

Abstract
Background
Transgenic mice with transient cardiac expression of constitutively active Galpha q (Gαq-TG) exhibt progressive heart failure and ventricular arrhythmias after the initiating stimulus of transfected constitutively active Gαq becomes undetectable. However, the mechanisms are still unknown. We examined the effects of chronic administration of olmesartan on heart failure and ventricular arrhythmia in Gαq-TG mice.
Methodology/Principal Findings
Olmesartan (1 mg/kg/day) or vehicle was chronically administered to Gαq-TG from 6 to 32 weeks of age, and all experiments were performed in mice at the age of 32 weeks. Chronic olmesartan administration prevented the severe reduction of left ventricular fractional shortening, and inhibited ventricular interstitial fibrosis and ventricular myocyte hypertrophy in Gαq-TG. Electrocardiogram demonstrated that premature ventricular contraction (PVC) was frequently (more than 20 beats/min) observed in 9 of 10 vehicle-treated Gαq-TG but in none of 10 olmesartan-treated Gαq-TG. The collected QT interval and monophasic action potential duration in the left ventricle were significantly shorter in olmesartan-treated Gαq-TG than in vehicle-treated Gαq-TG. CTGF, collagen type 1, ANP, BNP, and β-MHC gene expression was increased and olmesartan significantly decreased the expression of these genes in Gαq-TG mouse ventricles. The expression of canonical transient receptor potential (TRPC) 3 and 6 channel and angiotensin converting enzyme (ACE) proteins but not angiotensin II type 1 (AT1) receptor was increased in Gαq-TG ventricles compared with NTG mouse ventricles. Olmesartan significantly decreased TRPC6 and tended to decrease ACE expressions in Gαq-TG. Moreover, it increased AT1 receptor in Gαq-TG.
Conclusions/Significance
These findings suggest that angiotensin II type 1 receptor activation plays an important role in the development of heart failure and ventricular arrhythmia in Gαq-TG mouse model of heart failure.
Introduction
Our previous study showed that transient expression of a constitutively active the GTP-binding protein αq subunit in hearts of transgenic mice (Gαq-TG mice) is sufficient to induce cardiac hypertrophy and heart failure (HF) [1]. In fact, although the Gαq protein decreases at 4 weeks and is undetectable until 10 weeks, the mice develop cardiac hypertrophy and dilatation, leading to HF until 16 to 32 weeks of age [1]–[4]. When the cardiac hypertrophy and dilatation develop, endogenous but not transfected Gαq rises in the heart. Basal and Gq-coupled receptor agonist stimulated activity of phospholipase Cß (PLCß), leading to generatiton of inositol trisphosphate (IP3) and diacylglycerol (DAG), which is elevated in ventricles at 10 week age in Gαq-TG mice, presumably at least in part because of the rise in endogenous Gαq [1], [5]. Therefore, the pathological changes initiated by early transient constitutively active Gαq expression may be maintained by multiple and persistent changes in signal transduction pathways [1], [5]. Our more recent studies demonstrated that diacylglycerol kinase zeta, which catalyzes DAG, rescues HF [2] and inhibited atrial [3] and ventricular [4] arrhythmias in Gαq-TG mice, suggesting that DAG plays a critical role in the development of cardiac hypertrophy and HF in this mouse model. However, it is still unknown what factors act upstream of DAG. It is well known that the renin-angiotensin system, which increases the level of DAG, plays a critical role in the development of cardiac hypertrophy and HF [6]–[8]. We hypothesized that the renin-angiotensin system plays an important role in the development of cardiac hypertrophy and HF in this transgenic mouse model after the initiating stimulus of transfected constitutively active Gαq becomes undetectable. Olmesartan is an angiotensin II type 1 receptor antagonist, which can inhibit angiotensin II-induced cardiac remodeling and HF [9], [10]. In the present study, therefore, we investigated the inhibitory effects of olmesartan on ventricular remodeling, leading to HF and ventricular arrhythmias in Gαq-TG mice.
Materials and Methods
Ethics
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. This study was approved by the Animal Care Committee of the Iwate Medical University and Shinshu University. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Iwate Medical University (Permit Number: 22–39) and the Shinshu University (Permit Number: 200044). All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Experimental Animals
A transgenic mouse (Gαq-TG mouse) with transient, modest expression of HAα*q was used [1]. The genotypes of the non-transgenic (NTG) and Gαq-TG mice were identified by polymerase chain reaction (PCR) with the use of tail genomic DNA as a template, as previously reported [1]. Our previous studies demonstrated that Gαq-TG mice developed HF but not ventricular arrhythmias at the age of 16 weeks, whereas they developed ventricular arrhythmias by 32 weeks [4]. We measured the systemic blood pressure and heart rate using the tail-cuff method. (BP-98A Softron, Tokyo, Japan) and demonstrated that olmesartan at a dose of 1 mg/kg/day did not decrease the systemic blood pressure (Table 1). Therefore, to examine the effects of chronic olmesartan administration on HF and ventricular arrhythmias, olmesartan (1 mg/kg/day) was orally administered to Gαq-TG mice from 6 to 32 weeks of age. All experiments were performed in 32-week-old mice. As described in detail previously [11], all mice were anesthetized with sodium pentobarbital (30 mg/kg) applied intraperitoneally, and the adequacy of anesthesia was monitored by observing heart rate and the frequency and the degree of motion of the sternum as well as movement of the extremities.
Table 1. Systemic blood pressure (BP) and heart rates in NTG, Gαq-TG, and Gαq-TG+olmesartan mice.
| Parameters | NTG | Gαq-TG | Gαq-TG+olmesartan |
| HR (beats/min) | 613±23 | 566±45 | 651±20 |
| SBP (mmHg) | 102±6 | 94±3 | 103±3 |
| DBP (mmHg) | 62±3 | 60±4 | 64±2 |
| MBP(mmHg) | 75±4 | 72±5 | 77±1 |
Data are the mean ± SE obtained from 7 mice for each group. SBP, systolic BP; DBP, diastric BP; MBP, mean BP.
Echocardiography
Vehicle-treated NTG, vehicle-treated Gαq-TG, and olmesartan-treated Gαq-TG mice (n = 7 each) were anesthetized, and cardiac function was assessed by echocardiography (GE Yokogawa Medical System, Tokyo, Japan). As described in detail previously [12], the level of the papillary muscles along the short axis was used to view heart. The average of three consecutive beats in M-mode tracings was used to measure the following parameters: interventricular septum thickness, left ventricular end-diastolic dimension (LVEDd), end-systolic dimension (LVESd), and fractional shortening (LVFS), which was calculated as follows: (LVEDd - LVESd)/LVEDd×100%.
Electrocardiography (ECG) and Electrophysiological Measurement
Vehicle-treated NTG, vehicle-treated Gαq-TG, and olmesartan-treated Gαq-TG mice (n = 7 each) were anesthetized with sodium pentobarbital (30 mg/kg) applied intraperitoneally. Electrocardiography (ECG) lead II was recorded for 10 min in all mice. As described in detail previously [11], surface ECG was recorded and filtered (0.1 to 300 Hz), digitized with 12-bit precision at a sampling rate of 1000 Hz per channel (Microstar Laboratories Inc., Bellevue, WA, USA), transmitted into a microcomputer and saved on a CD-ROM.
In all mice examined, P, PR, QRS complex, QT, and RR intervals were measured from ECG lead II. The number of premature ventricular contractions (PVCs) per minute was calculated from ECG lead II. A high incidence of PVCs (High PVC) was defined as more than 20 beats/min of PVC.
Gross Anatomy and Histology
After vehicle-treated NTG, vehicle-treated Gαq-TG, and olmesartan-treated Gαq-TG mice (n = 10 each) were anesthetized, hearts were quickly excised. To examine gross anatomy and histology, the heart preparation was prepared. As described in detail previously [11], the hearts were fixed with a 30% solution of formalin in phosphate-buffered saline at room temperature for more than 24 hours, embedded in paraffin, and then cut serially from the apex to the base. Six sections were stained with hematoxylin/eosin or Masson’s trichrome for histopathological analysis. To measure the cross-sectional diameter of cardiomyocytes, the diameter of at least 20 cardiomyocytes in each section was measured using the image analyzing software MacSCOPE (MITANI Corporation, Tokyo) on a Macintosh computer. The measurements were performed on 3 sections in each preparation and averaged. The degree of fibrosis was assessed by digital microscopic images taken from the sections stained with Masson’s trichrome stain using light microscopy with a digital camera system. As described in detail previously [3], the measurements were performed on 3 images from different parts of the left ventricle in each preparation. The fibrosis fraction was obtained by calculating the ratio of total connective area to total myocardial area from 3 images in each preparation.
Western Blot Analysis
The ventricular myocardium of anesthetized NTG, vehicle-treated Gαq-TG, and olmesartan-treated Gαq-TG mice (n = 6 each) was prepared to extract the total protein using a lysis buffer (Cell Signaling Technology, Inc., Danvers, MA). The protein expression of canonical transient receptor potential (TRPC) and angiotensin-converting enzyme (ACE) isoforms was examined. As described in detail previously [11], protein concentrations were assayed, and equal amounts of the proteins were subjected to 10% SDS-PAGE and transferred to PVDF membranes. To ensure equivalent protein loading and to verify efficient protein transfer, membranes were stained with Ponceau S before incubating with primary isoform-specific antibodies against TRPC isoforms (TRPC 3 and 6; SIGMA, St. Louis, MO), ACE isoforms (ACE and ACE2; SIGMA, St. Louis, MO) and actin. [13] Immunoreactive bands were detected with an ECL kit (Amersham Biosciences Corp., Piscataway, NJ). The densitometric intensity of bands representing TRPC and ACE isoforms was normalized to that of actin. The protein expression levels of angiotensin II type 1 (AT1) receptor in the ventricular myocardium of anesthetized NTG, vehicle-treated Gαq-TG, and olmesartan-treated Gαq-TG mice (n = 6 each) were also examined, as described in detail previously [14]–[15]. Western blot analyses were performed using anti-GAPDH antibody (Chemicon International, Temecula, CA, USA) and anti-AT1 receptor antibody (Santa Cruz Biotechnology, Dallas, TX, USA).
Quantification of mRNA by Real-Time PCR
Total RNA was prepared from the ventricular myocardium of anesthetized NTG, vehicle-treated Gαq-TG and olmesartan-treated Gαq-TG mice (n = 7 each) with NueleoSpin RNA II (TAKARA Co. Ltd., Tokyo, Japan) according to the manufacturer’s instructions. The mRNA levels of atrial natriuretic factor (ANF), B-type natriuretic peptide (BNP), β-myosin heavy chain (β-MHC), connective tissue growth factor (CTGF), collagen type 1, and acidic ribosomal protein P0 (ARPP0) were examined. As described in detail previously [11], one microgram of total RNA was used as a template for reverse transcription with the SuperScript III First-Strand synthesis system for qRT-PCR (Invitrogen, Carlsbad, CA). Partial cDNA fragments of ANF, BNP, β-MHC, CTGF, collagen type 1, and ARPP0 were amplified from the heart cDNA by PCR with DNA polymerase Fast SYBR Green Master Mix (Takara Bio, Shiga, Japan) to generate a standard curve for mRNA quantification. Real-time PCR was performed with an ABI Step One Real-Time PCR System (Applied Biosystems, Foster City, CA). The PCR mixture (10 µl) contained Fast SYBR Green Master (Mix) (Roche Diagnostics), standard cDNA (5×102 ng per reaction), and 200 nM forward and reverse primers. All primers used are listed in Table S1. The expression of each gene was normalized to that of ARPP0 mRNA because the expression of ARPP0 mRNA was most consistent among the groups. The specificity of the method was confirmed by dissociation analysis according to the instructions supplied by Applied Biosystems.
Monophasic action potential (MAP) measurement
Vehicle-treated NTG, vehicle-treated Gαq-TG, and olmesartan-treated Gαq-TG mice (n = 6 each) were anesthetized with sodium pentobarbital (30 mg/kg) applied intraperitoneally, and then treated with sodium heparin (500 USP units/kg i.v.). After the hearts were quickly excised, we connected them to a Langendorff apparatus. We used a polyterafluoroethylene-coated silver bipolar electrode to stimulate the epicardial surface of the left ventricle at the twice diastolic threshold current with a duration of 1 ms. As described in detail previously [11], to measure the monophasic action potential (MAP) duration, we put on MAP electrode on the epicardial surface of the posterior left ventricle. MAPs were recorded for 5 sec at a basic cycle length of 200 ms. Each heart preparation was perfused under constant pressure conditions (65 mmHg) with oxygenated (95% oxygen, 5% CO2) Tyrode’s solution containing, in mM: NaCl, 141.0; KCl, 5.0; CaCl2, 1.8; NaHCO3, 25.0; MgSO4, 1.0; NaH2PO4, 1.2; HEPES, 5; and dextrose, 5.0 (pH of 7.4 at 36±1°C). The MAP signals were filtered (0.3 to 300 Hz), amplified (1,000×), and recorded. As described in detail previously [11], the MAP duration was calculated from MAP signals of all Langendorff hearts.
Data Analysis
All data are shown as the mean ± SE. The statistical analysis of multiple comparisons of data was calculated using An analysis of variance with Bonferroni’s test. The incidence of High PVC between different conditions was compared using Fisher’s exact test. P<0.05 was considered statistically significant.
Drug
Olmesartan was kindly provided by Daiichi Sankyo Pharmaceutical Co. (Tokyo, Japan).
Results
Effects of Olmesartan on the Development of Cardiomegaly and Contractile Dysfunction in Gαq-TG Mice
Effects of chronic administration of olmesartan on cardiac morphology was examined in NTG, vehicle-treated Gαq-TG, and olmesartan-treated Gαq-TG mice at the age of 32 weeks. All four-chambers were dilated in the vehicle-treated Gαq-TG heart compared with those in NTG and olmesartan-treated Gαq-TG hearts (Fig. 1A). The marked cardiomegaly was observed in the vehicle-treated Gαq-TG mouse. The heart/body weight ratio increased in vehicle-treated Gαq-TG mice compared with that in NTG mice. Olmesartan significantly reduced the ratio in Gαq-TG mice (Table 2). The left atrial size/tibial length ratio was also increased in vehicle-treated Gαq-TG compared with that in NTG hearts. Olmesartan also decreased the ratio in Gαq-TG hearts (Table 2). Representative M-mode echocardiograms are shown in Figure 1B. Compared with the NTG mice, vehicle-treated Gαq-TG mice showed the markedly reduced LVFS and the increased LVEDd (Fig. 1B and Table 3). Interestingly, olmesartan significantly improved the reduced LVFS and increased LVEDd in Gαq-TG mice (Fig. 1B and Table 3).
Figure 1. Effects of olmesartan on cardiac morphology and on the left ventricular contractile function.

Panel A: Gross examination of a heart and its four-chamber view histology stained with hematoxylin/eosin in NTG, Gαq-TG, and Gαq-TG+olmesartan mouse hearts. The four-chamber view histology revealed all chambers to be dilated in the vehicle-treated Gαq-TG heart compared with those in NTG and olmesartan-treated Gαq-TG hearts. Original magnification: 1.25×. Mice at the age of 32 weeks were used. Panel B: Representative M-mode echocardiograms of NTG, Gαq-TG, and Gαq-TG+olmesartan mice at the age of 32 weeks.
Table 2. General parameters and the incidence of premature ventricular contraction (PVC) in NTG, Gαq-TG, and Gαq-TG+olmesartan mice.
| Parameters | NTG | Gαq-TG | Gαq-TG+olmesartan |
| BW (g) | 26.5±1.9 | 27.8±2.0 | 32.0±1.9 |
| HW (mg) | 133±5.5 | 206±20.4c | 172±8.9a |
| HW/BW (mg/g) | 5.1±0.4 | 7.5±0.7b | 5.4±0.5+ |
| LA/TL (mm/mm) | 0.14±0.06 | 0.33±0.03c | 0.19±0.02$ |
| PVC (>20 beats/min) | 0/10 | 9/10c | 0/10+ |
Data are the mean ± SE obtained from 10 mice for each group. ap<0.05, bp<0.01, cp<0.001 vs. NTG, +p<0.01, $p<0.001 vs. values in corresponding parameters of Gαq-TG.
Table 3. Echocardiographic parameters in NTG, Gαq-TG, and Gαq-TG+olmesartan mice.
| Parameters | NTG | Gαq-TG | Gαq-TG+olmesartan |
| IVS (mm) | 0.73±0.03 | 0.62±0.05 | 0.75±0.07 |
| LVEDd (mm) | 2.6±0.2 | 3.5±0.1c | 2.8±0.2+ |
| LVFS (%) | 50.9±2.5 | 25.4±1.7c | 44.2±2.5a,$ |
Data are the mean ± SE obtained from 7 mice for each group. ap<0.01, cp<0.001 vs. NTG, +p<0.01, $p<0.001 vs. values in corresponding parameters of Gαq-TG. LVEDd, left ventricular end-diastolic dimension; IVS, intraventricular septum.
Olmesartan-induced Reduction of the Number of Premature Ventricular Contractions (PVCs) in Gαq-TG Mice
Figure 2 shows representative ECGs recorded form anesthetized NTG, vehicle-treated Gαq-TG, and olmesartan-treated Gαq-TG mice. The middle ECG shows ventricular arrhythmias recorded from vehicle-treated Gαq-TG mice. Premature ventricular contraction (PVC) and non-sustained ventricular tachyarrhythmia (VT) were frequently observed. In contrast, the upper and lower ECGs recorded from an NTG- and olmesartan-treated Gαq-TG mouse showed P waves and QRS complexes with regular RR intervals without any arrhythmia, indicating a sinus rhythm. Table 2 shows the overall data for ventricular arrhythmias. NTG mice did not induce ventricular arrhythmias such as a high PVC count (more than 20 beats/min). In contrast, a high number of PVCs was observed in 9 of 10 vehicle-treated Gαq-TG mice (Table 2). Moreover, non of olmesartan-treated Gαq-TG mice induced a high PVC count, indicating a significant reduction of ventricular arrhythmias in olmesartan-treated Gαq-TG mice compared with that in vehicle-treated Gαq-TG mice.
Figure 2. Electrocardiogram (ECG) lead II recordings from NTG, Gαq-TG, and Gαq-TG+olmesartan mice.
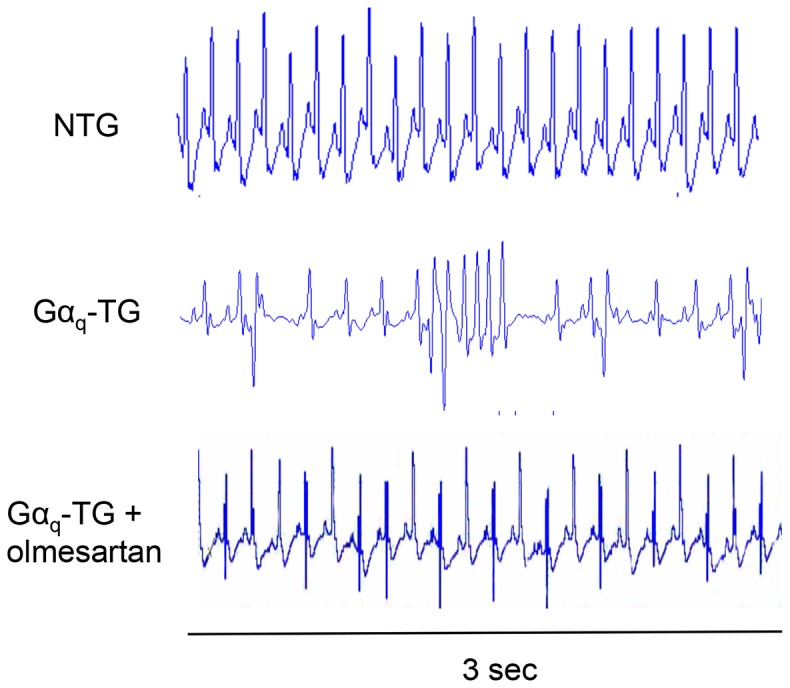
The middle ECG shows ventricular arrhythmias recorded from vehicle-treated Gαq-TG mice. PVC was frequently observed. In contrast, the upper and lower ECGs recorded from an NTG and olmesartan-treated Gαq-TG mouse showed P waves and QRS complexes with regular RR intervals without any arrhythmia, indicating a sinus rhythm. Mice at the age of 32 weeks were used.
Effects of Olmesartan on Changes in Electrocardiogram Parameters in Gαq-TG Mice
Overall data for the electrophysiological parameters in NTG, vehicle-treated Gαq-TG, and olmesartan-treated Gαq-TG mice at 32 weeks of age are shown in Table 4. P, PR, QRS, and QT interval were longer in vehicle-treated Gαq-TG mice than in NTG mice. Interestingly, while the prolonged QRS interval was still observed in olmesartan-treated Gαq-TG mice compared with NTG mice, the P, PR, and QT intervals were restored to normal levels in olmesartan-treated Gαq-TG compared with those in vehicle-treated Gαq-TG mice.
Table 4. Electrocardiographic parameters in NTG, Gαq-TG, and Gαq-TG+olmesartan mice.
| Parameters | NTG | Gαq-TG | Gαq-TG+olmesartan |
| P (msec) | 21±1 | 29±1b | 22±1$ |
| RR (msec) | 182±12 | 218±29 | 229±19 |
| PR (msec) | 49±5 | 87±8a | 50±4& |
| QRS (msec) | 15±0.6 | 20±1a | 21±1a |
| QT (msec) | 33±1 | 43±2b | 37±1a,+ |
Data are the mean ± SE obtained from 7 mice for each group. ap<0.05, bp<0.001 vs. WT, +p<0.05, &p<0.01, $p<0.001 vs. values in corresponding parameters of vehicle-treated Gαq-TG.
Effects of Olmesartan on Myocardial Fibrosis and the mRNA Expression of Profibrotic Genes in Gαq-TG Mice
The effects of chronic olmesartan administration on left ventricular myocardial fibrosis and profibrotic gene expressions of Gαq-TG mice at the age of 32 weeks are shown in figure 3. Vehicle-treated Gαq-TG hearts induced extensive interstitial fibrosis in the left ventricle compared with that in NTG and olmesartan-treated Gαq-TG hearts. The degree of myocardial fibrosis in the left ventricle was significantly greater in vehicle-treated Gαq-TG mice compared with that in NTG mice (Fig. 3B). Olmesartan-tretated Gαq-TG mice showed the reduced interstitial fibrosis compared with vehicle-treated Gαq-TG mice (Fig. 3B). Interestingly, compared with NTG mouse hearts CTGF and collagen type 1 mRNA expression levels were significantly upregulated in vehicle-treated Gαq-TG mouse hearts (Fig. 3C and D). Olmesartan significantly decreased the increased expression of those profibrotic genes in Gαq-TG hearts (Fig. 3C and D).
Figure 3. Effects of olmesartan on the left ventricular fibrosis and on connective tissue growth factor (CTGF) and collagen type 1 gene expression.

Panel A: Histology of the left ventricle stained with Masson’s trichrome in NTG, Gαq-TG, and Gαq-TG+olmesartan mice. Original magnification: 40×. Panel B: Comparison of the fibrosis fraction in the left ventricle in NTG, Gαq-TG, and Gαq-TG+olmesartan mice. Panels C and D: Quantitative analyses of CTGF (C) and collagen type 1 (D) gene expression by real-time reverse transcriptase-polymerase chain reaction (RT-PCR) in NTG, Gαq-TG, and Gαq-TG+olmesartan hearts. Data for CTGF and collagen type 1 were normalized to those for ARPP0. Data are the mean ± SE obtained from 6 mice for each group.
Effects of Olmesartan on Cardiomyocyte Hypertrophy, Fetal Gene Expression and TRPC 6 Channel Protein Levels in Gαq-TG Mice
The effects of olmesartan on the cardiomyocyte hypertrophy and the mRNA expression of fetal type genes such as ANF, β-MHC, and BNP in Gαq-TG mice are shown in figure 4. The cross-sectional diameter of cardiomyocytes in vehicle-treated Gαq-TG mice was longer than that in NTG mice (Fig. 4A). The increased cross-sectional diameter was significantly decreased in olmesartan-treated Gαq-TG mice (Fig. 4A). Moreover, the mRNA expression levels of ANF, BNP, and β-MHC were significantly upregulated in Gαq-TG hearts compared with that in NTG mouse hearts (Figs. 4B, C, and D). The increased gene expression of ANF, BNP, and β-MHC was decreased in olmesartan-treated Gαq-TG hearts (Figs. 4B, C, and D). Recent studies have suggested that the activation of TRPC channels plays important roles in the generation of cardiac hypertrophy and cardiac arrhythmia induction [4], [16]. Moreover, TRPC3 and 6 protein expression levels were increased in Gαq-TG mouse hearts [4]. Therefore, we examined the effects of olmesartan on the protein expression of TRPC3 and 6 channels in Gαq-TG hearts. Compared with NTG hearts, the vehicle-treated Gαq-TG hearts exhibited the increased TRPC 3 and 6 protein levels (Figs. 4E and F). The increased expression of TRPC 6 protein was decreased in olmesartan-treated Gαq-TG mouse hearts (Fig. 4F).
Figure 4. Effects of olmesartan on the left ventricular hypertrophy, on ANP, BNP, and β-MHC gene expression, and on protein expression of canonical transient receptor potential (TRPC) channel isoforms.

Panel A: Comparison of cardiomyocyte size in the left ventricle in NTG, Gαq-TG, and Gαq-TG+olmesartan mice. Panels B–D: Quantitative analyses of ANP (B), BNP (C), and β-MHC (D) gene expression by real-time RT-PCR in NTG, Gαq-TG, and Gαq-TG+olmesartan hearts. Data for ANP, BNP, and β-MHC were normalized to those for ARPP0. Data are the mean ± SE obtained from 6 mice for each group. Panel E: Expression of TRPC channel isoforms in NTG, Gαq-TG, and Gαq-TG+olmesartan hearts. TRPC isoform expression was normalized to actin expression and is expressed relative to wt (set at 1). Data are the mean ± SE obtained from 6 mice for each group. ANF, atrial natriuretic factor; BNP, B-type natriuretic peptide; β-MHC, β-myosin heavy chain; ARPP0, acidic ribosomal protein P0. Mice at the age of 32 weeks were used.
Effects of Olmesartan on Angiotensin Converting Enzyme (ACE) and Angiotensin II Type 1 (AT1) Receptor Protein Expression in Gαq-TG Mice
We examined the protein expression levels of ACE, ACE2, and AT1 receptor in NTG, vehicle-treated Gαq-TG, and olmesartan-treated Gαq-TG mice at the age of 32 weeks. The level of ACE but not ACE2 was significantly increased in Gαq-TG hearts compared with that in NTG hearts (Figs. 5A and B). Olmesartan tended to decrease the increased expression of ACE in Gαq-TG mouse hearts (Fig. 5A). The level of AT1 receptor was not changed in Gαq-TG hearts compared with that in NTG hearts (Fig. 5C). Olmesartan significantly increased the expression of AT1 receptor in Gαq-TG mouse hearts (Fig. 5C).
Figure 5. Effects of olmesartan on protein expression of angiotensin converting enzyme (ACE) isoforms and angiotensin II type 1 (AT1) receptor.

Expression of ACE (A), ACE2 (B), and AT1 receptor (C) in NTG, Gαq-TG, and Gαq-TG+olmesartan hearts. ACE isoform expression was normalized to actin expression and is expressed relative to NTG (set at 1). AT1 receptor was normalized to GAPDH and is expressed relative to NTG (set at 1). Data are the mean ± SE obtained from 6 mice for each group. Mice at the age of 32 weeks were used.
Effects of Olmesartan on Ventricular Monophasic Action Potential (MAP) in Gαq-TG Mice
Figure 6A showed examples of left ventricular MAPs in Langendorff-perfused NTG, Gαq-TG, and olmesartan-treated Gαq-TG mouse heart. The MAP duration prolonged in the Gαq-TG heart compared with that in the NTG and olmesartan-treated Gαq-TG mouse hearts. The overall data demonstrated that the chronic administration of olmesartan significantly shortened the ventricular MAP duration in Gαq-TG hearts.
Figure 6. Effects of chronic olmesartan treatment on ventricular monophasic action potential (MAP) duration.

Panel A: Representative examples of MAPs recorded from the posterior left ventricle in a Langendorff-perfused in NTG, Gαq-TG, and Gαq-TG+olmesartan hearts during steady state pacing at a cycle length of 200 msec. Panel B: Overall data of MAP duration in in NTG, Gαq-TG, and Gαq-TG+olmesartan hearts.
Discussion
In this study, we found that ACE but not ACE2 and AT1 receptor protein expression was increased in vehicle-treated Gαq-TG mouse hearts. Moreover, chronic administration of olmesartan for 26 weeks prevented the progression of heart failure and ventricular arrhythmia in Gαq-TG mice. We also found that olmesartan inhibited ventricular interstitial fibrosis and ventricular myocyte hypertrophy in Gαq-TG. CTGF, collagen type 1, ANP, BNP, and β-MHC gene expression was increased in vehicle-treated Gαq-TG. Olmesartan significantly decreased the expression of these genes in Gαq-TG mice. Electrocardiogram demonstrated that premature ventricular contraction (PVC) was frequently observed in 9 of 10 vehicle-treated Gαq-TG but in none of 10 olmesartan-treated Gαq-TG. These results suggest that angiotensin II type 1receptor activation plays crucial roles in cardiac remodeling and ventricular arrhythmia in Gαq-TG mice.
Clinical and experimental studies have demonstrated that the Gq-phosphoinositide signaling pathway plays important roles in the development of cardiac hypertrophy and heart failure [17]–[21]. It is well known that several bioactive factors such as angiotensin, endothelin, and norepinephrine activate the cardiac Gq-phosphoinositide signaling pathway. Our previous study showed that transient expression of a constitutively active mutant of Gαq in hearts of transgenic mice is sufficient to induce cardiac hypertrophy and dilatation. In fact, after the initiating stimulus of the transgenic constitutively active Gαq was not detected the cardiac hypertrophy and dilatation continued to progress [1]. We showed that the multiple and persistent changes in signal transduction pathways maintained cardiac pathological changes initiated by early transient expression of constitutively active Gαq [1], [5]. It is well known that the renin-angiotensin system, which increases the level of DAG, plays a critical role in the development of cardiac hypertrophy and HF [6]–[8]. In addition, cardiac renin-angiotensin system activation (i.e. local) is important in the development of cardiac hypertrophy [22]. Moreover, increased cardiac tissue ACE is known to play important roles in cardiac remodeling [23]. In this study, the protein expression of ACE was increased significantly in Gαq TG mouse hearts compared with that in NTG mouse hearts. In addition, left ventricular myocyte hypertrophy was observed and olmesartan significantly inhibited it (Fig. 4A), which was associated with the prevention of HF and ventricular arrhythmia induction in Gαq-TG mice (Fig. 4A). Moreover, mRNA expression of ANF, β-MHC, and BNP was significantly upregulated in Gαq-TG hearts compared with that in NTG mouse hearts and decreased by olmesartan in Gαq-TG hearts (Figs. 4B, C, and D). These results suggest that transient Gαq activation causes activation of the local renin-angiotensin system, leading to progressive heart failure and ventricular arrhythmias in Gαq-TG mice. These findings suggest that the cardiac renin-angiotensin system plays an important role in the development of cardiac hypertrophy and heart failure, even if the initiating stimulus of cardiac Gαq activation does not result from angiotensin II type I (AT1) receptor stimulation.
Several studies have demonstrated that cardiac remodeling is associated with increases in AT1 receptor protein expression [24]–[26]. Moreover, olmesartan suppressed cardiac AT1 receptor levels in hypertensive rats [24]. In this study, the protein expression levels of AT1 receptor were not changed in Gαq-TG hearts compared with those in NTG hearts (Fig. 5C). Moreover, olmesartan significantly increased the expression of AT1 receptor in Gαq-TG mouse hearts (Fig. 5C). The reason for the discrepancy between the previous and present results is uncertain. In fact, cardiac dysfunction is severe in this Gαq-TG mouse compared with that in animals used in previous studies [24], [26]. Moreover, the duration of olmesartan treatment was much longer in this study than in the previous study [24]. Those differences may explain the discrepancy. In any case, our present results suggest that AT1 receptor activation plays important roles in the development of heart failure and ventricular arrhythmias in this model.
It is known that myocardial ACE is a possible substrate for cardiac fibrosis [23]. In this study, the protein expression of ACE was increased significantly in Gαq TG mouse hearts compared with that in NTG mouse hearts. Moreover, the left ventricular fibrosis and mRNA expression of CTGF and collagen type I were also significantly increased in Gαq-TG mouse hearts. Olmesartan decreased the increased left ventricular fibrosis and the mRNA expression of CTGF and collagen type I, suggesting that the renin-angiotensin system participates in the development of cardiac fibrosis in this model. Importantly, together these findings suggest that the cardiac renin-angiotensin system plays an important role in the development of cardiac hypertrophy, fibrosis and heart even if the initiating stimulus of cardiac Gαq activation does not result from AT1 receptor stimulation.
It has been shown that mechanical stress activates AT1 receptor independently of angiotensin II, and this activation can be inhibited by an inverse agonist of the AT1 receptor [27]–[28]. Our previous study demonstrated that the left ventricular end-diastolic pressure was increased in Gαq-TG compared with that in NTG mice [2], suggesting that mechanical stretching of the myocardium was induced in Gαq-TG mice, leading to activation of AT1 receptors. Recent study has demonstrated that olmesartan has strong inverse agonist activities against the constitutively active AT1 receptor and the stretch-induced activation of AT1 receptor, respectively [28]. Therefore, olmesartan induced inhibition of ventricular myocyte hypertrophy and interstitial fibrosis in Gαq-TG may be caused in part through inverse agonistic action.
In this study, chronic administration of olmesartan prevented the progression of heart failure and ventricular arrhythmia in Gαq-TG mice. In fact, electrocardiogram demonstrated that PVC was frequently (more than 20 beats/min) observed in 9 of 10 vehicle-treated Gαq-TG mice but in none of 10 olmesartan-treated Gαq-TG mice. In addition, the QT interval was significantly shorter in olmesartan-treated Gαq-TG than in vehicle-treated Gαq-TG mice. Moreover, the MAP duration was also significantly shorter in olmesartan-treated Gαq-TG than in vehicle-treated Gαq-TG mice. It is well known that ventricular arrhythmias are common in heart failure. However, a recent study demonstrated that chronic angiotensin II stimulation in the heart directly induced QT prolongation through down-regulation of potassium channels, [29] which can induce triggered activity, leading to the production of PVC. Moreover, a recent study clearly demonstrated that AT1 receptor signaling in the heart directly contributed to the increased arrhythmogenicity in cardiac hypertrophy [30]. In fact, our previous study demonstrated that early-after depolarization by the prolongation of action potential duration caused triggered activity. Therefore, in addition to improvement of heart failure olmesartan might directly inhibit PVC induction because of the shortening of action potential duration. We previously demonstrated that the protein levels of TRPC3 and 6 are increased in Gαq-TG hearts [4] and suggested that the activation of TRPC channels participates in the generation of cardiac arrhythmia induction. Interestingly, olmesartan decreased the increased expression of TRPC 6 in Gαq-TG mouse hearts (Fig. 4F) in this study, suggesting that AT1 receptor activation contributes to an increase in TRPC6 expression, leading to ventricular arrhythmia induction.
Supporting Information
Primers used in this study.
(XLS)
Acknowledgments
We are grateful to Ms. Reiko Sakai for her secretarial assistance.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was supported in part by a Grant-in-Aid for Scientific Research from Ministry of Education, Culture, Sports, Science and Technology, Japan (No. 21590276) (M.H.) and the grant from KEIRYOKAI research foundation (M.H.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Mende U, Kagen A, Cohen A, Aramburu J, Schoen FJ, et al. (1998) Transient cardiac expression of constitutively active Gaq leads to hypertrophy and dilated cardiomyopathy by calcineurin-dependent and independent pathways. Proc Natl Acad Sci USA 95: 13893–13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Niizeki T, Takeishi Y, Kitahara T, Arimoto T, Koyama Y, et al. (2008) Diacylglycerol kinase zeta rescues G alpha q-induced heart failure in transgenic mice. Circ J 72: 309–317. [DOI] [PubMed] [Google Scholar]
- 3. Hirose M, Takeishi Y, Niizeki T, Shimojo H, Nakada T, et al. (2009) Diacylglycerol kinase ζ inhibits Gαq-induced atrial remodeling in transgenic mice. Heart Rhythm 6: 78–84. [DOI] [PubMed] [Google Scholar]
- 4. Hirose M, Takeishi Y, Niizeki T, Nakada T, Shimojo H, et al. (2011) Diacylglycerol kinase ζ inhibits ventricular tachyarrhythmias in a mouse model of heart failure: Roles of canonical transient receptor potential (TRPC) channels. Circ J 75: 2333–2342. [DOI] [PubMed] [Google Scholar]
- 5. Mende U, Kagen A, Meister M, Neer EJ (1999) Signal transduction in atria and ventricles of mice with transient cardiac expression of activated G protein alpha(q). Circ Res 85: 1085–1091. [DOI] [PubMed] [Google Scholar]
- 6. Granger CB, McMurray JJ, Yusuf S, Held P, Michelson EL, et al. (2003) Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin converting enzyme inhibitors: the CHARM-Alternative trial. Lancet 362: 772–776. [DOI] [PubMed] [Google Scholar]
- 7. The SOLVD Investigators (1991) Effect of enalapril on survival in patients with reduced left ventricular ejection fractions, and congestive heart failure. N Engl J Med 325: 293–302. [DOI] [PubMed] [Google Scholar]
- 8. Farmer JA, Torre-Amione G (2001) The renin angiotensin system as a risk factor for coronary artery disease. Curr Atheroscler Rep 3: 117–124. [DOI] [PubMed] [Google Scholar]
- 9. Nishio M, Sakata Y, Mano T, Yoshida J, Ohtani T, et al. (2007) Therapeutic effects of angiotensin II type 1 receptor blockerat an advanced stage of hypertensive diastolic heart failure. J Hypertens 25: 455–461. [DOI] [PubMed] [Google Scholar]
- 10. Yoshida K, Kohzuki M (2004) Clinical and experimental aspects of olmesartan medoxomil, a new angiotensin II receptor antagonist. Cardiovasc Drug Rev 22: 285–308. [DOI] [PubMed] [Google Scholar]
- 11.Hirose M, Takeishi Y, Nakada T, Shimojo H, Kashihara T, et al. Nicorandil prevents Gαq-induced progressive heart failure and ventricular arrhythmias in transgenic mice. PLoS One 7;e52667, 2012. [DOI] [PMC free article] [PubMed]
- 12. Kamiyoshi Y, Takahashi M, Yokoseki O, Yazaki Y, Hirose S, et al. (2005) Mycophenolate mofetil prevents the development of experimental autoimmune myocarditis. J Mol Cell Cardiol 39: 467–477. [DOI] [PubMed] [Google Scholar]
- 13. Niizeki T, Takeishi Y, Kitahara T, Arimoto T, Ishino M, et al. (2008) Diacylglycerol kinase-ε restores cardiac dysfunction under chronic pressure overload: a new specific regulator of Gαq signaling cascade. Am J Physiol 295: H245–H255. [DOI] [PubMed] [Google Scholar]
- 14. Sanbe A, Daicho T, Mizutani R, Endo T, Miyauchi N, et al. (2009) Protective effect of geranylgeranylacetone via enhancement of HSPB8 induction in desmin-related cardiomyopathy. PLoS One 4: e5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sanbe A, Marunouchi T, Yamauchi J, Tanonaka K, Nishigori H, et al. (2011) Cardioprotective effect of nicorandil, a mitochondrial ATP-sensitive potassium channel opener, prolongs survival in HSPB5 R120G transgenic mice. PLoS ONE 25: e18922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, et al. (2006) TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J 25: 5305–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harada K, Komuro I, Shiojima I, Hayashi D, Kudoh S, et al. (1998) Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation 97: 1952–1959. [DOI] [PubMed] [Google Scholar]
- 18. Schultz Jel J, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, et al. (2002) TGF-1 mediates mediates the hypertrophic cardio-myocyte growth induced by angiotensin II. J Clin Invest 109: 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Izumiya Y, Kim S, Izumi Y, Yoshida K, Yoshiyama M, et al. (2003) Apoptosis signal-regulating kinase 1 plays a pivotal role in angiotensin II–induced cardiac hypertrophy and remodeling. Circ Res 93: 874–883. [DOI] [PubMed] [Google Scholar]
- 20. Takeishi Y, Jalili T, Ball NA, Walsh RA (1999) Responses of cardiac protein kinase C isoforms to distinct pathological stimuli are differentially regulated. Circ Res 85: 264–271. [DOI] [PubMed] [Google Scholar]
- 21. Hunter JJ, Chien KR (1999) Signaling pathways for cardiac hypertrophy and failure. N Engl J Med 341: 1276–1283. [DOI] [PubMed] [Google Scholar]
- 22. Mazzolai L, Nussberger J, Aubert JF, Brunner DB, Gabbiani G, et al. (1998) Blood pressure-independent cardiac hypertrophy induced by locally activated renin-angiotensin system. Hypertension 31: 1324–1330. [DOI] [PubMed] [Google Scholar]
- 23. Sun Y, Weber KT (1994) Fibrosis and myocardial ACE: possible substrate and independence from circulating angiotensin II. J Card Fail 1: 81–89. [DOI] [PubMed] [Google Scholar]
- 24. Fukui S, Fukumoto Y, Suzuki J, Saji K, Nawata J, et al. (2009) Diabetes mellitus accelerates left ventricular diastolic dysfunction through activation of the renin-angiotensin system in hypertensive rats. Hypertens Res 32: 472–480. [DOI] [PubMed] [Google Scholar]
- 25. Wang XH, Wang WF, Cao YX, Ma AQ (2013) Ang II receptor expression and effect of Ang II receptor blockade in thyrotoxic rat myocardium. Eur Rev Med Pharmacol Sci 17: 2619–2627. [PubMed] [Google Scholar]
- 26.Lin L, Xu J, Ye Y, Ge J, Zou Y, et al.. (2014) Isosorbide dinitrate inhibits mechanical stress-induced cardiac hypertrophy and autophagy through downregulation of angiotensin II type 1 receptor. J Cardiovasc Pharmacol 2014Epub ahead of print. [DOI] [PubMed]
- 27. Zou Y, Akazawa H, Qin Y, Sano M, Takano H, et al. (2004) Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol 6: 499–506. [DOI] [PubMed] [Google Scholar]
- 28. Qin Y, Yasuda N, Akazawa H, Ito K, Kudo Y, et al. (2009) Multivalent ligand-receptor interactions elicit inverse agonist activity of AT1 receptor blockers against stretch-induced AT1 receptor activation. Hypertens Res 32: 875–883. [DOI] [PubMed] [Google Scholar]
- 29. Domenighetti AA, Boixel C, Cefai D, Abriel H, Pedrazzini T (2007) Chronic angiotensin II stimulation in the heart produces an acquired long QT syndrome associated with IK1 potassium current downregulation. J Mol Cell Cardiol 42: 63–70. [DOI] [PubMed] [Google Scholar]
- 30. Yasuno S, Kuwahara K, Kinoshita H, Yamada C, Nakagawa Y, et al. (2013) Angiotensin II type 1a receptor signalling directly contributes to the increased arrhythmogenicity in cardiac hypertrophy. Br J Pharmacol 170: 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used in this study.
(XLS)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.