

Abstract
In just seven years, next-generation technologies have reduced the cost and increased the speed of DNA sequencing by four orders of magnitude, and experiments requiring many millions of sequencing reads are now routine. In research, sequencing is being applied not only to assemble genomes and to investigate the genetic basis of human disease, but also to explore myriad phenomena in organismic and cellular biology. In the clinic, the utility of sequence data is being intensively evaluated in diverse contexts, including reproductive medicine, oncology and infectious disease. A recurrent theme in the development of new sequencing applications is the creative ‘recombination’ of existing experimental building blocks. However, there remain many potentially high-impact applications of next-generation DNA sequencing that are not yet fully realized.
The cost of DNA sequencing has plummeted since 2005 (refs. 1,2), from $1,000 per megabase down to a mere ten cents per megabase3,4. Next-generation technologies have also commoditized high-throughput DNA sequencing and rendered it broadly accessible to individual investigators outside of genome centers3,5 (J.S. and colleagues). For many applications, the cost of sequencing is already negligible in comparison to the costs of sample acquisition, library preparation and/or postsequencing data analysis (Box 1). For very large-scale applications, such as whole-genome sequencing of samples for entire cohort studies, the cost of sequencing remains substantial but only because we are undertaking projects that were out of reach just a few years ago in terms of scale and comprehensiveness.
Box 1. ‘Rate limiters’ of next-generation DNA sequencing experiments.
Until recently, the act of acquiring high-throughput molecular data (for example, DNA sequencing or DNA microarrays) was the primary cost associated with many experiments in genomics (for example, genome assembly, expression analysis and so on). For some projects, the emergence of next-generation sequencing has simply increased ambitions, such that DNA sequencing costs remain dominant. However, for most experiments, other ‘rate limiters’ are an increasing fraction of the overall cost and effort. These include the following: first, the cost of generating, acquiring and/or storing samples; second, the costs of constructing and indexing fragment libraries; third, the costs of building and maintaining infrastructure for large-scale data analysis, storage, exchange and deposition to public repositories; fourth, the time and labor costs of executing on both routine (for example, read mapping) and specialized (for example, data interpretation) tasks in large-scale data analysis; fifth, the costs of training personnel for the experimental and analytical skill sets associated with next-generation DNA sequencing; sixth, the costs associated with transient or persistent mismatches between the local capacity and local demand for next-generation DNA sequencing; and finally, for clinical samples, the costs associated with phenotyping subjects, obtaining consent from subjects and complying with regulations for working with human subjects.
As a consequence of these dramatic shifts in cost and accessibility, applications of DNA sequencing have proliferated (Table 1). Until 2005, the primary application of high-throughput DNA sequencing was to assemble reference genomes for humans and other high priority species. Today, sequencing has an increasingly fundamental role in the genetic analysis of human disease and model organism phenotypes as well as in addressing basic questions in organismic and cellular biology. For many researchers, the state of sequencing technology (for example, cost per genome, read-length constraints and so on) is already profoundly influencing the design and scope of their experiments. We predict that much of the agenda of biology in the coming decade will be driven in large part by the scientific opportunities afforded by next-generation DNA sequencing technologies. Understanding current applications of sequencing, the structure of sequencing experiments and principles for devising new sequencing applications will be useful for researchers seeking to effectively harness these opportunities.
Table 1.
Applications of next-generation DNA sequencing
| Method | Sequencing to determine: | Example reference | ‘Subway’ route as defined in Figure 3 |
|---|---|---|---|
| DNA-Seq | A genome sequence | 57 | Comparison, ‘anatomic’ (isolation by anatomic site), flow cytometery, DNA extraction, mechanical shearing, adaptor ligation, PCR and sequencing |
| Targeted DNA-Seq | A subset of a genome (for example, an exome) | 20 | Comparison, cell culture, DNA extraction, mechanical shearing, adaptor ligation, PCR, hybridization capture, PCR and sequencing |
| Methyl-Seq | Sites of DNA methylation, genome-wide | 34 | Perturbation, genetic manipulation, cell culture, DNA extraction, mechanical shearing, adaptor ligation, bisulfite conversion, PCR and sequencing |
| Targeted methyl-Seq | DNA methylation in a subset of the genome | 129 | Comparison, cell culture, DNA extraction, bisulfite conversion, molecular inversion probe capture, circularization, PCR and sequencing |
| DNase-Seq, Sono-Seq and FAIRE-Seq | Active regulatory chromatin (that is, nucleosome-depleted) | 113 | Perturbation, cell culture, nucleus extraction, DNase I digestion, DNA extraction, adaptor ligation, PCR and sequencing |
| MAINE-Seq | Histone-bound DNA (nucleosome positioning) | 130 | Comparison, cell culture, MNase I digestion, DNA extraction, adaptor ligation, PCR and sequencing |
| ChIP-Seq | Protein-DNA interactions (using chromatin immunoprecipitation) | 131 | Comparison, ‘anatomic’, cell culture, cross-linking, mechanical shearing, immunoprecipitation, DNA extraction, adaptor ligation, PCR and sequencing |
| RIP-Seq, CLIP-Seq, HITS-CLIP | Protein-RNA interactions | 46 | Variation, cross-linking, ‘anatomic’, RNase digestion, immunoprecipitation, RNA extraction, adaptor ligation, reverse transcription, PCR and sequencing |
| RNA-Seq | RNA (that is, the transcriptome) | 39 | Comparison, ‘anatomic’, RNA extraction, poly(A) selection, chemical fragmentation, reverse transcription, second-strand synthesis, adaptor ligation, PCR and sequencing |
| FRT-Seq | Amplification-free, strand-specific transcriptome sequencing | 119 | Comparison, ‘anatomic’, RNA extraction, poly(A) selection, chemical fragmentation, adaptor ligation, reverse transcription and sequencing |
| NET-Seq | Nascent transcription | 41 | Perturbation, genetic manipulation, cell culture, immunoprecipitation, RNA extraction, adaptor ligation, reverse transcription, circularization, PCR and sequencing |
| Hi-C | Three-dimensional genome structure | 71 | Comparison, cell culture, cross-linking, proximity ligation, mechanical shearing, affinity purification, adaptor ligation, PCR and sequencing |
| Chia-PET | Long-range interactions mediated by a protein | 73 | Perturbation, cell culture, cross-linking, mechanical shearing, immunoprecipitation, proximity ligation, affinity purification, adaptor ligation, PCR and sequencing |
| Ribo-Seq | Ribosome-protected mRNA fragments (that is, active translation) | 48 | Comparison, cell culture, RNase digestion, ribosome purification, RNA extraction, adaptor ligation, reverse transcription, rRNA depletion, circularization, PCR and sequencing |
| TRAP | Genetically targeted purification of polysomal mRNAs | 132 | Comparison, genetic manipulation, ‘anatomic’, cross-linking, affinity purification, RNA extraction, poly(A) selection, reverse transcription, second-strand synthesis, adaptor ligation, PCR and sequencing |
| PARS | Parallel analysis of RNA structure | 42 | Comparison, cell culture, RNA extraction, poly(A) selection, RNase digestion, chemical fragmentation, adaptor ligation, reverse transcription, PCR and sequencing |
| Synthetic saturation mutagenesis | Functional consequences of genetic variation | 93 | Variation, genetic manipulation, barcoding, RNA extraction, reverse transcription, PCR and sequencing |
| Immuno-Seq | The B-cell and T-cell repertoires | 86 | Perturbation, ‘anatomic’, DNA extraction, PCR and sequencing |
| Deep protein mutagenesis | Protein binding activity of synthetic peptide libraries or variants | 95 | Variation, genetic manipulation, phage display, in vitro competitive binding, DNA extraction, PCR and sequencing |
| PhIT-Seq | Relative fitness of cells containing disruptive insertions in diverse genes | 92 | Variation, genetic manipulation, cell culture, competitive growth, linear amplification, adaptor ligation, PCR and sequencing |
FAIRE-seq, formaldehyde-assisted isolation of regulatory elements–sequencing. MAINE-Seq, MNase-assisted isolation of nucleosomes-sequencing; RIP-Seq, RNA-binding protein immunoprecipitation-sequencing; CLIP-Seq, cross-linking immunoprecipitation-sequencing; HITS-CLIP, high-throughput sequencing of RNA isolated by cross-linking immunoprecipitation; FRT-Seq, on-flowcell reverse transcription–sequencing. NET-Seq, native elongating transcript sequencing. TRAP, translating ribosome affinity purification. PhIT-Seq, phenotypic interrogation via tag sequencing.
Details of next-generation DNA sequencing technologies are well described in recent reviews3,5–8. In this Review, we assume that future DNA sequencing technologies will yield large quantities of accurate DNA sequence at extremely low cost. This view does not seek to downplay the importance of the technical challenges particular to each sequencing platform (Table 2) or to suggest that DNA sequencing is a solved problem. There remains ample room for improvement with respect to almost every technical parameter. We also do not suggest that sequencers are interchangeable: specific applications are best supported by different platforms. Indeed, the next-generation sequencing market recently has begun to differentiate into ‘high-throughput’ instruments, ‘long-read’ instruments and ‘bench-top’ instruments (Box 1 and Table 2), a trend that is likely to continue.
Table 2.
Next-generation DNA sequencing instruments
| Cost per basea | Read length (bp)b | Speed | Capital costc | |||||
|---|---|---|---|---|---|---|---|---|
| Minimum cost per base | ||||||||
|
| ||||||||
| Complete Genomics | Low | Short | 3 months | None (service) | ||||
| HiSeq 2000 (Illumina) | Low | Mid | 8 days | +++++++ | ||||
| SOLiD 5500×l (Life Technologies) | Low | Short | 8 days | +++ | ||||
| Maximum read length | ||||||||
|
| ||||||||
| 454 GS FLX+ (roche) | High | Long | 1 day | +++++ | ||||
| RS (Pacific Biosciences) | High | Very long | <1 day | +++++++ | ||||
| Maximum speed, minimum capital cost and minimum footprint | ||||||||
|
| ||||||||
| 454 GS Junior (Roche) | High | Mid | <1 day | + | ||||
| Ion Torrent PGM (Life Technologies) | Mid | Mid | <1 day | + | ||||
| MiSeq (Illumina) | Mid | Long | 1 day | + | ||||
| Combined prioritization of speed and throughput | ||||||||
|
| ||||||||
| Ion Torrent Proton (Life Technologies) | Low | Mid | <1 day | ++ | ||||
| HiSeq 2500 (Illumina) | Low | Mid | 2 days | ++++++++ | ||||
‘Low’ is < $0.10 per megabase, ‘mid’ is in-between and ‘high’ is > $1 per megabase.
‘Short’ is < 200 bp, ‘mid’ is 200–400 bp, ‘long’ is > 400 bp and ‘very long’ is > 1,000 bp.
Each “+” corresponds to ~$100,000. We list only commercialized instruments that can be purchased and for which performance data are publically available (as opposed to a comprehensive list of companies developing next-generation sequencing technologies). The categorizations refer to the aspect of sequencing performance to which the technology and/or its implementation in a specific instrument are primarily geared. These estimates were made at the time of publication, and the pace at which the field is moving makes it likely that they will be quickly outdated.
In this Review, we consider applications that have been made possible by the recent advances in DNA sequencing technology and provide guidance for experimental design and the development of new sequencing applications. First, we propose a general framework for thinking about applications of next-generation DNA sequencing and discuss the most exciting applications and salient challenges in each area. Second, we identify building blocks that are common to many sequencing-based experimental strategies and consider how best to incorporate sequencing technologies into a variety of experimental approaches.
Applications of sequencing
The recent advances in sequencing technology are enabling researchers to consider questions at the level of the species, the organism, the cell and the biological mechanisms in a cell. Here we review applications of next-generation DNA sequencing at each of these levels (summarized in Fig. 1).
Figure 1.
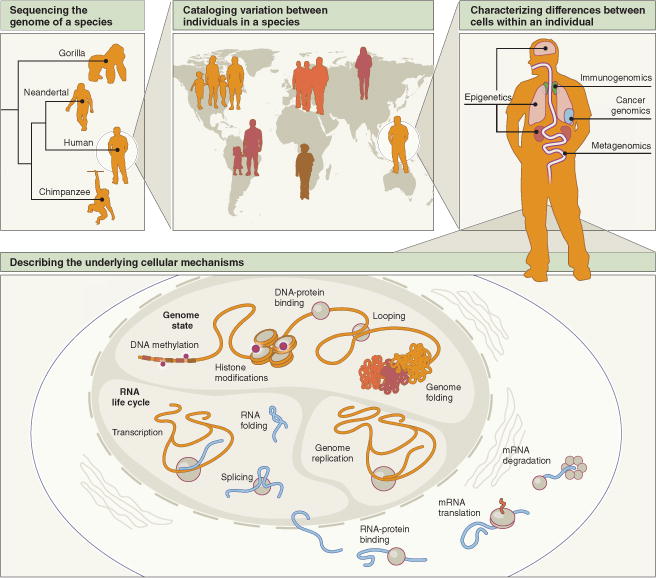
Where we are headed: a road map of sequencing science. The earliest sequencing projects focused on creating ‘reference’ genomes for individual species of interest. With new technologies, such projects can now be taken on for entire taxa, even when some of their members are extinct (top left). Next-generation sequencing is also enabling the study of biological systems at ever-finer scales. For example, we can explore genetic variation between individual members of a single species (top center), and the genetic and epigenetic differences between the cells of a single individual (top right). Sequencing can also provide a window into diverse processes in cells, including all of the phenomena shown (bottom).
Sequencing the genome of a species
Key milestones in genome sequencing include the Human Genome Project as well as other early projects directed at assembling reference genomes for prominent model organisms such as yeast, worm, fly and mouse. Subsequent projects have emphasized species most likely to inform evolutionary studies through comparative analysis—for example, the low-coverage sequencing of 29 mammalian genomes to broadly identify sequences under functional constraint9. As costs plummet, ambitions have skyrocketed: for instance, the Genome 10K project aims to produce a de novo assembly for each of over 10,000 vertebrate species10.
Nevertheless, the de novo sequencing of a complete genome (that is, a gapless, errorless, end-to-end assembly) is far from routine. New technologies are required to facilitate the sequencing of repetitive genomic regions (including transposons, satellite sequences, segmental duplications, ribosomal sequences and the like), which have largely confounded sequencing technologies to date. These regions are difficult to sequence even in relatively small genomes, such as that of Saccharomyces cerevisiae. For example, the current 12-Mb genome assembly of S. cerevisiae omits the 1-megabase rDNA locus.
Repetitive regions are particularly challenging for sequencing because identical reads may be generated from multiple locations in the genome. Despite advances in assembly algorithms11,12, the quality of de novo genome assemblies based purely on shotgun reads continues to fall short of the assemblies that can be achieved by hierarchical, clone-based Sanger sequencing13. It is unlikely that this gap can be overcome simply by increasing the amount of sequence data entering the assemblies. Instead, what are required are ‘next-generation’ methods of obtaining contiguity information at different scales. This might be as simple as including data from alternative sequencing platforms with a higher cost-per-base but substantially longer read lengths (Table 2 and Box 1). It could also include additional sources of contiguity information at a diversity of scales, such as long-distance mate-paired reads, hierarchical (that is, clone by clone) sequencing, dilution-pool sequencing, optical sequencing and genetic maps. As sequencing technologies continue to mature, a truly complete reference assembly of the human genome will be an increasingly realistic ambition (Table 3).
Table 3.
What is next for next-generation DNA sequencing?
| ‘End-to-end’ genome assembly or resequencing133 | Complete de novo assemblies are rare, and a substantial fraction of the human genome remains unfinished. Furthermore, there are particular regions as well as particular types of variation that are poorly detected owing to the limitations of current technology and algorithms. |
| Sequencing of nucleic acids within intact cells134 | In situ sequencing may enable applications directed at both DNA, for example, sequencing mitochondrial genomes or tumor genomes within single cells, and RNA, for example, massively parallel characterization of mRNA within single cells to quantify expression or assess subcellular localization. |
| Multiplex, single cell genomes, epigenomes and transcriptomes135 | The single-cell analysis of genomes and transcriptomes (but not yet epigenomes) is increasingly possible. Ideally this would not require individually isolating cells for processing. |
| Massively parallel assessment of synthetic double mutants136 | Large-scale analysis of double mutants can reveal functional relationships between genes, but double mutants are currently phenotyped one by one. |
| Massively parallel discovery of protein-protein interactions137 | The application of next-generation sequencing in the context of high-throughput interactome mapping (for example, yeast two-hybrid screens) will require methods that capture pairings of constructs expressing interacting products within individual cells, ideally in some massively multiplex way. |
| Quantitatively assaying cell-signaling cascades138 | Sequence-based reporters, that is, expressed barcode tags, can potentially be used to quantitatively monitor diverse aspects of cellular signaling in the context of a single cell. |
| Developmental fate-mapping139 | Sequencing can potentially be applied to enable the massively parallel assessment of cell lineage during the development of complex multicellular organisms. |
| Neuronal connectivity mapping140 | For example, ‘brainbow’ methods that use Cre-lox recombination to drive stochastic expression of fluorescent reporters potentially could be rendered more powerful by instead driving stochastic expression of mRNAs that were interrogated by in situ sequencing. |
| Massively parallel sequencing of polypeptides and post-translational modifications50 | Although translation can be interrogated with ribosome profiling48, we would ideally want to directly sequence polypeptides and their post-translational modifications in some massively parallel way analogous to next-generation sequencing of nucleic acids. |
References are to papers that are motivating or that describe important progress in a particular area.
Cataloging variation between individuals of a species
Genetic variation within a species underlies a substantial fraction of phenotypic variation—for example, the genetic contribution to human disease risk. Fortunately, using sequencing to identify genetic variation between individuals of the same species is considerably easier than assembling a reference genome for the species in the first place, as it only requires the mapping of reads to a reference assembly while allowing for differences owing to polymorphisms or sequencing errors1,14. With sufficient coverage, one can then identify certain types of genetic variation with high sensitivity and specificity, for example, single-nucleotide polymorphisms15.
One challenge for the future will be to develop techniques that enable more complete maps of genetic variation. For example, current methods have limited sensitivity and specificity for detecting small insertions and deletions, tandem repeat expansions, transposition events, copy-number variation, copy-neutral rearrangements and all types of variation within structurally complex regions of a genome. A separate aspect of completeness, effectively ignored by conventional shotgun genome sequencing, is haplotype information, for example, the combinations of alleles present on the same chromosome in a diploid human genome. Several methods have recently enabled the ascertainment of haplotypes at a genome-wide scale, albeit only locally16,17 or sparsely18,19. Combinations of these methods, or entirely new technologies, will be required fully resolve haplotypes in nonhaploid genomes.
Another challenge is how best to use sequencing to understand the genetic basis of human disease. Genome sequencing and exome sequencing (that is, the targeted sequencing of the ~1% of the human genome that is protein-coding) have substantially accelerated the determination of the genetic basis for single-gene (Mendelian) disorders20,21 and are being used to identify de novo mutations that may be risk factors for neuropsychiatric disorders such as intellectual disability22, autism23 (J.S. and colleagues) and schizophrenia24. For complex traits and common diseases, genome-wide association studies have already identified the most common risk alleles. Ongoing, sequencing-based studies may clarify the contribution of rare variants, common variants with small effect sizes and epistatic interactions to the so-called ‘missing heritability’25. However, these are likely to require enormous disease cohorts so as to have adequate statistical power.
In a clinical setting, many barriers still prevent the routine use of genome sequencing to inform patient care. Notably, the utility of genomic information to provide a personalized, asymptomatic prognosis for common diseases is inherently limited (exemplified by the simple fact that monozygotic twins do not usually die of the same disease)26,27. Instead, genome sequencing may be most useful for diagnosing rare, Mendelian disorders where the mutations are highly penetrant and thus more readily interpreted. For example, there are already case reports where exome or genome sequencing has led to a clear diagnosis of a known Mendelian disorder in a patient for whom that diagnosis had not been suspected, leading to substantial and sometimes life-saving changes in clinical management21,28,29.
Clinical sequencing of human genomes may also prove highly impactful in the context of reproductive health, as about 1% of new births are affected by a Mendelian disorder requiring specialized medical attention. Preconception screening of carrier status across hundreds of severe recessive disorders has been demonstrated and may eventually become routine30. After conception, an appreciable fraction of maternal cell–free DNA during pregnancy can be derived from the fetus. As such, aneuploidies can now be noninvasively detected by next-generation sequencing31,32, and clinical tests implementing this are gaining rapid adoption. Furthermore, by combining haplotype-resolved parental genome sequencing and maternal cell–free DNA sequencing, we have recently demonstrated noninvasive, whole-genome sequencing of a human fetus using samples obtained noninvasively from parents in the second trimester33 (J.S. and colleagues). Although improvements with respect to cost, accuracy and variant interpretation are necessary, such methods may eventually enable the noninvasive, prenatal diagnosis of many, if not most, Mendelian disorders.
Characterizing differences between cells within an individual
Recent studies have applied sequencing to reveal differences in regulatory state between different cell types in an organism, the somatic genetic variation that defines the immune repertoire, somatic differences between the genomes of cancerous and normal cells, and the microorganisms that colonize the human body.
Cell-to-cell differences in the regulatory state of a genome underlie differences in transcription, translation and cellular phenotype. Epigenetics traditionally refers to the study of biochemical changes in the immediate vicinity of DNA, including modifications of the DNA itself (for example, DNA methylation) and modifications of the histone proteins that package DNA into chromatin. Such epigenetic marks, as well as chromatin accessibility and transcription factor binding, directly involve DNA and are thus readily detected using sequencing-based assays34–37. Current challenges in applying sequencing to query the epigenome include specificity (for example, producing effective antibodies for each histone modification or transcription factor), adapting protocols to very low amounts of starting material and measuring the dynamics of epigenetic regulation rather than obtaining static snapshots38. Transcriptional processes have been interrogated in exquisite detail by sequencing-based methods for profiling steady-state expression39, allele-specific expression40, nascent transcription41, secondary structure formation42, alternative splicing43, RNA editing44, protein binding45,46 and degradation47. Translation of mRNA into protein can also be monitored via ribosomal profiling48. However, the methodological equivalent of massively parallel sequencing for amino acid polymers has yet to be developed (Table 3). Possible solutions are analogous to next-generation DNA sequencing technologies, for example, nanopores49 or identifying and counting single protein molecules spread on a two-dimensional surface50.
Immunogenomics uses massively parallel sequencing to characterize complex T-cell receptor and B-cell receptor repertoires in samples from individuals, with high depth and resolution51–54. Sequencing immune repertoires might make it possible to identify the acute response to diseases or malignancies, to gauge the status of the immune system in the context of immunodeficiency, transplantation or normal aging and to track malignancies of the hematopoietic system itself. Furthermore, profiling immune memory could identify markers of past exposures and successful vaccinations55. To achieve their potential, such methods must evolve toward reliably quantifying the abundances of an extraordinarily diverse population of immune receptor genes. Technical challenges to achieving this include the enormous dynamic range of the immune repertoire as well as the fact that the two chains of individual T-cell receptors and B-cell receptors are unlinked in the genome and transcriptome.
Cancer cells exhibit pronounced genomic instability56. Differences between an organism’s genome and a derivative cancer genome are readily determined by sequencing, albeit with the previously mentioned difficulties in detecting many relevant types of variation. Sequencing-based characterization of cancer genomes, epigenomes and transcriptomes is already informing the basic biology of specific cancer types, and the resulting insights may eventually provide a more powerful classification of human cancers than anatomy or histology. However, there is also remarkable heterogeneity among tumors ostensibly of the same type, and fully understanding the genetic basis of cancer is likely to require the characterization of extremely large numbers of cancer samples. Nonetheless, there is considerable enthusiasm about the use of genomic information to directly facilitate therapeutic decision-making for cancer patients57. For example, there are several reports of whole-genome sequencing of tumor DNA from individual patients yielding information that has altered clinical decisions58,59. Deep sequencing may also enable noninvasive cancer-screening methods, for instance, by examining circulating tumor cells60 or stool61. Detailed dissection of genetic heterogeneity in tumors through techniques such as ‘single-nucleus sequencing’ coupled with lineage analysis62 may also lead to a better understanding of cancer evolution as well as to improved diagnostics and prognostics. Of note, the needs of clinical laboratories in this area, for example, those seeking to implement next-generation sequencing of actionable cancer genes, are driving the market to offer instruments particularly geared to their requirements, for example, the development of ‘bench-top’ sequencers (Table 2 and Box 1).
Sequencing-based surveys of microbial communities (including shotgun metagenome sequencing and profiling of signature 16S rDNA), both in the human body63,64 and in environmental niches ranging from whale falls to acid mines, are overturning our world view of the number and breadth of species associated with humans and the environment. Genome sequencing of bacterial65,66 and viral67 pathogens (including of viral quasi-species68) has been used to detect the microorganisms responsible for disease outbreaks as well as to track the emergence and spread of antibiotic resistance. It is likely that these tools will become an increasingly important part of public health efforts in the coming decades. A key challenge for the future is to obtain a better understanding of the relationship between microbiome composition and human disease. For instance, obesity is associated with less microbial diversity in the gut microbiome, but the mechanisms underlying this association are not yet well-understood69. Although improved sequencing methods will enable more accurate quantification and content-determination of such complex microbial populations, new types of experiments (for example, perturbation instead of comparison; see below) may be required to study causal relationships.
Using sequencing to understand cellular mechanisms
Many of the techniques described above in the context of identifying cell-to-cell differences in regulatory state, when applied to a single cell or a single cell type, can be used to explore the underlying cellular circuitry (for example, chromatin immunoprecipitation with subsequent high-throughput sequence analysis (ChIP-Seq) for protein-DNA interactions, high-throughput RNA sequencing (RNA-Seq) for transcription and ‘Ribo-Seq’ for translation) or even molecular biophysics (for example, parallel analysis of RNA structure (PARS)42). In addition, new applications are emerging that are especially suited to this goal. For instance, methods based on in vivo proximity ligation, first used almost 20 years ago in the so-called nuclear ligation assay70, enable the exploration of the spatial arrangement of cellular components, for example, the proximity between genomic loci. Considerable improvements have been made to this technology, and recently, global mapping of DNA-DNA interactions using proximity ligation coupled with deep sequencing (Hi-C) has shed light on how the genome packs inside the nucleus (E.L.A. and colleagues71; J.S. and colleagues72). A related technique, chromatin interaction analysis by paired-end tag sequencing (ChIA-PET)73, focuses on the global interactions of specific transcription factors and may help shed light on the physical basis of enhancer activity.
Despite these challenges, such methods make sequencing a viable option for many research questions that would previously have required microscopy, for instance, the hypothesis that genome breakpoints in cancer tissue are spatially co-localized before rearrangement74. Cellular components can also be tracked over time, for example, by sequencing-based interrogation of the temporal pattern of DNA replication75. Eventually, new techniques may enable tracking of cellular dynamics through both space and time simultaneously (Table 3, in situ sequencing). Taken together, these methods highlight the dramatic transition that sequencing has undergone in biology: a technology originally designed to study genetics is rapidly becoming a mainstay of cell biology and biophysics.
Design of sequencing experiments
Wilhelm von Humboldt described language as a system that makes ‘infinite use of finite means’: despite a relatively small number of words and combinatorial rules, it is possible to express an infinite range of ideas. The space of contemporary sequencing applications has developed along similar lines: a relatively small number of experimental designs and protocols find use in a wide array of applications, and new applications often emerge by mixing and matching these building blocks. Our goal below is to capture the diversity of these techniques, to describe how they are combined in existing applications and to suggest how they might be combined to create new techniques.
We discuss sequencing experiments at two levels: experimental designs, which describe how biological systems are transformed into a collection of cell populations to be analyzed at the molecular level; and sequencing protocols, which describe the molecular steps by which specific information in these cells is captured and transformed into a population of adaptor-flanked DNA fragments for sequencing.
Below we discuss three approaches to experimental design (comparison, perturbation and variation) (Fig. 2a).
Figure 2.
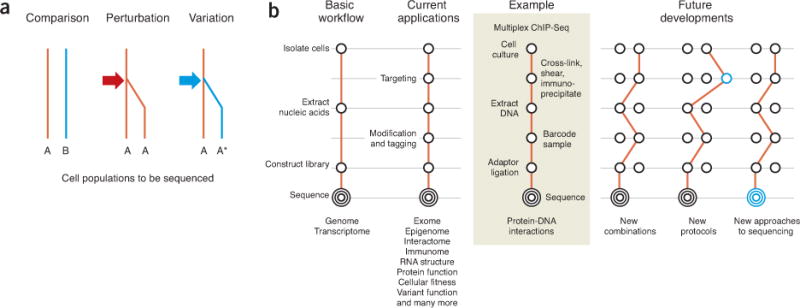
Structure of sequencing experiments. (a) Experimental designs describe the ways in which biological systems are transformed into a collection of cell populations to be analyzed at the molecular level. (b) Sequencing protocols are the molecular steps by which specific information in these cells is captured and transformed into a population of adaptor-flanked DNA fragments for sequencing. Future applications of sequencing may arise through new combinations of steps, the introduction of new steps or entirely new approaches to sequencing.
Comparison
This kind of study compares evolutionarily, developmentally, spatially, temporally or otherwise related samples to explore the differences that are present in natural environments. For example, comparison has been applied to identify differences between organisms or cell types76, to study spatiotemporal changes during development77 and to explore variation in cells78 or individuals79 that are presumed to be genetically identical.
One example of such a design is the comparison of phylogenetically related species, that is, comparative genomics. With the development of paleogenomic techniques, comparative genomics has even been extended to include species that are extinct80. Population genetics is essentially the comparison of patterns of natural variation between individuals in a species81. Similarly, the application of sequencing to identify genetic variations associated with disease-affected individuals (for germ-line variation) or cells (for somatic mutations) is also a comparative design. An additional paradigm, which might be termed comparative epigenetics, primarily studies epigenetic differences between related cell types. More recently, the comparative study of geographically related species, that is, natural ecosystems, has been gaining popularity. This ‘metagenomic’ approach includes the study of diverse ecological niches82 as well as site-to-site or individual-to-individual variation in the microbiomes associated with human body63,64,69.
The comparative strategy has been particularly common among large-scale consortia, for example, the International Human Genome Consortium, the HapMap project, Encyclopedia of DNA Elements (ENCODE), modENCODE, the 1000 Genomes project, the International Cancer Genome Consortium and the Human Microbiome project. These efforts have produced crucial infrastructure for genomics, for example, detailed genetic and epigenetic maps of the most common organism and cell models.
Advantages of comparative studies include that they exploit naturally occurring differences that cannot necessarily be generated by other means (for example, human genetic variation) and that these same natural differences may be of primary interest (for example, the contribution of human genetic variation to disease risk). However, this is also a constraint, as there is likely much variation or potential variation that is not readily manifest in accessible, naturally occurring samples. Examples include potential genetic variants that are simply not present in a population or cell types that are inaccessible because they occur very early during human development. A second disadvantage is that comparative studies are inherently observational, and therefore it can be difficult to establish causality. An example is genome-wide association studies, where linkage disequilibrium between nearby alleles makes it extremely difficult to specifically determine which genetic variant(s) causally underlie an observed association with a human disease.
Perturbation
In a perturbation experiment, an organism or cell culture is stimulated in a controlled manner (for instance, using heat shock), and the response is measured, with the aim being to more fully expose the underlying cellular program. In the short term, this response usually takes the form of changes to various aspects of cell state (for example, alterations in signaling cascades, epigenetic regulation or gene expression)—all of which are molecular phenotypes that can be interrogated by sequencing. When the perturbed cells are dividing, genetic changes and selection may occur, that is, experimental or directed evolution83,84. For example, experimental evolution accompanied by whole-genome sequencing can be used to explore the mechanisms by which pathogens or tumors become drug-resistant85. In a clinical context, the perturbation might consist of a therapy, where sequencing may be used to gauge the response86. Extremely specific perturbations have been achieved through the use of RNA interference–mediated knockdown of a particular gene(s)87 and optogenetic controls88.
Perturbation studies have the advantage of being truly controlled experiments—that is, comparing an experimental sample to a control sample with the only difference being the presence or absence of a specific perturbation. The use of high-dimensional, sequencing-based readouts for such experiments (for example, RNA-Seq to globally measure transcriptional changes in response to a perturbation) has both advantages and disadvantages. Although the comprehensiveness of the readout means that one is more likely to detect relevant biology in a global, unbiased way, the reality is that one is more often than not awash in such findings and it can be difficult to translate these into a meaningful biological interpretation. However, the solution may simply be more sophisticated experiments—for example, systematic, large-scale perturbation experiments that enable the de novo reconstruction of regulatory networks containing both known and new biology89.
Variation
Experimental designs in which the genetic program is directly modified seek to determine how specific functions are encoded by particular sequences. These include, for example, random mutagenesis, which is classically accomplished by a chemical agent (for example, N-ethyl-N-nitrosourea) or enzymatic process (for example, mutagenic PCR, transposase insertion90,91 and retroviral gene traps92). More recently, DNA synthesis has been applied to introduce structured93,94 or unstructured95,96 patterns of dense mutagenesis in sequences of interest.
An alternative to random mutagenesis is to engineer targeted genetic changes. Gene knockouts and knock-ins, viral transformation, and transfection by expression vectors are classic but still evolving techniques that remain extraordinarily powerful. The seminal discovery of cellular reprogramming, in which one cell type can be transformed into another (for example, via viral transformation97; viral, nonintegrating transfection98; nonviral, reversible transformation99; or episomal transient transfection100) is a particularly vivid example of the potential of targeted genetic manipulation. Sequencing is not only a powerful tool for monitoring cellular reprogramming and targeted genetic engineering but is also useful for comprehensively identifying the ‘off-target’ effects of these manipulations.
The manipulation of the genetic program is effectively a very defined type of perturbation, and using sequencing-based readouts for such experiments have the same advantages and disadvantages discussed above. However, a specific advantage of some designs invoking genetic variation is that they allow one to investigate many perturbations in one experiment, for example, genome-wide, random mutagenesis to identify genes in which mutations cause or rescue a specific phenotype. A related, emerging design involves the multiplex analysis of a single library of cells or molecules containing substantial nonrandom genetic variation. In such experiments, the population of cells or molecules is subjected to a single functional assay, and sequencing is used as a readout to reveal the relative activities of individual members of the library, each containing specific mutations. Recent examples of experiments in this vein include the massively parallel functional analysis of regulatory DNA93,94,96, protein domains95 and catalytic RNA101, all of which implemented high-density mutagenesis to achieve residue-by-residue perturbation of specific sequences. A related approach is to use multiplex functional assays to efficiently screen complex libraries in which individual cells contain single-gene knockouts90–92 or overexpress random genes102. Some of these methods rely on direct sequencing of the mutagenic event, whereas others require the introduction of synthetic ‘barcodes’ to capture this information93. Related methods are also enabling more complex explorations of the functional landscape via multiplex assays, for example, ‘designer’ regulatory elements103, fusion-protein libraries104, and the discovery of physical105 and genetic106,107 interactions.
Protocols for sequencing-based experiments
Sequencing protocols transform one or more cell populations defined by an experimental design into nucleic acids suitable for analysis at the molecular level by a sequencer (Fig. 2b). The conventional approach is to simply isolate DNA or RNA and construct a sequencing-compatible shotgun fragment library, that is, DNA-Seq or RNA-Seq. New protocols target specific subsets of nucleic acids. The first aspect of targeting is the enrichment of specific cells of interest from a heterogeneous cell population. Effective techniques for isolating subsets of cells include gross dissection, flow cytometry and laser-capture microdissection, and are dependent on the extent to which one can anatomically, molecularly or histologically delineate the cell type(s) of interest. For model organisms, these approaches can be facilitated by genetic manipulation to molecularly mark specific cell type(s). The second aspect of targeting is the extraction of desired subsets of DNA or RNA. The latter can be achieved by targeting the nucleic acids directly (based on primary sequence or accessibility), indirectly (by targeting neighboring molecular entities) or through modification (to capture noncanonical bases). Concurrently with targeting or after targeting, nucleic acids must be converted to a sequencing library, that is, a population of DNA fragments flanked by platform-specific adaptors. At various steps, the addition of sequence ‘tags’ can allow one to capture additional information (for example, sample indexing or molecular tagging).
Targeting nucleic acids directly
For both DNA and RNA, targeting methods usually achieve specificity through some combination of complementarity-mediated or otherwise specific hybridization, polymerization, ligation and/or cleavage. For DNA, methods for sequence-specific enrichment include standard PCR, multiplex PCR, molecular inversion probes108 (J.S. and colleagues), selective circularization109 and hybrid capture110,111. A somewhat distinct category of DNA targeting is to use endonuclease digestion, for example, for reduced representation112, or to define chromatin accessibility by DNase I hypersensitivity113. For RNA, one is usually interested in a particular subpopulation, such as mRNAs or small RNAs. For example, mRNAs can be enriched by poly(A)-mediated reverse transcription, oligo(dT) hybridization, ‘not-so-random’ amplification114 or BrdU incorporation (for recently synthesized mRNA). For RNA, preferred methods preserve strand information115, for example, by orientation-specific adaptor ligation, by the ordered incorporation of adaptors during cDNA synthesis or by strand-specific degradation after cDNA synthesis. Other RNA targeting methods are analogous to DNA-targeting methods, for example, reverse transcription–PCR of a specific target. A type of targeting aimed at capturing a different type of information, namely RNA secondary structure, involves digestion with one or more RNases42. The negative selection of nucleic acids is also possible, that is, enrichment via the subtraction of undesired material. This includes ‘footprinting’ techniques in which the target material is protected from nuclease activity, for example, by the presence of bound protein or by single-stranded or double-stranded status. Negative selection can also be accomplished by ‘subtractive’ hybrid capture, in which DNA or RNA sequences that bind a collection of ‘bait’ probes are removed.
Targeting nucleic acids indirectly
The alternative to direct targeting of nucleic acids is their indirect purification based on proximity to other molecular entities, for example, specific proteins or other nucleic acids. Such experiments usually begin with a cross-linking step; formaldehyde and UV light are the most common cross-linkers, but dimethyl suberimidate (DMS), dimethyl adipimidate (DMA), glutaradehyde, bis(sulfosuccinimidyl) suberate (BS3), spermine or spermidine and 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDAC) are alternatives. The choice of cross-linking agent is governed by the types of cross-links desired (for example, DNA-DNA, DNA-protein, RNA-protein and protein-protein), the tendency to damage substrates of interest and compatibility with downstream steps. The next step is usually immunoprecipitation, which enables extraction of nucleic acids of interest by exploiting their proximity to proteins of interest or to particular histone modifications, for example, the ChIP-Seq protocol35. More recently, a new protocol has been emerging that exploits the power of nucleic acid proximity ligation. In these methods, ligation is used to extract spatially co-localized nucleic acids, for example, to explore chromosome interactions in cells71–73.
Modifying nucleic acids to capture additional information
Both DNA and RNA molecules, in their in vivo context, can contain additional information beyond their four canonical bases. Recent work has explored an increasingly large collection of nucleotide modifications that are present in genomic DNA. A common approach to identifying such marks is to transform the four-base sequence of the DNA itself to encode the modification of interest. These techniques include bisulfite treatment (to detect cytosine methylation)34, and use of T4 bacteriophage b-glucosyltransferase and Huisgen cycloaddition (which is specific for 5-hydroxymethylcytosine)116. Post-transcriptional modifications of RNA might be detectable by identifying the characteristic error signatures that they cause in sequencing data117. Another creative example of this protocol is the use of specific polymerase error signatures secondary to cross-linking events to identify the precise RNA nucleotide involved in RNA-protein interactions118.
Library construction
Before sequencing, nucleic acids must be converted to a population of DNA fragments flanked by sequencing platform-specific adaptors. If this does not happen as a part of nucleic-acid targeting, it can be done afterward by any of a variety of methods for the in vitro construction of complex, shotgun sequencing libraries. Most common are ‘fragment libraries’, usually generated by random fragmentation (mechanical, chemical or enzymatic) followed by ligation of universal adaptor sequences and (optionally119) PCR amplification. A more recently developed alternative uses a hyperactive derivative of the Tn5 transposase to catalyze in vitro integration of the universal adaptor sequences into target DNA at a high density, usually followed by amplification120 (J.S. and colleagues). The resulting technique is faster, simpler and requires less input material. Of course, all methods that use amplification must contend with G+C bias and other sequence biases, although extensive efforts have been made to discover how such biases can be minimized121. PCR-free library preparation119 reduces sequence bias, as do sequencing technologies in which there is no amplification at any stage, for example, the Pacific Biosciences RS (Table 2)122.
Sample indexing or molecular tagging
The power of high-throughput sequencing has made it possible to do more than one experiment at a time, and an additional class of protocols enables many experiments to be efficiently multiplexed on a single sequencing lane. This is generally implemented by appending a synthetic index or barcode subsequence to all molecules in a given sequencing library, such that concurrent sequencing of the index can be used to assign reads in silico to the specific libraries from which they derived. But synthetic tags are proving to be increasingly useful in other contexts—for example, the tagging of individual molecules in subassembly123 (J.S. and colleagues), wherein the grouping of reads derived from the same nucleic acid enables more accurate quantification, robust error-correction and increased effective read length; the tagging of synthetic variants in synthetic saturation mutagenesis93 (J.S. and colleagues), wherein the synthetic tag is used as the functional readout; and the still unrealized possibility of associating tags with individual cells, to facilitate the ascertainment of genetic or epigenetic variability at single-cell resolution (Table 3).
Sequencing metrics
Although the conventional emphasis in the sequencing technology development field has been on ‘price per sequenced nucleotide’, costs have dropped to a point where other differentiators of specific technologies are increasingly relevant in a way that relates to the precise sequencing application (Table 2). The cost per base remains most important for large-scale resequencing projects, for example, sequencing large numbers of human genomes; the Illumina HiSeq is currently the most widely used instrument for such applications. For applications reliant on tag counting; for example, quantifying epigenetic phenomena via RNA-Seq or ChIP-Seq, the cost per read is more relevant than the cost per base. However, this is well correlated with the cost per base, provided that the read length is sufficient for accurate placement of reads to a reference genome.
There is usually an optimal tradeoff between the number of samples processed and the number of reads per sample that is highly dependent on the context of the experiment. For example, 30-fold coverage may be necessary to adequately ascertain single-nucleotide polymorphisms in an individual human genome, but with imputation one can get away with as little as 2× coverage per sample, albeit with tradeoffs with respect to rare and private variants124. For the transcriptome, <10 million reads may be sufficient for quantifying 80% of transcripts, but accurately quantifying alternative splicing in a similar number of transcripts would require >200 million reads125. Provided one is performing a sufficient volume of sequencing, sample indexing and pooling can allow one to maximize the value of individual sequencing runs in a way that is matched to the goals of the experiment.
However, cost per base and cost per read are not always the most important metrics. Technologies delivering better contiguity may be particularly relevant to applications such as de novo genome assembly or where local haplotype information is key, even when the cost per base is higher than that with other technologies. For example, despite the relatively high error rate and cost per base of the PacBio RS, its long reads can be combined with more cheaply generated short reads to facilitate de novo assembly of both small and large genomes126. Other differentiators may lend themselves to the deployment of DNA sequencing in clinical labs, for example, so-called ‘bench-top’ sequencers127. These include (i) speed: a rapid turnaround time, usually accompanied by some tradeoff with respect to cost per base; (ii) portability: a smaller instrument footprint (ideally, this will eventually be a hand-held device); (iii) low capital cost: lowering the barrier to entry for small-scale operations; and (iv) granularity: a low cost to perform one sequencing run. Several instruments, most prominently the Ion Torrent PGM and the Illumina MiSeq, were specifically designed to be appealing in these ways. Lastly, there are technologies that aim to find some middle ground between ‘bench-top’ characteristics and a low cost per base. These include the Ion Torrent Proton instrument as well as the HiSeq 2500 upgrade.
Future directions
A limited number of molecular and biochemical ‘building blocks’ have given rise to an extraordinary range of sequencing applications (Table 1). An analogy we introduce in this Review is a subway map, in which diverse routes (experiments) travel between stations (core techniques or building blocks) in myriad patterns (Fig. 3). Eventually, all the trains arrive at the hub: DNA sequencing.
Figure 3.
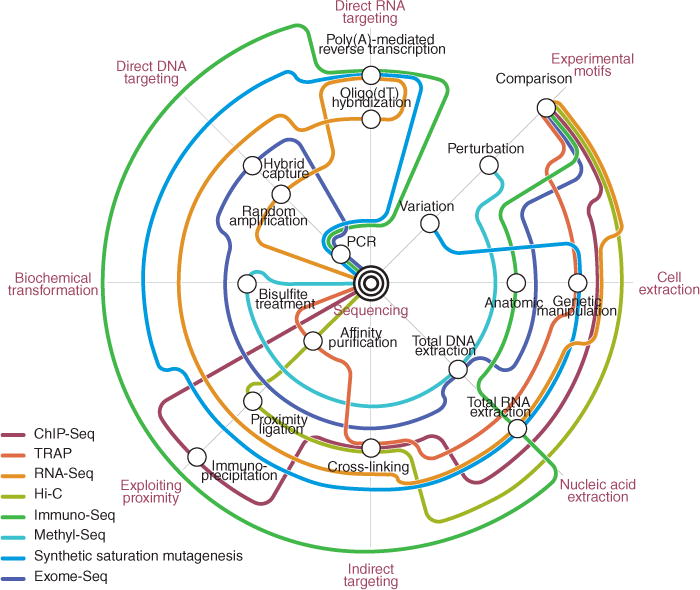
How we are getting there: a subway map of sequencing technology. Despite the disparate goals of different sequencing experiments, the great variety of sequencing experiments is a result of distinct combinations of a relatively small set of core techniques, which are represented as open circles or ‘stations’. Like subway lines, individual sequencing experiments move from station to station, until they ultimately arrive at a common terminal: DNA sequencing. For example, the initial demonstration of Hi-C71 was a comparative experiment that progressed through cell culture, cross-linking, proximity ligation, mechanical shearing, affinity purification, adaptor ligation and PCR amplification, before finally arriving at sequencing. Other examples shown correspond to sequencing applications in Table 1. For visual clarity, not all stations and routes are shown. New routes are being added regularly. TRAP, translating ribosome affinity purification.
Surveying the ‘subway map’ of present-day sequencing experiments provokes two observations. First, there are usually multiple routes for accomplishing the same task. For instance, DNA methylation can be mapped in a population of cells either by positive selection (capturing methylated DNA using the methyl-cytosine-binding domain of MeCP2), by negative selection (digesting with a methylation-sensitive restriction enzyme) or by direct transformation (bisulfite treatment). Such alternate pathways to the same end are inevitably differentiated by a variety of factors, such as comprehensiveness, cost, ease of implementation in a research or clinical setting, input requirements and compatibility with other protocols (for example, targeted capture). In designing a sequencing experiment, one should carefully consider these differentiating factors and ask whether a particular approach has advantages specific to one’s system. More generally, studies that systematically compare diverse protocols, for example, the many approaches for strand-specific RNA-Seq112, are extremely valuable to the community.
Second, new protocols tend to emerge through the reuse of existing techniques in new contexts. New building blocks are occasionally introduced. But more often than not, new sequencing applications simply borrow from other sequencing applications or from other existing methods. An example of a new technique emerging through such mixing and matching is the recently described use of oxidation mediated by Tet proteins in combination with beta-glucosyltransferase (bGT)115 and bisulfite treatment34 to distinguish 5-hydroxylmethylcytosines (5hmC) from 5-methylcytosines (5mC) genome-wide and at base-pair resolution128.
Given the remarkable proliferation of high-impact applications of next-generation DNA sequencing in the space of just a few years, we should be optimistic that additional applications—even particularly challenging applications such as those suggested in Table 3—are achievable with sufficient innovation and effort. The subway map analogy suggests that the development of new applications is likely to be best supported by a broad knowledge of existing and emerging sequencing protocols as well as a willingness to delve into the past 50 years of methods development in biochemistry and molecular biology. These sources effectively provide a toolbox that can be drawn on when evaluating potential routes to support new applications. For example, for any cellular phenomenon of interest, are there existing methods (or combinations of existing methods) that allow it to be coupled to nucleic acid sequence? In addition to actual protocols, there are also concepts that can be reapplied in new contexts. For example, ‘tagging’ is a general concept that can be applied to molecules, libraries, variants, cells, lineages or organisms. Although challenging, the development of new sequencing applications can be broadly impactful, in some cases opening up entirely new scientific territory to exploration.
Conclusions
The rapid maturation of massively parallel sequencing technology has been accompanied by a proliferation of exciting applications, each of which originated with an investigator asking: ‘can we solve this problem through sequencing?’ Sequencing is emerging as a ubiquitous, digital ‘readout’ for the deep, comprehensive exploration of genetics, molecular biology and cellular biophysics. In this Review, we attempted to develop a conceptual framework to describe this panoply of scientific applications as well as the underlying technical protocols that make them possible.
Though truly novel core techniques emerge from time-to-time, it is clear that most new sequencing applications have resulted from efforts to combine the building blocks of existing designs and protocols in different ways. We fully expect this trend to continue, as ‘recombination’ of these building blocks drives sequencing in new directions (Table 3). Like the cells that they study, biologists have learned to deploy a finite range of tools to meet an extraordinary array of challenges.
Supplementary Material
Acknowledgments
We thank L. Solomon and L. Gaffney of the Broad Institute for assistance with the design and preparation of figures; B. Wong and S. Arbesman for input on figure design; A.P. Aiden for valuable comments; and members of the Shendure lab and of the Laboratory at Large for discussions.
Footnotes
COMPETING FINANCIAL INTERESTS: The authors declare no competing financial interests.
References
- 1.Shendure J, et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309:1728–1732. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- 2.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 4.Wetterstrand KA. DNA sequencing costs: data from the NHGRI large-scale genome sequencing program. http://www.genome.gov/sequencingcosts/. Accessed 1 October 2012.
- 5.Shendure J, Mitra RD, Varma C, Church GM. Advanced sequencing technologies: methods and goals. Nat Rev Genet. 2004;5:335–344. doi: 10.1038/nrg1325. [DOI] [PubMed] [Google Scholar]
- 6.Fuller CW, et al. The challenges of sequencing by synthesis. Nat Biotechnol. 2009;27:1013–1023. doi: 10.1038/nbt.1585. [DOI] [PubMed] [Google Scholar]
- 7.Branton D, et al. The potential and challenges of nanopore sequencing. Nat Biotechnol. 2008;26:1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 9.Lindblad-Toh K, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Genome 10K Community of Scientists. Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. J Hered. 2009;100:659–674. doi: 10.1093/jhered/esp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pevzner PA, Tang H, Waterman MS. An Eulerian path approach to DNA fragment assembly. Proc Natl Acad Sci USA. 2001;98:9748–9753. doi: 10.1073/pnas.171285098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gnerre S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA. 2011;108:1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alkan C, Sajjadian S, Eichler EE. Limitations of next-generation genome sequence assembly. Nat Methods. 2011;8:61–65. doi: 10.1038/nmeth.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Durbin r. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitzman JO, et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol. 2011;29:59–63. doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peters BA, et al. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature. 2012;487:190–195. doi: 10.1038/nature11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan HC, Wang J, Potanina A, Quake SR. Whole-genome molecular haplotyping of single cells. Nat Biotechnol. 2011;29:51–57. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma L, et al. Direct determination of molecular haplotypes by chromosome microdissection. Nat Methods. 2010;7:299–301. doi: 10.1038/nmeth.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ng SB, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi M, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vissers LE, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 23.O’roak BJ, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Girard SL, et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat Genet. 2011;43:860–863. doi: 10.1038/ng.886. [DOI] [PubMed] [Google Scholar]
- 25.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohane IS, Shendure J. What’s a genome worth? Sci Transl Med. 2012;4:133fs113. doi: 10.1126/scitranslmed.3004208. [DOI] [PubMed] [Google Scholar]
- 27.Roberts NJ, et al. The predictive capacity of personal genome sequencing. Sci Transl Med. 2012;4:133ra158. doi: 10.1126/scitranslmed.3003380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Worthey EA, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–262. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 29.Bainbridge MN, et al. Whole-genome sequencing for optimized patient management. Sci Transl Med. 2011;3:87re83. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bell CJ, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3:65ra64. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci USA. 2008;105:16266–16271. doi: 10.1073/pnas.0808319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiu RW, et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci USA. 2008;105:20458–20463. doi: 10.1073/pnas.0810641105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitzman JO, et al. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med. 2012;4:137ra176. doi: 10.1126/scitranslmed.3004323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 36.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Hesselberth JR, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lickwar CR, Mueller F, Hanlon SE, McNally JG, Lieb JD. Genome-wide protein-DNA binding dynamics suggest a molecular clutch for transcription factor function. Nature. 2012;484:251–255. doi: 10.1038/nature10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 40.Montgomery SB, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kertesz M, et al. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang ET, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li JB, et al. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 45.Sanford JR, et al. Splicing factor SFrS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009;19:381–394. doi: 10.1101/gr.082503.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Licatalosi DD, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rabani M, et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol. 2011;29:436–442. doi: 10.1038/nbt.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Howorka S, Siwy ZS. Nanopores as protein sensors. Nat Biotechnol. 2012;30:506–507. doi: 10.1038/nbt.2264. [DOI] [PubMed] [Google Scholar]
- 50.Tessler LA, Reifenberger JG, Mitra RD. Protein quantification in complex mixtures by solid phase single-molecule counting. Anal Chem. 2009;81:7141–7148. doi: 10.1021/ac901068x. [DOI] [PubMed] [Google Scholar]
- 51.Boyd SD, et al. Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing. Sci Transl Med. 2009;1:12ra23. doi: 10.1126/scitranslmed.3000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Freeman JD, Warren RL, Webb JR, Nelson BH, Holt RA. Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res. 2009;19:1817–1824. doi: 10.1101/gr.092924.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang C, et al. High throughput sequencing reveals a complex pattern of dynamic interrelationships among human T cell subsets. Proc Natl Acad Sci USA. 2010;107:1518–1523. doi: 10.1073/pnas.0913939107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robins HS, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114:4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Price DA, et al. Public clonotype usage identifies protective Gag-specific CD8+ T cell responses in SIV infection. J Exp Med. 2009;206:923–936. doi: 10.1084/jem.20081127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 57.Ley TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Welch JS, et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. J Am Med Assoc. 2011;305:1577–1584. doi: 10.1001/jama.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones SJ, et al. Evolution of an adenocarcinoma in response to selection by targeted kinase inhibitors. Genome Biol. 2010;11:r82. doi: 10.1186/gb-2010-11-8-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leary RJ, et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med. 2010;2:20ra14. doi: 10.1126/scitranslmed.3000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li M, et al. Sensitive digital quantification of DNA methylation in clinical samples. Nat Biotechnol. 2009;27:858–863. doi: 10.1038/nbt.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grice EA, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gill SR, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gardy JL, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011;364:730–739. doi: 10.1056/NEJMoa1003176. [DOI] [PubMed] [Google Scholar]
- 66.Harris SR, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327:469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Codoner FM, et al. Added value of deep sequencing relative to population sequencing in heavily pre-treated HIV-1-infected subjects. PLoS ONE. 2011;6:e19461. doi: 10.1371/journal.pone.0019461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zagordi O, Klein R, Daumer M, Beerenwinkel N. Error correction of next-generation sequencing data reliable estimation of HIV quasispecies. Nucleic Acids Res. 2010;38:7400–7409. doi: 10.1093/nar/gkq655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cullen KE, Kladde MP, Seyfred MA. Interaction between transcription regulatory regions of prolactin chromatin. Science. 1993;261:203–206. doi: 10.1126/science.8327891. [DOI] [PubMed] [Google Scholar]
- 71.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duan Z, et al. A three-dimensional model of the yeast genome. Nature. 2010;465:363–367. doi: 10.1038/nature08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fullwood MJ, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berger MF, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hansen RS, et al. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci USA. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.ENCODE Project Consortium et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gerstein MB, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gore A, et al. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baranzini SE, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464:1351–1356. doi: 10.1038/nature08990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schacherer J, Shapiro JA, Ruderfer DM, Kruglyak L. Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature. 2009;458:342–345. doi: 10.1038/nature07670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hess M, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–467. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- 83.Kao KC, Sherlock G. Molecular characterization of clonal interference during adaptive evolution in asexual populations of Saccharomyces cerevisiae. Nat Genet. 2008;40:1499–1504. doi: 10.1038/ng.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gresham D, et al. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet. 2008;4:e1000303. doi: 10.1371/journal.pgen.1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Andries K, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 86.Logan AC, et al. High-throughput VDJ sequencing for quantification of minimal residual disease in chronic lymphocytic leukemia and immune reconstitution assessment. Proc Natl Acad Sci USA. 2011;108:21194–21199. doi: 10.1073/pnas.1118357109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bassik MC, et al. Rapid creation and quantitative monitoring of high coverage shRNA libraries. Nat Methods. 2009;6:443–445. doi: 10.1038/nmeth.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 89.Amit I, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Goodman AL, et al. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe. 2009;6:279–289. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gallagher LA, Shendure J, Manoil C. Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. MBio. 2011;2:e00315–e00310. doi: 10.1128/mBio.00315-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Carette JE, et al. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat Biotechnol. 2011;29:542–546. doi: 10.1038/nbt.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Patwardhan RP, et al. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat Biotechnol. 2009;27:1173–1175. doi: 10.1038/nbt.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Melnikov A, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 2012;30:271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fowler DM, et al. High-resolution mapping of protein sequence-function relationships. Nat Methods. 2010;7:741–746. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Patwardhan RP, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 2012;30:265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 98.Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kaji K, et al. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771–775. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu J, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pitt JN, Ferre-D’Amare AR. Rapid construction of empirical RNA fitness landscapes. Science. 2010;330:376–379. doi: 10.1126/science.1192001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yu Z, et al. Activators of the glutamate-dependent acid resistance system alleviate deleterious effects of YidC depletion in Escherichia coli. J Bacteriol. 2011;193:1308–1316. doi: 10.1128/JB.01209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gertz J, Siggia ED, Cohen BA. Analysis of combinatorial cis-regulation in synthetic and genomic promoters. Nature. 2009;457:215–218. doi: 10.1038/nature07521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nett JH, et al. A combinatorial genetic library approach to target heterologous glycosylation enzymes to the endoplasmic reticulum or the Golgi apparatus of Pichia pastoris. Yeast. 2011;28:237–252. doi: 10.1002/yea.1835. [DOI] [PubMed] [Google Scholar]
- 105.Di Niro R, et al. Rapid interactome profiling by massive sequencing. Nucleic Acids Res. 2010;38:e110. doi: 10.1093/nar/gkq052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Baryshnikova A, et al. Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat Methods. 2010;7:1017–1024. doi: 10.1038/nmeth.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Roguev A, et al. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science. 2008;322:405–410. doi: 10.1126/science.1162609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Turner EH, Lee C, Ng SB, Nickerson DA, Shendure J. Massively parallel exon capture and library-free resequencing across 16 genomes. Nat Methods. 2009;6:315–316. doi: 10.1038/nmeth.f.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dahl F, Gullberg M, Stenberg J, Landegren U, Nilsson M. Multiplex amplification enabled by selective circularization of large sets of genomic DNA fragments. Nucleic Acids Res. 2005;33:e71. doi: 10.1093/nar/gni070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bashiardes S, et al. Direct genomic selection. Nat Methods. 2005;2:63–69. doi: 10.1038/nmeth0105-63. [DOI] [PubMed] [Google Scholar]
- 111.Gnirke A, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Meissner A, et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.John S, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Armour CD, et al. Digital transcriptome profiling using selective hexamer priming for cDNA synthesis. Nat Methods. 2009;6:647–649. doi: 10.1038/nmeth.1360. [DOI] [PubMed] [Google Scholar]
- 115.Levin JZ, et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods. 2010;7:709–715. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Song CX, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Findeiss S, Langenberger D, Stadler PF, Hoffmann S. Traces of post-transcriptional RNA modifications in deep sequencing data. Biol Chem. 2011;392:305–313. doi: 10.1515/BC.2011.043. [DOI] [PubMed] [Google Scholar]
- 118.Zhang C, Darnell RB. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol. 2011;29:607–614. doi: 10.1038/nbt.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mamanova L, et al. FrT-seq: amplification-free, strand-specific transcriptome sequencing. Nat Methods. 2010;7:130–132. doi: 10.1038/nmeth.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Adey A, et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010;11:R119. doi: 10.1186/gb-2010-11-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Aird D, et al. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011;12:R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Eid J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 123.Hiatt JB, Patwardhan RP, Turner EH, Lee C, Shendure J. Parallel, tag-directed assembly of locally derived short sequence reads. Nat Methods. 2010;7:119–122. doi: 10.1038/nmeth.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Blencowe BJ, Ahmad S, Lee LJ. Current-generation high-throughput sequencing: deepening insights into mammalian transcriptomes. Genes Dev. 2009;23:1379–1386. doi: 10.1101/gad.1788009. [DOI] [PubMed] [Google Scholar]
- 126.Koren S, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol. 2012;30:693–700. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Loman NJ, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 128.Yu M, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Deng J, et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009;27:353–360. doi: 10.1038/nbt.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ponts N, et al. Nucleosome landscape and control of transcription in the human malaria parasite. Genome Res. 2010;20:228–238. doi: 10.1101/gr.101063.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Heiman M, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ribeiro F, et al. Finished bacterial genomes from shotgun sequence data. Genome Res. doi: 10.1101/gr.141515.112. Advance online publication 28 September 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Larsson C, et al. In situ genotyping individual DNA molecules by target-primed rolling-circle amplification of padlock probes. Nat Methods. 2004;1:227–232. doi: 10.1038/nmeth723. [DOI] [PubMed] [Google Scholar]
- 135.Ramskold D, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30:777–782. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Costanzo M, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yu H, et al. Next-generation sequencing to generate interactome datasets. Nat Methods. 2011;8:478–480. doi: 10.1038/nmeth.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Botvinnik A, Wichert SP, Fischer TM, Rossner MJ. Integrated analysis of receptor activation and downstream signaling with EXTassays. Nat Methods. 2010;7:74–80. doi: 10.1038/nmeth.1407. [DOI] [PubMed] [Google Scholar]
- 139.Carlson CA, et al. Decoding cell lineage from acquired mutations using arbitrary deep sequencing. Nat Methods. 2012;9:78–80. doi: 10.1038/nmeth.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Livet J, et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.