | nd |
| 7 | H | 4-NO2C6H4 | 8.44 (4.315–16.51) | na | 85.51 (79.36–92.14) | nd |
| 8 | H | 4-OHC6H4 | 189.91 (178.4–203.2) | 65.52 (61.10–70.26) | 173.62 (208.6–462.4) | 2.64 |
| 9 | H | Propyl | na | na | >200 | nd |
| 10 | C6H5 | 4-ClC6H4 | 20.72 (17.73–24.21) | 40.37 (13.16–123.80) | >200 | >4.95 |
| 11 | C6H5 | 4-FC6H4 | 28.82 (26.57–31.26) | 12.98 (8.59–19.58) | >200 | >15.4 |
| 12 | C6H5 | 4-CH3SC6H4 | 45.10 (40.72–49.95) | na | 138.2 (118.4–161.4) | nd |
| 13 | H | 2-CH3C6H4 | na | na | >200 | nd |
| 14 | H | 3-CH3C6H4 | na | na | >200 | nd |
| 15 | H | 2-Naphthyl | na | na | >200 | nd |
| 16 | C6H5 | 4-CH3C6H4 | 72.72 (58.20–90.87) | 29.54 (27.68–31.52) | 163.0 (143.2–185.7) | 5.51 |
| 17 | C6H5 | 4-OCH3C6H4 | 68.93 (62.24–76.33) | 57.0 (52.01–62.46) | 183.3 (48.09–698.7) | 3.21 |
| 18 | C6H5 | 4-OHC6H4 | 58.33 (50.84–66.91) | 49.35 (44.66–54.53) | 175.3 (81.14–378.9) | 3.55 |
| 19 | C6H5 | 4-NO2C6H4 | 9.06 (7.76–10.59) | na | 69.51 (53.99–89.50) | nd |
| 20 | C6H5 | 3-CH3C6H4 | 26.43 (23.78–29.38) | 14.70 (12.48–17.31) | 137.6 (118.3–160.0) | 9.36 |
| 21 | C6H5 | -C6H5 | 27.42 (24.53–30.65) | 16.60 (14.28–19.29) | 136.1 (115.4–160.4) | 8.20 |
| 22 | C6H5 | 2-CH3C6H4 | 30.94 (28.98–33.03) | 25.43 (19.78–32.69) | 45.11 (32.08–63.43) | 1.77 |
| 23 | C6H5 | 2-Naphthyl | 27.52 (24.19–31.30) | 46.89 (42.79–51.38) | 152.7 (139.7–167.0) | 3.25 |
| 24 | C6H5 | Propyl | na | na | >200 | nd |
| 25 | 4-NO2C6H4 | 4-CH3C6H4 | 107.8 (95.70–121.50) | na | 42.65 (34.23–53.14) | nd |
| 26 | 4-NO2C6H4 | 4-ClC6H4 | 49.17 (36.25–66.69) | na | 33.89 (28.20–40.73) | nd |
| 27 | 4-NO2C6H4 | 4-FC6H4 | 51.76 (27.65–96.90) | na | 42.11 (24.33–72.90) | nd |
| 28 | 4-NO2C6H4 | -C6H5 | 40.08 (14.50–110.80) | na | 94.18 (79.85–111.10) | nd |
| 29 | 4-NO2C6H4 | 4-CH3SC6H4 | 20.35 (7.99–51.80) | 39.15 (28.05–54.65) | 75.32 (52.08–108.90) | 1.92 |
| 30 | 4-NO2C6H4 | 4-NO2C6H4 | 120.5 (31.83–455.90) | 45.80 (40.77–51.45) | 81.56 (34.82–191.10) | 1.78 |
| 31 | 4-NO2C6H4 | 4-OHC6H4 | na | na | 134.8 (71.86–253.00) | nd |
| 32 | 4-NO2C6H4 | 4-OCH3C6H4 | 8.09 (3.55–18.46) | 39.11 (35.41–43.19) | 74.97 (45.40–123.80) | 1.91 |
| 33 | 4-NO2C6H4 | 3-CH3C6H4 | 55.94 (26.68–117.30) | 32.41 (30.62–34.31) | 47.93 (42.61–53.91) | 1.47 |
| 34 | 4-NO2C6H4 | 2-CH3C6H4 | 82.85 (45.93–149.50) | na | 96.04 (88.29–104.50) | nd |
| 35 | 4-NO2C6H4 | Propyl | na | na | 75.10 (54.27–103.90) | nd |
| 36 | 4-NO2C6H4 | 2-Naphthyl | na | na | 58.25 (34.63–98.00) | nd |
| miltefosine | - | - | 16.85 | 17.80 | 122 | 6.85 |
na: not active; nd: not determined; IC50: 50% inhibitory concentration; CC50: 50% cytotoxic concentration; 95% C.I.: 95% confidence interval; S.I.: selectivity index (CC50 mammalian cells/IC50 amastigotes).
Mammalian Cytotoxicity
The synthesized compounds were incubated with mammalian cells (NCTC clone 929) for 48 h to evaluate the in vitro cytotoxicity and the viability was detected by the colorimetric assay with MTT. Twenty-nine compounds showed 50% cytotoxic concentration (CC50) values in the range between 33.89 to 183.3 µM; compounds 9, 10, 11, 13, 14, 15 and 24 showed lack of toxicity, and among these compounds, 10 and 11 showed activity against intracellular amastigotes. Miltefosine was used as standard drug and resulted in CC50 value of 122 µM.
Analysis of Quantitative Structure-Activity Relationships (QSAR)
In order to gain information on relationships between the chemical structure and bioactivity, a QSAR analysis was undertaken with the activity data of twenty-seven compounds against L. (L.) infantum promastigotes. This set of activity data had to be chosen since the number of available data points for anti-amastigote activity and the variance within this set of data were limited.
QSAR aims at a mathematical description of the contributions of structural features and chemical properties, expressed in numerical form as descriptor variables, to bioactivity. Classically, linear regression methods are used to investigate the compounds for correlations between the descriptor and the biological activity data. Here, a set of 308 molecular descriptors were analyzed using a genetic algorithm as a means of variable selection for multiple linear regression (MLR) models, followed by analysis of the resulting QSAR equations of the best MLR models using partial least squares regression (PLS).
The best performance in PLS regression among all tested models was found with a QSAR equation consisting of four 3D descriptors. This model was characterized by an R2 (coefficient of determination for calibration vs experimental data) of 0.78 and a Q2 value (coefficient of determination for cross validation predictions vs. experimental data) of 0.68.
QSAR equation (1):
(R2 = 0.780, RMSE = 0.176, Q2 = 0.676, RMSE (cross validation) = 0.217; data were standardized to unit variance, i.e. each value was divided by the standard deviation of the descriptor column)
The descriptors encode the following properties: ASAP6 represents the accessible molecular surface area covered by atoms in a partial charge (qi) interval from +0.25 to +0.3 e [16] while FASA_P is the fraction of accessible molecular surface area covered by atoms with polar properties (i.e. |qi|≥0.2). ASAP6 within this series of compounds is mainly influenced by the partial charge and accessibility of the OH proton at the naphthoquinone core. In some compounds its calculated partial charge is below 0.25 e and apparently there is a correlation between activity and the degree of positive charge (or electron density) and accessibility of this acidic proton. Thus, it may be stated that the overall polarity of the molecules must not be too high for improving the antileishmanial activity (negative regression coefficient of FASA_P) while the OH proton in the quinone system has some influence, probably due to its H-bonding propensity (positive coefficient of ASAP6). Descriptors npr1 and glob are descriptors related to molecular mass distribution and shape. Npr1 is the normalized principal moment of inertia ratio pmi1/pmi3 where pmi1 and pmi3 are the first and third diagonal elements of the diagonalized moment of inertia tensor. The negative coefficient in the QSAR equation shows that a high ratio between these two elements would be detrimental to activity, i.e. that a more even mass distribution within the molecule has an enhancing effect on activity. Along the same lines, glob represents the molecular globularity, which is 1 in case of a perfect sphere and 0 in case of a purely 1 or 2-dimensional object. The positive regression coefficient in this case indicates that a more spherical shape appears to enhance activity in this series.
A plot of experimental data vs. calibration and cross validation data is shown in Figure 2. It becomes obvious in this plot that two compounds in the highest activity range, 32 and 7, are not well represented by the model, i.e. their activity values calculated by the QSAR equation are much lower than the experimental ones. In fact, compound 32 causes the most dramatic effect on the overall correlation, since eliminating it increased the R2 and Q2 values to 0.85 and 0.75, respectively (omission of both, 32 and 7 yielded 0.85 and 0.75). Based on these data, it can safely be assumed that the QSAR model presented here explains the variance of the biological data for most of the compounds very well and thus captures the major structural influences on the bioactivity under study.
Figure 2. Plot of experimental pIC50 values vs. values calculated by QSAR equation (1).

Blue rhombi: calibration data, red squares: leave one out cross validation predictions. Highlighted by ellipse: Data points for compounds 7 and 32 which are not well explained by the equation.
Discussion
Naphthoquinones have been considered promising scaffolds against protozoan parasites. Atovaquone, a naphtoquinone, has been used as a fixed-dose combination with proguanil for treating children and adults with uncomplicated malaria or as chemoprophylaxis for preventing malaria in travellers [17]. Other naphtoquinones as buparvaquone (2-((4-tert-Butylcyclohexyl)methyl)-3-hydroxy-1,4-naphthoquinone), a veterinary drug against the protozoan parasite Theileria spp., has been found to be a promising lead against Leishmania spp, with IC50 values in the range of 0.005 and 0.12 µM against L. (L.) donovani [18]–[19]. The synthesis of 2-hydroxy-3-phenylsulfanylmethyl-[1,4]-naphthoquinones and their activity against the protozoan parasite Plasmodium falciparum was recently reported by Sharma and co-workers [9] and directed us to evaluate their potential in vitro activity against the etiologic agent of visceral leishmaniasis in Brazil and the Mediterranean region, the L. (L.) infantum.
In this study, 2-hydroxy-3-phenylsulfanylmethyl-[1,4]-naphthoquinones were synthesized taking into consideration that the naphthoquinone core remained the same in all the assayed compounds. Their IC50 values were evaluated against promastigotes and intracellular amastigotes of L. (L.) infantum. Among the thirty-six tested compounds, twenty-seven showed activity against the extracellular form of Leishmania, the promastigotes, but only compounds 7, 10, 19, 29 and 32 showed similar effectiveness to the standard drug miltefosine. Considering the intracellular amastigotes as the clinically relevant forms, fourteen compounds showed activity; 10, 11, 16, 20 and 21 were the most selective compounds, with selectivity index above 5. Taking into account the mammalian cytotoxicity, compound 11 was the most promising candidate; it was at least 2-fold more selective than miltefosine, showing a selectivity index higher than 15. Despite the clinical use of miltefosine, it has a considerable in vitro cytotoxicity to mammalian cells; in our assays, it showed a selectivity index of 6.
Corroborating the study of Sharma and co-workers [9], our most promising compounds also demonstrated the two phenyl groups at R1 and R2, but in contrast to Plasmodium, the antileishmanial activity was not enhanced when the p-nitro group was used as a substituent at R1. Furthermore, the presence of the two phenyl groups at R1 and R2 was mandatory to the anti-amastigote effect in our 2-hydroxy-3-phenylsulfanylmethyl-[1,4]-naphthoquinones, since the increased hydrophobicity possibly enhanced the penetration into macrophages or affected specific enzymes of the intracellular stage of the parasite. Although nitro compounds have shown promising antileishmanial activity [20] with IC50 values below 100 nM against intracellular amastigotes, their elevated toxicity has also been described in literature [21]. Our data corroborates previous reports, demonstrating a higher mammalian toxicity when the nitro group was introduced to our 2-hydroxy-3-phenylsulfanylmethyl-[1,4]-naphthoquinones.
Among the most active compounds containing the phenyl groups at R1 and R2, it is noteworthy that fluorine as a substituent at the phenyl ring at R2 (compound 11), decisively improved the selectivity (S.I.>15) towards intracellular amastigotes, also reducing the mammalian toxicity to undetectable levels at the highest tested concentration. In contrast, the substitution of fluorine by a hydroxyl group in the same position at R2 reduced at least 4-fold the selectivity of compound 18. Despite the 3-fold increasing of the IC50 value, the substitution of fluorine (compound 11) by chlorine (compound 10) was also an important contribution, rendering a promising anti-amastigote activity with low mammalian toxicity. Both chlorine and fluorine possess some extreme properties, in particular, high electronegativity and oxidation potential, which may have contributed to the selectivity towards the intracellular amastigotes without affecting the host cell. Additionally, fluorine substitution can alter the chemical properties, disposition, and biological activity of drugs. Many fluorinated compounds are currently widely used in the treatment of diseases, including antimalarials, antibacterials, antifungals, antidepressants, antiinflammatory agents, antipsychotics, antivirals, steroids, and anesthetics [22]. Considering that fluorine is the most electronegative element in the periodic table, the replacement of a hydrogen atom for fluorine, can alter the pKa, the dipole moments, and even the chemical reactivity and stability of neighboring functional groups [23]. Sharma and co-workers [9] demonstrated the in vitro antimalarial activity against P. falciparum of a series of 2-hydroxy-3-phenylsulfanylmethyl-[1,4]-naphthoquinones, but conversely, the most active compounds (IC50<10 µM) had a p-nitro phenyl group as a substituent at R1.
Based on the substitution of methyl groups at different positions at R2, it was possible to note a considerable alteration in the biological effect. Changing the p-methyl group (compound 16) to a m-methyl group (compound 20) improved about 2-fold its anti-amastigote activity and its selectivity. On the other hand, the substitution of the m-methyl group (compound 20) by the o-methyl group (compound 22) increased about 3-fold the mammalian toxicity, while preserving the anti-amastigote activity at a similar level. Another important contribution could be observed by comparing compounds 16 and 17; it is notable that the substitution of a methyl group at R2 (16) improved by 2-fold the anti-amastigote activity when compared to compound 17, which displayed the methoxyl group at R2. Finally, the presence of the sulphur group at R2 (compound 12) negatively contributed to the anti-amastigote effect when compared to compounds 16 and 17.
Our synthesized series revealed moderate mammalian toxicity, as reflected by selectivity indexes ranging from 1.47 to >15. The cytotoxicity of naphthoquinones has been attributed to the induction of intrinsic apoptotic pathway in a manner associated with a significant reactive oxygen species (ROS) increase in the EL-4 mouse T lymphoma cells [24]. The capacity to produce free oxygen radicals is dramatically influenced by the nature and position of substituents and contributes to both therapeutic and toxic actions of these substances. The antitrypanosomal activity of other β-lapachol-derivatives has been attributed to the production of ROS and electrophilic metabolites, which bind to and inactivate Trypanosoma cruzi macromolecules [25]. Ribeiro and co-workers [26] also reported that pterocarpanquinone promotes apoptosis in L. (L.) amazonensis promastigotes through the formation of ROS, which cause oxidative stress, mitochondrial membrane depolarization and DNA fragmentation. It was found that diospyrin, a bisnaphthoquinonoid compound [1], and its derivative could induce apoptosis-like death in L. (L.) donovani promastigotes through depolarization of mitochondrial membrane potential [27]. Studies on the mechanism of antileishmanial activity showed diospyrin to be a specific inhibitor of type I DNA topoisomerase, an imperative therapeutic target for rational design of antiprotozoal drugs [28]. The antileishmanial activity of naphthoquinones against extracellular and intracellular L. (L.) donovani was also shown to be mediated by a nitric oxide-dependent mechanism [13].
Although the promastigote form is not the most relevant life stage with respect to the treatment of clinical infections, our QSAR analysis for the anti-promastigote activity reveals interesting general structural requirements for activity. The observation that descriptors of mass distribution and shape are of influence on the QSAR model indicates that the bioactivity under study depends on specific interactions with a particular biological target. Naphthoquinones may exert biological activity by mechanisms related to either oxidative modification or covalent interactions with target molecules. In the present case, due to the fact that the compounds do not have an unsubstituted carbon in the quinone ring, direct covalent reactions are unlikely. Quite interestingly, descriptors related to redox potential (i.e. HOMO and LUMO energies), which were also present in the variable set, did not yield significant contributions to the overall QSAR model. It may thus be assumed that the differences in activity observed between these molecules are not related to differences in the general mechanism of action but rather in subtle differences related to polarity, hydrogen-bonding and shape that influence non-covalent target binding.
It appears somewhat dissatisfactory that the two most active compounds in the series are not well described by the best QSAR equation we could find, which may indicate that these two compounds do not follow the common structure-activity relationship of the others. However, this may also mean that compounds 7 and 32 could act by a mechanism different from that of the other compounds of this series, which may be an interesting starting point for further studies. Compound 11, the most selective compound against the intracellular amastigotes, on the other hand, is very well predicted in the QSAR model and thus seems to be mechanistically related (at least concerning the mechanism addressing the promastigotes) with the majority of the data set. However, amastigotes being the more relevant life stage for drug targeting, determination of IC50 values for a larger set of compounds would be useful in order to enable us to derive a QSAR model also for this life stage which would be desirable for further optimization of this promising class of antileishmanials.
Considering the in vitro antileshmanial potential of 2-hydroxy-3-phenylsulfanylmethyl-[1,4]-naphthoquinones and their structure-activity relationships, these compounds appear promising novel lead candidates that could be exploited in drug design studies for leishmaniasis.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by grants from São Paulo Research Foundation (FAPESP project 2012/18756-1). The authors are grateful to FAPESP scholarship awarded to EGP (2011/23703-1) and the Conselho Nacional de Pesquisa e Desenvolvimento (CNPq) scientific award given to A.G.T. Fellowships granted to V.F.F., I.O.S. by CNPq (Brazil) and D.R.R. CAPES-FAPERJ are gratefully acknowledged. This work was partially supported by CNPq grant 471588/2009-1, FAPERJ-PRONEX grant number 110.574/2010. This publication is part of the activities of the Research Network Natural Products against Neglected Diseases (ResNetNPND) (http://www.uni-muenster.de/ResNetNPND). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Schmidt TJ, Khalid SA, Romanha AJ, Alves TM, Biavatti MW, et al. (2012) The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases - part I. Curr Med Chem 19: 2128–2175. [DOI] [PubMed] [Google Scholar]
- 2. Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, et al. (2012) Leishmaniasis worldwide and global estimates of its incidence. PLoS One 7: e35671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Croft SL, Olliaro P (2011) Leishmaniasis chemotherapy–challenges and opportunities. Clin Microbiol Infect 17: 1478–1483. [DOI] [PubMed] [Google Scholar]
- 4. Chirac P, Torreele E (2006) Global framework on essential health R&D. Lancet 367: 1560–1561. [DOI] [PubMed] [Google Scholar]
- 5. Su JC, Lin KL, Chien CM, Tseng CH, Chen YL, et al. (2010) Furano-1,2-naphthoquinone inhibits EGFR signaling associated with G2/M cell cycle arrest and apoptosis in A549 cells. Cell Biochem Funct 28: 695–705. [DOI] [PubMed] [Google Scholar]
- 6. Riffel A, Medina LF, Stefani V, Santos RC, Bizani D, et al. (2002) In vitro antimicrobial activity of a new series of 1,4-naphthoquinones. Braz J Med Biol Res 35: 811–818. [DOI] [PubMed] [Google Scholar]
- 7. de Rezende LC, Fumagalli F, Bortolin MS, de Oliveira MG, de Paula MH, et al. (2013) In vivo antimalarial activity of novel 2-hydroxy-3-anilino-1,4-naphthoquinones obtained by epoxide ring-opening reaction. Bioorg Med Chem Lett 23: 4583–4586. [DOI] [PubMed] [Google Scholar]
- 8. Schuck DC, Ferreira SB, Cruz LN, da Rocha DR, Moraes M, et al. (2013) Biological evaluation of hydroxynaphthoquinones as anti-malarials. Malar J 12: 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sharma A, Santos IO, Gaur P, Ferreira VF, Garcia CR, et al. (2013) Addition of thiols to o-quinone methide: new 2-hydroxy-3-phenylsulfanylmethyl[1,4]naphthoquinones and their activity against the human malaria parasite Plasmodium falciparum (3D7). Eur J Med Chem 59: 48–53. [DOI] [PubMed] [Google Scholar]
- 10. Ferreira SB, de Carvalho da Silva F, Bezerra FA, Lourenço MC, Kaiser CR, et al. (2010) Synthesis of alpha- and beta-pyran naphthoquinones as a new class of antitubercular agents. Arch Pharm (Weinheim) 343: 81–90. [DOI] [PubMed] [Google Scholar]
- 11. Camara CA, Silva TM, da-Silva TG, Martins RM, Barbosa TP, et al. (2008) Molluscicidal activity of 2-hydroxy-[1,4]naphthoquinone and derivatives. An Acad Bras Cienc 80: 329–334. [DOI] [PubMed] [Google Scholar]
- 12. Carneiro PF, do Nascimento SB, Pinto AV, Pinto Mdo C, Lechuga GC, et al. (2012) New oxirane derivatives of 1,4-naphthoquinones and their evaluation against T. cruzi epimastigote forms. Bioorg Med Chem 20: 4995–5000. [DOI] [PubMed] [Google Scholar]
- 13. Lezama-Dávila CM, Isaac-Márquez AP, Kapadia G, Owens K, Oghumu S, et al. (2012) Leishmanicidal activity of two naphthoquinones against Leishmania donovani. Biol Pharm Bull 35: 1761–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tada H, Shiho O, Kuroshima K, Koyama M, Tsukamoto K (1986) An improved colorimetric assay for interleukin 2. J Immunol Methods 93: 157–165. [DOI] [PubMed] [Google Scholar]
- 15. Tempone AG, Martins de Oliveira C, Berlinck RG (2011) Current approaches to discover marine antileishmanial natural products. Planta Med 77: 572–585. [DOI] [PubMed] [Google Scholar]
- 16. Schmidt TJ, Heilmann J (2002) Quantitative structure-cytotoxicity relationships of sesquiterpene lactones derived from partial charge (Q)-based fractional accessible surface area descriptors (Q_frASAs). Quant Struct Act Relat 21: 276–287. [Google Scholar]
- 17. Nixon GL, Moss DM, Shone AE, Lalloo DG, Fisher N, et al. (2013) Antimalarial pharmacology and therapeutics of atovaquone. J Antimicrob Chemother 68: 977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Croft SL, Hogg J, Gutteridge WE, Hudson AT, Randall AW (1992) The activity of hydroxynaphthoquinones against Leishmania donovani. J Antimicrob Chemother 30: 827–832. [DOI] [PubMed] [Google Scholar]
- 19. Reimão JQ, Colombo FA, Pereira-Chioccola VL, Tempone AG (2012) Effectiveness of liposomal buparvaquone in an experimental hamster model of Leishmania (L.) infantum chagasi. Exp Parasitol 130: 195–199. [DOI] [PubMed] [Google Scholar]
- 20. Voak AA, Seifert K, Helsby NA, Wilkinson SR (2014) Evaluating aziridinyl nitrobenzamide compounds as leishmanicidal prodrugs. Antimicrob Agents Chemother 58: 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kovacic P, Somanathan R (2014) Nitroaromatic compounds: Environmental toxicity, carcinogenicity, mutagenicity, therapy and mechanism. J Appl Toxicol doi:10.1002/jat.2980 [DOI] [PubMed] [Google Scholar]
- 22. Lu SF, Herbert B, Haufe G, Laue KW, Padgett WL, et al. (2000) Syntheses of (R)- and (S)-2- and 6-fluoronorepinephrine and (R)- and (S)-2- and 6-fluoroepinephrine: effect of stereochemistry on fluorine-induced adrenergic selectivities. J Med Chem 43: 1611–1619. [DOI] [PubMed] [Google Scholar]
- 23. Park BK, Kitteringham NR, O'Neill PM (2001) Metabolism of fluorine-containing drugs. Annu Rev Pharmacol Toxicol 41: 443–470. [DOI] [PubMed] [Google Scholar]
- 24. Di Rosso ME, Barreiro Arcos ML, Elingold I, Sterle H, Baptista Ferreira S, et al. (2013) Novel o-naphthoquinones induce apoptosis of EL-4 T lymphoma cells through the increase of reactive oxygen species. Toxicol In Vitro 27: 2094–2104. [DOI] [PubMed] [Google Scholar]
- 25. Salas CO, Faúndez M, Morello A, Maya JD, Tapia RA (2011) Natural and synthetic naphthoquinones active against Trypanosoma cruzi: an initial step towards new drugs for Chagas disease. Curr Med Chem 18: 144–161. [DOI] [PubMed] [Google Scholar]
- 26. Ribeiro GA, Cunha-Júnior EF, Pinheiro RO, da-Silva SA, Canto-Cavalheiro MM, et al. (2013) LQB-118, an orally active pterocarpanquinone, induces selective oxidative stress and apoptosis in Leishmania amazonensis. J Antimicrob Chemother 68: 789–799. [DOI] [PubMed] [Google Scholar]
- 27. Mukherjee P, Majee SB, Ghosh S, Hazra B (2009) Apoptosis-like death in Leishmania donovani promastigotes induced by diospyrin and its ethanolamine derivative. Int J Antimicrob Agents 34: 596–601. [DOI] [PubMed] [Google Scholar]
- 28. Ray S, Hazra B, Mittra B, Das A, Majumder HK (1998) Diospyrin, a bisnaphthoquinone: a novel inhibitor of type I DNA topoisomerase of Leishmania donovani. Mol Pharmacol 54: 994–999. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.