

Abstract
High-throughput gene expression microarrays can be examined by machine-learning algorithms to identify gene signatures that recognize the biological characteristics of specific human diseases, including cancer, with high sensitivity and specificity. A previous study compared 20 gastric cancer (GC) samples against 20 normal tissue (NT) samples and identified 1,519 differentially expressed genes (DEGs). In this study, Classification Information Index (CII), Information Gain Index (IGI), and RELIEF algorithms are used to mine the previously reported gene expression profiling data. In all, 29 of these genes are identified by all three algorithms and are treated as GC candidate biomarkers. Three biomarkers, COL1A2, ATP4B, and HADHSC, are selected and further examined using quantitative real-time polymerase chain reaction (qRT-PCR) and immunohistochemistry (IHC) staining in two independent sets of GC and normal adjacent tissue (NAT) samples. Our study shows that COL1A2 and HADHSC are the two best biomarkers from the microarray data, distinguishing all GC from the NT, whereas ATP4B is diagnostically significant in lab tests because of its wider range of fold-changes in expression. Herein, a data-mining model applicable for small sample sizes is presented and discussed. Our result suggested that this mining model may be useful in small sample-size studies to identify putative biomarkers and potential biological features of GC.
Keywords: gastric cancer, gene signature, microarray, machine-learning algorithm
Introduction
Gastric cancer (GC) is one of the most frequent malignant tumors, and caused 723,000 deaths in 2012.1 Almost two-thirds of these cancers occur in developing countries, and the incidence in China accounts for approximately 42% of all cases.2 Over the last few decades, cancer genomics and proteomics have been extensively used in biomedical research and clinical applications. After the gene chip and microarray technologies were introduced, many researchers used these techniques to find new subclasses in disease states,3,4 identify new biomarkers associated with diseases,5,6 classify subtypes of tumors,7 and predict the outcome of a disease.8 Gene expression profiling from microarray studies has been used to understand the development and mechanism of human diseases. However, most of the traditional statistical methods are not suitable for processing high dimensionality, and high noise, gene expression data.
Unsupervised classification algorithms, an unbiased approach to search for subgroups within the expression data, were one of the first statistical techniques applied to microarray and gene expression profiling data.9 While these techniques may be able to sufficiently cluster individuals with a common phenotype, the sensitivity and specificity is often significantly reduced when it is applied to individuals outside the training set.10 With the development of supervised classification algorithms and machine-learning algorithms, many researchers started to use these methods to identify feature gene sets that allow for the classification of the available samples. Compared to the unsupervised methods, the genes selected by supervised machine-learning algorithms have more stable expressed patterns both in training and testing samples, thereby allowing a more accurate classification. As no single algorithm is widely accepted as the optimal method for mining gene expression data,10 here we used several algorithms in combination to mine the gene expression data from a small study of GC.
Unfortunately, many of these studies suffer from the “curse of dimensionality”11–13 where the number of experimental observations, or features, greatly exceeds the number of samples. If the number of features gets too large, the classifier can simply fit the available data without providing insight into the underlying difference in the samples (eg sick vs. healthy). This is related to Ransohoff’s concept of chance.14,15 It has also been shown that a sufficiently flexible classifier can efficiently fit the available data, even if the number of features used in the classifier is much smaller than the number of samples in each group.16,17 Care must be taken in choosing the classifier because one with a sufficient number of adjustable parameters can obtain good results for the training set without containing features with biological information.
A previous study used the 22 K-oligonucleotide microarray with optimized experimental protocols and analytical tools to identify transcriptional expression profiles of GC from a Chinese cohort.18 In that study, GC and normal tissues (NTs) were obtained from two different sites. As this was not a paired analysis, the authors used a pool containing 20 normal samples as a reference and the fold change (FC) was determined for the GC and normal samples relative to this pool. Two unsupervised approaches, significant analysis of microarray (SAM) and Bayesian analysis of gene expression levels (BAGEL), were used to identify the differentially expressed genes (DEGs). A total of 1,519 DEGs were identified by comparing 20 intestinal-type GC samples against 20 NTs. This set of 1,519 DEGs provides significant research materials for biomarker identification that could be associated with the biological characteristics of GC.
In this study, three different algorithms are used to select feature genes based on differential expression profiling data of GC from the earlier study.18 A total of 29 genes are identified by all three algorithms and analyzed individually. Three genes (COL1A2, ATP4B, and HADHSC) are selected as candidate biomarkers from this GC study. Quantitative real-time polymerase chain reaction (qRT-PCR) is used to verify the expressed levels of these three genes in 30 new validation cases where each individual supplied a GC sample and normal adjacent tissue (NAT). Another 29 validation cases containing GC and NAT are also included for immunohistochemistry (IHC) staining.
Materials and Methods
GC tumor and the NAT samples
In addition to the 20 GC and normal samples used in the initial microarray study,18 a total of 59 GC and their corresponding NAT samples were obtained from the Beijing Tumor Hospital affiliated under Peking University School of Oncology. In the study, 30 tissues (GC and NAT) are used for the qRT-PCR validation assay, and the other 29 tissues are used in IHC.
Machine-learning algorithms
A basic concept of selecting feature genes is to examine a gene’s ability to divide samples with different phenotypes. Many different filtering algorithms are available, and three different algorithms are used in this study. Each filtering algorithm individually examines each of the 1,519 genes and determines their ability to distinguish the 20 GC samples from the 20 NTs. Unfortunately, this earlier study18 did not use technical replicates, and some of the expression levels were not measurable, and any missing expression levels were assigned the average of the expression across the remaining samples. In this investigation, any missing expression level is excluded in the analysis. Therefore, the significance of certain genes may be determined using less than 20 GC and 20 NT samples. The expression level of each gene in a given sample is given by the logarithm of the FC, log(FC), relative to the pooled set of 20 NT samples.
Classification Information Index (CII) algorithm
The calculation of divisibility can be computed by examining the similarity of sample properties in the same category (within-class distance), as well as the difference of the properties for samples in different categories (between-class distance). “Signal to noise ratio,” a statistical t-test proposed by Golub and co-workers,19 embodied in the CII algorithm,20 can better reflect the above-mentioned ideas and can be served as measures to estimate how much classification information each gene contains. The CII algorithm consists of two parts. The first part contains “signal to noise ratio” indicators, and the second reflects contributions caused by different expression levels of the distribution.
In this expression, μg+ and are the mean and variance of the log(FC) values for the GC samples, and μg− and are the corresponding values for the NT samples. The second term takes on a minimum value of zero whenever and increases as the differences in variance increase.
Information Gain Index (IGI) algorithm
IGI21 is used in many decision tree algorithms and measures the phenotypic homogeneity of the daughter nodes. As such, this metric does not depend on the value of the FC for a given gene, only the rank order of the log(FC) values. Given a total of S states, Ns being the total N samples in State s, the probability of being in this state is simply
The Information Entropy of the parent node containing all samples is then
For S states, (S − 1) cut points in the intensity range are selected to produce S daughter nodes. If daughter node d contains Nd samples and Ns,d samples from State s, the Information Entropy of this node is
The overall IGI for feature l is the Information Entropy of the parent node minus the Information Entropy of all daughter nodes.
Here, D is the number of daughter nodes and Pd is the probability of being in that node (Nd/N). In this application, there are only two states (GC and NT), so only a single log(FC) cut-value is used to construct two daughter nodes. All individuals with a log(FC) above the cut-value are placed in one daughter node and those below this cut-value are placed in the other daughter node. The cut-value is selected to maximize the Information Gain of each gene. The genes are then ranked from highest to lowest Information Gain.
RELIEF algorithm
The RELIEF algorithm evaluates the importance of attribute classification based on within-class and between-class distances.22 This algorithm starts from a random sample rather than from the statistical characteristics of the whole class to estimate sample class separability. For any learning sample S in the training set, the algorithm searches out K (K > 0) same-class samples closest to S (nearest hit) and K (nearest miss) heterogeneous samples. For the attribute Ai, if the difference between S and a heterogeneous sample is larger than the difference between S and same-class sample, the separability of sample S on attribute Ai is greater, and the classification weight of Ai is also greater.
The weight associated with the jth gene is the average difference in the squared distance between each sample point and the nearest sample in the same and a different phylogeny (K = 1).
In this equation, xnj is the FC of the jth gene for the nth sample, k is the number of samples with an expression level for this gene, and nearHitnj and nearMissnj are the log(FC) values for the nearest neighbor in the same and other philogenetic groups, respectively. The larger the weight, the better the gene is able to distinguish between the philogenetic groups. To be able to compare different genes, a squared Mahalanobis distance is used—meaning that the log(FC) values are divided by the standard deviation.
Real-time quantitative PCR
After approval by the Ethics Committee of the Beijing Cancer Hospital, 60 GC specimens comprising 30 GCs and matched NATs are used in the qRT-PCR validation assay. Total RNA is extracted from the tissue samples according to a standard Trizol protocol (Invitrogen, Carlsbad, CA, USA). In all, 5 μg of total RNA are reverse transcribed (RT) to cDNA with 200 U of Moloney Murine Leukemia Virus (MMLV) reverse transcriptase (Promega, Madison, WI, USA). The RT reaction uses the following conditions: 37 °C for 60 minutes and 72 °C for 10 minutes. The primer pairs for COL1A2 are 5′-CCTGGTGCCCCTGGTGAAAA-3′ (forward) and 5′-CCACACTTCCATCACTGCCACG-3′ (reverse); for ATP4B, they are 5′-TTCGCCCTGTGCCTCTATGT-3′ (forward) and 5′-TGTGAGGTCTGCCCAGGTT-3′ (reverse); for HADHSC, 5′-GCTAATGCCACCACCAGACAA-3′ (forward) and 5′-CGTCACCTCGTTCATACAGCC-3′ (reverse); and for β-actin, 5′-TTAGTTGCGTTACACCCTTTC-3′ (forward) and 5′-ACCTTCACCGTTCCAGTTT-3′ (reverse). qRT-PCRs are performed in a 20 μL mixture containing 2 μL of cDNA, 0.6 μL 20× EvaGreen (CapitalBio Corp., Beijing, China), 0.5 μL of each 10 μM forward and reverse primers, 0.5 μL of 2.5 mM dNTP, 1.5 U Cap Taq polymerase (Capital-Bio Corp., Beijing, China), 10 μL 2× PCR buffer for EvaGreen, and 6.1 μL of H2O. Using the RT-Cycler′ 466 system (Capital-Bio Corp., Beijing, China), PCRs are carried out with the following programmed parameters: heating at 95 °C for 5 minutes followed by 40 cycles of a three-stage temperature profile of 95 °C for 30 seconds, 57 °C for 30 seconds, and 72 °C for 30 seconds. All reactions are performed in triplicates, and the final Ct value is determined by the average Ct value of the three reactions. The melting curves for each PCR reaction are carefully analyzed to avoid nonspecific amplifications in PCR products. The expression of each gene is transformed using the 2−ΔΔCt formula and normalized with β-actin expression.23 Information about the primers, and the Ct values for each gene and β-actin in each sample are available as supplementary information (Supplemental Tables 3 and 4, respectively).
Tissue microarray (TMA) and IHC staining
TMA blocks are constructed in our laboratory. For each case, we sample five tissue cores at 1.0 mm in diameter, including two tumor and one matched-adjacent normal mucosa tissues to construct the TMA. A total of 29 human gastric specimens are obtained from the tumor bank of Beijing Cancer Hospital. The patients were fully informed and given consent for the collection of clinical samples. IHC staining is performed using EnVision + Kit (Dako, Denmark). Commercial antibodies are used in our study: anti-COL1A2 (ab72637), anti-ATP4B (ab2866), and anti-HADSHC (ab54477). Proper validation of the ATP4B, COL1A2, and HADSHC antibodies used the same procedure as was described previously.24 The ICH was semi-quantitatively scaled in a range from “−” to “+++” by evaluating the representative tumor with intensity and percentage of cells showing significantly higher immune staining than normal matched-adjacent tissues. Samples scoring “++” to “+++” were considered as “high expression,” and samples scoring “−” and “+” were termed as negative and no expression, respectively. Three pathological experts participated in the results’ evaluation of the IHC experiment. The section is incubated with the respective antibody at 4 °C overnight. More than 5% stained cells in the tissue is defined as positive reaction in this experiment. Unfortunately, some of the GC or NAT tissues were missing from the slice used in IHC staining; thus, the results for each gene did not necessarily contain 29 GC and 29 NAT samples.
Signature genes ontology analyses
To investigate the potential molecular function and associated pathway including the signature genes, we used an integrated gene ontology and pathway analysis database MAS3.0 (http://www.capitalbio.com) for this purpose.
Results
Identification of the DEGs based on gene expression data of GC
A 22 K-oligonucleotide microarray was previously used to measure the relative expression levels of human genes in 20 GC individuals and 20 NTs from different donors. That examination compared the expression levels to a common reference that was a mixture of 20 normal gastric mucosa tissues from non-tumor patients.18 BAGEL was used to analyze the DEGs with non-overlapping 95% confidence intervals from the Bayesian analysis dataset with P < 0.001. A total of 1,519 DEGs were identified consisting of 593 up-regulated genes and 926 down-regulated genes (Supplementary Table 1).
In this analysis, 1,182 of the 1,519 genes were characterized with respect to Gene Ontology (GO) terms, while 337 genes were undetectable. Among these 1,182 genes, 226 genes were cataloged into 60 pathways according to the gene ontology and pathway analysis while the remaining 956 genes were not related to cancer development (Supplementary Table 2). Our study shows that 114 of 226 genes are indeed associated with many human diseases including cancers.
Feature gene selection using three machine-learning algorithms
The 1,519 genes previously identified are further examined using three different machine-learning algorithms. In this analysis case-based exclusion is employed; so a sample is excluded from the analysis of a particular gene if no expression level was available from the previous study.18 The CII algorithm assigns each gene an information index. The genes are distributed into five intervals, and the 68 candidate genes with high CII value (i > 0.9) are selected (Table 1). An IGI is also used to select feature genes. The IGI of each gene is distributed into four intervals. The 99 candidate genes with the largest IGI values (g > 0.35) are selected (Table 1). Finally, the RELIEF algorithm employs the Mahalanobis square distance as a classifying index. All 1,519 genes are distributed into six intervals, and the 325 candidate genes with high classification weight (w > 0.3) are selected (Table 1). These thresholds are selected to ensure that a gene that scores the highest with a particular algorithm is also included in the list for the other two algorithms.
Table 1.
Feature gene selection using CII, IGI and Relief algorithm.
| ALGORITHMS | INTERVALS | GENE NUMBERS | PERCENTS |
|---|---|---|---|
| CII | i < 0.9 | 1451 | 95.52% |
| 0.9 < i < 1 | 24 | 1.58% | |
| 1 < i < 1.5 | 39 | 2.57% | |
| 1.5 < i < 2 | 4 | 0.26% | |
| i > 2 | 1 | 0.07% | |
| IGI | g < 0.35 | 1420 | 93.48% |
| 0.35 < g < 0.45 | 64 | 4.21% | |
| 0.45 < g < 0.6 | 33 | 2.17% | |
| 0.6 < g | 2 | 0.13% | |
| Relief | w < 0.3 | 1194 | 78.54% |
| 0.3 < w < 0.5 | 214 | 14.09% | |
| 0.5 < w < 0.6 | 47 | 3.09% | |
| 0.6 < w < 0.8 | 52 | 3.42% | |
| 0.8 < w < 1 | 11 | 0.72% | |
| w > 1 | 2 | 0.13% |
Abbreviations: i, Classification Information Index of each gene; g, Information Gain Index of each gene; w, Relief classification weight of each gene.
Combining the results from the intersection of the three algorithms described above, 29 putative biomarker genes are identified in all three machine-learning algorithms (Fig. 1). Of these, 23 genes are down-regulated and 6 genes are up-regulated in GC samples relative to their NAT (Table 2). The threshold column in Table 2 is threshold log(FC) that distinguishes the GC from NAT samples. This threshold is determined by finding a value that minimizes the GINI index of the two groups,25 a procedure that is used in many decision tree algorithms.
Figure 1.
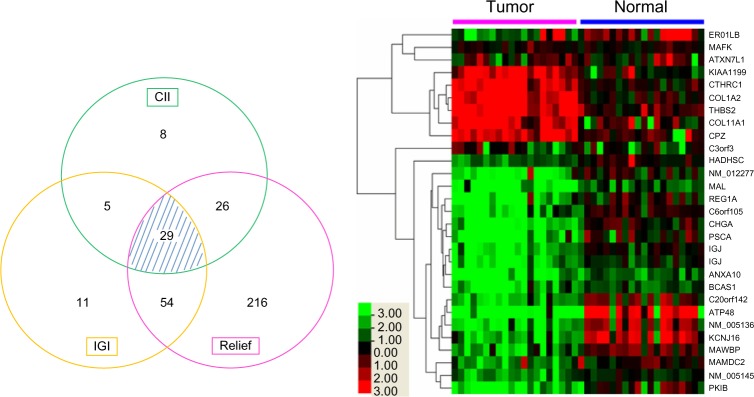
Venn diagram and cluster analysis of the selected genes by all of the filtering methods. The thresholds of CII, IGI, and RELIEF algorithms were set to 0.9, 0.35, and 0.3, respectively. In the cluster figure, columns represent samples and rows represent genes (black, green, and red correspond to unchanged, down-regulated, and up-regulated, respectively).
Table 2.
29 candidate feature genes selected by CII, IGI and relief algorithm.
| ACCESSION# | GENE | CHANGE | THRESHOLDa | SENSITIVITY | SPECIFICITY | RANGE[LOG(FC)] |
|---|---|---|---|---|---|---|
| NM_005327 | HADHSC | down | 0.763 | 100% | 100% | 7.792 |
| NM_000089 | COL1A2 | up | 2.394 | 100% | 100% | 4.359 |
| NM_001275 | CHGA | down | 0.317 | 95% | 100% | 4.491 |
| NM_019891 | ERO1LB | down | 0.626 | 100% | 94.7% | 5.648 |
| BC014245 | CTHRC1 | up | 2.112 | 95% | 95% | 4.343 |
| AK056767 | MAFK | down | 0.395 | 90% | 100% | 3.979 |
| NM_012277 | NM_012277 | down | 0.307 | 90% | 100% | 10.753 |
| AB033025 | KIAA1199 | up | 2.073 | 95% | 94.7% | 7.681 |
| NM_003247 | THBS2 | up | 2.570 | 100% | 85% | 3.819 |
| NM_002371 | MAL | down | 0.187 | 85% | 100% | 6.361 |
| NM_005672 | PSCA | down | 0.531 | 95% | 90% | 5.366 |
| NM_002909 | REG1A | down | 0.314 | 85% | 100% | 5.257 |
| NM_032744 | C6orf105 | down | 1.064 | 100% | 85% | 4.513 |
| NM_144646 | IGJ | down | 0.486 | 95% | 90% | 3.916 |
| AL117382 | C20orf142 | down | 0.369 | 85% | 100% | 4.559 |
| NM_020707 | C3orf3 | down | 0.640 | 85% | 100% | 4.774 |
| NM_003652 | CPZ | up | 3.155 | 85% | 100% | 11.824 |
| NM_000705 | ATP4B | down | 0.169 | 95% | 89.5% | 10.026 |
| NM_005136 | NM_005136 | down | 0.287 | 80% | 100% | 7.089 |
| NM_005145 | NM_005145 | down | 0.514 | 80% | 100% | 6.53 |
| BC015417 | MAMDC2 | down | 0.503 | 80% | 100% | 9.76 |
| BC003517 | ATXN7L1 | down | 0.755 | 80% | 100% | 10.797 |
| NM_007193 | ANXA10 | down | 0.149 | 75% | 100% | 6.131 |
| NM_003657 | BCAS1 | down | 0.301 | 75% | 100% | 4.044 |
| NM_018658 | KCNJ16 | down | 1.765 | 100% | 75% | 9.853 |
| NM_022129 | MAWBP | down | 0.558 | 75% | 100% | 4.693 |
| NM_032471 | PKIB | down | 0.617 | 100% | 75% | 9.477 |
| AA513382 | IGJ | down | 0.379 | 80% | 94.7% | 3.755 |
| NM_001854 | COL11A1 | up | 4.397 | 70% | 100% | 7.699 |
Note:
The threshold producing the sensitivity and specificity is selected to minimize the GINI index of the daughter nodes.
Gene ontology analyses
To investigate the potential molecular function of the 29 signature genes, we perform a GO analysis using MAS3.0. The results show that 18 of 29 genes are separated into 21 main GO terms, including some tumor-related functions such as cell adhesion, anti-apoptosis, TGF beta receptor signaling pathway, Wnt receptor signaling pathway, cell differentiation, cell proliferation, and others (Table 3). In addition, the GO network shows that COL1A2, ATP4B and HADHSC are separated into three sub-networks that are not related in molecular function.
Table 3.
Gene ontology analyses of the candidate signatures.
| GO TERMS | INPUT SYMBOL | P VALUE |
|---|---|---|
| Collagen fibril organization | COL1A2;COL11 A1 | 1.90E-06 |
| Cell adhesion | THBS2;COL1A2;COL11 A1 | 0.001965 |
| Anti-apoptosis | MAL | 0.01571 |
| Ion transport | ATP4B;KCNJ16;CPZ;ERO1 LB | 0.001751 |
| Protein binding | MAFK;THBS2;MAL;C3orf3;BCAS1 | 1.63E-06 |
| TGF beta receptor signaling pathway | COL1A2 | 0.009773 |
| Rho protein signal transduction | COL1A2 | 0.010276 |
| Wnt receptor signaling pathway | CPZ | 0.011365 |
| Cell differentiation | MAL | 0.106455 |
| Cell proliferation | REG1 A | 0.025169 |
| Metabolism | HADHSC | 0.016543 |
| Regulation of transcription | MAFK;MAL | 0.016968 |
| Protein thiol-disulfide exchange | ERO1 LB | 5.08E-04 |
| Sensory perception of sound | COL11 A1 | 5.92E-04 |
| Immune response | MAL;IGJ | 0.002569 |
| Negative regulation of protein kinase activity | PKIB | 0.005825 |
| ATP biosynthesis | ATP4B | 0.007507 |
| Blood vessel development | COL1A2 | 0.018041 |
| Nervous system development | MAFK | 0.067536 |
| Proteolysis | CPZ | 0.084789 |
| Calcium ion binding | CHGA;THBS2;ANXA10 | 2.03E-05 |
Clustering of signature genes
The 29 signature genes are then grouped using single linkage clustering based on the Pearson correlation of their log(FC) values across all available samples. The clustering is stopped at |r| = 0.70, meaning that each gene in a given cluster has log(FC) values that correlate (positively or negatively) with at least one other gene in the cluster to an |r| value of at least 0.70. This is done to ensure that candidate biomarkers are selected from different clusters. If two or more genes are selected from the same cluster, their log(FC) values correlate and only one of the genes contains unique information.
The clustering results are shown in Supplemental Figure S1. For each gene, the left column of “+” marks represents the log(FC) values for the GC samples; the log(FC) values for the NT samples are shown in the right column. The number below the columns represents the minimum log(FC) value for that gene, whereas the number at the top is the maximum log(FC) value. Figure S1A shows that one cluster contains 16 of the 29 signature genes. The next two clusters (Fig. S1B,C) each contained three signature genes, and seven signature genes reside in singleton clusters (Fig. S1D). This means that each of the genes listed in Figure S1D has log(FC) values that do not strongly correlate with any of the other 28 signature genes (|r| < 0.70).
Extracting putative biomarkers
An examination of Table 2 shows that two genes, HADHSC and COL1A2, have both sensitivities and specificities of 100%. HADHSC is a member of the second cluster (Fig. S1B) and distinguishes all 20 GC samples (log(FC) values vary from −1.620 to −0.305) from the 20 NT samples (log(FC) varies from −0.235 to 6.172). The Bonferroni corrected probability of randomly observing a gene where all 20 GC samples have a lower expression level than the 20 NT is 1.60 × 10−7. COL1A2 is a member of a singleton cluster (Fig. S1D) and also distinguishes the GC samples from the NT. log(FC) for the GC samples varies from 0.903 to 3.721, while the values for the NT vary from −0.638 to 0.844. The Bonferroni corrected probability of randomly observing a gene where all 20 GC samples have a higher expression level than the 19 NT samples (missing one expression data on one of the NT) is 3.20 × 10−7. These two genes obtained the top scores with the IGI and RELIEF algorithms, respectively (Table 4). ATP4B, also a member of a singleton cluster, obtains a significantly better score than all other genes using the CII algorithm, so it is also selected as a putative biomarker. Although ATP4B does not have as high of a sensitivity and specificity as HADHSC and COL1A2, its total range of log(FC) is definitely larger. It is important to note that the three selected genes have expression levels with a small Pearson correlation (|r| < 0.7) and therefore may represent independent aspects of GC.
Table 4.
Genes with the highest scores for each of the filtering methods.
| CII | IGI | RELIEF | |||
|---|---|---|---|---|---|
| Gene | Score | Gene | Score | Gene | Score |
| ATP4B | 2.41855 | HADHSC | 0.69315 | COL1A2 | 1.08879 |
| NM_012277 | 1.77127 | COL1A2 | 0.69282 | HADHSC | 1.01081 |
| ATP4 A | 1.69775 | SULF2 | 0.59264 | NM_005145 | 0.93394 |
| KCNJ16 | 1.58771 | CHGA | 0.59264 | CHGA | 0.86624 |
| COL4 A6 | 1.53349 | RDH12 | 0.59264 | ERO1 LB | 0.84882 |
| FAM3B | 1.39394 | CPZ | 0.58432 | ATXN7 L1 | 0.84856 |
| ANXA10 | 1.39052 | SPARC | 0.52560 | KIAA1199 | 0.84567 |
| SULT1C1 | 1.37157 | COL18 A1 | 0.52560 | NM_012277 | 0.84271 |
| PSCA | 1.35426 | CDC25B | 0.52560 | APBB1IP | 0.82600 |
| NM_005136 | 1.35310 | MAFK | 0.52560 | NQO3 A2 | 0.81854 |
Validation of the feature genes using real-time quantitative PCR and IHC staining
To validate the feature genes (COL1A2, ATP4B, and HADHSC) from our prediction model, qRT-PCR is used to measure the level of expression using an additional 30 validation cases containing 30 GC samples and their paired NAT. The results show that COL1A2 is up-regulated in GC samples comparatively to their NAT samples while ATP4B and HADHSC are low expressed in the GC samples and highly expressed in the NAT samples. When the 2−ΔΔCt values for the GC and NAT samples are treated as independent values, a GINI index25 can be used to separate the samples into two groups. Table 5 shows that for ATP4B, the optimum threshold value is 0.044. In all, 25 of the 30 GC samples have 2−ΔΔCt values below this threshold, whereas 27 of the 30 NAT samples have values above this threshold. This yields a sensitivity of 89.3% and a specificity of 84.4%; the positive and negative predictive values are 83.3 and 90.0%, respectively. For COL1A2, 23 GC samples have values above 1.027 and 22 NAT samples have 2−ΔΔCt values below this threshold. This yields a sensitivity and specificity of 74.2 and 75.9%, respectively, and a positive and negative predictive value of 76.6 and 73.3%, respectively. The threshold value for HADHSC is found to be 3.052, but this gene is not a good classifier for the NAT samples. In all, 17 samples have value below the threshold and 13 above. In contrast, 29 of the 30 GC samples have 2−ΔΔCt values below this threshold, yielding a positive predictive value of 96.7%, but a sensitivity of only 63.0%.
Table 5.
RT-PCR classification results when the 30 NAT and GC samples are treated as independent data.
| A | |||
|---|---|---|---|
| NAT | GC | ||
| <0.044 | 3 | 25 | Sensitivity = 89.3% |
| >0.044 | 27 | 5 | Specivifity = 84.4% |
| NPV = 90.0% | PPV = 83.3% | ||
| B | |||
| NAT | GC | ||
| <1.027 | 22 | 7 | Specificity = 75.9% |
| >1.027 | 8 | 23 | Sensitivity = 74.2% |
| NPV = 73.3% | PPV = 76.6% | ||
| C | |||
| NAT | GC | ||
| <3.052 | 17 | 29 | Sensitivity = 63.0% |
| >3.052 | 13 | 1 | Specivifity = 92.9% |
| NPV = 43.3% | PPV = 96.7% | ||
Note: The threshold values are determined by maximizing the GINI index. A, AT4B using a threshold of 0.044 for 2-DDCt. B, COL1A2 using a threshold of 1.027. C, HADHSC using a threshold of 3.052.
As each GC and NAT sample is obtained from the same individual, the ratio of their 2−ΔΔCt values can be used. For ATP2B, if the ratio of NAT-to-GC is set to 5.0, 23 of the 30 GC individuals (76.7%) are identified. If the ratio of GC-to-NAT is set to 2.0 for COL1A2, only 14 of the 30 GC individuals (46.7%) are identified, while for HADHSC a NAT-to-GC ratio of 2.0 identifies 18 of the 30 GC individuals (60.0%). If the rules for ATP4B and COL1A2 are both used so that at least one of the rules must be satisfied, then 28 of the 30 GC individuals (93.3%) are identified. Including HADHSC into the set of rules does not improve the results, meaning that two of the individuals failed all three tests.
An additional 29 GC and normal paired samples are used for IHC staining. The results (Fig. 2, Table 6) show thatCOL1A2 has a high expression in 17 of the 22 GC samples that show good results (77.3%) while only 9 of the 22 NAT samples show positive staining (40.9%). ATP4B is highly expressed in 20 of the 24 normal samples (83.4%), but positive staining is only seen in 11 of the 25 GC samples (44.0%). HADHSC shows 24 of the 26 normal samples with high expression (92.3%), whereas only 4 of the 25 GC samples show positive staining (16.0%). The IHC results are in agreement with the microarray and qRT-PCR results; COL1A2 has an increased expression, and ATP4B and HADHSC are low-expressed in GC samples.
Figure 2.
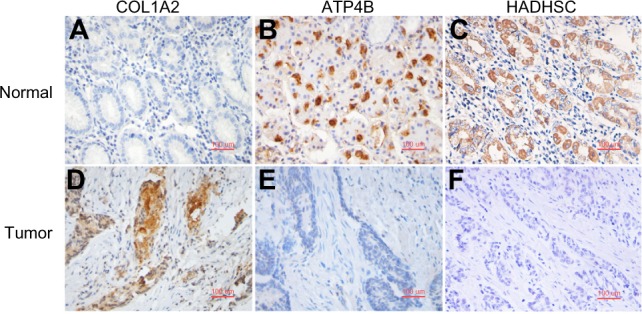
Validation of the feature genes using IHC staining. (A) and (B): positive staining of COL1A2 appeared in cancer but not in NT. COL1A2 was highly expressed in 17 GC samples with the positive rate of 77.3% (17/22). (C) and (D): negative staining of ATP4B appeared more often in cancer but positive in NT. ATP4B was highly expressed in 20 normal samples with the positive rate of 83.4% (20/24). (E) and (F). positive staining of HADHSC appeared in normal but not in cancer tissue. HADHSC showed 24 normal samples with high expression of 92.3% positivity (24/26).
Table 6.
IHC staining results for the three selected putative biomarkers.
| ANTIBODY | TYPES OF SAMPLES | POSITIVE | NEGATIVE | P VALUE |
|---|---|---|---|---|
| COL1A2 | T = 22 | 17(77.3%) | 5(22.7%) | 0.0305 |
| N = 22 | 9(40.9%) | 13(59.1%) | ||
| ATP4B | T = 25 | 11(44%) | 14(56%) | 0.0072 |
| N = 24 | 20(83.4%) | 4(16.6%) | ||
| HADHSC | T = 25 | 4(16.0%) | 21(84.0%) | 9.15 × 10−6 |
| N = 26 | 24(92.3%) | 2(7.7%) |
Discussion
Here we report an optimized data-mining and prediction model for biomarker identification based on gene expression profiling data of GC. We use three different machine-learning algorithms to select feature genes based on differentially expressed gene (DEG) profiling of GC from a previous investigation that contained 1,519 DEGs. The main point to note is that the three filtering algorithms use very different criteria in selecting putative biomarkers. The CII algorithm depends on the values of all the log(FC) values for the GC and NT samples, because the scoring metric depends on both the mean and standard deviation for each group. As such, this algorithm may be highly affected by outliers and an inspection of the FC values is warranted. In contrast, the IGI algorithm is independent of the magnitude of the log(FC) values and only depends on their rank order. Therefore, the IGI algorithm is independent of the FC value of an outlier. The RELIEF algorithm depends on the distance to the nearest neighbor from the GC and NAT phenotypes. As such, this algorithm will provide a good score to a feature that produces sub-clusters of each phenotype, and again an inspection of the log(FC) values is needed. This algorithm should only be affected to a minor extent by an outlier.
Our results show that though different algorithms selected different feature genes, there is a common set of 29 genes obtained by all algorithms. Examining these 29 candidate biomarkers show that two, COL1A2 and HADHSC, completely distinguished GC from NAT. They also have the highest scores for two of the three filtering algorithms (IGI and RELIEF algorithms). ATP4B has the highest score for the CII algorithm and has a larger range in its log(FC) values than either COL1A2 or HADHSC. All 29 genes listed in Table 2 may represent changes in cellular activity in GC. The fact that the three selected genes have uncorrelated expression levels suggests that they may be involved in different functions and pathways. If two or more of these genes have correlated expression levels, then only one of this set would represent unique information.
COL1A2 is located on human chromosome 7q22.1, encoding the pro-alpha2 chain of type I collagen, which belongs to the fibrillar collagen family. Serial analysis of gene expression (SAGE) results shows that it may be a new biomarker of GC.26 Inhibition of type I collagen synthesis has been shown to suppress angiogenesis and tumor growth.27 Moreover, we have presented a whole genome profile of copy number variant (CNV) and single-nucleotide polymorphisms (SNPs) in 10 GC samples. Our data show that 2 out of 10 samples with two-fold increase in copy number and two non-synonymous positions are detected in COL1A2 (manuscript in preparation). We propose that type I collagen is an important protein that participated in sustaining the stabilization of the physiological structure in normal cells, tissues, and organs.
HADHSC is located on human chromosome 4q22-q26 and is a member of the 3-hydroxyacyl-CoA dehydrogenase gene family. The encoded protein functions in the mitochondrial matrix to catalyze the oxidation of straight-chain 3-hydroxyacyl-CoAs as part of the beta-oxidation pathway. Its enzymatic activity is highest with medium-chain-length fatty acids. Gene ontology analyses’ results show that this gene is closely related to cellular metabolic process, including lipid metabolic process; response to hormone stimulus; fatty acid metabolic; and beta-oxidation process, and negatively regulates insulin secretion. Mutations in this gene cause one form of familial hyperinsulinemic hypoglycemia and hyperinsulinism.28–30 However, there is no research report so far about HADHSC in human cancer.
ATP4B is located on human chromosome 13q34, encoding the member of the P-type cation-transporting ATPases. This enzyme is a proton pump that catalyzes the hydrolysis of ATP coupled with the exchange of H+ and K+ ions across the plasma membrane, and is responsible for gastric acid secretion.31 In a mouse model, it was reported that ATP4B was required for normal function, development, and membrane structure of mouse parietal cells.32 No previous research has reported that this gene was associated with the development of GC, although our microarray results showed that it correctly identified 18 of the 19 GC samples.18 Meanwhile, the real-time PCR results show that ATP4B is down-regulated in 26 of 30 GC samples.
Here we use an optimized method of combined multiple machine-learning algorithms for data mining in small set of gene expression data. We have documented an integral and systematically data-mining model for biomarkers identification based on a small sample-size set of gene expression profiling data, and we identified COL1A2, HADHSC, and ATP4B as potential biomarkers of GC. These three genes are confirmed in 59 validation samples by real-time PCR and IHC staining, and are shown to be useful in recognizing the biological characteristics of GC.
The probability of a false positive, a gene that appears to be a biomarker but is not, increases as the sample size decreases. In initial studies with small sample sizes, the researcher can only hope to find putative biomarkers for further investigations. In this study, a gene had to be selected by all three filtering methods to be considered further, and only 29 out of the 1,519 genes with significant FC pass this criterion. In further investigations, only linearly independent genes should be examined because multiple genes with strongly correlated FC values (|r| > 0.7) do not increase the information content over a single gene from this set.
The results presented in Tables 2 and 4 as well as Supplemental Figure S1 suggest that other significant genes can be further tested as putative biomarkers. CHGA may represent a good candidate from the first cluster of genes (Fig. S1A). It is ranked fourth by both the IGI and RELIEF algorithms, and only misclassifies a single GC sample in the initial training set. Though the three genes in the third cluster are selected by all three filtering algorithms, Figure S1C suggests that there is a significant overlap in the log(FC) values between the GC and NT phenotypic groups. ATP4B is selected for further investigation because it has the best score with the CII algorithm and a large range in the log(FC) values, but three other genes have larger log(FC) ranges. CPZ and ATXN7L1 have the two largest ranges and represent singleton clusters (Fig. S1D). CPZ only misclassifies a single GC testing sample, but this figure shows that the log(FC) values have a high density in the intermediate region for both the GC and NT groups, suggesting that it may not be a good putative biomarker. This is reflected in the fact that it is in the top 10 genes of the IGI algorithm, which depends on ranking, and not the CII and RELIEF algorithms, which depend on log(FC) distributions (Table 4). ATXN7L1 should also be excluded as a putative biomarker because the log(FC) values for the GC group completely encompass the values for the NT group. A close inspection of the log(FC) values in Figure S1D shows that the NT samples have many values in regions with few log(FC) values from the GC group, and it is this type of sub-clustering that can produce a good score using the RELIEF algorithm. Finally, NM_012277 has a larger log(FC) range than ATP4B, but it is part of the same cluster as HADHSC and therefore yields no new information.
Of these 29 genes, COL1A2, HADHSC, and ATP4B are selected because their log(FC) values are uncorrelated (|r| < 0.7) and they score the highest by one of the filtering methods. Subsequent IHC and real-time PCR investigations of independent samples show that a gene with a large range in the log(FC) values performed better than the genes with better discriminating abilities but a smaller range, as measured by microarray, because the discrimination may be easier to distinguish with other methods and may be less dependent on experimental variability.
This study demonstrates that by combining the results of several independent screening methods, it is possible to obtain putative biomarkers from a small initial sample set. The IGI algorithm only depends on the rank order of each sample’s log(FC) value for a given gene, RELIEF depends on the local neighborhood of log(FC) values for each sample, and CII depends on the distribution of all log(FC) values for a given gene for the GC and NT sets. There is no requirement that these specific algorithms be used, or be limited to three, but only that all filtering algorithms must be independent. A putative biomarker should be a good discriminator by any reasonable filtering algorithm, and by requiring a gene to perform well using all three methods, the selection of a gene by chance is greatly diminished. This procedure is not limited to small samples sizes and will work just as efficiently with significantly larger datasets.
The initial set of 1,519 genes with significant FC values was reduced to 29 genes using the filtering algorithms, with 22 of them clustering into three groups based on correlated FC values across the samples. If two or more genes are selected from the same cluster, their log(FC) values will correlate and only one of the genes contains unique information. This reduces the initial set of 1,519 genes to a set of 10 independent genes.
The final point to stress is that the procedure outlined here represents a method to identify putative biomarkers. Any gene set from this type of analysis should be validated on an independent set of samples. The strength of the results presented here is that the putative biomarkers performed well on two independent sample sets using different experimental procedures to measure gene expression levels. Using a small set of microarray data to generate putative biomarkers that are validated using clinical procedures is very promising.
Conclusions
We use three different algorithms (CII, IGI, and RELIEF) to select feature genes based on differential expression profiling data of GC from the earlier study and identify 29 genes as GC candidate biomarkers. Furthermore, three putative biomarkers (COL1A2, ATP4B, and HADHSC) are selected and further examined using qRT-PCR and IHC staining in two independent sets of GC and NAT samples. The positive results show that our approach used in this study might be helpful in small sample-size studies to identify biomarkers, and the genes selected by our approach may act as candidate biomarkers for GC. Future studies should examine whether these putative biomarkers are also differentially expressed in the bile or urine, thereby affording a less-invasive means of identifying GC. In addition, further studies should examine a gene from cluster A such as CHGA (Supplemental Figure 1). It would also be informative if the change between GC and NAT expression levels correlates with the stage of the cancer because the early onset of a marker would be extremely useful in early diagnosis.
Supplementary Material
Supplementary Table 1. Details about the 1519 differentially expressed genes, consisting of 593 up-regulated genes and 926 down-regulated genes.
Supplementary Table 2. Details of the 226 genes that were catalogued into 60 pathways according to the gene ontology and pathway analysis.
Supplementary Table 3. Information about the primers for ATP4B, COL1A2, and HADHSC.
Supplementary Table 4. Ct values for ATP4B, COL1A2, and HADHSC for each sample and 2−ΔΔCt values relative to β-actin.
Supplementary Figure 1. log(FC) plots of the 29 signature genes after single linkage clustering using (1-|r|) as the distance metric, where r is the Pearson correlation coefficient.
Acknowledgments
We thank tissue bank of Beijing Cancer Hospital, and institutes for gastric specimens. The authors would also like to thank the reviewers and editor for their helpful comments and suggestions.
Footnotes
Author Contributions
Conceived and designed the experiments: ZY, BTL, JL, YL. Analyzed the data: ZY, BTL, SXT, JW, TG. Wrote the first draft of the manuscript: ZY, YL. Contributed to the writing of the manuscript: BTL, SXT, JL, JW, TG. Agree with manuscript results and conclusions: RX, YP, YL. Jointly developed the structure and arguments for the paper: BTL, SXT, YP, YL. Made critical revisions and approved final version: ZY, BTL, SXT, YP, YL. All authors reviewed and approved of the final manuscript.
Disclaimer
The content of this publication neither does necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
DISCLOSURES AND ETHICS
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.
ACADEMIC EDITOR: Karen Pulford, Associate Editor
FUNDING: This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E. This work was supported in part by the Ministry of Science and Technology of China (863 program, grants 2006AA02A302, 2009AA022707 and 2012AA02A203).
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
REFERENCES
- 1.World Health Organization Cancer (Fact sheet N°297) Updated February 2014; http://www.who.int/mediacentre/factsheets/fs297/en/
- 2.Yang L. Incidence and mortality of gastric cancer in China. World J Gastroenterol. 2006;12:17–20. doi: 10.3748/wjg.v12.i1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshihara K, Tajima A, Komata D, et al. Gene expression profiling of advanced-stage serous ovarian cancers distinguishes novel subclasses and implicates ZEB2 in tumor progression and prognosis. Cancer Sci. 2009;100:1421–8. doi: 10.1111/j.1349-7006.2009.01204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lau SK, Boutros PC, Pintilie M, et al. Three-gene prognostic classifier for early-stage non small-cell lung cancer. J Clin Oncol. 2007;25:5562–9. doi: 10.1200/JCO.2007.12.0352. [DOI] [PubMed] [Google Scholar]
- 5.Crispi S, Calogero RA, Santini M, et al. Global gene expression profiling of human pleural mesotheliomas: identification of matrix metalloproteinase 14 (MMP-14) as potential tumour target. PLoS One. 2009;4:e7016. doi: 10.1371/journal.pone.0007016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo RF, Zang SZ, Fang JY, et al. Identification of biomarkers for early detection in gastric cancer and its clinical biological significance. Beijing Da Xue Xue Bao. 2009;41:353–60. [PubMed] [Google Scholar]
- 7.Fèvre-Montange M, Champier J, Durand A, et al. Microarray gene expression profiling in meningiomas: differential expression according to grade or histopathological subtype. Int J Oncol. 2009;35:1395–407. doi: 10.3892/ijo_00000457. [DOI] [PubMed] [Google Scholar]
- 8.Zervakis M, Blazadonakis ME, Tsiliki G, Danilatou V, Tsiknakis M, Kafetzopoulos D. Outcome prediction based on microarray analysis: a critical perspective on methods. BMC Bioinformatics. 2009;10:53. doi: 10.1186/1471-2105-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quackenbush J. Microarray analysis and tumour classification. N Engl J Med. 2006;354:2463–72. doi: 10.1056/NEJMra042342. [DOI] [PubMed] [Google Scholar]
- 10.Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet. 2006;7:55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- 11.Bellman RE. Dynamic Programming. Princeton: Princeton University Press; 1957. [Google Scholar]
- 12.Bellman RE. Adaptive Control Processes: A Guided Tour. Princeton: Princeton University Press; 1961. [Google Scholar]
- 13.Bellman RE. Dynamic Programming. Mineola: Courier Dover Publications; 2003. [Google Scholar]
- 14.Ransohoff DF. Lessions from Controversy: Ovarian Cancer Screening and Serum Proteomics. J Natl Cancer Insit. 2005;97:315–9. doi: 10.1093/jnci/dji054. [DOI] [PubMed] [Google Scholar]
- 15.Ransohoff DF. Bias as a threat to the validity of cancer molecular-marker research. Nat Rev Cancer. 2005;5:142–9. doi: 10.1038/nrc1550. [DOI] [PubMed] [Google Scholar]
- 16.Luke BT, Collins JR. Examining the significance of fingerprint-based classifiers. BMC Bioinformatics. 2008;9:545. doi: 10.1186/1471-2105-9-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luke BT, Collins JR. A Comparison of Biomarker and Fingerprint-Based Classifiers of Disease. In: Khan TK, editor. Biomarker. InTech; 2012. pp. 179–200. Available from: http://www.intechopen.com/books/biomarker/a-comparison-of-biomarker-and-fingerprint-based-classifiers-ofdisease. [Google Scholar]
- 18.Zang S, Guo R, Zhang L, Lu Y. Integration of statistical inference methods and a novel control measure to improve sensitivity and specificity of data analysis in expression profiling studies. J Biomed Inform. 2007;40:552–60. doi: 10.1016/j.jbi.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: Class Discovery and class prediction by gene expression monitoring. Science. 1999;286:531–7. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 20.Li YX, Ruan XG. Feature selection for cancer classification based on support vector machine. J Comp Res Dev. 2005;42:1796–801. [Google Scholar]
- 21.Quinlan JR. C4.5 Programs for machine. Morgan Kaufmann; 1993. [Google Scholar]; Machine Learning. 1994;16:235–40. [Google Scholar]
- 22.Kira K, Rendell LA. A practical approach to feature selection. In: Sleeman D, Edwards P, editors. Proceedings of International Conference on Machine Learning. Morgan Kaufmann; Aberdeen, Scotland: 1992. pp. 249–56. [Google Scholar]
- 23.Kenneth JL, Thomas DS. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Zhao X, Kang B, Lu C, et al. Evaluation of p38 MAPK pathway as a molecular signature in ulcerative colitis. J Proteome Res. 2011;10:2216–25. doi: 10.1021/pr100969w. [DOI] [PubMed] [Google Scholar]
- 25.Gini C. Variabilità e mutabilità (Variability and Mutability) Bologna: C. Cuppini; 1912. [Google Scholar]
- 26.Yasui W, Oue N, Ito R, Kuraoka K, Nakayama H. Search for new biomarkers of gastric cancer through serial analysis of gene expression and its clinical implications. Cancer Sci. 2004;95:385–92. doi: 10.1111/j.1349-7006.2004.tb03220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang Y, Diehn M, Bollen AW, Israel MA, Gupta N. Type I collagen is overexpressed in medulloblastoma as a component of tumour microenvironment. J Neurooncol. 2008;86:133–41. doi: 10.1007/s11060-007-9457-5. [DOI] [PubMed] [Google Scholar]
- 28.Flanagan SE, Patch A-M, Locke JM, et al. Genome-wide homozygosity analysis reveals HADH mutations as a common cause of diazoxide-responsive hyperinsulinemic-hypoglycemia in consanguineous pedigrees. J Clin Endocrinol Metabol. 2011;96:E498–502. doi: 10.1210/jc.2010-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaur S, Kulkarni KP, Kochar IP. Severe dietary protein sensitivity and hyperinsulinemic hypoglycemia in a patient with heterozygous mutation in HADH gene. J Pediatr Endocrinol Metabol. 2010;23:953–5. doi: 10.1515/jpem.2010.152. [DOI] [PubMed] [Google Scholar]
- 30.Kapoor RR, Heslegrave A, Hussain K. Congenital hyperinsulinism due to mutations in HNF4 A and HADH. Rev Endocr Metab Disord. 2010;11:185–91. doi: 10.1007/s11154-010-9148-y. [DOI] [PubMed] [Google Scholar]
- 31.Göõz M, Hammond CE, Larsen K, Mukhin YV, Smolka AJ. Inhibition of human gastric H(+)-K(+)-ATPase alpha-subunit gene expression by Helicobacter pylori. Am J Gastrointest Liver Physiol. 2000;278:G981–91. doi: 10.1152/ajpgi.2000.278.6.G981. [DOI] [PubMed] [Google Scholar]
- 32.Scarff KL, Judd LM, Toh B-H, Gleeson PA, van Driel IR. Gastric H+, K+-adenosine triphosphatase β subunit is required for normal function, development, and membrane structure of mouse parietal cells. Gastroenterology. 1999;117:605–18. doi: 10.1016/s0016-5085(99)70453-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Details about the 1519 differentially expressed genes, consisting of 593 up-regulated genes and 926 down-regulated genes.
Supplementary Table 2. Details of the 226 genes that were catalogued into 60 pathways according to the gene ontology and pathway analysis.
Supplementary Table 3. Information about the primers for ATP4B, COL1A2, and HADHSC.
Supplementary Table 4. Ct values for ATP4B, COL1A2, and HADHSC for each sample and 2−ΔΔCt values relative to β-actin.
Supplementary Figure 1. log(FC) plots of the 29 signature genes after single linkage clustering using (1-|r|) as the distance metric, where r is the Pearson correlation coefficient.