

Abstract
Lymphangioleiomyomatosis (LAM) is an uncommon disease presented as diffuse thin-walled cystic changes in the lung. The main differential diagnoses include pulmonary Langerhans’ histiocytosis (PLCH), Birt-Hogg-Dubé syndrome (BHD), lymphoid interstitial pneumonia (LIP), and amyloidosis. A combination of clinical, radiological, and pathological approaches as well as genetic testing will clarify the diagnosis in most cases. LAM is a disease almost exclusively in women. Dyspnea, pneumothorax, and hemoptysis are common presentations in LAM patients. LAM is also a lymphatic disorder affecting lymphatic vessels and lymph nodes. Chylothorax, chylous ascites, and lymphangiomyomas are frequently seen. LAM can present sporadically as a single entity or as part of tuberous sclerosis complex (TSC). Angiomyolipoma (AML) is a characteristic extra-pulmonary lesion, either found in association with sporadic or TSC-related LAM. High-risk populations should be screened for LAM, including adult women with TSC and female patients with spontaneous pneumothorax, AMLs in the kidney, and diffuse cystic lung diseases. Definitive diagnosis of LAM is based on a high level of clinical suspicion on presentation supported by pathological findings or by a distinct feature, such as a history of TSC, AMLs in the kidney, chylothorax, or chylous ascites. Vascular endothelial growth factor-D (VEGF-D) in serum is a noninvasive and reliable diagnostic biomarker. In experienced centers, trans-bronchial lung biopsy (TBLB) provides a convenient and safe way to obtain lung specimens for diagnostic purposes. An effective treatment for LAM is now available, namely using a mechanistic target of rapamycin (mTOR) inhibitor such as sirolimus. Efficacy of sirolimus has been confirmed in clinical trials. Research in other molecular-targeted therapies is under investigation. A previously little-known rare disease with no cure is now better understood with regards to its pathogenesis, diagnosis, and management. In this review, current knowledge in diagnosis and differential diagnosis of LAM will be discussed, followed by the discussion of therapy with mTOR inhibitors.
Keywords: lymphangioleiomyomatosis, diffuse cystic lung diseases, tuberous sclerosis complex, vascular endothelial growth factor-D, sirolimus
Introduction
Substantial achievements have been made during the past two decades regarding the molecular mechanism involved in the pathogenesis, diagnosis, and management of a previously little-known rare lung disease, namely lymphangioleiomyomatosis (LAM). LAM is a condition causing diffuse cystic lung lesions seen on a high resolution computerized tomography scan of the lung (HRCT). It is regarded as a low-grade, destructive, and metastasizing neoplasma.1 LAM is almost exclusively seen among women, especially during child-bearing age. Symptoms may start insidiously or abruptly. Dyspnea develops gradually from mild, moderate, to severe. Hypoxemia, or respiratory failure, is common due to progressive deterioration of lung function and the presence of pneumothorax or chylothorax.
The diagnosis of LAM poses a challenge for pulmonary physicians. LAM is rare and hence unfamiliar to many physicians. There have been less than 3,000 patients registered in the international network of LAM clinics that are supported by the LAM Foundation (http://www.thelamfoundation.org). Nevertheless, LAM is frequently listed in differential diagnosis when diffuse cystic changes are present in the lung. Up until recently, HRCT has been commonly relied upon for the diagnosis of this condition. It is also difficult to prognosticate the long-term outcome of LAM because of its variable clinical features and variable rate of progression, as well as the lack of clear prognostic factors thus far.2 Hence, it is necessary to review the literature on the current knowledge in the diagnosis and differential diagnoses of LAM to aid understanding of this condition. Furthermore, as more clinical trials and scientific studies are conducted, there is now greater consensus about the management of patients with LAM. In particular, there is now greater understanding on various factors governing the pathogenesis of LAM leading to the advent of non-invasive treatment with mechanistic target of rapamycin (mTOR) inhibitors.
Diagnosis of LAM
Diagnostic criteria
Table 1 lists the diagnostic criteria for LAM by the European Respiratory Society (ERS) in 2010.2 Pathological diagnosis from lung tissue is not required for all definite LAM. Definite LAM can be diagnosed based on characteristic images on HRCT plus any one of the following clinical grounds (Figure 1): 1) radiological or pathological diagnosis of kidney angiomyolipoma (AML); 2) chylothorax or chylous ascites; 3) pathological diagnosis of lymphangioleiomyoma or lymph-node involvement of LAM; 4) tuberous sclerosis complex (TSC). It is important to carefully evaluate extra-pulmonary manifestations such as AMLs in the kidney, retroperitoneal tumor, or mixed solid and fluid containing cystic mass in the pelvis.
Table 1.
Diagnosis criteria for LAM
| Definite LAM | 1. Characteristic or compatible lung HRCT, and lung biopsy fitting the pathological criteria for LAM; or 2. Characteristic lung HRCT and any of the following: angiomyolipoma (kidney); thoracic or abdominal chylous effusion; lymphangioleiomyoma or lymph-node involved by LAM; Definite or probable TSC. |
| Probable LAM | 1. Characteristic HRCT and compatible clinical history; or 2. Compatible HRCT and any of following: angiomyolipoma (kidney); and thoracic or abdominal chylous effusion. |
| Possible LAM | Characteristic or compatible HRCT. |
Note: This table was created based on the diagnostic criteria of ERS, 2010.2
Abbreviations: LAM, lymphangioleiomyomatosis; ERS, European Respiratory Society; HRCT, high resolution computed tomography; TSC, tuberous sclerosis complex.
Figure 1.
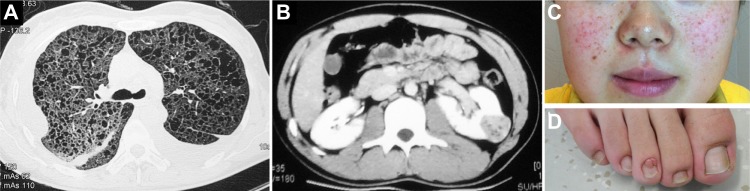
Typical presentations on lung, kidney, and skin in sporadic LAM or TSC. (A) Chest CT in a sporadic LAM patient showing diffuse pulmonary cystic changes and bilateral chylothorax; (B) renal angiomyolipomaseen in either sporadic LAM or TSC (TSC in this case); (C and D) angiofibromas on face and ungal fibromas in TSC.
Abbreviations: LAM, lymphangioleiomyomatosis; TSC, tuberous sclerosis complex; CT, computerized tomography.
There are two types of LAM – sporadic or TSC-related.2 TSC is a genetic disease with multiple organs involved. The onset of TSC may be found prenatally as evidenced by the presence of cardiac rhabdomyoma, subependymal giant cell astrocytoma, and by genetic testing. Epilepsy, an array of cutaneous abnormalities, and kidney AMLs are present in very young children. LAM had been found to be present only among adult female patients with TSC.3 For every patient diagnosed with LAM, it is important to clearly determine whether it is sporadic or associated with this genetic disease.
New diagnostic approaches
Vascular endothelial growth factor-D
Vascular endothelial growth factor-D (VEGF-D), a lymphangiogenic factor, has been found to be elevated in LAM.4 A serum VEGF-D level greater than 800 pg/mL yields high sensitivity and specificity for making the diagnosis of LAM.5 It was estimated that 90% of the probable LAM based on ERS guidelines could be upgraded to definite LAM if VEGF-D estimation is added to the diagnostic criteria. Similarly, invasive surgical biopsy could be decreased to less than 10% if VEGF-D is included in the diagnostic criteria.6 It would seem that adding VEGF-D in the initial evaluation is valuable for the diagnosis of LAM. Moreover, VEGF-D reflects lymphatic involvement and it correlates well with the severity shown on HRCT.6,7
It has been found that treatment with sirolimus stabilizes lung function and decreases the VEGF-D levels.8 Potentially VEGF-D blood level can also be used to gauge response to treatment with sirolimus.9 Further studies are needed to establish the value of VEGF-D in gauging the responsiveness to sirolimus treatment.
Procedural approaches in lung biopsy
Trans-bronchial lung biopsy (TBLB) has been routinely used in the diagnosis of diffuse parenchymal lung diseases but its application in LAM is limited. This is because the risk of producing pneumothorax may be high in cystic lung diseases. There is no prospective study to evaluate the yield and safety of TBLB in the diagnosis of LAM. From a recent survey conducted by Meraj et al, it was estimated that diagnostic yield from TBLB was about 60%.10 From this survey, the risk of pneumothorax and bleeding was reported to be 6% and 4% respectively.10 It would seem that TBLB is a convenient and safe procedure in experienced hands for the purpose of making the pathological diagnosis.10,11 As the sample size of tissue from TBLB is small, it was recommended that immunohistological staining of a specific marker HMB-45 be used as an adjunct to aid confidence in diagnosing LAM.10 Nevertheless, TBLB can provide valuable evidence for definitive diagnosis of LAM, especially when it is more widely applied in the investigation of this condition.11 If TBLB fails to provide meaningful results, video-assisted thoracoscopy can be used for pathological confirmation of LAM diagnosis. However, routine use of TBLB is not currently recommended outside experienced centers, eg, LAM clinics. The safety issue of this procedure warrants future prospective studies in order for it to be used in non-LAM clinics.
Circulating LAM cells
LAM cells have been found in blood, chylous effusion, bronchoalveolar lavage fluid, and urine.12–14 These circulating LAM cells can be used for further molecular study. Potentially, this is another non-invasive method that can be utilized to aid diagnosis.
Diagnosis algorithm
In addition to the diagnostic criteria of ERS 2010 guidelines for definite LAM, the inclusion of VEGF-D into the diagnostic algorithm is supported by recent data, as we discussed above. We therefore propose a revised algorithm (Figure 2).
Figure 2.

Proposed diagnostic algorithm for LAM.
Notes: + means LAM diagnosed; - means LAM not diagnosed.
Abbreviations: HRCT, high resolution CT; VEGF-D, vascular endothelial growth factor-D; LAM, lymphangioleiomyomatosis; TBLB, trans-bronchial lung biopsy; VATS, video-assisted thoracoscopy.
High-risk populations
Although LAM is mostly seen in women, there were cases reported among men in the literature.15,16 In practice LAM is usually excluded in the differential diagnosis in male patients with diffuse cystic lung diseases. There are several high-risk female populations for LAM. These include those patients with TSC, spontaneous pneumothorax, AMLs in kidney, and diffuse cystic lung diseases. Therefore, clinicians need to be aware of those conditions and undertake appropriate screening among these high-risk patients.
Adult women with TSC
On HRCT, more than 50% of TSC patients over the age of 15 were found to have multiple or diffuse cystic changes in their lungs.17 From the same study, in females over 40 years, the prevalence of multiple cysts is as high as 81%. ERS guidelines recommended that HRCT should be performed for female TSC patients at age 18 years old and, if negative, be repeated between 30–40 years old.2 TSC patients with unexplained respiratory symptoms should be screened at any age with HRCT. Measuring serum VEGF-D level is helpful in differentiating TSC with or without LAM.5
Spontaneous pneumothorax in women
Spontaneous pneumothorax is a common presentation in LAM. Conversely, the risk for LAM in a young, non-smoking woman with spontaneous pneumothorax is significantly increased. It is estimated that the prevalence of LAM in patients with spontaneous pneumothorax among the young and non-smoking population is about 5%.18 Therefore, screening for LAM with HRCT in young and non-smoking women with pneumothorax is recommended and cost-effective.
AMLs in kidney
There is a high prevalence of renal AMLs in patients with sporadic LAM or TSC. For patients presented with AMLs in the kidney, the possibility of TSC should be looked for from history and physical examination. In a retrospective study of AML without history of TSC or LAM, 5.1% of patients were found to have four or more cysts in the lung.19 Hence, it is recommended that patients with AMLs in the kidney should be screened with HRCT for LAM.2
Diffuse cystic lung diseases
LAM should be screened and evaluated in every female patient with diffuse cystic lung disease. Patients with characteristic history and radiographic changes of LAM are usually diagnosed without difficulties. However, there are a variety of diseases mimicking LAM (Table 2). To make a definitive diagnosis of LAM from these other diffuse cystic lung conditions can be a difficult task.20
Table 2.
Differential diagnosis of diffuse thin-walled cystic lung diseases
| LAM |
| Sporadic LAM |
| Tuberous sclerosis complex-related LAM |
| PLCH |
| BHD |
| LIP |
| Primary LIP |
| Secondary LIP* |
| Amyloidosis* |
| Light-chain deposition disease |
| Follicular bronchiolitis* |
| Metastatic malignancy |
| eg, sarcoma, meningioma, urothelial carcinoma |
| Pulmonary adenocarcinoma |
| Others |
Notes:
As found in Sjögren syndrome. Copyright © 2013. Higher Education Press. Modified with permission from Ryu JH, Tian X, Baqir M, Xu K. Diffuse cystic lung diseases. Front Med. 2013;7(3):316–327,20; Modified with permission of the American Thoracic Society. Copyright © 2014 American Thoracic Society. Imokawa S, Uehara M, Uto T, et al. 2013. Pulmonary metastasis from urothelial carcinoma showing progressive multiple cystic lesions. Am J Respir Crit Care Med. Volume 188(10):1267–1268. Official Journal of the American Thoracic Society.22
Abbreviations: LAM, lymphangioleiomyomatosis; PLCH, pulmonary Langerhans’ cell histiocytosis; BHD, Birt-Hogg-Dubé syndrome; LIP, lymphoid interstitial pneumonia.
Differential diagnosis of LAM
Diffuse cystic lung diseases consist of a group of heterogeneous lung diseases with similar cystic changes to LAM in the lung. Differential diagnosis of LAM has been excellently reviewed previously.20,21 Table 2 lists common thin-walled diffuse cystic lung diseases.20,22 The five most common diseases are LAM, pulmonary Langerhans’ cell histiocytosis (PLCH), Birt-Hogg-Dubé syndrome (BHD), lymphoid interstitial pneumonia (LIP), and amyloidosis. Diffuse thin-walled cystic lung disease is a common presentation in Sjögren syndrome. Sjögren syndrome is not listed in Table 2 because of a wide range of pathological manifestations which include LIP, amyloidosis, follicular bronchiolitis, nonspecific interstitial pneumonia, and mucosa-associated lymphoid tissue lymphoma.23–25 Nevertheless, primary or secondary Sjögren syndrome should be considered in the investigation of diffuse cystic lung diseases.
History and physical examination
Thorough history taking is critical in differentiating LAM from other similar lesions. Important information includes sex and age, pulmonary symptoms, and dermatological/renal signs.
Sex and age
It had been generally thought that cystic lung disease found in men is not LAM.2 However, this is not entirely the case as LAM in men has been reported in the past.15,16 Nevertheless the diagnosis of LAM is mostly made among women. LAM is also a late presentation in TSC women after adolescence. PLCH and other diseases are seen in male or female patients. LAM is frequently diagnosed around 30–40 years of age. Similar age distribution is seen in PLCH and BHD.20
Pulmonary symptoms
Pneumothorax is a very frequent and recurrent symptom in LAM. It is found in over 50% of LAM in contrast to BHD and PLCH (10%–25%).20,26 Pneumothorax is less common in LIP and amyloidosis. Chylothorax is typically seen in LAM. Frequent hemoptysis with a small amount of blood is common in LAM. Despnea is common in LAM and PLCH. On the other hand, BHD patients rarely present with dyspnea.
Skin and kidney involvements
Skin and kidney involvements are very frequently found in TSC-LAM and BHD. Dermatological manifestations in TSC include angiofibromas, hypo-melanotic macules, shagreen patch, and ungal fibromas. Tumors in the kidney are usually benign in TSC, typically AMLs. AMLs are also found in sporadic LAM, but dermatological findings are absent. The presence of dermatological and renal manifestations in TSC is very high (>80%) as well as being very specific. This contributes towards supporting the diagnosis of LAM. In BHD, multiple benign cutaneous neoplasms, including fibro-folliculomas on the face and neck, are also frequently seen. There is a high risk of renal cell carcinoma in BHD.
Other extra-pulmonary symptoms
In patients with TSC-LAM, epilepsy is common due to cerebral involvement of TSC. Bone lesions and diabetes insipidus are frequently found in patients with PLCH. For connective tissue disease-related LIP or amyloidosis, Sjögren syndrome is a most common finding. However, LIP could be idiopathic or secondary to Sjögren syndrome, AIDS, autoimmune thyroid disease, and other lympho-proliferative disorders. Systemic amyloidosis may have other clinical findings depending on the sites of involvement.
Cigarette smoking
Cigarette smoking or heavily exposed passive smoking is seen in the majority (>90%) of adult PLCH patients.27 Complete or partial remission of PLCH lesions could be achieved after smoking has ceased. Cigarette smoking has not been reported to be related to LAM, BHD, or LIP.20
Family history
TSC and BHD are genetic diseases. Similar clinical presentations in a family are pointers toward an inherited disease. Family history of pneumothorax and presence of bullae are suggestive of BHD.
Laboratory and genetic testing
A number of tests can be used to differentiate LAM from other conditions. Serum VEGF-D is now a well established biomarker for diagnosing, measuring severity, and potentially predicting the treatment response in LAM, as we discussed. Anti-nuclear antibodies, including anti-SSA and anti-SSB antibodies, are used to screen Sjögren syndrome. The presence of light chain immunoglobulin in blood is suggestive of amyloidosis or light-chain deposition disease. Gene tests for TSC1 or TSC2 gene mutations are recommended for those who are diagnosed with TSC.3 Gene tests of FLCN gene mutations are used for diagnosis of BHD.28
Radiological examination
HRCT is recommended for all patients with cystic lung diseases because it can provide information on the characteristics of the cysts. LAM is described as a thin-walled round cystic lung disease with walls less than 3 mm. It differs from thick-walled cysts, cavities, emphysema, bronchiectasis, or honeycomb changes.29 To diagnose LAM in the lung from radiological images, ERS guidelines for LAM suggested that there should be more than two cysts present on HRCT.2 Less than ten but more than two cysts is regarded as consistent with probable LAM. Lung cysts are usually 2 to 5 mm diameter in size, but larger cysts are commonly seen. LAM on CT is further graded into mild (Grade 1), moderate (Grade 2), and severe (Grade 3).30 An area of cyst less than one-third of the lung field is defined as Grade 1, larger than two-thirds as Grade 3, between one third and two-thirds as Grade 2. Images with Grade 2 or Grade 3 are usually quite diagnostic for LAM on HRCT.
Obviously not all diffuse thin-walled cystic lung diseases are LAM. HRCT images provide valuable clues in aiding a diagnosis. The distribution of cysts in PLCH is predominately in the upper and middle fields of the lung with sparing of the costo-phrenic angles. Cysts in PLCH are irregular in size with the wall thicker than that in LAM. Distribution of asymmetrical large cysts under the pleura or peri-mediastinum in the lower lung field is suggestive of BHD. A combination of cysts and nodules is suggestive of PLCH.
Multiple fine nodules are seen also in TSC with micro-nodular pneumocyte hyperplasia shown on histology.31 LIP has features of interstitial lung disease, such as reticulo-nodular opacities and alveolar infiltrates. Nodules, interlobular septal thickening, and ground-glass opacity are seen in amyloidosis.
Pathological examination
Surgical lung biopsy is the gold standard for diagnosing LAM. However, since the publication of ERS guidelines for LAM diagnosis, pathological evidence for a definite diagnosis is no longer mandatory.2 Pathological examination, however, does provide a solid basis in the differential diagnosis for cystic lung diseases.
Pathologically, there is a prominent presence of proliferative smooth muscle-like cells (LAM cells) as well as cystic formation in the lung. It is generally accepted that TSC2 gene mutations and over-activation of a down-stream protein mTOR are major mechanisms involved at the molecular level.32 The role of estrogen in the pathogenesis was not clear until recently when estrogen was found to be involved in the growth and metastasis of LAM cells.33,34
The origin of LAM cells is unknown. Microscopically, LAM cells are present as clusters of immature smooth muscle cell-like spindle cells, staining positive on smooth muscle actin and a melanocytic marker HMB-45. Likewise, estrogen or progesterone receptors are frequently positive in immunochemical staining. This may be one of the potential usages in future molecular-targeted therapeutic options.35 Similar pathological findings are seen in lung and kidney AMLs with the same TSC2 mutations.36 For this reason, pulmonary LAM is generally believed to be metastatic lesion originated from the kidney. There are also evidences of a potential uterine origin of LAM cells.1 Estrogen is believed to be one of the major forces in tumor growth and metastasis.34 The metastatic model is also supported by the presence of circulating LAM cells found in blood, urine, chylous effusion, bronchioalveolar lavage fluid, lymphatic vessels, and lymph nodes.13,14 LAM is referred to as a low-grade, destructive, and metastasizing neoplasm.1 In the World Health Organization classification of tumors, LAM is listed as one of the mesenchymal tumors within the malignant epithelial tumor category.37
Management of LAM
Evaluation of severity
Severity of LAM in patients should be evaluated every 6–12 months in local LAM clinics or as needed depending on other associated conditions. The patients are assessed with regards to their lung function, cardiopulmonary status, and quality of life. Assessments or tests include presence of symptoms and signs, pulmonary function tests, including diffusion capacity of lung, 6-minute walking test, pulse oximetry, and health-related quality of life assessment based on St George’s Respiratory Questionnaire.6,9,26,30,38–42 Arterial blood gas analysis might be needed under special conditions, such as rapid deterioration of pulmonary function. Other tests, including serum VEGF-D, electrocardiogram, cardiac ultrasound, HRCT of the lung, as well as radiological evaluation of abdomen and pelvis with a CT scan or magnetic resonance imaging for extra-pulmonary lesions, are recommended. During the follow-up, disease progression and the effect of medication should also be evaluated from history and clinical examination. Table 3 lists the suggested evaluation of severity and timing for patients with LAM. Test items selected and their periodical review should be adjusted according to the individual need of the patients.
Table 3.
Evaluation of severity and timing for patients with LAM
| At baseline or prior to therapy | Every 6–12 months | Six months after sirolimus therapy | In case of clinical worsening | |
|---|---|---|---|---|
| Clinical | ✓ | ✓ | ✓ | ✓ |
| Pulse oximetry | ✓ | ✓ | ✓ | ✓ |
| 6MWT | ✓ | ✓ | ✓ | ✓ |
| SGRQ | ✓ | ✓ | ✓ | ✓ |
| PFT | ✓ | ✓ | ✓ | ✓ |
| ABG | ✓ | ✓a | ✓a | ✓a |
| VEGF-D | ✓ | ✓a | ✓a | ✓a |
| ECG | ✓ | ✓a | ✓b | ✓a |
| UCG | ✓ | ✓a | ✓b | ✓a |
| HRCT of chest | ✓ | ✓c | No evaluation is necessary | ✓a |
| CT/MRI of abdomen and pelvis | ✓ | ✓b | ✓b | ✓a |
Notes:
Optional depending on the clinical requirements
if abnormal at baseline
HRCT of chest can be assessed every 12 months or as necessary.
Abbreviations: LAM, lymphangioleiomyomatosis; 6MWT, 6-minute walking test; SGRQ, St George Respiratory Questionnaire; PFT, pulmonary function test; ABG, arterial blood gas analysis; VEGF-D, vascular endothelial growth factor-D; ECG, electrocardiogram; UCG, ultrasonic cardiogram; HRCT, high resolution computerized tomography scan of the lung; CT, computerized tomography; MRI, magnetic resonance imaging.
General management
General management of patients with LAM is recommended (Table 4). Management should be tailored individually depending on the severity, rate of disease progression, complications, and social-economical conditions. Through the LAM foundation, a network of over 40 LAM clinics has been setup in the USA and other countries. Any patients who are diagnosed with LAM are recommended to be referred to those LAM clinics for assessments and follow-ups. Patient groups in different countries and districts also provide valuable information and support for patients and families.
Table 4.
General recommendations for the management of LAM
| Recommendations | |
|---|---|
| Vaccine | Influenza and pneumococcal vaccinations are recommended |
| Pregnancy | Pregnancy is associated with worsening of disease and high risk of developing pneumothorax, chylothorax, and bleeding from AMLs |
| Travel | Patients can travel safely by air when symptoms are mild. Patients should avoid air travel in the presence of current pneumothorax, or pneumothorax within the past one month |
| Pulmonary rehabilitation | Patients are recommended to take part in a pulmonary rehabilitation program |
| Oxygen | Oxygen is recommended for those with hypoxemia |
| Pneumothorax | Chemical pleurodesis is recommended at first onset of pneumothorax to decrease the risk of recurrent pneumothorax |
| Chylothoraxa | Fat-free diet supplemented with mid-chain triglycerides is advised |
| AMLsa | Options include observation, embolization, or nephro-sparing surgery |
| Lung transplantations | Should be recommended to patients who have severe impairment in lung function and exercise capacity reaching the NYHA functional class III or IV |
Notes:
Sirolimus for chylothorax and AMLs is now an option. Data adapted from ERS, 2010.2
Abbreviations: LAM, lymphangioleiomyomatosis; ERS, European Respiratory Society; AML, angiomyolipoma; NYHA, New York Heart Association.
Recent advances
Sirolimus: the first effective medication for LAM
Fascinating progress has also been made in the specific treatment of LAM. In 2000, TSC2 gene mutations were found in LAM.36 Subsequently, the role of TSC1 and TSC2 complex in relation to inhibition mechanism of mTOR was demonstrated.43 This mechanism resulted in an over-activation of mTOR. Sirolimus (an mTOR inhibitor) used as an immunosuppressant post kidney transplantation was tested in LAM and TSC. In 2008, the effectiveness of sirolimus for treating renal AMLs was reported.44 The size of AMLs was found to have dramatically shrunk to around 50% in volume after 12 months of treatment. Analysis of eleven LAM patients in this trial revealed encouraging results of improvement in lung function. Subsequently, a randomized placebo-controlled trial using sirolimus for LAM on 89 cases found that sirolimus significantly stabilized the decline of forced expiratory volume in 1 second (FEV1).8 After 12 months of treatment, there was an additional gain of 19±124 mL in FEV1 among the sirolimus group compared to a loss of 139±182 mL in the placebo group. The biomarker VEGF-D also decreased significantly in the treatment group. Since this report, sirolimus is used frequently in treating LAM patients.
There are several issues on sirolimus treatment that remain to be addressed: 1) when should one initiate sirolimus therapy and on what type of patients?; 2) what are the optimal doses?; 3) how long should it be used?; and 4) what is the long-term safety effect? Theoretically, sirolimus should be prescribed once the diagnosis is established. Clinically, patients with reduced lung function or loss of lung function,8 chylothorax,45 and AMLs in the kidney44 would benefit from sirolimus therapy. For mild or asymptomatic patients, whether early treatment of sirolimus will improve the prognosis is yet unknown.
The dosage of sirolimus for LAM was quoted from recommendations for kidney transplantation, ie, with a target trough blood concentration of 5–15 ng/mL. We found that trough blood concentration of 5–10 ng/mL is effective in stabilizing the lung function (unpublished personal record). However, sirolimus at a lower dosage, less than 5 ng/mL, has been found to be similarly effective.46 One can therefore surmise that the use of a lower dosage should be a reasonable approach for long-term therapeutic maintenance. Further clinical trials are required to establish an optimal dosage. The studies also showed that when sirolimus stops, the size of AMLs may increase and pulmonary function may worsen again.8,44 More clinical trials are needed to ascertain optimal doses and treatment duration with sirolimus. The common adverse effects are oral ulcers, dyslipidemia, acneiform rash, irregular menses, etc.8,47 Patients are advised to stop sirolimus temporarily when pulmonary infections happen. For selective surgery, patients are advised to stop sirolimus 2 weeks before surgery as sirolimus may inhibit wound healing. The long-term safety is still unknown in LAM patients.
Everolimus, a novel mTOR inhibitor
Everolimus, another mTOR inhibitor, had also been shown to be effective in the treatment of kidney AMLs associated with TSC or sporadic LAM,48 subependymal giant cell astrocytomas associated with TSC,49 and many other presentations in TSC such as skin lesions and epilepsy. A recent study has reported that everolimus is also effective for treating LAM.50 In this open-label study, treatment of everolimus for 26 weeks in 24 subjects brought significant increase in FEV1 of 114 mL. Thus, everolimus provides an alternative option when the use of mTOR inhibitors is considered. Future randomized studies are necessary to establish the role of everolimus on lung functions for LAM patients. A similar safety profile was observed for both everolimus and sirolimus.
Other clinical trials in LAM
Doxycycline, a matrix metalloproteinase inhibitor, has been tested in LAM following a successful case report.51 In another open-label observation when doxycycline was used (100 mg/d over a 12-month period), 31 subjects showed mean decrease of FEV1 of 70 mL/year. Patients who would benefit from doxycycline treatment are those with mild impairment of FEV1 and forced vital capacity.52 In a randomized placebo-controlled trial, 23 LAM patients were given doxycycline (100 mg/d, 3 months, then 200 mg/d, 21 months).53 Although matrix metallopeptidase 9 in urine was decreased, no benefits on lung function were observed. Generally, doxycycline is not routinely recommended to LAM patients.
An anti-estrogen strategy was examined in a prospective study using Triptorelin, a gonadotrophin releasing hormone analogue, in eleven premenopausal LAM patients for 36 months.54 No beneficial effects were observed. Retrospective studies did not show effectiveness of treating with progesterone and simvastatin or atorvastatin in LAM.55,56 As LAM is a disease in women with a high number of estrogen receptors and progestogen receptors in the lesions, anti-ER, and anti-PR approaches should be investigated.35 Letrozole, an aromatase inhibitor, is currently being investigated in an ongoing clinical trial for the treatment of LAM (ClinicalTrials.gov Identifier: NCT 01353209).
Likewise, several other ongoing clinical trials using simvastatin and sirolimus plus hydroxychloquine for the treatment of LAM are being undertaken (ClinicalTrials. gov Identifier: NCT02061397, NCT01687179). These new clinical trials reflect the novel understandings regarding mechanisms of LAM and the rapid attempt at applying knowledge from laboratory to clinical usage.
Conclusion
Amazing achievements have been made in the last 2 decades in every aspect in LAM. Serum VEGF-D is now a useful component, in combination with HRCT, in diagnosis and differential diagnosis of LAM. So far, data from several important clinical trials with sirolimus and everolimus in LAM and TSC have established a concrete basis for TOR inhibitors being used as the treatment of choice in LAM. Many other clinical trials are ongoing or in planning. There remains a great need for more placebo-controlled trials to be conducted to prove the value of a definite therapeutic intervention with these drugs. Collaborative effort among medical scientists, physicians within a network of global LAM clinics, and patients worldwide will facilitate the rapid progress of this rare disease efficiently.
Acknowledgments
The authors thank Dr Verne Caradus, retired Developmental Physician in Sydney, Australia, for proofreading this manuscript.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.McCormack FX, Travis WD, Colby TV, Henske EP, Moss J. Lymphangioleiomyomatosis: calling it what it is: a low-grade, destructive, metastasizing neoplasm. Am J Respir Crit Care Med. 2012;186(12):1210–1212. doi: 10.1164/rccm.201205-0848OE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson SR, Cordier JF, Lazor R, et al. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J. 2010;35(1):14–26. doi: 10.1183/09031936.00076209. [DOI] [PubMed] [Google Scholar]
- 3.Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 international Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):243–254. doi: 10.1016/j.pediatrneurol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seyama K, Kumasaka T, Souma S, et al. Vascular endothelial growth factor-D is increased in serum of patients with lymphangioleiomyomatosis. Lymphat Res Biol. 2006;4(3):143–152. doi: 10.1089/lrb.2006.4.143. [DOI] [PubMed] [Google Scholar]
- 5.Young LR, Vandyke R, Gulleman PM, et al. Serum vascular endothelial growth factor-D prospectively distinguishes lymphangioleiomyomatosis from other diseases. Chest. 2010;138(3):674–681. doi: 10.1378/chest.10-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu KF, Zhang P, Tian X, et al. The role of vascular endothelial growth factor-D in diagnosis of lymphangioleiomyomatosis (LAM) Respir Med. 2013;107(2):263–268. doi: 10.1016/j.rmed.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 7.Glasgow CG, Avila NA, Lin JP, Stylianou MP, Moss J. Serum vascular endothelial growth factor-D levels in patients with lymphangioleiomyomatosis reflect lymphatic involvement. Chest. 2009;135(5):1293–1300. doi: 10.1378/chest.08-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCormack FX, Inoue Y, Moss J, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364(17):1595–1606. doi: 10.1056/NEJMoa1100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young L, Lee HS, Inoue Y, et al. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med. 2013;1(6):445–452. doi: 10.1016/S2213-2600(13)70090-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meraj R, Wikenheiser-Brokamp KA, Young LR, Byrnes S, McCormack FX. Utility of transbronchial biopsy in the diagnosis of lymphangioleiomyomatosis. Front Med. 2012;6(4):395–405. doi: 10.1007/s11684-012-0231-5. [DOI] [PubMed] [Google Scholar]
- 11.Harari S, Torre O, Cassandro R, Taveira-DaSilva AM, Moss J. Bronchoscopic diagnosis of Langerhans cell histiocytosis and lymphangioleiomyomatosis. Respir Med. 2012;106(9):1286–1292. doi: 10.1016/j.rmed.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crooks DM, Pacheco-Rodriguez G, DeCastro RM, et al. Molecular and genetic analysis of disseminated neoplastic cells in lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2004;101(50):17462–17467. doi: 10.1073/pnas.0407971101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai X, Pacheco-Rodriguez G, Fan QY, et al. Phenotypic characterization of disseminated cells with TSC2 loss of heterozygosity in patients with lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2010;182(11):1410–1418. doi: 10.1164/rccm.201003-0489OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumasaka T, Seyama K, Mitani K, et al. Lymphangiogenesis-mediated shedding of LAM cell clusters as a mechanism for dissemination in lymphangioleiomyomatosis. Am J Surg Pathol. 2005;29(10):1356–1366. doi: 10.1097/01.pas.0000172192.25295.45. [DOI] [PubMed] [Google Scholar]
- 15.Miyake M, Tateishi U, Maeda T, et al. Pulmonary lymphangioleiomyomatosis in a male patient with tuberous sclerosis complex. Radiat Med. 2005;23(7):525–527. [PubMed] [Google Scholar]
- 16.Kang HW, Kim CJ, Kang SK, Lee KS, Lee CS, Kim YH. Pulmonary lymphangioleiomyomatosis in a male. J Korean Med Sci. 1991;6(1):83–85. doi: 10.3346/jkms.1991.6.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cudzilo CJ, Szczesniak RD, Brody AS, et al. Lymphangioleiomyomatosis screening in women with tuberous sclerosis. Chest. 2013;144(2):578–585. doi: 10.1378/chest.12-2813. [DOI] [PubMed] [Google Scholar]
- 18.Hagaman JT, Schauer DP, McCormack FX, Kinder BW. Screening for lymphangioleiomyomatosis by high-resolution computed tomography in young, nonsmoking women presenting with spontaneous pneumothorax is cost-effective. Am J Respir Crit Care Med. 2010;181(12):1376–1382. doi: 10.1164/rccm.200910-1553OC. [DOI] [PubMed] [Google Scholar]
- 19.Ryu JH, Hartman TE, Torres VE, Decker PA. Frequency of undiagnosed cystic lung disease in patients with sporadic renal angiomyolipomas. Chest. 2012;141(1):163–168. doi: 10.1378/chest.11-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryu JH, Tian X, Baqir M, Xu K. Diffuse cystic lung diseases. Front Med. 2013;7(3):316–327. doi: 10.1007/s11684-013-0269-z. [DOI] [PubMed] [Google Scholar]
- 21.Trotman-Dickenson B. Cystic lung disease: achieving a radiologic diagnosis. Eur J Radiol. 2014;83(1):39–46. doi: 10.1016/j.ejrad.2013.11.027. [DOI] [PubMed] [Google Scholar]
- 22.Imokawa S, Uehara M, Uto T, et al. Pulmonary metastasis from urothelial carcinoma showing progressive multiple cystic lesions. Am J Respir Crit Care Med. 2013;188(10):1267–1268. doi: 10.1164/rccm.201305-0887LE. [DOI] [PubMed] [Google Scholar]
- 23.Baqir M, Kluka EM, Aubry MC, et al. Amyloid-associated cystic lung disease in primary Sjögren’s syndrome. Respir Med. 2013;107(4):616–621. doi: 10.1016/j.rmed.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Lohrmann C, Uhl M, Warnatz K, et al. High-resolution CT imaging of the lung for patients with primary Sjogren’s syndrome. Eur J Radiol. 2004;52(2):137–143. doi: 10.1016/j.ejrad.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Shi JH, Liu HR, Xu WB, et al. Pulmonary manifestations of Sjögren’s syndrome. Respiration. 2009;78(4):377–386. doi: 10.1159/000214841. [DOI] [PubMed] [Google Scholar]
- 26.Ryu JH, Moss J, Beck GJ, et al. The NHLBI lymphangioleiomyomatosis registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med. 2006;173(1):105–111. doi: 10.1164/rccm.200409-1298OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suri HS, Yi ES, Nowakowski GS, Vassallo R. Pulmonary langerhans cell histiocytosis. Orphanet J Rare Dis. 2012;7:16. doi: 10.1186/1750-1172-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furuya M, Nakatani Y. Birt-Hogg-Dube syndrome: clinicopathological features of the lung. J Clin Pathol. 2013;66(3):178–186. doi: 10.1136/jclinpath-2012-201200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koo HK, Yoo CG. Multiple cystic lung disease. Tuberc Respir Dis (Seoul) 2013;74(3):97–103. doi: 10.4046/trd.2013.74.3.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Avila NA, Dwyer AJ, Rabel A, Moss J. Sporadic lymphangioleiomyomatosis and tuberous sclerosis complex with lymphangioleiomyomatosis: comparison of CT features. Radiology. 2007;242(1):277–285. doi: 10.1148/radiol.2421051767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franz DN, Brody A, Meyer C, et al. Mutational and radiographic analysis of pulmonary disease consistent with lymphangioleiomyomatosis and micronodular pneumocyte hyperplasia in women with tuberous sclerosis. Am J Respir Crit Care Med. 2001;164(4):661–668. doi: 10.1164/ajrccm.164.4.2011025. [DOI] [PubMed] [Google Scholar]
- 32.McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest. 2008;133(2):507–516. doi: 10.1378/chest.07-0898. [DOI] [PubMed] [Google Scholar]
- 33.Gu X, Yu JJ, Ilter D, Blenis N, Henske EP, Blenis J. Integration of mTOR and estrogen-ERK2 signaling in lymphangioleiomyomatosis pathogenesis. Proc Natl Acad Sci U S A. 2013;110(37):14960–14965. doi: 10.1073/pnas.1309110110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu JJ, Robb VA, Morrison TA, et al. Estrogen promotes the survival and pulmonary metastasis of tuberin-null cells. Proc Natl Acad Sci U S A. 2009;106(8):2635–2640. doi: 10.1073/pnas.0810790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El-Chemaly S, Henske EP. Towards personalised therapy for lymphangioleiomyomatosis: lessons from cancer. Eur Respir Rev. 2014;23(131):30–35. doi: 10.1183/09059180.00008813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2000;97(11):6085–6090. doi: 10.1073/pnas.97.11.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.World Health Organization . In: World Health Organization Classification of Tumors. Pathology and Genetics. Tumours of the Lung, Pleura, Thymus and Heart. Travis WD, Brambilla E, Muller-Hermelink HK, Harris CC, editors. Lyon: IARC Press; 2004. [Accessed July 16, 2014]. Available from: http://www.iarc.fr/en/publications/pdfs-online/pat-gen/bb10/BB10.pdf. [Google Scholar]
- 38.Taveira-DaSilva AM, Hedin C, Stylianou MP, et al. Reversible airflow obstruction, proliferation of abnormal smooth muscle cells, and impairment of gas exchange as predictors of outcome in lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2001;164(6):1072–1076. doi: 10.1164/ajrccm.164.6.2102125. [DOI] [PubMed] [Google Scholar]
- 39.Xu KF, Wang L, Tian XL, et al. The St George’s Respiratory Questionnaire in lymphangioleiomyomatosis. Chin Med Sci J. 2010;25(3):140–145. doi: 10.1016/s1001-9294(10)60038-7. [DOI] [PubMed] [Google Scholar]
- 40.Swigris JJ, Lee HS, Cohen M, et al. St George’s Respiratory Questionnaire has longitudinal construct validity in lymphangioleiomyomatosis. Chest. 2013;143(6):1671–1678. doi: 10.1378/chest.12-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cottin V, Harari S, Humbert M, et al. Pulmonary hypertension in lymphangioleiomyomatosis: characteristics in 20 patients. Eur Respir J. 2012;40(3):630–640. doi: 10.1183/09031936.00093111. [DOI] [PubMed] [Google Scholar]
- 42.Taveira-DaSilva AM, Hathaway OM, Sachdev V, Shizukuda Y, Birdsall CW, Moss J. Pulmonary artery pressure in lymphangioleiomyomatosis: an echocardiographic study. Chest. 2007;132(5):1573–1578. doi: 10.1378/chest.07-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goncharova EA, Goncharov DA, Eszterhas A, et al. Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation. A role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM) J Biol Chem. 2002;277(34):30958–30967. doi: 10.1074/jbc.M202678200. [DOI] [PubMed] [Google Scholar]
- 44.Bissler JJ, McCormack FX, Young LR, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358(2):140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taveira-DaSilva AM, Hathaway O, Stylianou M, Moss J. Changes in lung function and chylous effusions in patients with lymphangioleiomyomatosis treated with sirolimus. Ann Intern Med. 2011;154(12):797–805. doi: 10.1059/0003-4819-154-12-201106210-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ando K, Kurihara M, Kataoka H, et al. The efficacy and safety of low-dose sirolimus for treatment of lymphangioleiomyomatosis. Respir Investig. 2013;51(3):175–183. doi: 10.1016/j.resinv.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 47.Braun M, Young J, Reiner CS, et al. Low-dose oral sirolimus and the risk of menstrual-cycle disturbances and ovarian cysts: analysis of the randomized controlled SUISSE ADPKD trial. PloS One. 2012;7(10):e45868. doi: 10.1371/journal.pone.0045868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9869):817–824. doi: 10.1016/S0140-6736(12)61767-X. [DOI] [PubMed] [Google Scholar]
- 49.Franz DN, Belousova E, Sparagana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381(9861):125–132. doi: 10.1016/S0140-6736(12)61134-9. [DOI] [PubMed] [Google Scholar]
- 50.Goldberg HJ, Harari S, McCormack FX, et al. Efficacy and safety of everolimus for the treatment of lymphangioleiomyomatosis: a phase II study; The LAM Foundation 17th Annual International Lymphangioleiomyomatosis Research Conference and Patient and Family Educational LAMposium; March 27–29, 2014; Chicago. [Google Scholar]
- 51.Moses MA, Harper J, Folkman J. Doxycycline treatment for lymphangioleiomyomatosis with urinary monitoring for MMPs. N Engl J Med. 2006;354(24):2621–2622. doi: 10.1056/NEJMc053410. [DOI] [PubMed] [Google Scholar]
- 52.Pimenta SP, Baldi BG, Kairalla RA, Carvalho CR. Doxycycline use in patients with lymphangioleiomyomatosis: biomarkers and pulmonary function response. J Bras Pneumol. 2013;39(1):5–15. doi: 10.1590/S1806-37132013000100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang WY, Cane JL, Kumaran M, Lewis S, Tattersfield AE, Johnson SR. A 2-year randomised placebo-controlled trial of doxycycline for lymphangioleiomyomatosis. Eur Respir J. 2014;43(4):1114–1123. doi: 10.1183/09031936.00167413. [DOI] [PubMed] [Google Scholar]
- 54.Harari S, Cassandro R, Chiodini I, Taveira-DaSilva AM, Moss J. Effect of a gonadotrophin-releasing hormone analogue on lung function in lymphangioleiomyomatosis. Chest. 2008;133(2):448–454. doi: 10.1378/chest.07-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.El-Chemaly S, Taveira-DaSilva A, Stylianou MP, Moss J. Statins in lymphangioleiomyomatosis: a word of caution. Eur Respir J. 2009;34(2):513–514. doi: 10.1183/09031936.00012709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taveira-DaSilva AM, Stylianou MP, Hedin CJ, Hathaway O, Moss J. Decline in lung function in patients with lymphangioleiomyomatosis treated with or without progesterone. Chest. 2004;126(6):1867–1874. doi: 10.1378/chest.126.6.1867. [DOI] [PubMed] [Google Scholar]