

Abstract
Background
Human pluripotent stem cells (PSCs) play an important role in disease modeling and drug testing. However, the current methods are time consuming and lack an isogenic control.
Objectives
To establish an efficient technology to generate human PSCs-based disease models with isogenic control.
Methods
The ion channel genes KCNQ1 and KCNH2 with dominant negative mutations causing long QT syndrome (LQTS) type-1 and -2, respectively, were stably integrated into a safe harbor AAVS1 locus using zinc finger nuclease (ZFN) technology.
Results
Patch-clamp recording revealed that the edited induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) displayed characteristic LQTS phenotype and significant prolongation of the action-potential duration (APD) compared with the un-edited control cells. Finally, addition of nifedipine (L-type calcium channel blocker) or pinacidil (KATP-channel opener) shortened the APD of iPSC-CMs, confirming the validity of isogenic iPSC lines for drug testing in the future.
Conclusions
Our study demonstrates that PSC-based disease models can be rapidly generated by overexpression of dominant negative gene mutants.
Keywords: stem cells, disease models, genome editing, drug testing, long QT syndrome
Introduction
Predictive disease models, such as animal models and cell lines, play an important role in studying the pathophysiological mechanisms of human disease and developing targeted therapies. However, many human phenotypes fail to be successfully recapitulated in these models owing to species differences or a lack of synteny. Human pluripotent stem cell (PSC) biology has opened a new avenue for disease modeling. There are 2 main types of human PSCs: embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) (1-4). Human PSCs can self-renew indefinitely and can be differentiated to any cell types present in the human body. Due to their properties and origin, human PSCs are considered to be an inexhaustible, scalable, and physiologically native material for study.
For human disease modeling, the first step is to obtain the PSCs with disease-causing genetic mutations, and subsequently differentiate them into the disease relevant cell types. Disease-specific iPSCs can be obtained through the conversion of somatic cells isolated from patients with genetic mutations. By using this approach, several groups have recently modeled familial dilated cardiomyopathy (DCM) (5), familial hypertrophic cardiomyopathy (HCM) (4), Timothy syndrome (6), LEOPARD syndrome (7), arrhythmogenic right ventricular dysplasia (ARVD) (8), and long QT syndrome (LQTS) (3,9). However, the limitation of this approach is that obtaining patient samples with the desired disease-causing mutation is unpredictable, the subsequent iPSC generation from skin fibroblasts or peripheral blood mononuclear cells is time consuming, and the disease models generated from various individuals lack an isogenic unaffected control. Most current iPSC studies use age-matched, unaffected cells within the same family pedigree as control (4,9,10), but these are suboptimal due to differences in the genetic background and other confounders.
Recent advances in genome editing technology allow the direct introduction of specific genetic mutations into the human PSCs to make disease models, with the un-edited cells serving as an isogenic control. This technology relies on artificially engineered nucleases to cut and create specific double-stranded breaks (DSBs) at predetermined locations in the genome. The DSBs can be repaired by the cell's endogenous DNA repair system through either homologous recombination (HR) or non-homologous end-joining (NHEJ) to produce desired mutations. Several diseases have been modeled using this technique by introducing mutations into the human PSCs (11-13). Although the genome editing technology is powerful for disease modeling, the technology remains a challenge for the non-specialist.
Long QT syndrome (LQTS) is an inherited cardiac arrhythmic disease, predisposing the patient to life-threatening ventricular arrhythmias and sudden cardiac death. Mutations in the potassium channels, KCNQ1 (LQTS1) and KCNH2 (LQTS2), account for the 2 most common clinically definite LQTSs (14). Since KCNQ1 and KCNH2 function as tetramers, a substantial number of KCNQ1 and KCNH2 mutants display dominant-negative effect, because they interact with wild-type monomer and impair tetramerization (9,10,14). In this study, we successfully modeled LQTS by overexpression of dominant negative mutations in human PSCs, using un-edited human PSCs as isogenic controls. We further showed that the LQTS models generated by this approach can be used for drug screening. Our study demonstrates an easy and efficient strategy to generate human disease models.
Methods
An extended methods section is available in the Online Data Supplement.
Cell culture and maintenance of human pluripotent stem cells
Human ESCs (WA09, Wicell, Madison, WI) and iPSCs were cultured on Matrigel-coated plates (ESC qualified, BD Biosciences, San Diego, CA) using hESC mTeSR-1 cell culture medium (StemCell Technologies, Vancouver, Canada) under conditions of 37°C, 95% air, and 5% CO2 in a humidified incubator, as previously described (15). Results for subsequent experiments are based on 1 hESC line (WA09), 4 un-edited iPSC lines (2 from healthy individuals, 1 from patient with G269S mutation, 1 from patient with A614V mutation), 4 edited iPSC lines (with mutations R190Q, G269S, and G345E on KCNQ1, and A614V on KCNH2, respectively), and 2 edited ESC lines (with mutations G269S on KCNQ1 and A614V on KCNH2, respectively).
Vector construction
The DNA fragment containing EF1a promoter was polymerase chain reaction (PCR)-amplified from the pCDH_EF1_MCS_T2A_copGFP vector (System Biosciences, Mountain View, CA) and digested with restriction enzymes MluI and NcoI (NEB). The fragment was then inserted into the MluI/NcoI-cut site of the donor plasmid AAV-CAGGS-eGFP backbone (Addgene, Cambridge, MA) (16). The resultant plasmid was designated AAVS1-EF1a. Human cDNA clones containing KCNQ1 and KCNH2 were obtained from GeneCopoeia (Rockville, MD). The coding regions were PCR amplified and cloned into the AAVS1-EF1a KpnI/AgeI site for the KCNQ1, and AflII/EcoRV site for the KCNH2. The mutations G569A (R190Q on KCNQ1), G805A (G269S on KCNQ1), G1034A (G345E on KCNQ1), and C821T (A614V on KCNH2) were generated by Mutagenex Inc. (Piscataway, NJ).
Gene targeting using AAVS1 ZFNs Refer to the Online Data Supplement.
Patch-clamp electrophysiology Refer to the Online Data Supplement.
Imaging of calcium dynamics in genome edited iPSC-CMs Refer to the Online Data Supplement.
Statistical analysis
Results are expressed as mean ± SEM. We used Student's t-test for the comparison between 2 normally distributed groups of data. One-way or two-way analysis of variance (ANOVA) followed by all pairwise multiple comparison procedures, where appropriate, was used for the comparison of multiple groups of data. A p-value of <0.05 was considered significant.
Results
ZFN-mediated targeted gene addition into AAVS1 safe harbor locus in ESCs and iPSCs
To test the hypothesis that the disease models can be created by overexpression of genes with dominant negative mutations, we chose 3 mutations on KCNQ1 (R190Q, G269S, and G345E) (3,10,17) and 1 mutation on KCNH2 (A614V) (9) to model LQTS1 and LQTS2, respectively (Figure 1A). To allow a direct comparison of different disease models and avoid mutagenic random integration or epigenetic silencing via viral insertions, the transgenes were inserted into a “safe harbor” AAVS1 locus using zinc finger nuclease (ZFN) technology (Figure S1A) (18). These ion gene mutants were subcloned into the donor vector driven by the EF1a promoter flanked by short (800 bp) stretches of homology to the ZFN target site (16) on chr 19 in exon 1 of the PPP1R12C gene (16). The donor construct and the ZFN expression vector were introduced into iPSCs by electroporation. The ZFN-edited iPSC lines with mutations R190Q, G269S, G345E, and A614V are designated as ziR190Q, ziG269S, ziG345E, and ziA614V, respectively. In addition to these 4 ZFN-edited iPSC lines, we also introduced G269S-KCNQ1 and A614VKCNH2 mutants into human ESCs (WA09); these lines are designated as H9-G269S and H9-A614V, respectively.
Figure 1.

Overexpression of the KCNQ1 and KCNH2 genes with dominant negative mutation at the AAVS1 locus.
(A) Schematic representation of the KCNQ1 and KCNH2 proteins, with the R190Q mutation located in the cytoplasmic loop between transmembrane segments S2 and S3; G269S mutation located in the S5; G345E mutation located in the S6 of the KCNQ1 protein; and A614V mutation in the pore region of the KCNH2 protein. The six transmembrane domains (S1 to S6) are flanked by amino (NH2)–terminal and carboxyl (COOH)–terminal regions. (B) Transgene expression in the ZFN-edited iPSC lines (ziR190Q, ziG269S, ziG345E, and ziA614V) and control cell lines was confirmed by immunostaining. The upper left un-edited iPSCs were stained by anti-KCNQ1 antibody; the upper right un-edited iPSCs were stained by anti-KCNH2 antibody. (C) ziG269S expressed pluripotency markers that included Oct4, Tra-1–60, Sox2, Tra- 1–81, Nanog, and SSEA4. Blue stainings (right) are 4′-6-diamidino-2-phenylindole (DAPI) staining of nuclei. (D) ziG269S formed embryoid bodies (EBs) in vitro as shown by brightfield (BF, top left) microscopic appearance and by expression of the neuroectoderm marker Nestin (top right), mesoderm marker smooth muscle actin (SMA) (bottom left), and endoderm marker Sox17 (bottom right). (E) ziG269S formed teratomas in vivo in immunodeficient mice. The teratomas contain all 3 germ layers, identified here as neural element ectoderm (left), cartilage mesoderm (middle), and epithelium endoderm (right).
Following puromycin selection, 23 clones for each construct were screened by genomic PCR, 95.2% (197/207) of which carried the transgenic cassette at the ZFN-specified location (Figure S1B and Table 1). Of these PCR-positive clones, six clones for each mutation were further screened by Southern blotting (Figure S1C and Table 1). The results revealed that all the clones contained 1 or 2 copies of targeted gene addition, except 1 from ziG345E. We next genotyped a panel of putative ZFN off-target sites for each mutation (Figure S2), and data demonstrate all to be wild type, in agreement with previous studies on the robust specificity of this ZFN set (16,18). After extensive genetic characterization, immunostaining confirmed the transgene expression (Figure 1B and Figure S3).
Table 1. Efficiency of the gene addition to the AAVS1 locus.
| Cell line | Donor | PCR-positive clones / total screened clones | Clones screened by Southern blot | Targeted additional integrations | + Correct clones | targeted |
|---|---|---|---|---|---|---|
|
| ||||||
| Het. | Homo. | |||||
| iPSC | R190Q | 22/23 | 6 | 2 | 3 | 1 |
| iPSC | G269S | 22/23 | 6 | 4 | 2 | 0 |
| iPSC | G345E | 23/23 | 6 | 3 | 1 | 1 |
| iPSC | A614V | 22/23 | 6 | 4 | 0 | 2 |
| H9 | G269S | 21/23 | 6 | 4 | 2 | 0 |
| H9 | A614V | 21/23 | 6 | 3 | 1 | 2 |
Genome editing does not affect pluripotent stem cell characteristics
To investigate whether the genetic engineering and transgene overexpression would affect stemness, we tested the pluripotency of our cells after ZFN editing and transgene overexpression and observed that all the transgenic cell lines displayed normal morphology relative to control unmodified iPSCs (Figure S4). Additional testing revealed that transgenic cells maintained their pluripotent state, as indicated by the expression of pluripotency markers, such as Oct4, Tra-1–60, Sox2, Tra-1–81, Nanog, and SSEA4 (Figure 1C and Figure S5). Functionally, these cells were capable of differentiation into all 3 germ layers both in vitro (Figure 1D and Figure S6A) and in vivo (Figure 1E and Figure S6B). The transgenic cells exhibited normal karyotypes (Figure S7). Taken together, these results show that transgenic iPSCs maintained their pluripotent potential after genetic engineering and transgene overexpression.
ZFN editing and transgene expression does not influence whole genome expression profile
We next differentiated the transgenic iPSCs into cardiomyocytes (iPSC-CMs) in vitro using a modified monolayer protocol, as previously described (19). After 12 days of differentiation, cell beating was observed for both transgenic and un-edited iPSC-CMs (Video S1-5). The cardiac marker expression (α-actinin, TNNT2, MLC2a, and MLC2v) in the transgenic iPSCs was similar to those seen in control un-edited iPSCs (Figure 2A). To study whether the genetic engineering and ion channel gene overexpression influence the expression of other genes in iPSC-CMs, gene expression array (Affymetrix, Santa Clara, CA) was performed with genome-edited ziG269S iPSC-CMs, patient11 derived G269S iPSC-CMs (piG269S) (3) and un-edited iPSC-CMs. As shown in Figure 2B, 20 genes that were closely related to the action potential forming, calcium handling, and conductivity of the iPSC-CMs were compared among the 3 groups. As expected, the KCNQ1 gene is up-regulated in ziG269S group, yet no obvious changes were shown in the expression level of other genes. Overall, our microarray analysis showed very similar gene expression patterns in these 3 groups, and the correlation between any 2 groups are no less than 97.5% (Figure 2C, 2D). In addition, we also tested the expression of a panel of ion channel gene expression by quantitative PCR, and their expression was similar to that for the un-edited iPSC-CMs, except the transgenes were overexpressed (Figure S8).
Figure 2.

Transgene overexpression did not change the whole genome gene expression profile. (A) The ZFN-edited iPSC-CMs expressed normal cardiac markers that included TNNT2 (top red), actinin (top green), MLC2a (bottom red), and MLC2v (bottom green). (B) Microarray analysis of gene expression pattern for un-edited, patient-specific piG269S, and ZFN-edited ziG269S iPSC-CMs. The expression profiles of 20 key genes in action potential formation and calcium handling were plotted as a heat map. Note that KCNQ1 was over-expressed in ziG269S iPSC-CMs. (C) Pearson's correlation analysis of expression profile from 3 lines of the tested iPSC-CMs. (D) Scatter plot comparison of the whole-genome wide gene expression pattern of the un-edited, piG269S, and ziG269S iPSC-CMs.
Transgenic cell lines recapitulated LQTS phenotypes
To assess whether the genome-edited iPSC-CMs recapitulated the LQTS phenotype, patch clamp recordings were performed from single cells, and cardiac action potentials were obtained. We first recorded the action potentials in parallel using iPSC-CMs derived from un-edited iPSC, ziG269S, and piG269S lines. Consistent with previous reports, cardiac action potentials were recorded from single iPSC-CMs and all three subtypes of myocytes (e.g., ventricular-like, atrial-like, and nodal-like) were detected in the 3 groups (Figure 3A). Key parameters of action potentials were collected and assessed (Table S1). There were no significant changes observed in key parameters except action potential duration at 90% repolarization (APD90). The ventricular-like and atrial-like myocytes derived from ziG269S exhibited significantly longer APD compared to un-edited iPSC-CMs, which are similar to the data obtained from piG269S iPSC-CMs.
Figure 3.

Whole-cell patch-clamp recording of genome edited LQTS iPSC-CMs. (A) Actionpotential recordings from un-edited, piG269S, and ziG269S iPSC-CMs showing ventricular-like and atrial-like morphologies. All the LQTS iPSC-CMs displayed the marked APD prolongation in both ventricular-like and atrial-like iPSC-CMs. (B) APD50, APD70, and APD90 values in unedited control and LQTS iPSC-CMs. **P<0.01 when compared to control cells. Error bars show mean ± s.e.m. (C) Development of triggered activity in LQTS iPSC-CMs, manifested as EADs or even single and multiple triggered beats. The EADs or aberrant beats were indicated by red arrows.
More interestingly, both the ZFN-edited ziG269S iPSC-CMs and patient-specific piG269S iPSC-CMs displayed early afterdepolarizations (EADs), which were not present in un-edited iPSC-CMs (Figure 3C). In addition, we also recorded action potentials from ziR190Q, ziG345E, ziA614V, and patient-derived A614V (piA614V) iPSC-CMs (9), all of which showed significantly prolonged APDs compared with the un-edited iPSC-CMs (Figure S9 and Table S1), consistent with previous studies (9,20). In conclusion, ZFN-edited LQTS iPSC lines can faithfully recapitulate the disease phenotype, which is similar to the one obtained from the patient-specific mutation-matched iPSC lines.
Genome-edited iPSC-CMs as a new platform for drug testing
To evaluate the suitability of the ZFN-edited iPSC disease model as a tool for drug screening, we next tested nifedipine (a specific Ca2+ blocker) on both ziG269S and piG269S iPSC-CMs. Consistent with previous studies (9), when tested on piG269S iPSC-CMs with a long APD baseline, nifedipine (100 nM) can shorten the APD due to its Ca2+ blockade effect (Figure 4A). Interestingly, nifedipine also can significantly shorten the APD when tested in ziG269S iPSC-CMs (Figure 4B), indicating that the genome-edited iPSC-CMs can be used as a new platform for drug testing. To confirm the conclusion above, we recorded spontaneous calcium transient in iPSC-CMs (Table S2). Compared to un-edited control iPSC-CMs, both ziG269S and piG269S iPSC-CMs exhibited greater prolongation of transient duration 90 by 63.6±7.9% and 60.8±8.6%, respectively (Table S2). After treatment with nifedipine, the transient duration 90 in both ziG269S and piG269S iPSC-CMs were significantly shortened, similar to the results obtained from patch clamping recording. We further tested the effect of pinacidil, a KATPchannel opener that can augment outward potassium currents. Interestingly, pinacidil (100 nM) also can shorten the transient duration 90 of both ziG269S and piG269S iPSC-CMs (Figure S10, Table S2). Taken together, these data suggest that both genome-edited iPSC models and patient13 derived iPSC models can be used for drug screening, with the added advantage that the former approach obviates patient recruitment, which can be time-consuming and unpredictable (Central Illustration).
Figure 4.
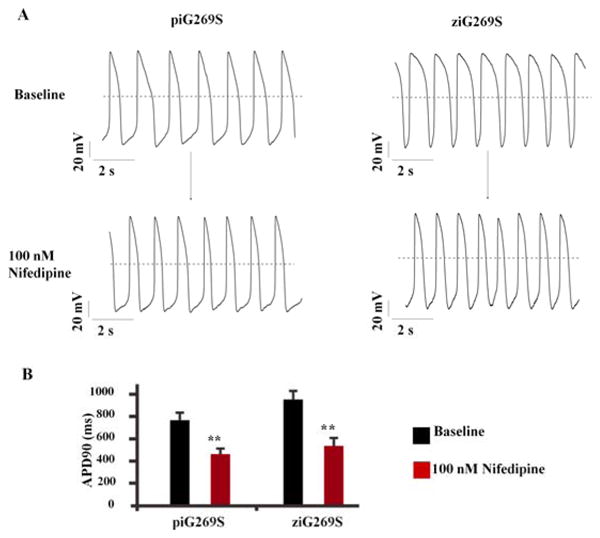
Drug evaluation using genome-edited LQTS iPSC-CMs. (A) APD shortening was induced by 100 nM nifedipine in LQTS iPSC-CMs. The left panel shows that nifedipine shortens patient-specific piG269S iPSC-CMs. The right panel shows nifedipine shortens ZFN-edited ziG269 iPSC-CMs. (B) APD90 before and after nifedipine treatment was quantified. **P<0.01 vs baseline by student's T-test.
Figure 5. (Central Illustration): Generation of Isogenic Human iPSC Lines for Cardiac Disease Modeling and Drug Testing.
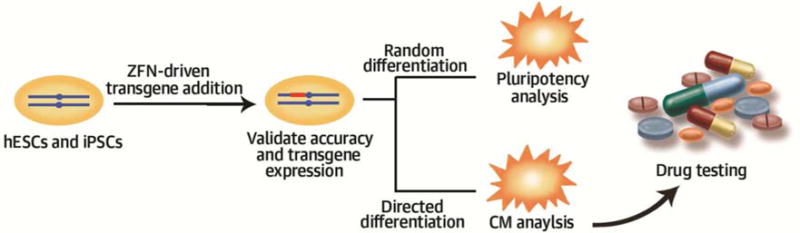
A rapid and efficient strategy was developed to generate human pluripotent stem cell-based disease models with isogenic control. First, ion channel genes, KCNQ1 or KCNH2, with dominant negative mutations causing long QT syndrome (LQTS) were introduced into human ESCs or iPSCs. To achieve stable transgene expression, the ion channel genes were integrated into AAVS1 locus using ZFN technology. The specificity of the addition process was validated by genomic PCR and Southern Blot; the transgene expression was validated by immunostaining; the pluripotency of the transgenic cells was confirmed by in vivo teratoma analysis and in vitro embryoid body differentiation. Next, the transgenic cells were differentiated into cardiomyocytes (CMs), with the un-edited cells serving as an isogenic control. The CMs was confirmed by immunostaining of the marker gene expression. Gene expression array analysis revealed that the transgene expression did not influence the whole-genome gene expression profile. The electrophysiology analysis showed that the ZFN-edited iPSC-CMs displayed prolonged action potential duration (APD), a typical phenotype of the LQTS-CMs. Finally, the application of ZFN-edited iPSC-CMs for drug screening was investigated. The addition of nifedipine (L-type calcium channel blocker) or pinacidil (KATP-channel opener) shortened the APD of iPSC-CMs, confirming the validity of isogenic iPSC lines for drug testing in the future.
Discussion
In the present work, we demonstrated a new strategy to create human PSC-based disease modeling by overexpression of genes with dominant negative mutations. This strategy is easy and highly efficient. Using this strategy, we successfully generated 6 cell lines representing LQTS1 and LQTS2 in a short period of time. Importantly, the un-edited parent cell lines can serve as an isogenic control. We further showed that this strategy can be used as an effective platform for drug screening (Central Illustration).
Human PSCs represent an excellent system for disease modeling, because they can be potentially differentiated to any cell types present in the human body. However, obtaining human PSCs with specific disease-causing genetic mutations can be unpredictable and time-consuming. By contrast, genetic engineering strategy to generate iPSC-based disease models is much easier and faster. The reagents and methods to engineer the AAVS1 locus already exist and have been optimized (16,18). An investigator with basic molecular biology skills can generate appropriate cell lines in 3 steps: identifying a gene with desired dominant negative mutations, cloning the gene into the targeting vector, and integrate the construct into the AAVS1 locus following the provided protocol. One person can generate several disease models in 3 months using this strategy.
The quality of the studies depends on the availability of appropriate controls, and hence, any phenotypes observed in the PSC-derived cells should only be interpreted via comparison with control cells, which ideally would have the same genetic background as the mutant PSCs and would differ only in the disease-linked gene. The usual practice is to generate control iPSC lines from a close relative of the patient who is age-matched and unaffected by the disease within the same pedigree, but they are often unavailable and represent only an approximate control. Differences in genetic background are another major concern. Even in studies in which healthy siblings have been used as controls for disease patients, only ∼50% of the genome is shared between any siblings. Furthermore, phenotypic differences could be the result of DNA variants in the other ∼50% of the genome, rather than the disease-associated mutations (21). By contrast, the strategy we developed in this study can use un-modified parental cell line as an isogenic control, which genetically differs from the transgenic cell lines only in the introduced gene. In our study, the ion channel genes with dominant negative mutations were overexpressed in the transgenic cells. The iPSC-CMs derived from transgenic PSCs showed normal cardiac gene expression, without changing the whole genome gene expression profile of the resultant cells. These iPSC-CMs recapitulated LQTS phenotypes and had prolonged APD. Importantly, the iPSC-CMs derived from both patient-specific and genome-edited iPSCs showed similar response to drugs that affect ion channels, demonstrating that transgenic iPSCs can be used for drug screening in the future.
It is widely recognized that human PSC-CMs are immature. Previous studies have shown that human ESC-CMs expressed cardiac genes at levels similar to those found in 20-week fetal heart cells (22,23). Mummery et al. have shown that the upstroke velocities for the ventricular-like cells were low but comparable to those in cultured human fetal ventricular cardiomyocytes (24). Nevertheless, several publications have revealed that these cells are capable of capturing specific traits of an electrical disease of the heart, including LQTS1 (3,10), LQTS2 (9), LQTS3 (25), and LQTS8 (6,26).
Finally, disease-specific iPSCs have enormous potential for drug screening. Proof of principle for iPSC models as a platform for small-molecule and chemical testing already exists. For example, neurons from spinal muscular atrophy (27) and familial dysautonomia (28) displayed in vitro defects that could be corrected by candidate drugs, and the effect of β-blocking drugs has been evaluated for LQTS iPSC-CMs (10). However, the possibility that the genetically edited PSCs could be used as a platform for the drug screening was not investigated until now.
In this study, we demonstrate that the LQTS phenotype of the genetic edited iPSCs-CMs can be corrected by the nifedipine and pinacidil. We further showed that the genetically edited LQTS models worked equally well on the patient LQTS iPSC models. In summary, our study provides investigators a robust and reliable system to generate iPSC-based disease models.
Perspectives
Competency in Medical Knowledge
Introducing ion channel genes with dominant negative mutations causing long QT syndrome into the human induced pluripotent stem cells can generate cardiomyocytes that display the long QT phenotype including prolonged action potential duration.
Translational Outlook 1
The integration of transgenes into specific loci using zinc finger nuclease technology avoids random mutagenic integration and epigenetic silencing via viral insertions and may be applicable to modeling of other diseases caused by dominant negative mutations.
Translational Outlook 2
This strategy has the potential to accelerate laboratory modeling of genetic diseases for identification of novel drug targets and testing pharmacological strategies in vitro to improve the likelihood of clinical efficacy.
Supplementary Material
Acknowledgments
This work was supported in part by grants from Leducq Foundation, American Heart Association 13EIA14420025, National Institutes of Health (NIH) R01 HL113006, NIH U01 HL099776, and NIH P01 GM099130 (JCW).
Abbreviations
- APD
action potential duration
- CM
cardiomyocyte
- ESC
embryonic stem cell
- iPSC
induced pluripotent stem cell
- LQTS
long QT syndrome
- PSC
human pluripotent stem cell
- ZFN
zinc finger nuclease
Footnotes
Conflict of interests: F.D. Urnov is a full-time employee of Sangamo Biosciences, Inc. Wu is a co-founder of Stem Cell Theranostics. The other authors have no conflicts to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Yan B, Singla DK. Transplanted induced pluripotent stem cells mitigate oxidative stress and improve cardiac function through the akt cell survival pathway in diabetic cardiomyopathy. Molecular Pharmaceutics. 2013;10(9):3425–3432. doi: 10.1021/mp400258d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liang P, Lan F, Lee AS, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation. 2013;127:1677–91. doi: 10.1161/CIRCULATIONAHA.113.001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lan F, Lee AS, Liang P, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–13. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun N, Yazawa M, Liu J, et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4:130ra47. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yazawa M, Hsueh B, Jia X, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–4. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carvajal-Vergara X, Sevilla A, D'Souza SL, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–12. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim C, Wong J, Wen J, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494:105–10. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Itzhaki I, Maizels L, Huber I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–9. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 10.Moretti A, Bellin M, Welling A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 11.Horii T, Tamura D, Morita S, et al. Generation of an ICF syndrome model by efficient genome editing of human induced pluripotent stem cells using the CRISPR system. Int J Mol Sci. 2013;14:19774–81. doi: 10.3390/ijms141019774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding Q, Lee YK, Schaefer EA, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–51. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bellin M, Casini S, Davis RP, et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. 2013;32:3161–75. doi: 10.1038/emboj.2013.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang T, Moss A, Cong P, et al. LQTS gene LOVD database. Hum Mutat. 2010;31:E1801–10. doi: 10.1002/humu.21341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson KD, Sun N, Huang M, et al. Effects of ionizing radiation on self-renewal and pluripotency of human embryonic stem cells. Cancer Res. 2010;70:5539–48. doi: 10.1158/0008-5472.CAN-09-4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeKelver RC, Choi VM, Moehle EA, et al. Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res. 2010;20:1133–42. doi: 10.1101/gr.106773.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Tristani-Firouzi M, Xu Q, et al. Functional effects of mutations in KvLQT1 that cause long QT syndrome. J Cardiovasc Electr. 1999;(10):817–26. doi: 10.1111/j.1540-8167.1999.tb00262.x. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Zhang WY, Hu S, et al. Genome editing of human embryonic stem cells and induced pluripotent stem cells with zinc finger nucleases for cellular imaging. Circ Res. 2012;111:1494–503. doi: 10.1161/CIRCRESAHA.112.274969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu S, Wilson KD, Ghosh Z, et al. MicroRNA-302 increases reprogramming efficiency via repression of NR2F2. Stem Cells. 2013;31:259–68. doi: 10.1002/stem.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moretti A, Bellin M, Welling A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 21.Musunuru K. Genome editing of human pluripotent stem cells to generate human cellular disease models. Dis Model Mech. 2013;6:896–904. doi: 10.1242/dmm.012054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao F, Wagner RA, Wilson KD, et al. Transcriptional and functional profiling of human embryonic stem cell-derived cardiomyocytes. PloS One. 2008;3:e3474. doi: 10.1371/journal.pone.0003474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beqqali A, Kloots J, Ward-van Oostwaard D, et al. Genome-wide transcriptional profiling of human embryonic stem cells differentiating to cardiomyocytes. Stem Cells. 2006;24:1956–67. doi: 10.1634/stemcells.2006-0054. [DOI] [PubMed] [Google Scholar]
- 24.Mummery C, Ward-van Oostwaard D, Doevendans P, et al. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003;107:2733–40. doi: 10.1161/01.CIR.0000068356.38592.68. [DOI] [PubMed] [Google Scholar]
- 25.Ma D, Wei H, Zhao Y, et al. Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int J Cardiol. 2013;168:5277–86. doi: 10.1016/j.ijcard.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 26.Terrenoire C, Wang K, Tung KW, et al. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol. 2013;141:61–72. doi: 10.1085/jgp.201210899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ebert AD, Yu J, Rose FF, Jr, et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–80. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee G, Studer L. Modelling familial dysautonomia in human induced pluripotent stem cells. Philosophical Transactions of the Royal Society of London, Series B. Biological Sciences. 2011;366:2286–96. doi: 10.1098/rstb.2011.0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.