

Abstract
Most available knowledge on fungal arginine metabolism is derived from studies on Saccharomyces cerevisiae, in which arginine catabolism is initiated by releasing urea via the arginase reaction. Orthologues of the S. cerevisiae genes encoding the first three enzymes in the arginase pathway were cloned from Kluyveromyces lactis and shown to functionally complement the corresponding deletion in S. cerevisiae. Surprisingly, deletion of the single K. lactis arginase gene KlCAR1 did not completely abolish growth on arginine as nitrogen source. Growth rate of the deletion mutant strongly increased during serial transfer in shake-flask cultures. A combination of RNAseq-based transcriptome analysis and 13C-15N-based flux analysis was used to elucidate the arginase-independent pathway. Isotopic 13C15N-enrichment in γ-aminobutyrate revealed succinate as the entry point in the TCA cycle of the alternative pathway. Transcript analysis combined with enzyme activity measurements indicated increased expression in the Klcar1Δ mutant of a guanidinobutyrase (EC.3.5.3.7), a key enzyme in a new pathway for arginine degradation. Expression of the K. lactis KLLA0F27995g (renamed KlGBU1) encoding guanidinobutyrase enabled S. cerevisiae to use guanidinobutyrate as sole nitrogen source and its deletion in K. lactis almost completely abolish growth on this nitrogen source. Phylogenetic analysis suggests that this enzyme activity is widespread in fungi.
Introduction
Arginine metabolism has been subject of intensive biochemical studies. After discovery of the urea cycle for use of arginine as a nitrogen source (Krebs and Henseleit, 1932; Krebs, 1973), attention focused on its role as a precursor for the synthesis of polyamine and the signalling compounds γ-aminobutyrate (GABA) and nitric oxide (Knowles and Moncada, 1994; Pitkanen et al., 2001). The most widely distributed pathway for arginine degradation that occurs across all three kingdoms (Abdelal, 1979) is initiated by arginase (EC 3.5.3.1), a ureohydrolase that converts arginine to ornithine and urea. Its active site, which contains several Mn2+ binding sites, is also conserved in other ureohydrolases such as agmatinase (EC 3.5.3.11), formiminoglutamase (EC 3.5.3.8), proclavaminate amidinohydrolase (EC 3.5.3.22), guanidinobutyrase (EC 3.5.3.7) and guanidinopropionase (EC 3.5.3.17) (Ouzounis and Kyrpides, 1994). Genes encoding these enzymes are assumed to have emerged early in evolution (Hartman, 1975) and have been used as markers in phylogenetic studies (Ouzounis and Kyrpides, 1994; Sekowska et al., 2000).
In eukaryotes, only two types of ureohydrolase have hitherto been described. In addition to arginase, higher eukaryotes express agmatinase (Coleman et al., 2004), which participates in an alternative pathway for arginine catabolism (Fig. 1). In this pathway, arginine is first decarboxylated to agmatine, which is converted to putrescine and urea by agmatinase. Putrescine can then be converted either to GABA or to the polyamines spermine and spermidine (Pegg, 2009). The rapidly increasing number of whole-genome sequences has enabled the putative identification of arginase and agmatinase genes in many eukaryotes. However, since such annotation is based on sequence homology only, it does not enable definitive conclusions on the catalytic function of the encoded proteins.
Fig. 1.

Overview of the key reactions in eukaryotic arginine metabolism. Thick lines indicate ureohydrolase reactions. EC 1.2.1.3: aldehyde dehydrogenase:, EC 3.5.3.11: agmatinase, EC 4.1.1.19: arginine decarboxylase, EC 4.3.2.1: argininosuccinate lyase, EC 6.3.4.5: argininosuccinate synthase, EC.1.14.13.39: nitric oxide synthase, EC 1.4.3.10: putrescine oxidase, EC 2.1.3.3: ornithine carbamoyltransferase, EC 3.5.3.1: arginase, EC 4.1.1.17: ornithine decarboxylase, EC 2.6.1.13: ornithine aminotransferase, EC 1.5.1.2: pyrroline-5-carboxylate reductase, EC 1.5.99.8: proline dehydrogenase, EC 1.5.1.12: l-Δ1-pyrroline-5-carboxylate dehydrogenase.
Much of the knowledge on fungal arginine metabolism is based on studies with the model organism Saccharomyces cerevisiae. In S. cerevisiae, arginine is transported into the cell and subsequently hydrolysed by arginase (Car1) to yield ornithine and urea (Cooper et al., 1992; Sumrada and Cooper, 1992; Shima et al., 2003). An ATP-dependent amidolyase (Dur1,2) then converts urea into ammonia and carbon dioxide. Ornithine is further converted by an ornithine-specific transaminase (Car2) into glutamate-γ-semialdehyde (GSA), which spontaneously forms 1-pyrroline-5-carboxylate (P5C) (Martin et al., 2003). Due to subcellular compartmentation, S. cerevisiae is unable to convert cytosolic P5C directly to glutamate (Davis, 1986). Instead, P5C is reduced to proline using pyroline-5-carboxylate reductase (Pro3). Proline is then transported into the mitochondria (Brandriss and Falvey, 1992), converted back to P5C by an oxidase (Put1) and, finally converted to glutamate by mitochondrial P5C dehydrogenase (Put2) (Davis, 1986). Since only very few physiological studies have been conducted on arginine metabolism in non-Saccharomyces yeasts, it is unknown whether the arginase pathway, which is essential for growth of S. cerevisiae on arginine as sole nitrogen source (Bossinger and Cooper, 1977), is the only fungal pathway for arginine catabolism.
Saccharomyces cerevisiae and Kluyveromyces lactis both belong to the Saccharomycetaceae family. These two related yeasts are considered to have genetically separated before the whole-genome duplication (WGD) event that reshaped the genome of S. cerevisiae, furthermore K. lactis is regarded as resembling a pre-WGD ancestor of S. cerevisiae (Dujon, 2010). While many studies have been conducted on the differences in sugar metabolism between these two species, the differences in amino acid metabolism have not been studied in detail. Nonetheless, the complete genome sequence of K. lactis revealed many putative orthologues of S. cerevisiae genes involved in arginine metabolism (Dujon et al., 2004; Souciet et al., 2009; Dias et al., 2012).
The goal of the present study was to characterize arginine catabolism in K. lactis. To this end, CAR1, CAR2 and PRO3 orthologues in K. lactis were identified and functionally analysed by deletion, expression in S. cerevisiae and enzyme activity assays. Since deletion of the arginase gene in K. lactis was found not to completely abolish growth on arginine as a sole nitrogen source, the alternative pathway for arginine catabolism operating in this yeast was studied by a combination of transcriptome analysis, 13C and 15N isotope-based flux analysis and enzyme activity assays in cell extracts.
Results
Arginine catabolism in K. lactis occurs primarily via the arginase pathway
To investigate arginine metabolism in K. lactis, Blastp searches were performed to identify orthologues of the S. cerevisiae l-arginase (Car1), (Brandriss and Magasanik, 1980; Sumrada and Cooper, 1992), ornithine transaminase (Car2) (Dubois et al., 1978; Park et al., 1999), and pyrroline-5-carboxylate reductase (Pro3) (Brandriss and Falvey, 1992) in the K. lactis genome. The predicted proteins KLLA0F00704p, KLLA0B05247p and KLLA0D10736p showed 65, 71 and 71% identity at the amino acid level to the S. cerevisiae proteins and were therefore tentatively named KlCAR1, KlCAR2 and KlPRO3 respectively. Single-deletion strains for each of the three S. cerevisiae genes and for their putative K. lactis orthologues were constructed. Moreover, KlCAR1, KlCAR2 and KlPRO3 were cloned in a S. cerevisiae expression vector and transformed to the corresponding S. cerevisiae deletion strains to test heterologous complementation.
To verify whether deletion resulted in elimination of the corresponding enzyme activity, enzyme activity assays were performed on the mutant strains. After an initial growth phase on ammonium as sole nitrogen source [with the exception of the cells harvested for measurement of the P5C reductase activity which required addition of proline due to their proline auxotrophy (Tomenchok and Brandriss, 1987)], cells were washed and incubated for 2 h on arginine or ornithine as sole nitrogen source to induce arginine-related enzymes. Deletion of CAR1, CAR2 and PRO3 orthologues led to complete elimination of the corresponding enzyme activity in both K. lactis and S. cerevisiae (Table 1). Moreover, S. cerevisiae deletion strains expressing KlCAR1, KlCAR2 and KlPRO3 orthologues showed a high activity for each of the corresponding enzymes (Table 1).
Table 1.
Arginase, ornithine transaminase (OTA) and P5C-reductase enzyme activities measured in cell extracts of the yeast strains K. lactis GG1632 (KlCAR1, KlCAR2, KlPRO3), S. cerevisiae IMZ312 (CAR1, CAR2, PRO3), K. lactis IMK432 (Klcar1Δ), S. cerevisiae IMK435 (car1Δ), S. cerevisiae IMZ310 (car1Δ KlCAR1↑), K. lactis IMK433 (Klcar2Δ), S. cerevisiae IMK436 (car2Δ), S. cerevisiae IMZ311 (car2Δ KlCAR2↑), K. lactis IMK434 (Klpro3Δ), S. cerevisiae IMK445 (pro3Δ) and S. cerevisiae IMK342 (pro3Δ KlPRO3↑)
| Strain | Activity (μmol min−1 mg protein−1) | ||
|---|---|---|---|
| Arginase | GG1632 (KlCAR1) | K. lactis | 1.45 ± 0.3 |
| IMK432 (Klcar1Δ) | K. lactis | B.D. | |
| IMZ312 (CAR1) | S. cerevisiae | 3.73 ± 0.8 | |
| IMK435 (car1Δ) | S. cerevisiae | B.D. | |
| IMZ310 (car1Δ KlCAR1↑) | S. cerevisiae | 12.59 ± 3.0 | |
| Ornithine transaminase | GG1632 (KlCAR2) | K. lactis | 30.18 ± 2.1 |
| IMK433 (Klcar2Δ) | K. lactis | B.D. | |
| IMZ312 (CAR2) | S. cerevisiae | 421.93 ± 3.4 | |
| IMK436 (car2Δ) | S. cerevisiae | B.D. | |
| IMZ311 (car2Δ KlCAR2↑) | S. cerevisiae | 606.28 ± 37.8 | |
| P5C reductase | GG1632 (KlPRO3) | K. lactis | 0.58 ± 0.1 |
| IMK434 (Klpro3Δ) | K. lactis | B.D. | |
| IMZ312 (PRO3) | S. cerevisiae | 0.096 ± 0.0 | |
| IMK445 (pro3Δ) | S. cerevisiae | B.D. | |
| IMZ342 (pro3Δ KlPRO3↑) | S. cerevisiae | 25.71 ± 2.0 |
Cells were harvested after incubation with arginine, ornithine or proline. Data represent the average ± mean deviation of independent biological duplicates. B.D.: below detection limit of the enzyme assays, estimated at 0.07, 0.03 and 0.02 μmol min−1 mg protein−1 for the arginase, OTA or and P5C-reductase measurement respectively.
Phenotypes of the deletion strains and of the heterologously complemented S. cerevisiae strains were first analysed on spot plates containing arginine, ornithine, proline or ammonia as a sole nitrogen source (Fig. 2). Consistent with earlier studies (Brandriss and Magasanik, 1980; Brandriss and Falvey, 1992), the S. cerevisiae strains IMK435 (car1Δ), IMK436 (car2Δ) and IMK445 (pro3Δ) were unable to grow on arginine as a sole nitrogen source. Furthermore, growth on ornithine was abolished for IMK436 (car2Δ) and also for IMK445 (pro3Δ), consistent with the proline auxotrophy of pro3 mutants (Tomenchok and Brandriss, 1987). On spot plates, the same pattern was observed for K. lactis strains IMK432 (Klcar1Δ) and IMK434 (Klpro3Δ) (Fig. 2). In contrast, the strain IMK433 (Klcar2Δ) exhibited a very slow but noticeable growth on ornithine plates. While unexpected this results might reflect a difference of specificity and higher substrate promiscuity of the K. lactis transaminases. Furthermore KlCAR1, KlCAR2 or KlPRO3 in the S. cerevisiae deletion mutants showed similar growth characteristics to the prototrophic empty-vector reference strain S. cerevisiae IMZ312, indicating that the K. lactis orthologues fully complemented the corresponding null mutations in S. cerevisiae.
Fig. 2.
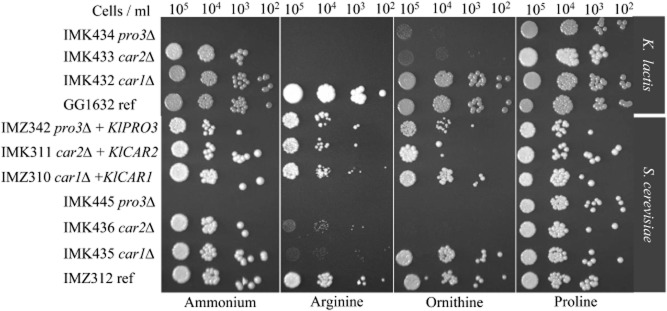
Phenotypic analysis of K. lactis GG1632 (KlCAR1, KlCAR2, KlPRO3), IMK432 (Klcar1Δ), IMK433 (Klcar2Δ), IMK434 (Klpro3Δ) and S. cerevisiae IMZ312 (CAR1, CAR2, PRO3), IMZ310 (car1Δ KlCAR1↑), IMZ311 (car2Δ KlCAR2↑), IMK342 (pro3Δ KlPRO3↑), IMK435 (car1Δ), IMK436 (car2Δ), IMK445 (pro3Δ) for growth on various nitrogen sources. The strains were plated on synthetic medium with glucose and either ammonium, arginine, ornithine or proline. The plates were incubated at 30°C and scored after 72 h.
Growth of arginase-negative K. lactis on arginine as sole nitrogen source
In contrast to the apparent absence of arginase activity and growth on solid medium, the K. lactis strain IMK432 (Klcar1Δ) was still able to grow in liquid synthetic medium containing arginine as sole nitrogen. Growth in these cultures was characterized by an extended lag phase (estimated specific growth rate of 0.03 h−1), and it took about 6 days to reach stationary phase (Fig. 3). During four consecutive serial transfers the specific growth rate gradually increased up to 0.1 h−1 and the fully grown culture reached a final OD600 of 30 in stationary phase. No growth was observed in serial transfer experiments with S. cerevisiae IMK435 (car1Δ). To investigate whether the observed increase in growth rate was due to adaptation of the culture or was genetically stable, cells were transferred to a shake flask containing glucose synthetic medium with ammonium sulphate as the nitrogen source and grown for approximately 20 generations. After reintroduction into arginine medium, this culture did not show any delay or decrease in growth rate compared to the fourth transfer on arginine, suggesting that a mutation contributed to the fast growth of the arginine-adapted cultures. For further characterization, a single-colony isolate was obtained from the final transfer on arginine. The resulting K. lactis strain was named IMS0367 (Klcar1Δ Arg+) and exhibited a specific growth rate of 0.09 h−1. Deletion of the KlCAR1 gene in this strain was confirmed by diagnostic PCR.
Fig. 3.
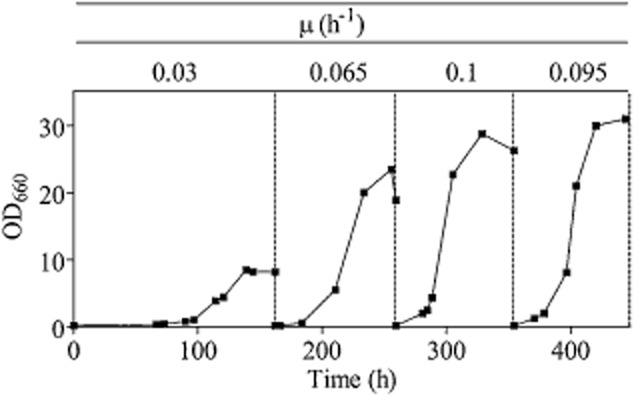
Adaptive growth profile of the strain IMK432 (Klcar1Δ Arg+) in sequential shake flask cultivations. The strain was grown in four successive shake flaks with chemically defined medium with glucose and arginine as nitrogen source. A single colony isolate from the last shake flask was selected, and named IMS0367.
Physiological and transcriptome analysis of an arginase-negative K. lactis strain
To further investigate arginine metabolism in the evolved arginase-negative K. lactis strain, strains GG1632 (Klku80Δ KlCAR1 reference strain) and IMS0367 (Klcar1Δ Arg+) were grown in aerobic bioreactor batch cultures on chemically defined medium with glucose and arginine as sole nitrogen source (Fig. 4). The specific growth rate of the arginase-negative strain was around fourfold lower than that of the reference strain (Table 2). The lower apparent biomass yield on glucose of the mutant strain (0.41 g·g−1 compared to 0.60 g·g−1 for the reference strain) was at least partially due to the fact that, in contrast to the reference strain; it did not completely consume the available arginine (Table 2). Moreover, no extracellular urea and/or ammonia were found during growth of IMS0367. For both strains, approximately 98% of the carbon initially present in the form of arginine and glucose could be recovered at the end of cultivation as CO2, and biomass for GG1632 and as CO2, biomass and residual arginine, in case of IMS0367 (Fig. 4). Similar observations could be drawn from the nitrogen balance indicating that, also in the evolved mutant strain, all four nitrogen atoms in arginine were utilized (Fig. 4).
Fig. 4.
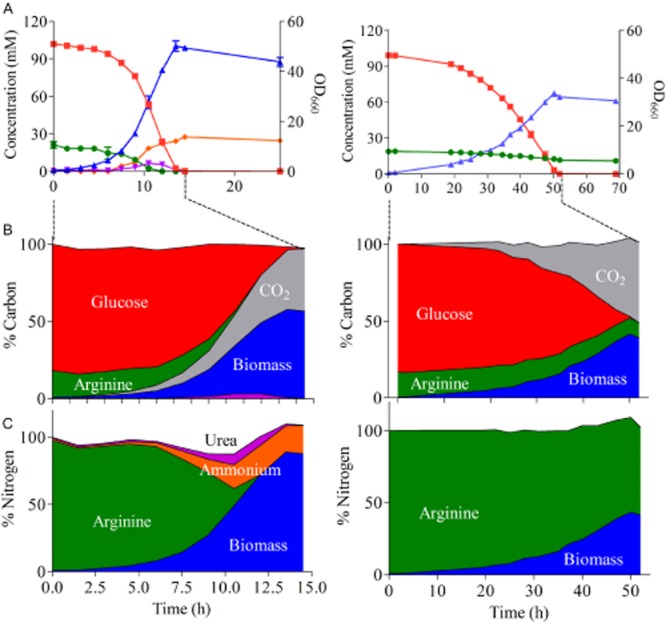
Growth kinetics of K. lactis GG1632 (left) and IMS0367 Klcar1Δ (right).A. Optical density at 660 nm (blue) and extracellular concentration of arginine (green), glucose (red), urea (purple) and ammonia (orange) were followed in aerobic batch cultures on glucose synthetic medium with arginine as nitrogen source.B and C. The total Carbon (B) and Nitrogen (C) pools were determined by HPLC and off-gas measurements taken during the exponential growth phase (15 h for GG1632 and 52 h for IMS0367) based on standard biomass composition estimation (Supplemental information) and are shown as a percentage of the initial pool.
Table 2.
Physiological data of K. lactis strains GG1632 and IMS0367 (Klcar1Δ Arg+) grown in batch cultures on synthetic glucose medium with arginine as sole nitrogen source
| GG1632 | IMS0367 | |
|---|---|---|
| Growth rate (h−1) | 0.38 ± 0.01 | 0.087 ± 0.001 |
| YSX.gluc (gdw·g−1) | 0.60 ± 0.04 | 0.41 ± 0.01 |
| Arginine consumed (mM): | 19.8 ± 0.0 | 8.2 ± 0.1 |
| Dry Weight (g l−1): | 10.8 ± 0.5 | 7.2 ± 0.36 |
| Approx. qs.gluc (g · h−1·gdw−1) | 0.57 ± 0.07 | 0.22 ± 0.07 |
| Approx qs.arg (g·h−1·gdw−1) | 0.19 ± 0.02 | 0.012 ± 0.003 |
| Arginase activity ( μmol min−1 mg protein−1) | 0.516 ± 0.15 | B.D. |
The batch cultures were sampled in mid-exponential phase, cell extracts were prepared and arginase activity was measured. Data represent the average ± mean deviation of independent biological duplicate cultures and enzymatic measurements. B.D. denotes below the detection limit, which was estimated at 0.07 μmol min−1 mg protein−1.
RNA sequencing of samples taken during the exponential phase of growth on glucose-arginine media of the reference strain and the evolved arginase-negative strain revealed 430 genes whose transcripts levels differed by more than twofold between the two strains. Functional category enrichment analysis (Kanehisa et al., 2008; Knijnenburg et al., 2009; Hunter et al., 2012) using KEGG and InterPro database annotations identified significant over-representation of genes involved in arginine and proline metabolism, amino acid transporters and alanine, aspartate and glutamate metabolism (Table 3). The transcriptional data mimicked the cellular response to nitrogen limitation and/or growth on a non-preferred nitrogen source. This is illustrated by the upregulation of the K. lactis orthologues of several genes that, in S. cerevisiae are under nitrogen catabolite repression, amino acid transporters, and amino acid catabolism-related genes (Boer et al., 2005; 2007). Interestingly, the K. lactis orthologues of S. cerevisiae PUT1 and PUT2 genes involved in proline degradation (KLLA0D1692g and KLLA0C10549g), were significantly downregulated in IMS0367.
Table 3.
KEGG and INTERPRO categories overrepresented in the set of genes that showed significantly different (P-value < 0.005) transcript levels in K. lactis IMS0367 (Klcar1Δ Arg+) relative to the reference strain K. lactis GG1632 1(FC > |2|, FDR 1%)
| Arginine and proline metabolism (KEGG, P-value: 5E-7, 15 of 65 genes) | FC | |
|---|---|---|
| KLLA0F27995g | Conserved hypothetical protein/putative arginase | 43.4 |
| KLLA0B08800g | Similar to S. cerevisiae YDR242W AMD2 putative amidase | 14.7 |
| KLLA0F04235g | Conserved hypothetical protein/putative arginase | 13.5 |
| KLLA0E08119g | Highly similar to S. cerevisiae YBR208C DUR1 2 urea amidolyase | 6.1 |
| KLLA0F14366g | Similar to S. cerevisiae ARG3 ornithine carbamoyltransferase | 4.1 |
| KLLA0F00594g | Highly similar to S. cerevisiae GDH3 NADP(+)-dependent glutamate dehydrogenase | 3.9 |
| KLLA0B05247g | Highly similar to S. cerevisiae CAR2 l-ornithine transaminase (OTAse) | 2.9 |
| KLLA0E21077g | Highly similar to S. cerevisiae ARG8 acetylornithine aminotransferase | 2.8 |
| KLLA0F26268g | Similar to S. cerevisiae mitochondrial ornithine acetyltransferase catalyses | 2.2 |
| KLLA0C07997g | Highly similar to S. cerevisiae ARG5 enzyme | 2.2 |
| KLLA0A08492g | Conserved hypothetical protein | 2.0 |
| KLLA0D16962g | Similar to S. cerevisiae PUT1 proline oxidase | −2.5 |
| KLLA0C10549g | Highly similar to S. cerevisiae PUT2 delta-1-pyrroline-5-carboxylate dehydrogenase | −7.3 |
| Amino acid transporters (INTERPRO, P-value: 0.003, 7 of 323 genes) | ||
| KLLA0B14685g | Weakly similar to S. cerevisiae PUT4 proline-specific permease | 6.4 |
| KLLA0C02343g | Similar to similar to S. cerevisiae CAN1 plasma membrane arginine permease | 6.3 |
| KLLA0A06930g | Similar to S. cerevisiae GAP1 general amino acid permease | 4.0 |
| KLLA0F01012g | Similar to S. cerevisiae GNP1 high-affinity glutamine permease | 2.8 |
| KLLA0A10813g | Similar to S. cerevisiae TAT2 tryptophan permease high affinity | 2.2 |
| KLLA0C01606g | Similar to S. cerevisiae GNP1 high-affinity glutamine permease | −6.9 |
| KLLA0A06886g | Similar to S. cerevisiae GAP1 general amino acid permease | −15.7 |
| Alanine, aspartate and glutamate metabolism (KEGG, P-value: 0.005, 6 of 65 genes) | ||
| KLLA0F20548g | Similar to S. cerevisiae UGA1 gamma-aminobutyrate (GABA) transaminase | 4.7 |
| KLLA0F24882g | Similar to S. cerevisiae CPA1 small subunit of carbamoyl phosphate synthetase | 4.1 |
| KLLA0F00594g | Similar to S. cerevisiae GDH3 NADP(+)-dependent glutamate dehydrogenase | 3.9 |
| KLLA0F13640g | Similar to S. cerevisiae mitochondrial aspartate aminotransferase | 3.4 |
| KLLA0D12980g | Highly similar to S. cerevisiae ASP1 cytosolic l-asparaginase | −2.2 |
| KLLA0C10549g | Highly similar to S. cerevisiae PUT2 delta-1-pyrroline-5-carboxylate dehydrogenase | −7.3 |
Both strains were grown on batch cultures with glucose and arginine as sole N-source. P-values were calculated using Fischer exact statistics.
Among the genes that were transcriptionally upregulated in K. lactis IMS0367, two open reading frames (KLLA0F27995g and KLLA0F04235g) shared sequence similarity with ureohydrolase encoding genes. As no arginase activity was detected in K. lactis IMS0367 (Table 2), it was hypothesized that the proteins encoded by these two genes might catalyse a similar reaction but with a different substrate. Several genes related to GABA metabolism were also upregulated in K. lactis IMS0367. In addition to the upregulation of KLLA0E14675g, which is similar to the S. cerevisiae PUT4 gene encoding the GABA-accepting permease, the transcription of a putative γ-aminobutyrate transaminase, which in S. cerevisiae is responsible for the initial step in GABA catabolism (Vissers et al., 1989; Bach et al., 2009), was 4.7-fold upregulated in K. lactis IMS0367 (Table 3).
Carbon and nitrogen isotopologue labelling using 13C15N l-arginine
To investigate whether the alternative route of arginine degradation was active in K. lactis IMS0367 (Klcar1Δ Arg+), a 13C15N labelling experiment was performed in shake flask cultures with fully labelled 13C615N4 arginine (M+10) as a sole nitrogen source. Intracellular metabolite concentrations and 13C15N enrichment levels for each metabolite were determined over the course of 64 min of cultivation for strain GG1632 cultivation and 300 min for strain IMS0367 (Figs S1 and S2). The already high intracellular arginine concentration in strain GG1632 [370 μmol g−1 (biomass dry weight), corresponding to an intracellular concentration of 185 mM assuming an intracellular volume of c. 2 ml g−1] was found to be even higher in IMS0367 at 660 μmol g−1 (biomass dry weight). Combined with the effect of vacuolar storage (Davis, 1986) this resulted in an overall arginine enrichment that only reached 38% and 26% for strains GG1632 and IMS0367 after 64 and 300 min respectively (Table 4 and Table S1).
Table 4.
Intracellular concentrations, and corresponding final 13C15N enrichments (%) of arginine catabolism intermediates measured in K. lactis IMS0367 (Klcar1Δ Arg+) and K. lactis GG1632 (reference strain, KLCAR1 Arg+)
| Intracellular conc. (μmol gdw−1) | Labelled isotopologue species (final overall 13C15N enrichment) | |||
|---|---|---|---|---|
| Metabolite | GG1632 | IMS0367 | GG1632 | IMS0367 |
| Arginine | 370 ± 80 | 660 ± 72 | 38% | 26% |
| Proline | 7.24 ± 1.92 | 0.06 ± 0.02 | 83% | 9% |
| Glutamate | 158 ± 47 | 48 ± 17 | 86% | 44% |
| α-Ketoglutarate | 2.97 ± 0.81 | 2.28 ± 0.62 | 55% | 3% |
| Succinate | 3.01 ± 1.2 | 6.61 ± 0.93 | 49% | 5% |
| GABA | 0.01 ± 0.01 | 0.41 ± 0.33 | 3% | 42% |
| Putrescine | 2.56 ± 0.89 | 0.03 ± 0.02 | 27% | B.D. |
Cellular extracts were prepared from batch cultures grown on glucose synthetic medium with fully (13C15N) labelled arginine as sole nitrogen source. Data represent the average ± mean deviation of independent biological duplicate cultures. B.D. denotes below the detection limit, N.D. denotes not determined.
In the reference strain K. lactis GG1632 (KlCAR1), the enrichment found for fully labelled 13C15N isotopologue species of 13C515N proline, 13C515N glutamate and 13C515N2 glutamine were in agreement with the presence of the arginase pathway described for S. cerevisiae as they could only derive directly from the 13C615N4 labelled arginine (Fig. 5). In contrast, the enrichment of the carbon labelled isotopologue species in strain IMS0367 (Klcar1Δ Arg+) were found to reach a maximum level of maximally 4%, thereby confirming that the arginase pathway was inactive. A substantial enrichment of only the 15N glutamate isotopologue species confirmed that the cells were able to mobilize the nitrogen contained in 13C15N arginine. While for K. lactis GG1632 a distinct enrichment in fully labelled 13C5 α-ketoglutarate was observed, the absence of such an isotopologue species, combined with the identification of fully labelled succinate, fumarate and malate, indicated that, in K. lactis IMS0367, carbon originating from arginine entered the TCA cycle at a different point. Isotopologue measurements of GABA subsequently revealed that, unlike the situation in K. lactis GG1632, a substantial increase in fully labelled 13C515N GABA was found in strain IMS0367. In eukaryotes, GABA is an intermediate of an arginine degradation pathway that also involves the polyamine putrescine. In K. lactis GG1632, enrichment of fully 13C415N2 labelled putrescine was observed, most likely originating from decarboxylation of 13C515N2 ornithine. Intracellular putrescine levels in K. lactis IMS0367 were 10-fold lower than in K. lactis GG1632. Moreover, we were unable to quantify any of the isotopologue species of putrescine, as the signal was below the detection limit. For arginine, a slight increase in the 13C615N3 (M+9) isotopologue species was observed in both GG1632 (3%) and IMS0367 (6%), probably caused by reversible arginine transamination.
Fig. 5.

Enrichment of intracellular metabolites and flux distribution of K. lactis GG1632 (KlCAR1) and K. lactis IMS0367 (Klcar1Δ Arg+) grown on 13C615N4 arginine. Enrichment profiles are shown next to the corresponding compounds in plots with light blue background for strain GG1632 and green for the IMS0367 strains. Red lines refer to the 15N species only. The fluxes are indicated in boxes (expressed in μmol gdw−1 min−1), fluxes indicated in blue correspond to strain GG1632 (KlCAR1 Arg+) and those indicated in green to IMS0367 (Klcar1Δ Arg+).
To determine whether arginine catabolism in K. lactis IMS0367 indeed involved GABA as an intermediate, a metabolic flux model of the hypothetical pathway was constructed (Fig. 5). In this model, an alternative catabolic route was evaluated, in which arginine is first transaminated, yielding ketoarginine and glutamate. Ketoarginine is then transformed into GABA via a decarboxylation and subsequent hydrolysis, releasing CO2 and urea respectively. The model was not solely based on a 13C-calculated flux analysis, because the co-consumption of unlabelled glucose prevented accurate and unequivocal quantification of all intracellular metabolic fluxes. Consequently, the model consisted of two separate parts, in which metabolic fluxes were quantified based on data obtained from nitrogen (Fig. S3 and Table S2) and carbon labelling (Fig. S4 and Table S3) of arginine respectively. To account for the impact of vacuolar storage of arginine on label enrichment patterns, separate cytosolic and vacuolar pools were introduced in the model. The observed increases in flux through TCA cycle reactions were in agreement to the previously observed arginine and glucose uptake rates during the batch cultivations. For K. lactis GG1632, the metabolic flux analysis demonstrated that nitrogen originating from arginine was not solely incorporated into biomass. Instead, ‘excess’ nitrogen was excreted in the form of ammonium, as observed experimentally during batch cultivation (Fig. 4). The flux analysis of K. lactis IMS0367 cultivation showed a different pattern. The model was only able to accurately describe the observed enrichment profiles when it was assumed that all nitrogen contained in the arginine molecule was mobilized in form of urea or glutamate. Flux calculations focusing on the fate of the carbon skeleton of arginine revealed that the rate of enrichment of the TCA cycle intermediates of the arginase-negative strain K. lactis IMS0367 strain was fully compatible with the degradation of GABA into succinate (Fig. 5, Figs S2 and S4).
Identification of a functional guanidinobutyrase in K. lactis
Transcriptome data and 13C15N flux analysis identified GABA as a key intermediate in arginine catabolism in the arginase-negative strain K. lactis IMS0367 (Klcar1Δ Arg+). Combined with increased transcript levels of two putative ureohydrolase genes (KLLA0F27995g and KLLA0F04235g) in this strain, this strongly suggested involvement of a ureohydrolase in the alternative arginine catabolic pathway. To test this hypothesis, we measured the ureohydrolase activity with both agmatine and guanidinobutyrate as substrate in cell extracts of the K. lactis strains IMS0367 and GG1632, grown on glucose with arginine as sole nitrogen source (Table 5). No activity with either substrate was measured in the reference strain GG1632, while the arginase-negative strain IMS0367 exhibited a high activity exclusively with guanidinobutyrate (Table 5). Additionally, in line with these enzyme activity measurements, shake flask experiments revealed that IMS0367 grew on media containing guanidinobutyrate as sole nitrogen source. The K. lactis reference strain GG1632 was also able to use guanidinobutyrate as nitrogen source, indicating that the expression of the guanidinobutyrase activity was tightly controlled and at least induced in presence of guanidinobutyrate in KlCAR1 strains. In contrast, the prototrophic S. cerevisiae strain CEN.PK113-7 D was unable to use guanidinobutyrate as nitrogen source.
Table 5.
Guanidinobutyrase activities measured in cell extracts of K. lactis and S. cerevisiae strains GG1632, IMS0367, IME215, IME216 and CEN.PK113-7 D grown in batch cultures with arginine as sole nitrogen source
| Strain | Description | Activity (μmol min−1 mg protein−1) |
|---|---|---|
| K. lactis GG1632 | Prototrophic reference KlCAR1 KlGBU1 | B.D. |
| K. lactis IMS0367 | Klcar1Δ Arg+ | 0.14 ± 0.004 |
| S. cerevisiae IME215 | MATa ura3-52 pUDE264 (TDH3pr-KlGBU1-CYC1ter URA3) | 0.17 ± 0.006 |
| S. cerevisiae IME216 | MATa ura3-52 pUDE265 (TDH3pr- KLLA0F04235g-CYC1ter URA3) | B.D. |
| S. cerevisiae CEN.PK113-7D | Prototrophic reference | B.D. |
The K. lactis strains GG1632 and IMS0367, as well as the S. cerevisiae strains IME216 and IME215 were pre-grown in synthetic medium with glucose and ammonium as sole nitrogen source. B.D. denotes below detection limit, which was estimated at 0.005 μmol min−1 mg protein−1. Data represent the average ± mean deviation of independent biological duplicate cultures.
To identify which of the two putative ureohydrolase genes in K. lactis contributed to the measured guanidinobutyrase activity, KLLA0F27995g and KLLA0F04235g were cloned under the control of the strong constitutive TDH3 promoter (TDH3pr) in an expression vector and transformed to S. cerevisiae. Cell extracts of the resulting strains did not exhibit any agmatinase activity but only cell extracts of S. cerevisiae IME215 (TDH3pr::KLLA0F27995g) showed a substantial activity with guanidinobutyrate, cell extracts of S. cerevisiae IME216 (TDH3pr-KLLA0F04235g-CYC1ter) did not exhibit any measurable activity (Table 5). In agreement with these results, S. cerevisiae IME215 (TDH3pr::KLLA0F27995g) was able to grow on guanidinobutyrate as sole nitrogen source (Fig. 6), resulting in a specific growth rate of approximately 0.1 h−1 in chemically defined glucose medium. Therefore, the gene KLLA0F27995g was renamed KlGBU1.
Fig. 6.
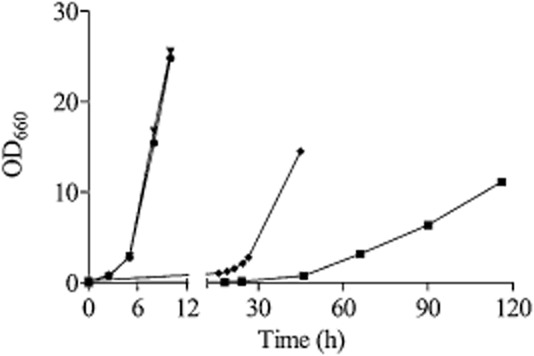
Typical growth profile of K. lactis and S. cerevisiae strains on guanidinobutyrate: The K. lactis strains GG1632 (KlCAR1) (•), IMS0367 (Klcar1Δ) (▼), IMK560 (Klcar1ΔKlgbu1Δ) (▪) and the S. cerevisiae strain IME215 (TDH3pr::KlGBU1::CYC1ter) (♦) were grown in shake flasks on glucose synthetic medium containing guanidinobutyrate as sole nitrogen source.
To further confirm the function of KLLA0F27995g/KlGBU1 and its role in an arginase-independent arginine catabolism pathway, KLLA0F27995g was deleted in IMS0367. While the K. lactis reference strain GG1632 and the Klcar1Δ Arg+ strain IMS0367 both grew at a specific growth rate of 0.45 h−1 on medium containing guanidinobutyrate as nitrogen source, remarkably the K. lactis Klcar1Δ KLLA0F27995g/Klgbu1Δ mutant (IMK560) exhibited a 22.5-fold lower specific growth rate on guanidinobutyrate as sole nitrogen source (c. 0.02 h−1) (Fig. 6). Additionally, the strain IMK560 (Klcar1Δ KLLA0F27995g/Klgbu1Δ) was still able to grow on medium with arginine as sole nitrogen source at a specific growth rate of 0.026 h−1 (versus 0.065 h−1 for IMS0367 in shake flask culture). In line with the labelling experiment, these results reinforced the predominant role of a pathway that involves an early transamination and the mobilization of the peptidic amino group which can still supply nitrogen for growth. The reduced specific growth rate of the strain IMS0367 on arginine (0.065 h−1) relative to growth on guanidinobutyrate (0.45 h−1) also suggests that the flux through the alternative arginine degradation pathway is limited by one or more reactions upstream the guanidinobutyrase (arginine transamination, ketoarginine decarboxylase and/or oxidation of guanidinobutanal) (Fig. 5).
Phylogenetic analysis of ureohydrolases
While guanidinobutyrase activity had previously been measured in Aspergillus niger (Kumar and Punekar, 2014), no gene encoding this activity has been identified in eukaryotes, a phylogenetic analysis was performed by searching for KlCar1 (arginase) and KlGbu1 (guanidinobutyrase) orthologues among the predicted protein sequences encoded in the genomes of yeasts belonging to the Saccharomycotina group, a subphylum of the Ascomycota (Kurtzman, 2003; Dujon, 2010). Using a phylogenetic tree topology proposed by Dujon (2010) for the Saccharomycotina group, orthologues of KlCar1 were found in all interrogated yeast proteomes. In contrast, KlGbu1 (guanidinobutyrase) orthologues were not found in Saccharomytacea genera such as Saccharomyces sensu stricto, Kazachstania, Naumovozyma, Nakaseomyces and Vanderwaltozyma, proposed to have arisen after the WGD event that played a major role in the evolution of this subphylum. Remarkably, one out of the 34 annotated S. cerevisiae genome sequences, available from the SGD database (http://www.yeastgenome.org/) belonging to strain EC1118, did harbour a gene whose predicted protein sequence shared 62% similarity with KlGbu1 (Fig. 7). A similar sequence occurs in the S. cerevisiae wine strain VIN7. Further inspection of the genome structure of the wine strain EC1118 revealed that the KlGbu1 orthologue was found on a genomic region acquired by horizontal transfer, most probably from Torulaspora delbrueckii. This gene origin fits the phylogenetic distribution of KlGbu1 orthologues in Saccharomycatae genera (Zygosaccharomyces, Lachancea, Torulaspora, Kluyveromyces and Eremothecium), and in genera belonging to the CTG group and Dipodascacae (Kurtzman, 2003; Dujon, 2010) (Fig. 7).
Fig. 7.

Phylogenetic tree of Saccharomycotina yeast species and analysis of the distribution of K. lactis KlGBU1/KLLA0F27995g and KLLA0F04235g orthologues within this subphylum. The tree is based on Kurtzman (2003) and Dujon (2010). K. lactis orthologous amino acid sequences were identified by Blastp or tBlastn in genomics data of each individual species mentioned. The branch length is arbitrary. The reported species are divided in several groups: The subphylum Saccharomycetaceae (in blue) includes the following clades: 1 – Saccharomyces sensu stricto, 2 – Kazachstania, 3 – Naumovozyma, 4 – Nakaseomyces, 5 – Vanderwaltozyma, 6 – Zygosaccharomyces, 7 – Lachancea, 8 – Kluyveromyces, 9 – Eremothecium, 10 – Torulaspora, 11 – Ogataea, The CGT group (in green) includes the clades: 12 – Debaryomyces, 13 – Clavispora, 14 – Candida I, 15 – Candida II; The Dipodascaceae and related families (*) (in orange) includes: 16 – Komagataella, 17 – Yarrowia. Label accession indicates the specific amino acid sequence accession number and E-value indicates the significance of the comparison, the significance threshold was set at 1.0E-70. The K. lactis sequences used as query for the Blast analysis were indicated in a box.
To broaden the search for possible fungal orthologous of KlGbu1, we explored sequence data from the fungal phyla Basidiomycota, Ascomycota (excluding the Saccharomycotina already presented above) and Zygomycota which, together, comprise more than 85% of the characterized fungi. Blastp analysis using the KlGbu1 amino acid sequence as query confirmed that the presence of orthologous genes was widespread among fungi. This screen was based on stringent sequence similarity criteria (Blastp score > 250 and E-value < 1.0E-70); as a reference, the KlCar1 amino acid sequence queried with KlGbu1 sequence exhibited Blastp scores of 58 and an E-value of 8.0E-10, much lower than for the KlGbu1 orthologues identified in species reported in Table S4. These results suggest that guanidinobutyrase and, possibly, the newly identified alternative arginine degradation pathway are not limited to K. lactis but may be widespread among fungi.
Discussion
Characterization of an arginase independent arginine degradation pathway
The identification of an alternative, arginase-independent pathway for arginine metabolism in K. lactis could only be achieved by a systems biology approach that involved inspecting and integrating data from various cellular levels (Mustacchi et al., 2006; Kim et al., 2012). Together these approaches provided a strong basis for proposing a new fungal pathway for arginine degradation (Fig. 8). This pathway starts with a transamination of arginine into ketoarginine. Biochemical characterization of this enzymatic step was limited by the fact that ketoarginine is not commercially available and by the promiscuous substrate specificity of the numerous transaminases in K. lactis (Dujon et al., 2004). However, detection of a specific labelling pattern corresponding to 13C615N3 (M+9) arginine in the labelling experiments is fully consistent with removal and reincorporation of a labelled/unlabelled primary amino group via a reversible transamination reaction. This arginine isotopologue was observed in strain K. lactis IMS0367 (Klcar1Δ Arg+) as well as in the reference strain GG1632, which suggested that the reaction involved one or more constitutively expressed transaminases. In the second and third steps, ketoarginine is decarboxylated to guanidinobutanal and oxidized to guanidinobutyrate respectively. This coincided with the moderate transcriptional upregulation, (+2.7-fold) of KLLA0E02707g, a gene that is highly similar to S. cerevisiae ARO10, which encodes a broad substrate TPP-dependent 2-oxo acid decarboxylase (Vuralhan et al., 2005; Romagnoli et al., 2012; Bolat et al., 2013), a key component of the Erhlich pathway (Hazelwood et al., 2008). There are no literature data on activity of fungal TPP-dependent 2-oxo acid decarboxylases with ketoarginine as a substrate in yeasts. However, ketoarginine decarboxylase activity has been demonstrated in Pseudomonas species (Stalon and Mercenier, 1984; Yang and Lu, 2007b). The next step corresponds to a ureohydrolase reaction that converts guanidinobutyrate into γ-aminobutyrate and participates in the mobilization of nitrogen from the arginine skeleton by releasing urea. The role of the KlGBU1 encoded protein has been demonstrated both in vitro and in vivo. Finally, γ-aminobutyrate can be converted into succinate by the sequential activity of a GABA transaminase and succinate semialdehyde dehydrogenase. A transcript corresponding to KLLAF20548g gene, which shows strong sequence similarity with the S. cerevisiae GABA transaminase, was significantly upregulated in K. lactis IMS0367 (+4.7-fold). The topology of this proposed arginine catabolic pathway in K. lactis is similar to the arginine:pyruvate transaminase pathway, one of the several arginine degradation routes described in Pseudomonas aeruginosa (Yang and Lu, 2007a,b).
Fig. 8.

Updated arginine catabolism in K. lactis. Thick lines indicate the new guanidinobutyrase pathway linking arginine to succinate. The grey lines represent the reactions absent in K. lactis but present in other eukaryotes (as shown in Fig. 1). Reactions: EC 3.5.3.1: arginase, EC 4.1.1.17, ornithine decarboxylase, EC 2.6.1.13: ornithine aminotransferase, EC 1.5.1.2: pyrroline-5-carboxylate reductase, EC 1.5.99.8: proline dehydrogenase, EC 1.5.1.12: 1-pyrroline-5-carboxylate dehydrogenase, EC 2.6.1.: aminotransferase, EC 4.1.1.75: 2-oxo acid decarboxylase, EC 1.2.1.54: gamma-guanidinobutyraldehyde dehydrogenase, EC 3.5.3.7: guanidinobutyrate, EC 2.6.1.19: GABA transaminase, EC 1.2.1.16: succinate-semialdehyde dehydrogenase.
Guanidinobutyrase is widely distributed in Fungi
Most of the components of the proposed new pathway for arginine catabolism in K. lactis are also involved in other parts of metabolism: the transaminases are primarily involved in amino acid biosynthesis, the 2-oxo acid decarboxylases are involved in degradation of certain amino acids (Hazelwood et al., 2008), while γ-aminobutyrate transaminase and succinate semialdehyde dehydrogenase are involved in metabolism of GABA, which can be formed independently of guanidinobutyrate through decarboxylation of glutamate (Bach et al., 2009). The only enzyme activity that appears to be specific to this alternative arginine degradation pathway is the guanidinobutyrase. We identified KlGbu1 orthologues in a wide diversity of fungal genomes. Together, these sequences form a distinct group of putative guanidino-acid ureohydrolase genes that is well separated from genes encoding guanidino-amino acid hydrolase (arginase) and guanidino-amide (agmatinase) ureohydrolase groups. Several members (Table S4) of the new guanidino-acid ureohydrolase group were previously annotated as agmatinase [e.g. Scheffersomyces (Pichia) stipitis (SPEB2) agmatine ureohydrolase XM_001385297]. The present study indicates that functional annotation of these genes may have to be revised (Jeffries et al., 2007; Smith et al., 2008). Heterologous expression in a car1Δ strain of S. cerevisiae, followed by growth tests on media with either arginine or guanidinobutyrate as sole nitrogen source, offers a relatively simple approach to investigate whether these genes indeed encode guanidinobutyrases.
To further explore the diversity of fungal arginine catabolism, we investigated whether arginine decarboxylase genes could be identified in fungi. This would enable an alternative to the pathway described above, since arginine decarboxylase yields agmatine, the substrate for agmatinase, whose decarboxylation yields putrescine. This pathway is found in bacteria (Szumanski and Boyle, 1990; Sekowska et al., 2000), plants (Klein et al., 1999) and animals (Iyer et al., 2002). A Blastp search using the Escherichia coli arginine decarboxylase SpeA amino acid sequence as a query failed to identify predicted protein sequences in the fungal kingdom (thresholds E-value < 1.0E-20, Blast score > 100). We could hypothesize that the occurrence of a guanidino-acid ureohydrolase is correlated with the absence of arginine decarboxylase and consequently of a guanidino-amide ureohydrolase. This would indicate that fungi synthesize putrescine directly by decarboxylation of ornithine exclusively. Therefore the presence of guanidino acid ureohydrolase pathway would then represent an alternative to arginase in absence of an agmatinase pathway.
This new pathway described in this study, links arginine to succinate and demonstrates different entry points in central carbon and nitrogen metabolisms. It definitely increased the nitrogen source spectrum of K. lactis enabling this yeast to grow on medium containing guanidinobutyrate as sole nitrogen source; however, the physiological significance of this pathway remains unclear. Dedicated studies to investigate transcriptional regulation, localization of the enzymes comprising the pathway and connection to the arginine metabolism (arginase pathway and the polyamine synthesis) are necessary to understand the metabolic, regulatory and evolutionary meanings of the guanidinobutyrase pathway deeper.
Guanidinobutyrase has been lost after the WGD event
Although widespread in fungi, guanidinobutyrase genes were absent in Saccharomycetaceae clades (Saccharomyces sensu stricto, Vanderwaltozyma, Nakasoemyces, Naumovozyma) (Fig. 7) that arose after the WGD, an iconic event in the recent evolutionary history of the Saccharomycetaceae that occurred nearly 100 million year ago (Wolfe and Shields, 1997; Seoighe and Wolfe, 1999). The WGD is proposed to have involved the transient formation of a tetraploid ancestor, which gradually returned to a normal ploidy (2n) through a combination of a large number of deletion and epigenetic silencing (Sankoff et al., 2012; Zheng and Sankoff, 2012). The current model proposes that S. cerevisiae originates from an ancestor with a genome of ∼ 10 000 genes, a number that has been reduced to less than 6000 genes in S. cerevisiae, of which 1104 form 552 so-called ohnolog pairs (Scannell et al., 2006; Byrne and Wolfe, 2007). The guanidinobutyrase example is consistent with the notion that, for some genes, both ohnologs may have been lost during post-WGD genome size reduction. The observation that this particular reaction has been lost during genome size reduction may, after further physiological analysis of the enzymology and regulation of different fungal pathways for arginine catabolism, provide clues on the environmental conditions under which this event took place.
Interestingly, a few S. cerevisiae strains appear to have re-acquired KlGbu1 orthologue (Novo et al., 2009). Recent genome sequence analysis of the wine strains EC1118 (Novo et al., 2009) and VIN7 (Borneman et al., 2012) demonstrated that these strains have, by horizontal gene transfer, acquired DNA segments from microorganisms that share their natural habitats. In both strains, the KlGBU1 orthologous genes were found in an introgression region (region A, 38 kb) on chromosome VI (Novo et al., 2009) that most probably originated from T. delbrueckii (Gordon et al., 2011). Arginine is together with proline, the most abundant nitrogenous compound in grape must (Beltran et al., 2005). Under anaerobic conditions, arginine catabolism via the arginase pathway enables utilization of three of its four nitrogen atoms, two as a result of the arginase reaction and one from the ornithine transaminase step (Figs 1 and 8). The fourth nitrogen atom ends up in proline which, under anaerobic conditions cannot be assimilated by S. cerevisiae, as proline catabolism requires molecular oxygen. In contrast, arginine catabolism through the guanidinobutyrase pathway should enable the mobilization of all four nitrogen atoms, one issued from the arginine transamination, two from the guanidinobutyrase reaction, and the last one from GABA transamination. Therefore, the ‘guanidinobutyrase pathway’ could provide a selective advantage under the anaerobic and nitrogen limited conditions of wine fermentation (Bisson, 1999). The present study therefore provides a clear lead for exploring the significance of a horizontal gene transfer event in wine yeasts that can be explored by functional analysis of the wine-yeast KlGBU1 orthologue.
The present study clearly demonstrates that in K. lactis and more generally in Fungi arginine degradation was not limited to the ubiquitous arginase pathway. By using a top-down approach we could identify a new fungal metabolic pathway involved in arginine degradation implicating a guanidinobutyrase, an enzyme activity that could be widespread in Fungi.
Experimental procedures
Yeast strains and maintenance
The S. cerevisiae and K. lactis strains used in this study are listed in Table 6. The S. cerevisiae strains were constructed in the CEN.PK background (Entian and Kötter, 2007; Nijkamp et al., 2012) and the K. lactis strains were all derived from the strain CBS2359 (Wesolowski-Louvel et al., 1996; Dujon et al., 2004; Kooistra et al., 2004). Yeast strains were maintained on YPD medium (demineralized water; 10 g l−1 yeast extract; 20 g l−1 peptone; 20 g l−1 glucose). Culture stocks were prepared from shake flask cultures incubated at 30°C and stirred at 200 r.p.m., by addition of 20% (v/v) glycerol and were stored at −80°C.
Table 6.
Kluyveromyces lactis and Saccharomyces cerevisiae strains used in this study
| Strain | Organism | Genotype | Reference |
|---|---|---|---|
| CBS2359 | K. lactis | Prototrophic reference strain | Dujon et al. (2004) |
| GG1632 | K. lactis | Klku80Δ::loxP | Kooistra et al. (2004) |
| IMK432 | K. lactis | Klku80Δ::loxP KLLA0F00704Δ (Klcar1Δ)::loxP-kanMX-loxP | This study |
| IMK433 | K. lactis | Klku80Δ::loxP KLLA0B05247Δ (Klcar2Δ)::loxP-kanMX-loxP | This study |
| IMK434 | K. lactis | Klku80Δ::loxP KLLA0D10736Δ (Klpro3Δ)::loxP-kanMX-loxP | This study |
| IMS0367 | K. lactis | Klku80Δ::loxP KLLA0F00704Δ (Klcar1Δ)::loxP-kanMX-loxP Arg+ | This study |
| IMK560 | K. lactis | Klku80Δ::loxP KLLA0F00704Δ (Klcar1Δ)::loxP-kanMX-loxP Klgbu1Δ::loxP-amdsYM-loxP | This study |
| CEN.PK113-7 D | S. cerevisiae | Prototrophic reference strain MATa | Nijkamp et al. (2012) |
| CEN.PK113-5 D | S. cerevisiae | MATa ura3-52 | Entian and Kötter (2007) |
| IMK435 | S. cerevisiae | MATa ura3-52 car1Δ::loxP-kanMX-loxP | This study |
| IMK436 | S. cerevisiae | MATa ura3-52 car2Δ::loxP-kanMX-loxP | This study |
| IMK445 | S. cerevisiae | MATa ura3-52 pro3Δ::loxP-kanMX-loxP | This study |
| IMZ310 | S. cerevisiae | MATa ura3-52 car1Δ::loxP-kanMX-loxP pUDE168 (TDH3pr-KLCAR1-CYC1ter URA3 2μ) | This study |
| IMZ311 | S. cerevisiae | MATa ura3-52 car2Δ::loxP-kanMX-loxP pUDE169 (TDH3pr-KLCAR2-CYC1ter URA3 2μ) | This study |
| IMZ342 | S. cerevisiae | MATa ura3-52 pro3Δ::loxP-kanMX-loxP pUDE170 (TDH3pr-KLPRO3-CYC1ter URA3 2μ) | This study |
| IMZ312 | S. cerevisiae | MATa ura3-52 pAG426GPD-ccdB (TDH3pr-CYC1ter URA3 2μ) | This study |
| IME215 | S. cerevisiae | MATa ura3-52 pUDE264 (TDH3pr-KlGBU1-CYC1ter URA3 2μ) | This study |
| IME216 | S. cerevisiae | MATa ura3-52 pUDE265 (TDH3pr-KLLA0F04235g-CYC1ter URA3 2μ) | This study |
Deletion of CAR1, CAR2, PRO3 and KLLA0F27995g (KlGBU1) in S. cerevisiae and K. lactis
Gene deletions in S. cerevisiae were performed by integration of loxP-kan-loxP (kanMX) cassettes (Guldener et al., 1996) via the short-flanking-homology PCR method (Wach et al., 1994). Sequences of oligonucleotide primers are shown in Table S5. Deletion cassettes for CAR1, CAR2 and PRO3 were amplified using Phusion Hot-Start polymerase (Finnzymes, Landsmeer, the Netherlands) and the template plasmid pUG6 (Guldener et al., 1996) using primers ScCAR1Fw/ScCAR1Rv, ScCAR2Fw/ScCAR2Rv and ScPRO3Fw/ScPRO3Rv respectively. The transformation of S. cerevisiae CEN.PK113-5 D with the CAR1, CAR2 or PRO3 deletion cassettes were performed using the LiAc method as previously described in Gietz and Woods (2002) resulting in strains IMK435, IMK436 and IMK445 respectively. Cassettes for deletion of the corresponding K. lactis genes were similarly obtained from pUG6 using primers KlCAR1Fw/KlCAR1Rv, KlCAR2Fw/KlCAR2Rv and KlPRO3Fw/KlPRO3Rv. The resulting deletion cassettes were transformed into K. lactis GG1632 resulting in strains IMK432, IMK433 and IMK434 respectively (Kooistra et al., 2004). The deletion of the gene KLLA0F27995g (KlGBU1) was performed by the integration of a loxP-amdsYM-loxP (Solis-Escalante et al., 2013) cassette by homologous recombination at the locus of the corresponding gene using a cassette with long flanking homology region. The initial cassette was amplified from plasmid pUGamdsYM plasmid (Solis-Escalante et al., 2013) using primer KlGBU1del1 and KlGBU1del2, while the two flanking regions were amplified directly from genomic DNA using primers KlGBU1del5/KlGBU1del3 for the 5′ UTR and primers KlGBU1del4/KlGBU1del7 for the 3′ UTR. The three resulting PCR products were purified and used as template for a fusion PCR with the nested primers KlGBU1del6/KlGBU1del8. The fragment with the appropriate length was gel purified and used to transform the strain IMS0367 resulting in strain IMZ449 (Kooistra et al., 2004).
Correct integration of the kanMX or amdsYM cassettes and replacement of the gene of interest was, in all cases, verified by diagnostic PCR using two sets of primers: a forward primer specific for the 5′ UTR (untranslated region) of the gene (Table S5 labelled with conFw) and two reverse primers, one specific for the original gene (labelled with conRv) and one for the deletion cassette (tagged with KanA or amdSY).
Cloning and overexpression of KlCAR1, KlCAR2, KlPRO3, KLLA0F27995g/KlGBU1 and KLL0F04235g in S. cerevisiae
Genomic DNA of the prototrophic reference strains S. cerevisiae CEN.PK113-7D and K. lactis CBS2359 was prepared as described previously (Burke et al., 2007). ORFs KLLA0F00704g (KlCAR1), KLLA0B05247g (KlCAR2), KLLA0D10736g (KlPRO3) KLLA0F27995g (KlGBU1) and KLLA0F04235g were cloned from genomic DNA using Phusion Hot-Start polymerase (Finnzymes) and primers KlCAR1clFw/KlCAR1clRv, KlCAR2clFw/KlCAR2clRv, KlPRO3clFw/KlPRO3clRv, KlGBU1clFw/KlclGBU1Rv and KLLA0F04235gclFw/KLLA0F04235gclRv respectively.
The first three PCR products were cloned into the pCR-zero-blunt vector (Invitrogen, Breda, the Netherlands) resulting in plasmid pUD180, pUD181 and pUD186 respectively. These plasmids as well as the PCR product deriving from the KlGBU1 and KLLA0F04235g genes and the p426GPDccdB (Alberti et al., 2007) plasmid were digested with SpeI and XhoI, purified from gel and ligated into the expression vectors to yield pUDE168, pUDE169, pUDE170, pUDE264 and pUDE265 respectively (Table 7). The pUDE168 plasmid was transformed in strain IMK435 resulting in IMZ310; the pUD169 in strain IMK436 resulting in strain IMZ311; plasmid pUD170 in strain IMK445 resulting in strain IMZ342 and finally plasmid pUDE264 and pUDE265 in strain CEN.PK113-5 D resulting in strain IME215 and IME216 respectively.
Table 7.
Plasmids used in this study
| Plasmid | Characteristic | Reference |
|---|---|---|
| pUG6 | PCR template for loxP-kanMX4-loxP cassette | Guldener et al. (1996) |
| pUG-hphNT1 | PCR template for loxP-hphNT1-loxP cassette | Gueldener et al. (2002) |
| pUG-AmdsY | PCR template for loxP-amdSYM-loxP cassette | Solis-Escalante et al. (2013) |
| pCR-zero-blunt | Gateway entry clone | Invitrogen |
| pUD180 | Gateway entry clone, KlCAR1 | This study |
| pUD181 | Gateway entry clone, KlCAR2 | This study |
| pUD186 | Gateway entry clone, KlPRO3 | This study |
| pAG426GPDccdB | 2μ ori URA3 TDH3pr-ccdB-CYC1ter | Alberti et al. (2007) |
| pUDE168 | 2μ ori URA3 TDH3pr-KlCAR1-CYC1ter | This study |
| pUDE169 | 2μ ori URA3 TDH3pr-KlCAR2-CYC1ter | This study |
| pUDE170 | 2μ ori URA3 TDH3pr-KlPRO3-CYC1ter | This study |
| pUDE264 | 2μ ori URA3 TDH3pr-KlGBU1-CYC1ter | This study |
| pUDE265 | 2μ ori URA3 TDH3pr- KLLA0F04235-CYC1ter | This study |
Media and culture conditions
Growth experiments were conducted in synthetic medium containing salts, trace elements and vitamins, prepared and sterilized as described previously (Verduyn et al., 1992). Glucose was added to a final concentration of 20.0 g l−1. When ammonium sulphate was not the nitrogen source in the synthetic medium, it was replaced by l-arginine, l-ornithine, l-proline or l-guanidinobutyrate, these compounds were filter sterilized and added to sterile medium to concentrations of 3.3 g l−1, 5.0 g l−1, 8.7 g l−1 and 2.9 g l−1 respectively. Moreover, 3.3 g l−1 potassium sulphate was added to compensate for the removal of ammonium sulphate. If required, 0.15 g l−1 uracil and/or 200 mg l−1 of G418 were added to complete media. Selection Agar plates were made by adding 20.0 g l−1 agar to these synthetic media. Agar plates for selection of the amdsYM marker were prepared as described previously (Solis-Escalante et al., 2013).
Shake flask cultures were conducted in 500 ml or 100 ml shake flasks containing 100 ml or 20 ml of liquid medium respectively and incubated in an orbital shaker (New Brunswick Scientific, Edison, NJ) at 200 r.p.m. at 30°C. Controlled aerobic batch cultures were grown in 2 l laboratory bioreactors (Applikon, Schiedam, the Netherlands) with a working volume of 1.2 l. Culture pH was maintained at pH 6.0 by automatic addition of 2 M KOH (ADI 1030 Biocontroller, Applikon) and temperature was kept constant at 30°C (Lauda E300 thermostat, Lauda DR. R. Wosber Gmbh & Co. KG Lauda-Königshofen, Germany). Aeration was achieved by sparging air through the cultures at a flow rate of 0.5 l min−1 continuously stirring at 800 r.p.m. The exhaust gas of the bioreactor was cooled in a condenser using a cryostat set at 4°C (Lauda E100 cryostat). CO2 and O2 concentrations in the off gas were measured with an NGA 2000 analyser (Rosemount Analytical, Solon, OH). For spot plate assays, pre-cultures in synthetic medium with glucose diluted to 106 cells ml−1, which corresponded to an OD660 of 0.1 (Hazelwood et al., 2006). Serial dilutions (106–102 cells ml−1) were made and 5 μl of each dilution was spotted on agar plates and incubated at 30°C for 3 days.
Preparation of cell extracts
For preparation of cell extracts, culture samples were harvested by centrifugation, washed twice with 10 mM potassium phosphate buffer (pH 7.5) containing 2 mM EDTA and stored at −20°C. Before cell disruption, samples were thawed at room temperature, washed, and resuspended in 100 mM potassium phosphate buffer (pH 7.5) containing 2 mM MgCl2 and 2 mM dithiothreitol. Extracts were prepared by sonication with 0.7 mm glass beads at 0°C for 2 min at 0.5 min intervals with an MSE sonicator (Wolf Laboratories Limited, Pocklington, UK) (150 W output; 8 μm peak-to-peak amplitude) (Luttik et al., 2008). Unbroken cells and debris were removed by centrifugation at 4°C (20 min; 36 000 g). The resulting cell extract was used for enzyme assays.
Enzyme activity assays
For the arginase enzymatic assay 50 μl cell extract were activated in 950 μl manganese maleate buffer (50 mM manganese sulphate, 50 mM maleic acid, pH 7) for 1 h at 37°C (Messenguy et al., 1971). The reaction mixture for arginase assays, prepared in dark Eppendorf tubes, contained 60 μl of activated cell extract, 400 μl of 713 mM arginine solution (pH 9.5) and demineralized water up to 1 ml. The reaction mixture was incubated for 30 min at 37°C. To stop the reaction, 0.7 ml sulphuric-phosphoric acid mixture (20% v/v concentrated sulphuric acid and 60% v/v syrupy phosphoric acid in demineralized water) was added to the reaction mixture. The amount of urea produced was quantified using the Archibald method (Archibald, 1945) with a calibration line ranging from 0 until 0.6 mM of urea. 0.06 ml of a 4% v/v α-isonitrosopropiophenone in ethanol solution was added and samples were thoroughly mixed before boiling for 1 h in a 100°C water bath to develop the colour. The samples were cooled at room temperature for 15 min and the absorbance at 540 nm was measured in a Libra S11 spectrophotometer (Biochrom, Cambridge, UK).
The reaction mixture for ornithine amino transferase (OAT) was prepared in dark eppendorf tubes, containing, in a 1 ml final volume: 50 mM ornithine, 20 mM α-ketoglutarate, 1 mM pyridoxal 5-phosphate and 100 mM potassium phosphate buffer pH 8 (Vogel and Kopac, 1960) and 2.5 to 50 μl cell extract. The mixture was incubated for 30 min at 37°C before the reaction was stopped by addition of 0.5 ml 10% trichloroacetic acid. The amount of pyrroline-5-carboxylate produced was determined by the O-amino-benzaldehyde assay using a calibration line ranging from 0 to 0.5 mM. Colour was developed by addition of 0.5 ml 0.5% o-aminobenzaldehyde in 95% ethanol and incubation at room temperature for 1 h. Samples were then centrifuged and the clear supernatant was used to measure absorbance at 440 nm with a Libra S11 spectrophotometer (Biochrom).
The reaction mixture for pyrroline-5-carboxylate (P5C) reductase contained 50 mM N-[Tris(hydroxymethyl)methyl]-3-aminopropanesulphonic acid (TAPS), 0.14 mM NADH and cell extract in a final volume of 300 μl (Brandriss and Magasanik, 1979). Enzyme activity was monitored as NADH consumption at a wavelength of 340 nm using a GENios Microplate Reader (Tecan, Giessen, the Netherlands), after addition of pyrroline-5-carboxylate at pH 7 to a final concentration of 1 mM. Δ1-pyrroline-5-carboxylate (P5C) was synthesized through oxidation of 5-hydroxylysine as previously described in Williams and Frank (1975). To start, 71.4 mM hydroxylysine was prepared in a brown bottle. 50 mM sodium metaperiodate was prepared in dim light, neutralized with 1 M NaOH and 4.4 ml of the neutralized pre-cooled periodate solution were quickly added to 2.8 ml of ice-cold hydroxylysine solution and incubated for 8 min at 4°C. Remaining periodate was destroyed by addition of 70 μl of 1 M glycerol and incubation for 2 min. The mixture was then acidified by the addition of 60 μl of a 6 M HCl solution. The complete reaction mixture was applied to a 5 ml Dowex 50 column, which was equilibrated with water after thorough washing with a 1 M HCl solution. Elution of the P5C was performed by applying 100 ml of 1 M HCl and collecting 2 ml fractions. For qualitative detection of P5C 20 μl of each elution fraction were mixed with 20 μl of 3 M sodium acetate and 500 μl 0.15% ninhydrin in glacial acetic acid. The mixture was incubated on a hot plate at 100°C. If more than 0.5 μg of P5C were present in the test tube the mixture turned cherry-red shortly before reaching the boiling point of the solution. Elution fractions showing presence of sufficient amounts of P5C were then analysed quantitatively. Aliquots of the elution fractions containing P5C were mixed with HCl to a final concentration of 1 M HCl. Subsequently 200 μl of this mixture were added to 100 μl of 0.5% o-aminobenzaldehyde solution in 100% ethanol. After incubation for 40 min at room temperature the absorbance was measured at 444 nm. Concentrations were calculated using the molar absorption coefficient of 2940 M−1 cm−1.
The reaction mixture for guanidinobutyrase (GBU) enzyme assays was prepared in dark eppendorf tubes, containing in a 1 ml final volume: 50 mM glycine buffer (pH 9), 5 mM MnSO4 and 50 μl to 100 μl cell extract. The reaction was started by addition of 50 mM guanidinobutyric acid. After 30 min of incubation at 37°C, the reaction was stopped by addition of 700 μl of sulphuric-phosphoric acid mixture (20% v/v concentrated sulphuric acid and 60% v/v syrupy phosphoric acid in demineralized water). The amount of urea produced was quantified using the Archibald method (Archibald, 1945) with a calibration line ranging from 0 until 0.6 mM of urea. 0.06 ml of a 4% v/v α-isonitrosopropiophenone in ethanol solution was added and samples were thoroughly mixed before boiling for 1 h in a 100°C water bath to develop the colour. The samples were cooled at room temperature for 15 min and the absorbance at 540 nm was measured with a Libra S11 spectrophotometer (Biochrom).
Isotopomer labelling
The biomass was pre-cultivated in 500 ml shake flasks containing 120 ml of glucose synthetic medium with 3.3 g l−1 of non-labelled arginine as sole N-source. During the exponential growth phase the cells were harvested, washed with 10 mM potassium phosphate buffer (pH 7.5) and subsequently suspended in 2 separate shake flasks containing 50 ml minimal medium without any nitrogen source. Immediately after the addition of the labelled and unlabelled arginine to the 2 shake flasks, an initial sample of 800 μl was taken for intracellular metabolite measurement as described below. Samples were taken over the course of the experiment for half cell-duplication time. Intracellular metabolite samples obtained from cultivations without labelled material were used to determine intracellular metabolite concentrations. Isotopomer measurement data derived from the shake flasks containing the 13C615N4 labelled arginine was used for metabolic flux analysis.
Sampling and analytical techniques
Specific growth rates were estimated from the increase in optical density at 660 nm (OD660), as measured with a Novaspec II spectrophotometer (GE Healthcare Europe GmbH, Diegem, Belgium) after dilution of culture samples to OD660 values below 0.3. Biomass dry weight was determined by filtrating 10 ml culture samples over dry, pre-weighed nitrocellulose filters (Gelman laboratory, Ann Arbor, MI). After washing with demineralized water, filters were dried using a 360 W microwave oven and immediately weighed. Using multiple OD and corresponding dry weight measurements, a stoichiometric relation between these two parameters was determined for each of the batch cultures.
Extracellular metabolite samples were obtained after centrifugation of the culture broth. The supernatant was subsequently analysed using the analytical technology described below.
Glucose and ethanol concentration were determined using HPLC (Waters Alliance 2695 Separation Module combined with a Waters 2487 Dual Absorbance Detector and a Waters 2410 Refractive Index Detector, Waters, Milford, MA). Elution was achieved using a Bio Rad HPX87H column (Bio-Rad) at 60°C with 0.5 mM H2SO4 at a flow rate of 0.6 ml·min−1. Free amino acid concentrations were determined using the AccQ-Tag Ultra Derivatization Kit (Waters) and measurement by HPLC (Waters Alliance 2695 Separation Module combined with a Waters 474 fluorescent detector, Waters) according to the procedure described in vanWandelen and Cohen (1997). Extracellular urea and ammonia was measured using an enzyme essay kit (K-URAMR, Megazyme, Ireland). Using this kit the amount of both compounds was quantified by measuring the decrease in absorbance at 340 nm in a GENios Microplate Reader (Tecan).
Intracellular metabolite sampling requires rapid quenching of the cells using −40°C 80%/20% methanol/ethanol mixture. After extraction the 800 μl samples were added directly to 5 ml tubes of pre-cooled methanol. Subsequently cold methanol was used to wash the samples and the metabolites were extracted with boiling ethanol as previously described by Canelas et al. (2008). Accurate intracellular metabolite quantification required the addition of an internal standard in the form of uniformly labelled 13C-cell-extract (Wu et al., 2005). Using GC-MS, the concentration and mass shift of 13C15N samples was measured for, pyruvate (Pyr), alanine (Ala), glycine (Gly), valine (Val), leucine (Leu), iso-leucine (Ile), proline (Pro), serine (Ser), threonine (Thr), methionine (Met), aspartate (Asp), phenylalanine (Phe), cysteine (Cys), glutamate (Glu), lysine (Lys), asparginine (Asp), glutamine (Gln), tyrosine (Tyr), histidine (His), ornithine (Orn) and tryptophan (Trp) using the protocol described by de Jonge et al. (2011). GC-MS was also used to quantify iso-citrate (i-Cit), citrate (Cit), malate (Mal), fumarate (Fum), α-ketoglutarate (aKg), putrescine (Put) and γ-aminobutyrate (GABA) concentrations and 13C15N enrichment (Cipollina et al., 2009). Succinate (Suc) was measured using LC-MS according to methods previously developed by van Dam et al. (2002). Arginine and ornithine was measured by ion pair reversed phase liquid chromatography combined with tandem mass spectrometry (Seifar et al., 2009) after derivatization using o-phetalaldehyde/isobutyryl-l-cysteine (OPA-IBLC) in accordance to Hess (2012). The data derived from the isotopomer measurements were corrected for naturally occurring isotopes as well as mass-shifts caused by derivatization and non-carbon isotopes.
13C15N metabolic flux analysis
All calculations were done using the gPROMS software suite (Process Systems Enterprise Limited, London, UK) in which all fluxes and intracellular concentration were assumed to be in steady state. The set of differential equations consisted of two distinct parts; initially the fate of the 15N in nitrogen containing compounds was determined assuming a sole influx of labelled material through arginine. For each measured amino acid the corresponding incorporation into new biomass was calculated assuming the biomass composition as reported by Duarte et al. (2004). The 13C15N metabolic model established in this study contains 30 metabolites and 46 metabolic reactions. A more elaborate description of model parameters, data fitting and assumptions can be found in the supplemental data (Tables S2 and S3; Figs S1 and S2).
RNA-seq transcriptome analysis
Biomass samples for RNA extractions were sampled from mid-exponential phase of K. lactis strains GG and IMS0367 grown in batch cultivations in bioreactors with chemically defined medium with glucose and arginine as sole N-source. The sampling, the extraction and the cDNA synthesis was performed as previously described in Hazelwood et al. (2010).
Sequencing was performed using Illumina Genome Analyser II and carried out by Baseclear (Leiden, the Netherlands). Data sets of 50 bp single reads of at least 1-Gb were generated. RNA-seq data generated in this study were submitted to Genome Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) under Accession No. GSE56060.
The genome sequence of K. lactis strain CBS2359 and its annotations were retrieved from the genolevures database (Martin et al., 2011) and used for all analysis. The data were aligned to the reference using Burrows-Wheeler Alignment (BWA) tool (Li and Durbin, 2009) and the resulting Sequence Alignment/Map (SAM) file was converted to a sorted and indexed Binary Alignment/Map (BAM) format using samtools (Li and Durbin, 2009; Li et al., 2009). Gene expression levels were estimated using FPKM values by the Cufflinks software (Trapnell et al., 2010). To identify differential gene expression between strains GG1632 and IMS0367 grown in bioreactor batch cultures with arginine as sole nitrogen source, RNAseq data comparison was performed and statistically assessed using Cuffdiff (Trapnell et al., 2010).
Phylogenetic distribution analysis
The phylogenetic tree of the Saccharomycotina was based on the previously postulated structure in Kurtzman (2003) and Dujon (2010). The sequence of KlGBU1 orthologous genes were identified by Blastp and tBlastn in the genome sequences of the microorganisms mentioned in Fig. 8. The Blast significance thresholds were set at an E-value < 1.0E-70 and a Blast score > 250.
Acknowledgments
The research group of J.T.P. is part of the Kluyver Centre for Genomics of Industrial Fermentation (http://www.kluyvercentre.nl/pro1/general/home.asp), which is supported by The Netherlands Genomics Initiative (http://www.genomics.nl/). J.T.P. and J. M.D. were also supported by the ‘Platform Green Synthetic Biology’ programme (http://www.pgsb.nl) funded by NGI. This research was supported by BIOFLAVOUR, COST Action FA0907 (http://www.bioflavour.insa-toulouse.fr). The authors declare that they have no conflict of interests.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
Supporting Information
References
- Abdelal AT. Arginine catabolism by microorganisms. Annu Rev Microbiol. 1979;33:139–168. doi: 10.1146/annurev.mi.33.100179.001035. [DOI] [PubMed] [Google Scholar]
- Alberti S, Gitler AD, Lindquist S. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast. 2007;24:913–919. doi: 10.1002/yea.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archibald RM. Colorimetric determination of urea. J Biol Chem. 1945;157:507–518. [Google Scholar]
- Bach B, Meudec E, Lepoutre JP, Rossignol T, Blondin B, Dequin S, Camarasa C. New insights into {gamma}-aminobutyric acid catabolism: evidence for {gamma}-hydroxybutyric acid and polyhydroxybutyrate synthesis in Saccharomyces cerevisiae. Appl Environ Microbiol. 2009;75:4231–4239. doi: 10.1128/AEM.00051-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran G, Esteve-Zarzoso B, Rozes N, Mas A, Guillamon JM. Influence of the timing of nitrogen additions during synthetic grape must fermentations on fermentation kinetics and nitrogen consumption. J Agric Food Chem. 2005;53:996–1002. doi: 10.1021/jf0487001. [DOI] [PubMed] [Google Scholar]
- Bisson LF. Stuck and sluggish fermentations. Am J Enol Vitic. 1999;50:107–119. [Google Scholar]
- Boer VM, Daran JM, Almering MJ, de Winde JH, Pronk JT. Contribution of the Saccharomyces cerevisiae transcriptional regulator Leu3p to physiology and gene expression in nitrogen- and carbon-limited chemostat cultures. FEMS Yeast Res. 2005;5:885–897. doi: 10.1016/j.femsyr.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Boer VM, Tai SL, Vuralhan Z, Arifin Y, Walsh MC, Piper MD, et al. Transcriptional responses of Saccharomyces cerevisiae to preferred and nonpreferred nitrogen sources in glucose-limited chemostat cultures. FEMS Yeast Res. 2007;7:604–620. doi: 10.1111/j.1567-1364.2007.00220.x. [DOI] [PubMed] [Google Scholar]
- Bolat I, Romagnoli G, Zhu F, Pronk JT, Daran JM. Functional analysis and transcriptional regulation of two orthologs of ARO10, encoding broad-substrate-specificity 2-oxo-acid decarboxylases, in the brewing yeast Saccharomyces pastorianus CBS1483. FEMS Yeast Res. 2013;13:505–517. doi: 10.1111/1567-1364.12051. [DOI] [PubMed] [Google Scholar]
- Borneman AR, Desany BA, Riches D, Affourtit JP, Forgan AH, Pretorius IS, et al. The genome sequence of the wine yeast VIN7 reveals an allotriploid hybrid genome with Saccharomyces cerevisiae and Saccharomyces kudriavzevii origins. FEMS Yeast Res. 2012;12:88–96. doi: 10.1111/j.1567-1364.2011.00773.x. [DOI] [PubMed] [Google Scholar]
- Bossinger J, Cooper TG. Molecular events associated with induction of arginase in Saccharomyces cerevisiae. J Bacteriol. 1977;131:163–173. doi: 10.1128/jb.131.1.163-173.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandriss MC, Falvey DA. Proline biosynthesis in Saccharomyces cerevisiae: analysis of the PRO3 gene, which encodes delta 1-pyrroline-5-carboxylate reductase. J Bacteriol. 1992;174:5176. doi: 10.1128/jb.174.15.5176b.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandriss MC, Magasanik B. Genetics and physiology of proline utilization in Saccharomyces cerevisiae: mutation causing constitutive enzyme expression. J Bacteriol. 1979;140:504–507. doi: 10.1128/jb.140.2.504-507.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandriss MC, Magasanik B. Proline: an essential intermediate in arginine degradation in Saccharomyces cerevisiae. J Bacteriol. 1980;143:1403–1410. doi: 10.1128/jb.143.3.1403-1410.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke D, Dawson D, Stearns T. Methods in Yeast Genetics: Edition 2000. New York: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Byrne KP, Wolfe KH. Consistent patterns of rate asymmetry and gene loss indicate widespread neofunctionalization of yeast genes after whole-genome duplication. Genetics. 2007;175:1341–1350. doi: 10.1534/genetics.106.066951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canelas AB, Ras C, ten Pierick A, van Dam JC, Heijnen JJ, van Gulik WM. Leakage-free rapid quenching technique for yeast metabolomics. Metabolomics. 2008;4:226–239. [Google Scholar]
- Cipollina C, ten Pierick A, Canelas AB, Seifar RM, van Maris AJ, van Dam JC, Heijnen JJ. A comprehensive method for the quantification of the non-oxidative pentose phosphate pathway intermediates in Saccharomyces cerevisiae by GC-IDMS. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3231–3236. doi: 10.1016/j.jchromb.2009.07.019. [DOI] [PubMed] [Google Scholar]
- Coleman CS, Hu G, Pegg AE. Putrescine biosynthesis in mammalian tissues. Biochem J. 2004;379:849–855. doi: 10.1042/BJ20040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TG, Kovari L, Sumrada RA, Park HD, Luche RM, Kovari I. Nitrogen catabolite repression of arginase (CAR1) expression in Saccharomyces cerevisiae is derived from regulated inducer exclusion. J Bacteriol. 1992;174:48–55. doi: 10.1128/jb.174.1.48-55.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam JC, Eman MR, Frank J, Lange HC, vanDedem GWK, Heijnen JJ. Analysis of glycolytic intermediates in Saccharomyces cerevisiae using anion exchange chromatography and electrospray ionization with tandem mass spectrometric detection. Anal Chem Acta. 2002;460:209–218. [Google Scholar]
- Davis RH. Compartmental and regulatory mechanisms in the arginine pathways of Neurospora crassa and Saccharomyces cerevisiae. Microbiol Rev. 1986;50:280–313. doi: 10.1128/mr.50.3.280-313.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias O, Gombert AK, Ferreira EC, Rocha I. Genome-wide metabolic (re-) annotation of Kluyveromyces lactis. BMC Genomics. 2012;13:517. doi: 10.1186/1471-2164-13-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte NC, Herrgard MJ, Palsson BO. Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model. Genome Res. 2004;14:1298–1309. doi: 10.1101/gr.2250904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois E, Hiernaux D, Grennon M, Wiame JM. Specific induction of catabolism and its relation to repression of biosynthesis in arginine metabolism of Saccharomyces cerevisiae. J Mol Biol. 1978;122:383–406. doi: 10.1016/0022-2836(78)90417-5. [DOI] [PubMed] [Google Scholar]
- Dujon B. Yeast evolutionary genomics. Nat Rev Genet. 2010;11:512–524. doi: 10.1038/nrg2811. [DOI] [PubMed] [Google Scholar]
- Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, et al. Genome evolution in yeasts. Nature. 2004;430:35–44. doi: 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- Entian KD, Kötter P. Yeast genetic strain and plasmid collections. Methods Microbiol. 2007;36:629–666. [Google Scholar]
- Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- Gordon JL, Armisen D, Proux-Wera E, OhEigeartaigh SS, Byrne KP, Wolfe KH. Evolutionary erosion of yeast sex chromosomes by mating-type switching accidents. Proc Natl Acad Sci USA. 2011;108:20024–20029. doi: 10.1073/pnas.1112808108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002;30:e23. doi: 10.1093/nar/30.6.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 1996;24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman H. Speculations on the evolution of the genetic code. Orig Life. 1975;6:423–427. doi: 10.1007/BF01130344. [DOI] [PubMed] [Google Scholar]
- Hazelwood LA, Tai SL, Boer VM, de Winde JH, Pronk JT, Daran JM. A new physiological role for Pdr12p in Saccharomyces cerevisiae: export of aromatic and branched-chain organic acids produced in amino acid catabolism. FEMS Yeast Res. 2006;6:937–945. doi: 10.1111/j.1567-1364.2006.00094.x. [DOI] [PubMed] [Google Scholar]
- Hazelwood LA, Daran JM, van Maris AJ, Pronk JT, Dickinson JR. The Ehrlich pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae metabolism. Appl Environ Microbiol. 2008;74:2259–2266. doi: 10.1128/AEM.02625-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazelwood LA, Walsh MC, Pronk JT, Daran JM. Involvement of vacuolar sequestration and active transport in tolerance of Saccharomyces cerevisiae to hop iso-alpha-acids. Appl Environ Microbiol. 2010;76:318–328. doi: 10.1128/AEM.01457-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess S. A universal HPLC–MS method to determine the stereochemistry of common and unusual amino acids. Methods Mol Biol. 2012;828:63–75. doi: 10.1007/978-1-61779-445-2_7. [DOI] [PubMed] [Google Scholar]
- Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 2012;40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer RK, Kim HK, Tsoa RW, Grody WW, Cederbaum SD. Cloning and characterization of human agmatinase. Mol Genet Metab. 2002;75:209–218. doi: 10.1006/mgme.2001.3277. [DOI] [PubMed] [Google Scholar]
- Jeffries TW, Grigoriev IV, Grimwood J, Laplaza JM, Aerts A, Salamov A, et al. Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis. Nat Biotechnol. 2007;25:319–326. doi: 10.1038/nbt1290. [DOI] [PubMed] [Google Scholar]
- de Jonge LP, Buijs NA, ten Pierick A, Deshmukh A, Zhao Z, Kiel JA, et al. Scale-down of penicillin production in Penicillium chrysogenum. Biotechnol J. 2011;6:944–958. doi: 10.1002/biot.201000409. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IK, Roldao A, Siewers V, Nielsen J. A systems-level approach for metabolic engineering of yeast cell factories. FEMS Yeast Res. 2012;12:228–248. doi: 10.1111/j.1567-1364.2011.00779.x. [DOI] [PubMed] [Google Scholar]
- Klein RD, Geary TG, Gibson AS, Favreau MA, Winterrowd CA, Upton SJ, et al. Reconstitution of a bacterial/plant polyamine biosynthesis pathway in Saccharomyces cerevisiae. Microbiol-SGM. 1999;145:301–307. doi: 10.1099/13500872-145-2-301. [DOI] [PubMed] [Google Scholar]
- Knijnenburg TA, Daran JM, van den Broek MA, Daran-Lapujade PA, de Winde JH, Pronk JT, et al. Combinatorial effects of environmental parameters on transcriptional regulation in Saccharomyces cerevisiae: a quantitative analysis of a compendium of chemostat-based transcriptome data. BMC Genomics. 2009;10:53. doi: 10.1186/1471-2164-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J. 1994;298((Part 2)):249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra R, Hooykaas PJ, Steensma HY. Efficient gene targeting in Kluyveromyces lactis. Yeast. 2004;21:781–792. doi: 10.1002/yea.1131. [DOI] [PubMed] [Google Scholar]
- Krebs H. The discovery of the ornithine cycle of urea synthesis. Biochem Educ. 1973;1:19–23. [Google Scholar]
- Krebs H, Henseleit K. Untersuchungen über die Harnstoffbildung im Tierkörper I. Wien Klin Wochenschr. 1932;11:757–759. [Google Scholar]
- Kumar S, Punekar NS. High-throughput screening of dye-ligands for chromatography. Methods Mol Biol. 2014;1129:53–65. doi: 10.1007/978-1-62703-977-2_6. [DOI] [PubMed] [Google Scholar]
- Kurtzman CP. Phylogenetic circumscription of Saccharomyces, Kluyveromyces and other members of the Saccharomycetaceae, and the proposal of the new genera Lachancea, Nakaseomyces, NaumoviaVanderwaltozyma and Zygotorulaspora. FEMS Yeast Res. 2003;4:233–245. doi: 10.1016/S1567-1356(03)00175-2. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttik MA, Vuralhan Z, Suir E, Braus GH, Pronk JT, Daran JM. Alleviation of feedback inhibition in Saccharomyces cerevisiae aromatic amino acid biosynthesis: quantification of metabolic impact. Metab Eng. 2008;10:141–153. doi: 10.1016/j.ymben.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Martin O, Brandriss MC, Schneider G, Bakalinsky AT. Improved anaerobic use of arginine by Saccharomyces cerevisiae. Appl Environ Microbiol. 2003;69:1623–1628. doi: 10.1128/AEM.69.3.1623-1628.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin T, Sherman DJ, Durrens P. The Genolevures database. C R Biol. 2011;334:585–589. doi: 10.1016/j.crvi.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Messenguy F, Penninckx M, Wiame JM. Interaction between arginase and ornithine carbamoyltransferase in Saccharomyces cerevisiae. The regulatory site for ornithine. Eur J Biochem. 1971;22:277–286. doi: 10.1111/j.1432-1033.1971.tb01542.x. [DOI] [PubMed] [Google Scholar]
- Mustacchi R, Hohmann S, Nielsen J. Yeast systems biology to unravel the network of life. Yeast. 2006;23:227–238. doi: 10.1002/yea.1357. [DOI] [PubMed] [Google Scholar]
- Nijkamp JF, van den Broek MA, Datema E, de Kok S, Bosman L, Luttik MA, et al. De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7 D, a model for modern industrial biotechnology. Microb Cell Fact. 2012;11:36. doi: 10.1186/1475-2859-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novo M, Bigey F, Beyne E, Galeote V, Gavory F, Mallet S, et al. Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proc Natl Acad Sci USA. 2009;106:16333–16338. doi: 10.1073/pnas.0904673106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouzounis CA, Kyrpides NC. On the evolution of arginases and related enzymes. J Mol Evol. 1994;39:101–104. doi: 10.1007/BF00178255. [DOI] [PubMed] [Google Scholar]
- Park HD, Scott S, Rai R, Dorrington R, Cooper TG. Synergistic operation of the CAR2 (Ornithine transaminase) promoter elements in Saccharomyces cerevisiae. J Bacteriol. 1999;181:7052–7064. doi: 10.1128/jb.181.22.7052-7064.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg AE. Mammalian polyamine metabolism and function. IUBMB Life. 2009;61:880–894. doi: 10.1002/iub.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen LT, Heiskala M, Andersson LC. Expression of a novel human ornithine decarboxylase-like protein in the central nervous system and testes. Biochem Biophys Res Commun. 2001;287:1051–1057. doi: 10.1006/bbrc.2001.5703. [DOI] [PubMed] [Google Scholar]
- Romagnoli G, Luttik MA, Kotter P, Pronk JT, Daran JM. Substrate specificity of thiamine pyrophosphate-dependent 2-oxo-acid decarboxylases in Saccharomyces cerevisiae. Appl Environ Microbiol. 2012;78:7538–7548. doi: 10.1128/AEM.01675-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankoff D, Zheng C, Wang B. A model for biased fractionation after whole genome duplication. BMC Genomics. 2012;13(Suppl. 1):S8. doi: 10.1186/1471-2164-13-S1-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell DR, Byrne KP, Gordon JL, Wong S, Wolfe KH. Multiple rounds of speciation associated with reciprocal gene loss in polyploid yeasts. Nature. 2006;440:341–345. doi: 10.1038/nature04562. [DOI] [PubMed] [Google Scholar]
- Seifar RM, Ras C, van Dam JC, van Gulik WM, Heijnen JJ, van Winden WA. Simultaneous quantification of free nucleotides in complex biological samples using ion pair reversed phase liquid chromatography isotope dilution tandem mass spectrometry. Anal Biochem. 2009;388:213–219. doi: 10.1016/j.ab.2009.02.025. [DOI] [PubMed] [Google Scholar]
- Sekowska A, Danchin A, Risler JL. Phylogeny of related functions: the case of polyamine biosynthetic enzymes. Microbiol-SGM. 2000;146:1815–1828. doi: 10.1099/00221287-146-8-1815. [DOI] [PubMed] [Google Scholar]
- Seoighe C, Wolfe KH. Updated map of duplicated regions in the yeast genome. Gene. 1999;238:253–261. doi: 10.1016/s0378-1119(99)00319-4. [DOI] [PubMed] [Google Scholar]
- Shima J, Sakata-Tsuda Y, Suzuki Y, Nakajima R, Watanabe H, Kawamoto S, Takano H. Disruption of the CAR1 gene encoding arginase enhances freeze tolerance of the commercial baker's yeast Saccharomyces cerevisiae. Appl Environ Microbiol. 2003;69:715–718. doi: 10.1128/AEM.69.1.715-718.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Quinlan AR, Peckham HE, Makowsky K, Tao W, Woolf B, et al. Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res. 2008;18:1638–1642. doi: 10.1101/gr.077776.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis-Escalante D, Kuijpers NG, Bongaerts N, Bolat I, Bosman L, Pronk JT, et al. amdSYM, a new dominant recyclable marker cassette for Saccharomyces cerevisiae. FEMS Yeast Res. 2013;13:126–139. doi: 10.1111/1567-1364.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souciet JL, Dujon B, Gaillardin C, Johnston M, Baret PV, Cliften P, et al. Comparative genomics of protoploid Saccharomycetaceae. Genome Res. 2009;19:1696–1709. doi: 10.1101/gr.091546.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalon V, Mercenier A. L-arginine utilization by Pseudomonas species. J Gen Microbiol. 1984;130:69–76. doi: 10.1099/00221287-130-1-69. [DOI] [PubMed] [Google Scholar]
- Sumrada RA, Cooper TG. The arginase (CAR1) gene is situated near MF alpha 1 on the right arm of chromosome XVI. Yeast. 1992;8:311–314. doi: 10.1002/yea.320080408. [DOI] [PubMed] [Google Scholar]
- Szumanski MB, Boyle SM. Analysis and sequence of the speB gene encoding agmatine ureohydrolase, a putrescine biosynthetic enzyme in Escherichia coli. J Bacteriol. 1990;172:538–547. doi: 10.1128/jb.172.2.538-547.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomenchok DM, Brandriss MC. Gene-enzyme relationships in the proline biosynthetic pathway of Saccharomyces cerevisiae. J Bacteriol. 1987;169:5364–5372. doi: 10.1128/jb.169.12.5364-5372.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vanWandelen C, Cohen SA. Using quaternary high-performance liquid chromatography eluent systems for separating 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate-derivatized amino acid mixtures. J Chromatogr A. 1997;763:11–22. [Google Scholar]
- Verduyn C, Postma E, Scheffers WA, van Dijken JP. Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast. 1992;8:501–517. doi: 10.1002/yea.320080703. [DOI] [PubMed] [Google Scholar]
- Vissers S, Andre B, Muyldermans F, Grenson M. Positive and negative regulatory elements control the expression of the UGA4 gene coding for the inducible 4-aminobutyric-acid-specific permease in Saccharomyces cerevisiae. Eur J Biochem. 1989;181:357–361. doi: 10.1111/j.1432-1033.1989.tb14732.x. [DOI] [PubMed] [Google Scholar]
- Vogel RH, Kopac MJ. Some properties of ornithine delta transaminase from Neurospora. Biochim Biophys Acta. 1960;37:539–540. [Google Scholar]
- Vuralhan Z, Luttik MA, Tai SL, Boer VM, Morais MA, Schipper D, et al. Physiological characterization of the ARO10-dependent, broad-substrate-specificity 2-oxo acid decarboxylase activity of Saccharomyces cerevisiae. Appl Environ Microbiol. 2005;71:3276–3284. doi: 10.1128/AEM.71.6.3276-3284.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Wesolowski-Louvel M, Breuning KD, Fukuhara H. Kluyveromyces lactis. In: Wolf K, editor. Non Conventional Yeasts in Biotechnology. A Hanbook. Heidelberg: Springer-Verlag; 1996. pp. 139–201. [Google Scholar]
- Williams I, Frank L. Improved chemical synthesis and enzymatic assay of delta-1-pyrroline-5-carboxylic acid. Anal Biochem. 1975;64:85–97. doi: 10.1016/0003-2697(75)90408-x. [DOI] [PubMed] [Google Scholar]
- Wolfe KH, Shields DC. Molecular evidence for an ancient duplication of the entire yeast genome. Nature. 1997;387:708–713. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]
- Wu L, Mashego MR, van Dam JC, Proell AM, Vinke JL, Ras C, et al. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal Biochem. 2005;336:164–171. doi: 10.1016/j.ab.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Yang Z, Lu CD. Characterization of an arginine:pyruvate transaminase in arginine catabolism of Pseudomonas aeruginosa PAO1. J Bacteriol. 2007a;189:3954–3959. doi: 10.1128/JB.00262-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Lu CD. Functional genomics enables identification of genes of the arginine transaminase pathway in Pseudomonas aeruginosa. J Bacteriol. 2007b;189:3945–3953. doi: 10.1128/JB.00261-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Sankoff D. Gene order in rosid phylogeny, inferred from pairwise syntenies among extant genomes. BMC Bioinformatics. 2012;13(Suppl. 10):S9. doi: 10.1186/1471-2105-13-S10-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information