

Abstract
Tumor suppressor retinoblastoma-associated protein (Rb) is an important cell cycle regulator, arresting cells in early G1. It is commonly inactivated in cancers and its level is maintained during the cell cycle. Rb is regulated by various post-translational modifications such as phosphorylation, acetylation, ubiquitination and so on. Several E3 ligases including murine double minute 2 (MDM2) promote the degradation of Rb. This study focuses on the role of HAUSP (herpes virus associated ubiquitin specific protease) on Rb. Here, we show that HAUSP colocalizes and interacts with Rb to stabilize it from proteasomal degradation by removing wild-type and K48-linked ubiquitin chains in human embryonic kidney 293 (HEK293) cells. HAUSP deubiquitinates Rb in vivo and in vitro, leading to an increased cell population in the G1 phase. Hence, HAUSP is a novel deubiquitinase for Rb. Immunohistochemistry, western blotting and cell-based assays show that HAUSP is overexpressed in glioma and contributes towards glioma progression. However, HAUSP activity on Rb is abrogated in glioma (cancer), where these two proteins show an inverse relationship. MDM2 (a known substrate of HAUSP) serves as a better target for HAUSP-mediated deubiquitination in cancer cells, facilitating degradation of Rb and oncogenic progression. This novel regulatory axis is proteasome mediated, p53 independent, and the level of MDM2 is critical. The shift in equilibrium by differential deubiquitination in regulation of Rb explains a subtle difference existing between normal and cancer cells. This leads to speculation about a new possibility for distinguishing cancer cells from normal cells at the molecular level, which may be investigated for therapeutic intervention in the future.
Structured digital abstract
HAUSP and Rb colocalize by fluorescence microscopy (View interaction)
HAUSP binds to Rb by pull down (View interaction)
HAUSP physically interacts with Rb by anti bait coip (1, 2, 3, 4, 5, 6)
Keywords: deubiquitination, glioma, herpes virus associated ubiquitin specific protease, murine double minute 2, Rb
Introduction
Retinoblastoma-associated protein (Rb) was the first reported tumor suppressor [1] and is a nuclear phosphoprotein [2]. It restricts the cell cycle in normal cells and its function is often impaired in several cancers. Rb is a multifunctional protein exhibiting distinct roles in growth suppression, differentiation and apoptosis [3,4]. Rb is conserved in flies and mammals and knockout mice show embryonic lethality, emphasizing its role in development and cancer [5]. Inactivation of the Rb pathway by deletion or mutation in the RB gene or deregulation of its upstream regulators – cyclin D1, cdk4 or p16 in cancer cells – partially accounts for their capacity for uncontrolled growth [6]. Rb expression is maintained throughout the cell cycle and the variation in its activity is mainly due to changes in its phosphorylation state [7]. Under genotoxic stress, p38 mitogen activated protein kinase (p38-MAPK) preferentially phosphorylates Rb in a cell cycle independent manner, facilitating Rb–murine double minute 2 (MDM2) interaction and subsequent Rb degradation [8]. Apart from phosphorylation, Rb activity is modulated by a number of other post-translational modifications such as acetylation [9], methylation [10], sumoylation [11] and ubiquitination [12].
The process of ubiquitination is extremely dynamic and regulated. A new class of proteins, the deubiquitinating enzymes, can reverse the action of E3 ligases by specifically removing the ubiquitin (Ub) tags from proteins. The importance of these enzymes lies in the fact that these are critical factors, maintaining the overall cellular signaling [13]. Rb degradation is associated with E3 ligases such as human papilloma virus E7, Epstein–Barr virus nuclear antigen 3C, human cytomegalovirus pp71, hepatocellular carcinoma associated protein gankyrin, human T-lymphotropic virus I Tax and MDM2 [12]. MDM2 degrades Rb via both Ub-dependent 26S proteasome [14] and Ub-independent 20S proteasome [15]. MDM2 also induces degradation of the other pocket proteins p107 and p130, upon 5-aza-2′-deoxycytidine treatment [16]. Although we have some knowledge of the mechanisms of Rb degradation, very little is known about stabilization of Rb. Recent research focuses on proteins increasing Rb stability. Lamin A acts as a scaffolding protein for Rb by interacting with and tethering Rb to the nuclear matrix. Cells lacking A-type lamin show reduced levels of Rb which is degraded by the proteasomal system [17,18]. Paired box protein 8 (Pax8) also stabilizes Rb and consequently regulates E2F1 transactivation [19].
Herpes virus associated ubiquitin specific protease (HAUSP or USP7) has varied roles in a number of biological processes ranging from genome stability, epigenetic regulation, cell cycle and apoptosis to viral infection, immunity and even stem cell maintenance and hence emerges as a very important candidate with implications in cancer and other pathologies [20]. In this study, we report for the first time that HAUSP stabilizes Rb in human embryonic kidney 293 (HEK293) cells by deubiquitination, but this activity is abrogated in glioma cells. MDM2 directs Rb degradation via Ub-dependent as well as Ub-independent mechanisms. In addition to stabilization of MDM2 by HAUSP, it might be possible that HAUSP reverses Rb ubiquitination by MDM2 in normal cells but is overwhelmed by abundant MDM2 in the case of tumor tissues or cancer cells. Clinical reports suggest deregulated Rb pathways in glioma: RB deletions in low-grade gliomas (including oligodendroglioma and ependymoma), RB1 mutation in high-grade astrocytomas (∼ 25%), loss of heterozygosity in RB1 in malignant glioma (54%) [21,22], amplification of CDK4 (134%) and CDK6 (2%) [23] or p16 loss-of-function [24] in ∼ 15% high-grade gliomas. These indicate Rb inactivation to be an early genetic event responsible for the development and progression of glioma [25,26] and also that inactivation of the Rb pathway is essential for glioblastoma multiforme (GBM), although it may not serve as the sole strategy to block cell cycle and proliferation. MDM2 is also known to be amplified and overexpressed (both gene and protein) in GBM [27] and is associated with short-term survival of patients [28]. Certain MDM2 splice variants are found in some GBM cases [29]. Here, we show that HAUSP is upregulated in glioma and its regulation of Rb is MDM2 dependent. This indicates the tumorigenic potential of HAUSP, which is partially fulfilled by decrease in Rb levels in cancer cells due to stabilization of MDM2. This may be yet another mechanism for Rb loss-of-function in the context of glioma.
Results
HAUSP stabilizes Rb protein levels
To find the role of HAUSP in Rb stabilization, HAUSP was exogenously expressed in HEK293 cells. Upon HAUSP overexpression, there was an increase in the Rb protein level (Fig. 1A). Similar data were also found in the normal monkey kidney cell line COS-7 (Fig. 1B). As Rb is a member of the pocket protein family with the other two members, p107 and p130 [30], we analyzed whether HAUSP had any effect on the other Rb family proteins. Under similar conditions, we examined the levels of p107 and p130 and their levels were found to remain unaltered (Fig. 1A). Additionally, there was no significant change found in Rb transcript levels (Fig. 1C).
Fig. 1.

HAUSP stabilizes Rb. (A) GFP-HAUSP was overexpressed in HEK293 cells and Rb, p107 and p130 protein levels were determined by WB. Lamin B serves as the loading control (HAUSP, upper band in lane 2 indicates GFP-tagged HAUSP; Rb, two bands may represent differentially phosphorylated forms; p130, lowermost band at 130 kDa represents the specific form). (B) GFP-HAUSP was overexpressed in COS-7 cells and Rb protein levels were determined by WB. Actin serves as the loading control. (C) Rb transcript levels were quantified by quantitative real-time PCR (qRT-PCR) upon HAUSP overexpression. Data represent the mean ± SEM from three biological repeats in triplicate; P = 0.96 (ns). (D) WB was done to verify the effect of siRNA-mediated partial knockdown of HAUSP on Rb protein expression using specific siRNAs for 72 h. (E) WB was performed to verify the effect of shRNA-mediated HAUSP partial knockdown on Rb protein expression. Two rounds of transfections were performed and cells were harvested after 72 h. (F) WB was performed to verify HAUSP knockdown and the effect on Rb protein expression using two different shRNAs. The values have been estimated by densitometry and normalized against the loading control actin (left panel). Graphical representation of HAUSP and Rb expression levels in HAUSP knockdown conditions from two independent repeats; P = 0.0073 (**), 0.0226 (*), 0.9 (ns) and 0.022 (*) (right panel). (G) Under similar conditions of HAUSP knockdown, Rb transcripts were quantified by qRT-PCR. Data represent mean ± SEM from three biological repeats in triplicate; P = 0.56 (ns). (H) For the rescue experiment, specific siRNA was used to knockdown HAUSP and the protein level of Rb was estimated by WB (left panel). Under similar conditions HAUSP was exogenously overexpressed and Rb level was estimated by WB (middle panel). The values have been estimated by densitometry and normalized against the loading control actin. Densitometric analysis of protein levels from three repeats of the rescue experiment show significant decrease in Rb level with knockdown of HAUSP [P = 0.004 (*)] while overexpression of HAUSP leads to rescue of Rb levels [P = 0.057 (ns)] (right panel). (I) Cells were transfected either with empty vector (left panel) or GFP-HAUSP (right panel) and treated with CHX (50 μg·mL−1) for a CHX chase assay to estimate the Rb protein levels in the presence or absence of HAUSP for the indicated time points in hours. (J) Graph showing Rb half-life in the absence or presence of HAUSP. Data represent mean ± SD from three independent experiments. ns, not significant; *, significant; **, very significant; ***, extremely significant.
Partial knockdown of HAUSP using specific small interfering RNA (siRNA) (Fig. 1D) or small hairpin RNA (shRNA) (Fig. 1E,F) resulted in decreased Rb protein levels. Again, upon gene knockdown, no significant change was observed in the Rb transcript level (Fig. 1G). To verify whether the effect of HAUSP knockdown was specific, we performed a rescue experiment by overexpressing HAUSP in the knockdown background. It was found that Rb expression was significantly rescued (Fig. 1H) proving the effect to be specific with respect to HAUSP activity.
When protein synthesis was blocked using cycloheximide (CHX) in either the absence or presence of HAUSP for the indicated time points, it was found that HAUSP stabilized Rb (Fig. 1I). The assessment of Rb turnover showed that HAUSP indeed increased the Rb protein half-life (Fig. 1J). Altogether these results clearly indicate that HAUSP stabilizes the Rb protein level.
HAUSP and Rb colocalize and physically interact with each other
As HAUSP is a deubiquitinating enzyme, we speculated that the stabilization of Rb could be an outcome of HAUSP-mediated deubiquitination for which interaction is necessary. First, we performed immunocytochemical (ICC) analysis to verify whether the two proteins colocalize within the cell or not. Confocal images from ICC in HEK293 cells show abundant yellow spots revealing that indeed HAUSP and Rb colocalize (Fig. 2A, upper panel). Again, similar data were found when ICC was done in COS-7 cells (Fig. 2A, lower panel).
Fig. 2.
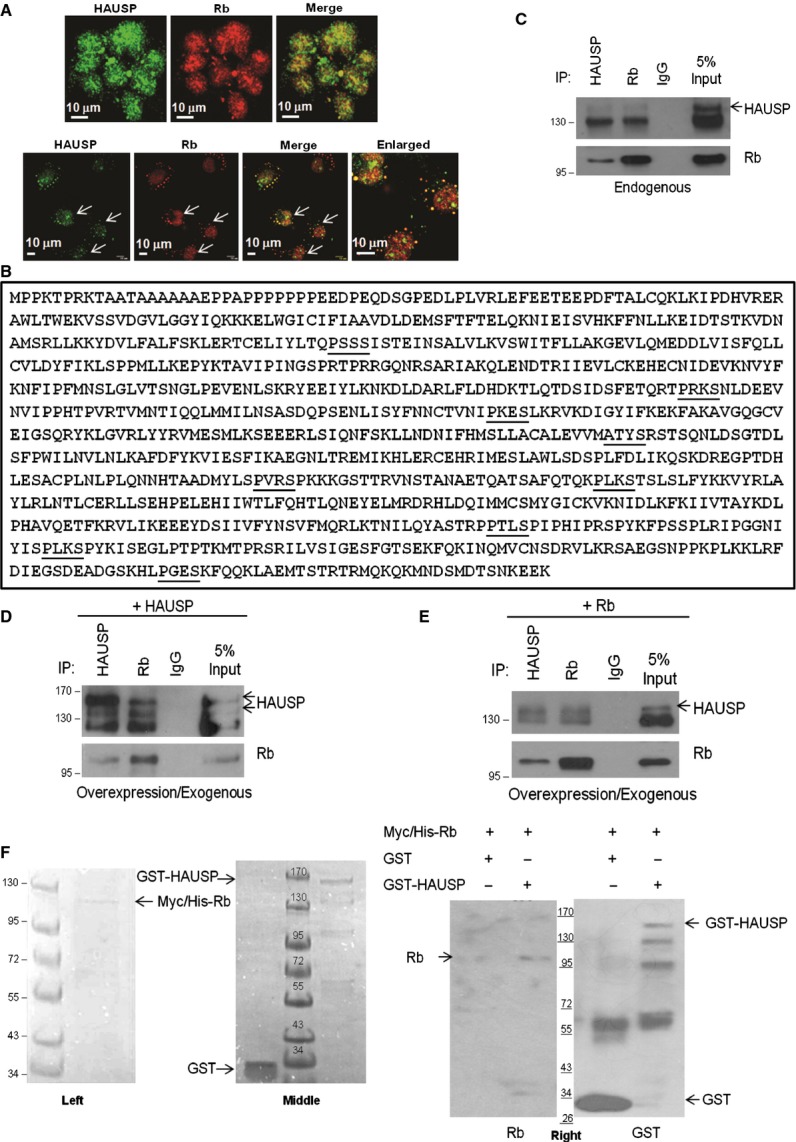
HAUSP physically interacts with Rb. (A) Confocal images of HEK293 (upper panel) and COS-7 (lower panel) cells showing stains for HAUSP (green), Rb (red) and HAUSP-Rb merge (yellow). Images were taken at 60 × optical magnification and scale bars are marked. (B) Protein sequence of Rb showing putative HAUSP binding motifs in bold and underlined characters. (C) Equal amounts of proteins were immunoprecipitated with either HAUSP or Rb antibodies followed by WB for HAUSP and Rb, showing their interaction in endogenous forms. (D), (E) Equal amounts of proteins from cells overexpressing either (D) HAUSP or (E) Rb were immunoprecipitated with HAUSP and Rb antibodies followed by WB for HAUSP and Rb, showing interaction of (D) endogenous Rb with exogenous HAUSP and (E) endogenous Rb with exogenous HAUSP. All immunoprecipitations were carried out using nuclear lysates from HEK293 cells. (F) Coomassie stained gels showing His-tagged Rb (Myc/His-Rb) after elution with 250 mm imidazole from Ni–nitrilotriacetic acid (Ni-NTA) beads (left panel) and the GST-tagged proteins (GST and GST-HAUSP) after elution with reduced glutathione from Glutathione HiCap Matrix beads (middle panel). In vitro interaction of Rb and HAUSP is shown and purified Rb is pulled down by GST-HAUSP (lane 2) and not GST only (lane 1) using Glutathione HiCap Matrix beads (right panel).
Second, we analyzed the protein sequence of Rb which revealed a number of putative HAUSP binding motifs (P/AxxS) [31] (Fig. 2B) and, to examine whether the two proteins physically interact, immunoprecipitation (IP) assays were performed. HAUSP is mainly a nuclear protein present in nuclear promyelocytic leukemia (PML) bodies [32] and Rb is also a nuclear protein; hence the IPs were done with the nuclear lysates obtained from HEK293 cells. IP with antibody against HAUSP showed endogenous Rb to co-immunoprecipitate while the reverse IP with antibody against Rb revealed that endogenous HAUSP was pulled down (Fig. 2C). Therefore, HAUSP and Rb were found to interact with each other in endogenous conditions.
Third, to further confirm interactions of these two proteins in an overexpressed system, HAUSP was transiently expressed in HEK293 cells and the lysate was subjected to IP using antibodies against both HAUSP and Rb; again, HAUSP and Rb were found to co-immunoprecipitate (Fig. 2D). A similar exogenous system was obtained by transient transfection of Rb in HEK293 cells and IP with antibody against HAUSP and reverse IP with antibody against Rb revealed similar results to those above (Fig. 2E). Therefore, exogenously expressed proteins were also capable of interacting with endogenous proteins.
In an in vitro set-up, purified His-tagged Rb was pulled down with purified GST-HAUSP but not with glutathione S-transferase (GST) (Fig. 2F, left panel, purified Rb; middle panel, purified GST and GST-HAUSP; right panel, in vitro interaction), indicating that the proteins interact in vitro (the GST purified protein is seen as multiple bands because of degradation due to its large size ∼ 155 kDa). Hence, HAUSP and Rb colocalize and physically interact with each other.
HAUSP deubiquitinates Rb
The obvious question that arises at this point is whether this interaction leads to HAUSP-mediated Rb deubiquitination. To verify the phenomenon, HAUSP (wild-type, WT) or its catalytically inactive mutant C223S (CI) [33] were cotransfected with Ub-WT in HEK293 cells. Western blotting (WB) showed Rb deubiquitination in the presence of WT but not with CI HAUSP (Fig. 3A, WB, left panel; graphical representation, right panel). An in vivo deubiquitination assay in the presence of Ub-WT showed a distinct decrease in Ub laddering of Rb with WT but not CI HAUSP, proving HAUSP to be a deubiquitinase for Rb (Fig. 3B, left panel). This is the first report suggesting Rb to be a novel target for HAUSP-mediated deubiquitination. As lysine 48 ubiquitin (K48) linkage is implicated in proteasome-mediated degradation [34], to evaluate the role of HAUSP in removing K48-linked Ub chains the above experiment was repeated with Ub-K48 (all the Lys residues except K48 are mutated to Arg) instead of Ub-WT. The results clearly showed that HAUSP could also remove K48-linked Ub chains, rescuing Rb from proteasomal degradation (Fig. 3B, middle panel). Both sets of data were represented graphically (Fig. 3B, right panel). Hence, deubiquitination by HAUSP accounts for the stabilization of Rb. An in vitro deubiquitination assay was next carried out in a cell-free system, where it was found that in the presence of purified GST-HAUSP poly-Ub Rb becomes deubiquitinated, while the presence of the inhibitor N-ethylmaleimide (NEM) inhibits HAUSP enzyme activity (Fig. 3C, WB, left panel; graphical representation, right panel).
Fig. 3.

HAUSP deubiquitinates Rb both in vivo and in vitro, leading to increased cell population in G1 in HEK293 cells. (A) WB of Rb cotransfected with WT or CI HAUSP in the presence or absence of Ub-WT (left panel). Graphical representation of data obtained from WB (right panel). Data represent mean ± SEM from three independent experiments; P values are 0.05 (*), 0.02 (*), 0.3 (ns) and 0.007 (**) respectively. (B) In vivo deubiquitination assay. IP was done with Rb antibody from lysates of HEK293 cells cotransfected with either WT or CI HAUSP along with Ub-WT (left panel) or Ub-K48 (middle panel). WB with Rb and Ub antibodies show HAUSP-mediated deubiquitination of Rb. Graphical representation of the WB data (right panel). Data represent mean ± SEM from three independent experiments; P values are 0.0198 (*), 0.0007 (***), 0.006 (**) and 0.023 (*) respectively. (C) In vitro deubiquitination assay. Poly-Ub Rb (substrate) in the presence of 20 nm GST-HAUSP (purified enzyme) in lane 1 is deubiquitinated compared with lane 2, when inhibitor NEM (10 mm) is present, or lane 4, in the absence of purified enzyme. Lane 3 shows the input. Poly-Ub Rb was obtained from HEK293 cell lysates cotransfected with Rb and Ub-WT by IP with Rb antibody. Purified HAUSP was obtained from E. coli BL21 cells. The deubiquitination reaction was set up with the beads bound to the substrate for 1 h at 37 °C. The ubiquitination pattern was analyzed by WB with Ub antibody. IgG-Hc indicates equal pull-down in all the lanes and lamin B in the input lane alone indicates the absence of any non-specific pull-down (left panel). Graphical representation showing comparison of lanes 1 and 2 from the WB data (right panel). Data represent mean ± SEM from three independent experiments; P = 0.0316 (*). (D) FACS analysis of cells cotransfected with Rb and empty GFP vector (middle panel) or GFP-HAUSP (right panel) compared with only vector transfected negative control (left panel). Data represent one of three separate experiments. (E) Graphical representation of cells distributed in the various phases of the cell cycle as shown in (D). (F) Graphical representation showing the changes in the G1 population. Data represent mean ± SEM from three separate experiments; P = 0.0145 (*), 0.0001 (***) and 0.028 (*) respectively.
The net effect of Rb stabilization by HAUSP was further assessed by a flow cytometric analysis (FACS) of the cell cycle profile. To emphasize the regulation of Rb specifically, Rb was overexpressed, which led to an increase in the G1 population as reported earlier [35]. Upon cotransfection with HAUSP, increase in the G1 population was much more prominent (Fig. 3D). The distribution of cells in the different phases of the cell cycle is represented in Fig. 3E and the statistical significance of the changes in G1 phase population is shown in Fig. 3F. Therefore, HAUSP-mediated deubiquitination of Rb leads to its stabilization manifested by an increased cell population in the G1 phase of the cell cycle.
Role of HAUSP in glioma
The Rb pathway is generally inactivated in most gliomas as discussed earlier in the Introduction section. As HAUSP stabilizes Rb and increases its half-life in HEK293 cells, we sought to identify the role of HAUSP in regulating Rb in the context of glioma. For this purpose, we perturbed the HAUSP levels in glioma cells to investigate first the role of HAUSP in glioma. Two pairs of stable cell lines, overexpression and knockdown systems, were generated as described in Materials and methods. Interestingly, when we monitored the rate of cell proliferation by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay in the knockdown stable cell lines, the data revealed reduced proliferation upon HAUSP knockdown (sh-H2) (Fig. 4A). Supporting these data, similar results were obtained when a scratch assay was performed and the HAUSP knockdown cells again showed significantly slower wound healing capacity (Fig. 4B). In a colony formation assay, the number of colonies formed significantly increased in the constitutively HAUSP overexpressing cell line (Neo-H) (Fig. 4C), while HAUSP knockdown decreased the number of colonies formed (Fig. 4D). Neo-H showed larger sized colonies compared with Neo-V (Fig. 4E) and knockdown of HAUSP resulted in highly impaired growth (Fig. 4F). The average diameter of sh-H2 colonies was ∼ 55 μm, which was five times smaller than the sh-Scr colonies with an average diameter of ∼ 265 μm. The maximum diameter of sh-H2 colonies recorded was 200 μm. All the above data suggest an oncogenic role of HAUSP in glioma cell proliferation.
Fig. 4.
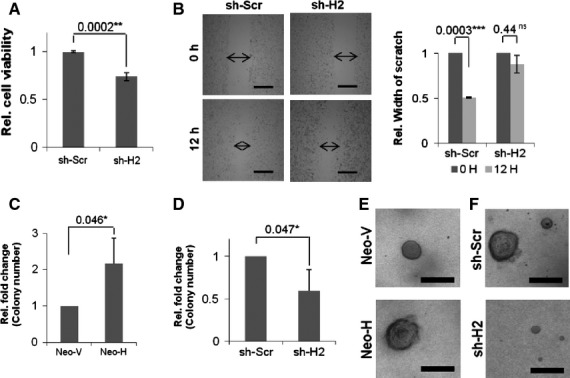
Role of HAUSP in glioma. (A) Graphical representation of the relative number of viable cells obtained from MTT assay as a measure of cell proliferation in HAUSP knockdown (sh-H2) compared with the control cell line (sh-Scr). Data represent mean ± SD from three independent experiments in triplicate; P = 0.0002 (**). (B) Scratch assay with HAUSP knockdown cell line (sh-H2) showing reduced proliferation compared with the control cell line (sh-Scr) (left) and a graphical representation showing a comparative analysis of the wound healing capacity of the respective cell lines from three different experiments (right). P values: sh-Scr, 0.0003 (**); sh-H2, 0.44 (ns). (C) Graph representing a comparative analysis of the relative number of colonies formed upon HAUSP overexpression (Neo-H) versus the control cell line (Neo-V). Data represent the mean ± SD from three independent experiments; P = 0.046 (*). (D) Graph representing a comparative analysis of the relative number of colonies formed upon HAUSP knockdown (sh-H2) versus the control cell line (sh-Scr). Data represent mean ± SD from three independent experiments; P = 0.047 (*). (E) C6-stable cell lines showing larger sized colonies in HAUSP overexpressing cells (Neo-H) compared with the control cell line (Neo-V). (F) C6-stable cell lines showing colony size reduced in HAUSP knockdown cells (sh-H2) compared with the control cells (sh-Scr). All images were taken at 4 × optical magnification using an Olympus IX81 and scale bars (500 μm) are shown.
HAUSP and Rb status in glioma
Earlier reports suggest that there are profound changes in the HAUSP expression levels in various cancers [36] and that HAUSP acts both as a tumor suppressor and as an oncogenic protein in a context-specific manner by exerting its deubiquitinating activity on several proteins such as p53 [33], MDM2 [37], PTEN [38], FOXO4 [39] and so on. To validate the role of HAUSP in the proliferation and progression of glioma that we found in the glioma stable cell lines with the status in human patients, we procured formalin-fixed paraffin-embedded (FFPE) sections from human glioma patients (n = 38) and analyzed them by immunohistochemical studies (IHC). Corroborating the earlier data, HAUSP was found to be overexpressed in glioma patient samples (Fig. 5A). But, if HAUSP expression is increased in glioma, what would be the Rb status? So, to study the expression of Rb, a subset of these patient samples (n = 15) was analyzed by IHC using Rb antibody. Also, its E3 ligase MDM2, which is mostly upregulated in glioma, was analyzed in the same set of samples. The results indicated that the overall Rb expression was low, while there was varied expression of MDM2, although the level of expression remained mostly high (Fig. 5B). Both results were found to be similar to previously established facts. The comparative analysis of the expression patterns of HAUSP, MDM2 and Rb was represented using the mean H-scores obtained from each of the 15 samples (Fig. 5C). To re-validate the results found from the analysis of samples from human glioma patients in glioma cell lines, the three protein levels were compared in normal rat astrocytes (NRA) versus C6 rat glioma and human GBM cell lines by WB (Fig. 5D). These data were further supported by ICC studies in U87MG (Fig. 5E, upper panel) and DBTRG-05MG (Fig. 5E, lower panel) cells, where it is clearly observed that cells showing higher HAUSP expression have reduced Rb expression levels and vice versa. The results indicate, first, an increase in HAUSP expression in glioma with respect to NRA; second, that the Rb level is low while expression of both HAUSP and MDM2 is higher in glioma; and third, that there was a negative correlation in the expression patterns of HAUSP and Rb.
Fig. 5.

Differential regulation of Rb in glioma. (A) IHC of human glioma patient samples for HAUSP expression. Data show representative images of each grade at 10 × optical magnification (n = 38). (B) IHC of a subset of the above glioma samples for Rb and MDM2 expression. Data show representative images at 10 × optical magnification (n = 15). (C) Dot plot representation of the mean H-scores calculated for each sample to estimate the overall levels of expression of HAUSP, MDM2 and Rb (n = 15). (D) WB of HAUSP, MDM2 and Rb in NRA versus C6 rat glioma and other human glioma cell lines. (E) Confocal images of GBM cell lines U87MG (upper panel) and DBTRG-05MG (lower panel) (from ICC) showing a comparison of HAUSP and Rb expression. Images were taken at 60 × optical magnification and scale bars are marked.
Differential regulation of Rb in glioma
HAUSP stabilizes Rb in normal (HEK293, COS-7) cells, but in human glioma samples and cancer cells there is a negative correlation between the two proteins. Also HAUSP is important for glioma cell proliferation and progression. This raises a number of questions. Is Rb level enhanced upon HAUSP overexpression in glioma, leading to G1 cell cycle arrest? What would be the fate of cancer cells in such a scenario? What are the factors determining the effect? and so on. To answer these questions, HAUSP was overexpressed in the human GBM cell line U87MG and, surprisingly, we found a decrease in Rb level. By blocking the proteasome machinery using the inhibitor MG132, Rb level was restored (Fig. 6A), while knockdown of HAUSP using specific siRNA resulted in increased Rb levels (Fig. 6B). An in vivo deubiquitination assay in U87MG cells showed increased ubiquitination of Rb in the presence of WT compared with CI HAUSP (Fig. 6C). Therefore, although HAUSP overexpression in normal cells stabilizes Rb, it cannot do so in glioma cells, where Rb becomes ubiquitinated and degraded. This clearly indicates the involvement of other players in a context-dependent manner.
Fig. 6.

Mechanism of Rb regulation in glioma. (A) WB of Rb upon HAUSP overexpression in U87MG cells either with (right panel) or without (left panel) MG132 treatment (10 μm) for 14 h. (B) WB was performed to verify HAUSP knockdown and the effect on Rb protein expression using specific siRNA in U87MG cells. Actin serves as the loading control. (C) In vivo deubiquitination assay in U87MG cells in the presence of WT or CI HAUSP; IP was done with Rb and WB with Rb and Ub. (D) WB of Rb in H1299 (p53 null) cells upon HAUSP (WT or CI) overexpression with Ub cotransfected. (E) ICC for HAUSP and Rb in a panel of cancer cells with varied p53 background. H1299, p53 null (top); HCT116, p53 WT (middle); HT29, p53 mutant (lower). (F) WB of Rb in HEK293 cells cotransfected with HAUSP and MDM2. (H) Fluorimetric estimation of Rb expression in the presence of HAUSP alone or cotransfected with MDM2, represented as relative fold change with respect to GFP-Rb alone. P = 0.02 (*) and 0.007 (**). (G) Fluorescence emission spectra of cells transfected with either GFP-Rb alone (red graph, red line represents the mean value) or along with HAUSP (blue graph, blue line represents the mean value) or by increasing the load of MDM2 transiently (green graph, green line represents the mean value) keeping a negative control without GFP transfection (grey graph, grey line represents the mean value). (I) Graphical representation of cells distributed in the various phases of the cell cycle from FACS analysis (extended from the experiment shown in Fig. 3D,E,F). (J) WB of Rb upon HAUSP overexpression in U87MG cells with or without MDM2 inhibitor (10 μm) for 24 h (left panel) and in the absence or presence of siMDM2 (right panel). The values in (A), (F) and (J) have been estimated by densitometry and normalized against the loading control actin/GAPDH.
Does p53 have any role to play?
The previous reports, MDM2 degrades Rb [14,15], MDM2 itself is stabilized/destabilized by HAUSP in a context-dependent manner [40], and Rb exists in a trimeric complex with p53 and MDM2 wherein Rb regulates the p53 function [41]; when considered in this context provide several possible mechanistic explanations. This difference observed in cancer cells compared with HEK293 could be because of p53, MDM2 or several other factors, including those yet unknown. To verify the involvement of p53, the experiments were repeated in the p53 null cell line H1299 (Fig. 6D) and the results were found to be similar to those for U87MG cells. Further ICC studies were performed in a panel of cells with varied p53 backgrounds (H1299, p53 null; HCT116, p53 WT; HT29, p53 mutant), as shown in Fig. 6E, and also shown earlier in the human glioma cell lines U87MG (p53 WT) and DBTRG-05MG (p53 mutant) in Fig. 5E. All the data show the inverse relationship between HAUSP and Rb expression patterns in cancer cell lines irrespective of the p53 background. Thus, this regulation of Rb by HAUSP is proteasome dependent and p53 independent.
MDM2 is the critical regulator
To explore the role of MDM2, the other common node in this axis, the available cellular level of MDM2 was increased by ectopic expression of MDM2 using transient transfection in HEK293 cells to simulate a situation similar to cancer cells. We found enhanced Rb ubiquitination even when HAUSP was present (Fig. 6F). GFP-Rb (with green fluorescent protein tag) was cotransfected along with only HAUSP or together with MDM2 and the fluorescence emission of GFP-Rb was monitored by FACS (Fig. 6G) and fluorimetry (Fig. 6H) to determine the changes in the expression levels of Rb. In both experiments, an increase in the fluorescence emission of GFP-Rb was recorded in the case when HAUSP alone was overexpressed while there was a significant decrease in the fluorescence emission when MDM2 was cotransfected along with HAUSP. To corroborate these data, we repeated the FACS experiment shown earlier (Fig. 3D,E) by incorporating one more parameter, i.e. cotransfection of MDM2 along with Rb and HAUSP. It was found that there was a significant decrease in the G1 population (Fig. 6I, graphical representation of the distribution of cells in various phases of the cell cycle; Fig. 3F, last bar, change in G1 population). This indicates that, in the presence of an increased amount of MDM2, HAUSP can no longer rescue Rb. When MDM2 activity was inhibited using a specific inhibitor, upon HAUSP overexpression the Rb protein level was regained in U87MG cells (Fig. 6J, left panel). Again, when HAUSP was overexpressed under MDM2 knockdown conditions (using a specific siRNA), the Rb level was restored (Fig. 6J, right panel). All these data suggest that the level of MDM2 in the cell is a critical factor involved in regulating HAUSP activity on Rb (Fig. 7).
Fig. 7.
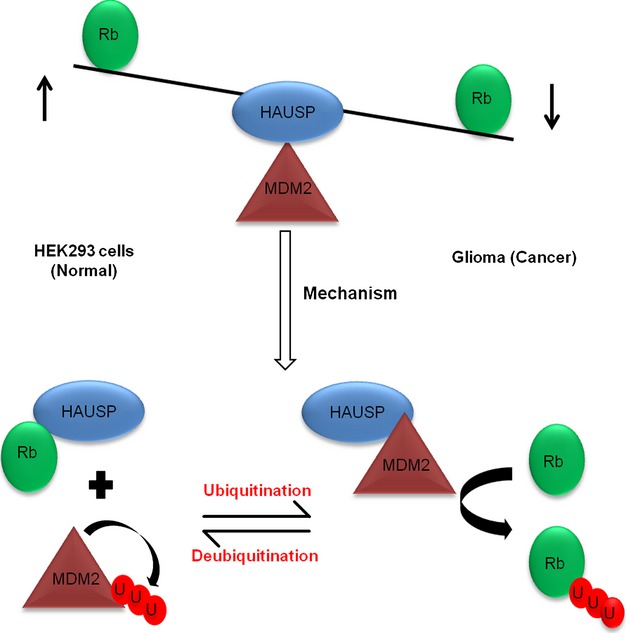
Model showing HAUSP-mediated deubiquitination in normal versus cancer cells determining the fate of Rb. The equilibrium is biased towards Rb stabilization under normal conditions while in cancer, when the MDM2 load in the cells increases, the balance shifts to destabilize Rb, which deregulates the normal cell cycle and helps in enhanced proliferation of cells. Hence, showing MDM2 level to be the critical factor for regulation of HAUSP activity on Rb.
Discussion
Cell division is an outcome of the coordinated activity of several cell cycle regulators. These exhibit an oscillatory pattern which is determined by the protein turnover, maintained by the ubiquitin proteasome system, restricting their activity to specific phases of the cell cycle where it is essential. Rb activity in cell cycle regulation is unique, as its level is maintained throughout the various phases of the cell cycle and, depending on the stimuli, can be reactivated or deactivated to regulate the cell cycle [5]. Association of Rb with several E3 Ub ligases [12] indicates that the protein is degraded, but there is a huge lacunae in information for proteins involved in Rb stabilization to maintain the required equilibrium in cells. As discussed earlier, recent findings show that lamin A and Pax8 are involved in stabilizing Rb. USP4 has been reported to be associated with Rb and the other pocket proteins [42], although no specific role in deubiquitination has been demonstrated as an outcome of such interactions. Here, HAUSP has been identified as a novel deubiquitinase for Rb and to our knowledge this is the first report of any deubiquitinase involved in Rb stability. Here we also find a specificity of HAUSP for Rb and not the other two pocket proteins, which can be attributed to two facts. First, Rb shares only 25% sequence homology to p107 and p130 and, second, although they share a conserved pocket domain and have similar regulation of the cell cycle by binding to distinct E2F family members, yet they have non-overlapping and discrete roles in the perspective of cellular physiology [5,43].
HAUSP interacts with Rb and stabilizes it from K48-linked ubiquitination and proteasomal degradation, increasing its half-life in normal (HEK293) cells. The HAUSP-mediated stabilization of Rb leads to changes in the cell cycle by increasing the G1 population. As different Ub linkages are implicated in various cellular activities [44] it would be intriguing to explore the role of HAUSP in deconjugating any other types of Ub linkages found on Rb and investigating what could be the possible physiological relevance of the process.
Contrary to normal cells, deubiquitinating activity of HAUSP on Rb is abrogated in glioma cell lines, where Rb undergoes degradation. This effect is independent of p53, and the level of MDM2, which is often found to be high in GBM, is the crucial factor in this regulation. Hence, inhibiting MDM2 could be devised as a strategy for therapy.
Several studies indicate the role of phosphorylation in the degradation of Rb. Some suggest phosphorylated Rb [8], while others suggest that hypo-phosphorylated Rb [45] can interact with MDM2. We know that hypo-phosphorylated Rb is the active form and that there is extensive crosstalk between phosphorylation, ubiquitination/deubiquitination and other post-translational modifications [46]. Hence, it could be interesting to dissect out whether the HAUSP-mediated deubiquitination also has any preference for the differentially phosphorylated forms of Rb in different contexts in the future.
Our IHC data from glioma patient samples as well as in vitro data from cell lines go hand in hand corroborating each other and the results demonstrate that HAUSP is highly upregulated in glioma, as also shown recently in patient samples from a Chinese population [47]. This facilitates glioma progression and cellular proliferation, emphasizing the oncogenic role of HAUSP. This is an indication that targeting HAUSP could prove to be a potential strategy for glioma therapy. Several pharmacological inhibitors, peptides and broad spectrum cysteine protease inhibitors [48,49] and very recently a natural pyrrole alkaloid [50] have been the focus of research. However, with the present treatment modalities and taking into consideration the potential problems with GBM, the most deadly brain cancer, which is associated with low survival rate and poor prognosis, recurrence, surgical inadequacies and difficulties in targeting drugs to the brain across the blood–brain barrier, research has to be streamlined more towards small molecules and newer technologies, and a major break-through is yet to come.
Here we have shown that in normal HEK293 cells HAUSP deubiquitinates and stabilizes Rb, while in cancer cells HAUSP and Rb protein expression show a negative correlation. Hence, a major difference in activity of HAUSP with respect to Rb regulation exists in normal versus cancer cells. This has important physiological relevance and can be used as a potential molecular tool to distinguish cancer cells from normal cells. Mechanistic elucidation of further details could provide ideas for reactivating Rb to restore its tumor suppressive activity to arrest the growth of fast dividing cancer cells or to trigger apoptosis. It would also be very interesting to check for Rb regulation by HAUSP in brain tumor stem cells which are quiescent in nature, and this could provide new insights into targeting these cells [51]. These ideas open up several future directions of research on mechanisms of cellular transformation, tumorigenesis and cancer progression, and for targeting HAUSP in cancer therapy, in a context-specific manner.
Materials and methods
Cell lines and culture reagents
Human cell lines GBM, U87MG and DBTRG-05MG; lung adenocarcinoma H1299 (p53 null); colon carcinoma HT29 (p53 mutated) and HCT116 (p53 WT); normal embryonic kidney HEK293; rat glioma cell line C6; and monkey normal kidney cell line COS-7 were procured from American Type Culture Collection (Manassas, VA, USA). Cells were cultured in McCoy's 5A, RPMI-1640, DMEM or MEM as required, supplemented with 10% heat-inactivated fetal bovine serum, 2000 units·L−1 penicillin, 2 mg·L−1 streptomycin and gentamycin (Invitrogen, Carlsbad, CA, USA). NRA were isolated from 0-day-old rat pups and cultured for 14 days. All cells were maintained at 37 °C in a humid incubator with 5% CO2. HEK293 cells were transfected by calcium phosphate, GBM cell lines with Attractene reagent (Qiagen, Valencia, CA, USA) and other cell lines with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. For siRNA-mediated knockdown experiments Lipofectamine RNAi MAX (Invitrogen) was used. For shRNA-mediated knockdown experiments two rounds of transfection were performed. To generate HAUSP overexpressing stable cell lines, C6 cells were transfected either with vector (Neo-V) or HAUSP (Neo-H) and selected with 1 mg·mL−1 G418 (Calbiochem, Darmstadt, Germany). For knockdown stable cells, scrambled (sh-Scr) or HAUSP (sh-H2) shRNAs were transfected and selected with 1 μg·mL−1puromycin (Calbiochem).
DNA constructs, cloning and quantitative real-time PCR
Human HAUSP and Rb genes were cloned according to the procedure described earlier [52]. HAUSP was subcloned in pGZ-21dx vector (a gift from K. N. Yamada, Bethesda, MD, USA) with N-terminal GFP tag (GFP-HAUSP) and pGEX-4T1 vector (GE Healthcare, Uppsala, Sweden) with N-terminal GST tag (GST-HAUSP). Rb was subcloned in pcDNA3.1-Myc/His B(−) (Invitrogen) (Rb) and pGZ-21dx (GFP-Rb). Catalytically inactive mutant of HAUSP (CI) was generated by a point mutation of the residue 223 from Cys to Ser using the Quik Change II XL Kit (Stratagene, Stockport, Cheshire, UK) according to the manufacturer's protocol. pMKO.1-Puro p53 shRNA1 (10671), pRK5-HA-Ub-WT (17608), pRK5-HA-Ub-K48 (17605), pcI-Neo-USP7 (16655) and pcDNA3-MDM2-WT (16233) plasmids were obtained from Addgene (Cambridge, MA, USA). Two different shRNAs targeting HAUSP and a control (scrambled) shRNA were subcloned in pMKO.1-Puro p53 shRNA1 according to Addgene's protocol. All shRNAs were tested for knockdown and the best one (shRNA2, referred to as sh-H2) was used for all experiments. All constructs were verified by sequencing. qRT-PCR was done using SYBR Green Master Mix (Applied Biosystems, ABI, Foster City, CA, USA) on a 7500 Fast machine (ABI) [53]. All primers and synthetic oligonucleotides were synthesized from IDT (Integrated DNA Technologies, Coralville, IA, USA) and the sequences are available in Table 1.
Table 1.
List of primers
| Primer name | 5′-3′ sequence |
|---|---|
| C-HAUSP-F | 5′-ATG AAC CAC CAG CAG CAG CAG CAG-3′ |
| C-HAUSP-R | 5′-GTT ATG GAT TTT AAT GGC CTT TTC AAG GTA AGT GTA GCG-3′ |
| C-RB1-F | 5′-ATG CCG CCC AAA ACC CCC-3′ |
| C-RB1-R | 5′-TTT CTC TTC CTT GTT TGA GGT ATC CAT GC-3′ |
| C223S-HAUSP-F | 5′-GAA TCA GGG AGC GAC TAG TTA CAT GAA CAG CCT G-3′ |
| C223S-HAUSP-R | 5′-CAG GCT GTT CAT GTA ACT AGT CGC TCC CTG ATT C-3′ |
| RT-RB1-F | 5′-CAG AAG GTC TGC CAA CAC CAA C-3′ |
| RT-RB1-R | 5′-TTG AGC ACA CGG TCG CTG TTA C-3′ |
| RT-18S rRNA-F | 5′-GCT TAA TTT GAC TCA ACA CGG GA-3′ |
| RT-18S rRNA-R | 5′-AGC TAT CAA TCT GTC AAT CCT GTC-3′ |
| C-Scrambled_shRNA_F | 5′-CCG GCC TAA GGT TAA GTC GCC CTC GCT CGA GCG AGG GCG ACT TAA CCT T AG GTT TTT G-3′ |
| C-Scrambled_shRNA_R | 5′-AAT TCA AAA ACC TAA GGT TAA GTC GCC CTC GCT CGA GCG AGG GCG ACT T AA CCT TAG G-3′ |
| C-HAUSP-shRNA1-F | 5′-CCG GAA TGT GGC CCT GAG TGA TGG ACT CGA GTC CAT CAC TCA GGG CCA CAT TTT TTT G-3′ |
| C-HAUSP-shRNA1-R | 5′-AAT TCA AAA AAA TGT GGC CCT GAG TGA TGG ACT CGA GTC CAT CAC TCA GGG CCA CAT T-3′ |
| C-Hausp-shRNA2-F | 5′-CCG GAA GTC TTT GTA CAG GCG GAT GCT CGA GCA TCC GCC TGT ACA AAG ACT TTT TTT G-3′ |
| C-Hausp-shRNA2-R | 5′-AAT TCA AAA AAA GTC TTT GTA CAG GCG GAT GCT CGA GCA TCC GCC TGT ACA AAG ACT T-3′ |
Western blotting (WB) and immunoprecipitation (IP)
Whole cell and nuclear lysates were prepared as described earlier [54]. For IP experiments, cells were lyzed in RIPA buffer with 2 × protease inhibitor cocktail (PIC from Calbiochem). Lysates were pre-cleared by incubating with protein A Sepharose beads (GE Healthcare) for 1 h at 4 °C. Equal amounts of lysates were then incubated with specific antibodies overnight at 4 °C. The immunocomplexes were pulled down using protein A Sepharose beads for 2 h at 4 °C. Beads were washed twice with RIPA buffer. All samples were separated on SDS/PAGE and transferred onto poly(vinylidene difluoride) membrane (Millipore, Billerica, MA, USA). Membranes were blocked with 5% BSA in Tris-buffered saline containing 0.1% Tween 20, incubated with specific antibodies and detected by ECL reagent (GE Healthcare).
Antibodies and other reagents
Antibodies against HAUSP, Ub and horseradish peroxidase-tagged secondary antibodies against rabbit and mouse antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). GFP, Rb, p107, p130, HAUSP, actin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), lamin B and α-tubulin antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Horseradish peroxidase-tagged secondary antibody against goat antibodies was purchased from Sigma (Gillingham, Dorset, UK). IHC specific antibodies for HAUSP and MDM2 were obtained from Abcam (Cambridge, MA, USA). Alexa Fluor-tagged secondary antibodies AF488 or AF594 were obtained from Molecular Probes (Invitrogen). Control, HAUSP and MDM2 siRNAs were purchased from Santa Cruz Biotechnology. NEM, MG132 and MDM2 inhibitor II were purchased from Calbiochem and CHX from Sigma.
Rescue experiment
Two sets of cells (HEK293) were transfected with either control or HAUSP siRNAs on day 1 using Lipofectamine RNAi MAX (Invitrogen). On day 3, 48 h post transfection the second set of cells was transfected with GFP-HAUSP using Lipofectamine (Invitrogen). All cells were harvested on day 4 after 72 h post siRNA transfection and 24 h post gene transfection. Set 1 served as the control set for HAUSP knockdown and set 2 showed the specific rescue by HAUSP.
In vivo deubiquitination assay
WT or CI HAUSP was cotransfected with Ub-WT or Ub-K48 and cells were treated with MG132 for 14 h before harvesting. Lysates were immunoprecipitated with antibody against Rb as described above and subjected to WB (protocol based on the work of Mahul-Mellier et al. [55]).
Protein purification using GST tag and glutathione HiCap matrix
Protein purification was done according to the manufacturer's protocol (Qiagen). Briefly, competent Escherichia coli BL21 cells were transformed with either vector control (GST) or GST-HAUSP and cultured until mid-log phase, followed by isopropyl thio-β-d-galactoside (IPTG) induction (1 mm) at 22 °C for about 4–6 h. Cells were pelleted, washed with Tris-buffered saline, incubated in lysis buffer (50 mm Tris/HCl, pH 7.5; 150 mm NaCl; 1 mm EDTA) with 1% Triton-X-100 and lysozyme, 1 mg·mL−1 for 30 min, and frozen overnight at −80 °C. Mild sonication was performed and the supernatant was used for further purification processes. Glutathione HiCap Matrix (Qiagen) beads were pre-equilibrated in GST binding buffer (50 mm Tris/HCl, pH 7.5; 150 mm NaCl; 1 mm EDTA), added to the lysate and left for binding at 4 °C for 2 h, followed by two washes in wash buffer. GST tagged protein was eluted with elution buffer (50 mm Tris/HCl, pH 8.0; 5% glycerol; 10 mm reduced glutathione; 1 mm EDTA). Lysates were analyzed on SDS/PAGE followed by Coomassie staining.
Protein purification using His tag and Ni–nitrilotriacetic acid (Ni-NTA) system
Protein purification was done according to the manufaturer's protocol (Invitrogen and Qiagen). Briefly, HEK293 cells were transfected with Myc/His-Rb in pcDNA 3.1 B(−) and cells were harvested 48 h post transfection. Cell lysate was incubated with Ni–NTA beads for 1 h at room temperature and washed with increasing amounts of imidazole and finally eluted in buffer containing 250 mm imidazole. Lysates were analyzed on SDS/PAGE followed by Coomassie staining.
In vitro deubiquitination assay
Polyubiquitinated substrate (poly-Ub Rb) was obtained by IP using Rb antibody from lysates of HEK293 cells cotransfected with Rb and Ub-WT as described above. Deubiquitination reaction was set up with the beads bound to the substrate and 20 nm of purified enzyme (GST-HAUSP), in either the absence or the presence of the inhibitor NEM (10 mm) in deubiquitination buffer (50 mm Tris/HCl, pH 7.5; 150 mm NaCl; 1 mm EDTA) for 1 h at 37 °C as described earlier [55,56]. The ubiquitination pattern was analyzed by WB with Ub antibody.
In vitro protein–protein interaction
Purified Rb was obtained as described above and 1 μg was incubated with 1 μg of either purified GST or GST-HAUSP overnight at 4 °C in GST binding buffer. The complex was captured using Glutathione HiCap Matrix (Qiagen) beads as described above.
FACS analysis
Cells were harvested by trypsinization, washed with phosphate-buffered saline, resuspended in chilled 70% ethanol for fixation and kept on ice for 30 min. After two washes in phosphate-buffered saline, cells were resuspended in PI/RNase staining buffer (Becton Dickinson, BD Biosciences, San Jose, CA, USA) and incubated in the dark for 15 min at 37 °C. Cell cycle distribution was examined using FACSVerse (BD Biosciences) and analyzed by BD facsuite software (BD Biosciences). For fluorimetric analysis, cells were harvested similarly but FACS was performed without PI/RNase staining using the 488 laser on LSRFortessa (BD Biosciences) and analyzed using the softwares BD facsdiva version 6.2 (BD Biosciences) and fcs express version 4 (De Novo Software, Los Angeles, CA, USA).
Fluorimetry
Cell lysates were prepared as described above and the fluorescence emission of GFP-Rb was monitored by Varioskan Flash Spectral Scanning Multimode Reader, Thermo Scientific (West Palm Beach, FL, USA). Emission from GFP-Rb only transfected cells was considered as unity and the rest were estimated as relative fold changes. The results were analyzed by skanit software (Thermo Scientific).
Immunohistochemistry and immunocytochemistry
IHC analysis and H-scoring of the FFPE tissue sections was performed as described earlier [57]. Images were captured using an Olympus BX61 microscope using image pro plus software (Olympus, Center Valley, PA, USA) at 10 × and 20 × optical magnifications. The human samples contained a collection of grade II, III and IV tumor sections (n = 38) with adjacent normal portions wherever possible.
For ICC, cells were seeded on coverslips and processed as described earlier [52]. Epifluorescence images were captured using an Olympus BX61 microscope and image pro plus software (Olympus) and confocal images with an Andor spinning disc confocal microscope (Andor Technology, Belfast, UK) and Andor iXon 8797 EMCCD camera using andor iq2 software, both at 60 × optical magnification.
Colony formation and scratch assay
For colony formation in soft agar 2 × 103 cells were suspended in the top agar and grown for 2 weeks and for scratch assay 2 × 104 cells were seeded and maintained up to 24 h in 35 mm culture dishes, as described earlier [52].
Proliferation assay
Cells were plated at a concentration of 104 per well in a 96-well plate in triplicate sets and allowed to grow to ∼ 80% confluency. The MTT assay was performed as described earlier [58]. The results were obtained by statistical analysis from three separate experiments.
Statistical analysis
At least three independent experiments were performed to calculate means, standard deviations and standard errors of the mean. Statistical significance was analyzed by unpaired Student's t test. Two-tailed P values were calculated and values < 0.05 were considered significant. All statistical calculations were done using graphpad (QuickCalcs, http://www.graphpad.com/quickcalcs/index.cfm) calculator.
Acknowledgments
The authors would like to thank Dr Sandip Chatterjee (Neurosurgeon, Park Clinic, Kolkata) for providing post-operative and biopsy samples from glioma patients and Dr Uttara Chatterjee (Pathologist, Park Clinic, Kolkata) for the pathology reports, grading and helping in analysis of patient samples. The authors also acknowledge the technical support from Dr Sanjaya Mallick (Nano Centre, CU, Kolkata) for operating the BD FACSVerse instrument, Ms Anushila Gangopadhyay (CSIR-IICB, Kolkata) for operating the BD LSRFortessa instrument and Mr Diptadeep Sarkar (CSIR-IICB, Kolkata) for operating the spinning disc confocal microscope. The work was supported by CSIR (EMPOWER: OLP-2 and MEDCHEM: BSC0108) and DST (SR/SO/HS-0150/2010).
Glossary
- CHX
cycloheximide
- CI
catalytically inactive mutant
- FACS
fluorescence activated cell sorting
- FFPE
formalin-fixed paraffin-embedded
- GBM
glioblastoma multiforme
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- HAUSP
herpes virus associated ubiquitin specific protease
- HEK293
human embryonic kidney 293
- ICC
immunocytochemistry
- IHC
immunohistochemistry
- IP
immunoprecipitation
- K48
lysine 48 ubiquitin
- MDM2
murine double minute 2
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
- NEM
N-ethylmaleimide
- Neo
neomycin
- NRA
normal rat astrocyte
- Pax8
paired box protein 8
- Puro
puromycin
- Rb
retinoblastoma-associated protein
- shRNA
small hairpin RNA
- siRNA
small interfering RNA
- Ub
ubiquitin
- WB
western blotting
- WT
wild-type
Author contributions
SB: concept, experiments, analysis and manuscript writing; MKG: concept, manuscript and experimental analysis, correspondence.
References
- 1.Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–646. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- 2.Lee WH, Bookstein R, Hong F, Young LJ, Shew JY, Lee EY. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science. 1987;235:1394–1399. doi: 10.1126/science.3823889. [DOI] [PubMed] [Google Scholar]
- 3.Fattman CL, An B, Dou QP. Characterization of interior cleavage of retinoblastoma protein in apoptosis. J Cell Biochem. 1997;67:399–408. [PubMed] [Google Scholar]
- 4.Yee AS, Shih HH, Tevosian SG. New perspectives on retinoblastoma family functions in differentiation. Front Biosci. 1998;3:D532–D547. doi: 10.2741/a301. [DOI] [PubMed] [Google Scholar]
- 5.Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14:297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medema RH, Herrera RE, Lam F, Weinberg RA. Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc Natl Acad Sci USA. 1995;92:6289–6293. doi: 10.1073/pnas.92.14.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeCaprio JA, Ludlow JW, Lynch D, Furukawa Y, Griffin J, Piwnica-Worms H, Huang CM, Livingston DM. The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell. 1989;58:1085–1095. doi: 10.1016/0092-8674(89)90507-2. [DOI] [PubMed] [Google Scholar]
- 8.Delston RB, Matatall KA, Sun Y, Onken MD, Harbour JW. p38 phosphorylates Rb on Ser567 by a novel, cell cycle-independent mechanism that triggers Rb-Hdm2 interaction and apoptosis. Oncogene. 2011;30:588–599. doi: 10.1038/onc.2010.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat Cell Biol. 2001;3:667–674. doi: 10.1038/35083062. [DOI] [PubMed] [Google Scholar]
- 10.Saddic LA, West LE, Aslanian A, Yates JR, Rubin SM, Gozani O, Sage J. Methylation of the retinoblastoma tumor suppressor by SMYD2. J Biol Chem. 2010;285:37733–37740. doi: 10.1074/jbc.M110.137612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ledl A, Schmidt D, Müller S. Viral oncoproteins E1A and E7 and cellular LxCxE proteins repress SUMO modification of the retinoblastoma tumor suppressor. Oncogene. 2005;24:3810–3818. doi: 10.1038/sj.onc.1208539. [DOI] [PubMed] [Google Scholar]
- 12.Ying H, Xiao Z-XJ. Targeting retinoblastoma protein for degradation by proteasomes. Cell Cycle. 2006;5:506–508. doi: 10.4161/cc.5.5.2515. [DOI] [PubMed] [Google Scholar]
- 13.Ventii KH, Wilkinson KD. Protein partners of deubiquitinating enzymes. Biochem J. 2008;414:161. doi: 10.1042/BJ20080798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uchida C, Miwa S, Kitagawa K, Hattori T, Isobe T, Otani S, Oda T, Sugimura H, Kamijo T, Ookawa K, et al. Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. EMBO J. 2005;24:160–169. doi: 10.1038/sj.emboj.7600486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sdek P, Ying H, Chang DLF, Qiu W, Zheng H, Touitou R, Allday MJ, Xiao Z-XJ. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell. 2005;20:699–708. doi: 10.1016/j.molcel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 16.Zheng Z, Li L, Liu X, Wang D, Tu B, Wang L, Wang H, Zhu W-G. 5-Aza-2′-deoxycytidine reactivates gene expression via degradation of pRb pocket proteins. FASEB J. 2012;26:449–459. doi: 10.1096/fj.11-190025. [DOI] [PubMed] [Google Scholar]
- 17.Nitta RT, Smith CL, Kennedy BK. Evidence that proteasome-dependent degradation of the retinoblastoma protein in cells lacking A-type lamins occurs independently of gankyrin and MDM2. PLoS One. 2007;2:e963. doi: 10.1371/journal.pone.0000963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nitta RT, Jameson SA, Kudlow BA, Conlan LA, Kennedy BK. Stabilization of the retinoblastoma protein by A-type nuclear lamins is required for INK4A-mediated cell cycle arrest. Mol Cell Biol. 2006;26:5360–5372. doi: 10.1128/MCB.02464-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li CG, Nyman JE, Braithwaite AW, Eccles MR. PAX8 promotes tumor cell growth by transcriptionally regulating E2F1 and stabilizing RB protein. Oncogene. 2011;30:4824–4834. doi: 10.1038/onc.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicholson B, Suresh Kumar KG. The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem Biophys. 2011;60:61–68. doi: 10.1007/s12013-011-9185-5. [DOI] [PubMed] [Google Scholar]
- 21.James CD, Carlbom E, Dumanski JP, Hansen M, Nordenskjold M, Collins VP, Cavenee WK. Clonal genomic alterations in glioma malignancy stages. Cancer Res. 1988;48:5546–5551. [PubMed] [Google Scholar]
- 22.Henson JW, Schnitker BL, Correa KM, von Deimling A, Fassbender F, Xu HJ, Benedict WF, Yandell DW, Louis DN. The retinoblastoma gene is involved in malignant progression of astrocytomas. Ann Neurol. 1994;36:714–721. doi: 10.1002/ana.410360505. [DOI] [PubMed] [Google Scholar]
- 23.Rao SK, Edwards J, Joshi AD, Siu I-M, Riggins GJ. A survey of glioblastoma genomic amplifications and deletions. J Neurooncol. 2010;96:169–179. doi: 10.1007/s11060-009-9959-4. [DOI] [PubMed] [Google Scholar]
- 24.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 25.Lee SH, Kim JH, Rhee CH, Kang YS, Lee JH, Hong SI, Choi KS. Loss of heterozygosity on chromosome 10, 13q(Rb), 17p, and p53 gene mutations in human brain gliomas. J Korean Med Sci. 1995;10:442–448. doi: 10.3346/jkms.1995.10.6.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Louis DN, Gusella JF. A tiger behind many doors: multiple genetic pathways to malignant glioma. Trends Genet. 1995;11:412–415. doi: 10.1016/s0168-9525(00)89125-8. [DOI] [PubMed] [Google Scholar]
- 27.Biernat W, Kleihues P, Yonekawa Y, Ohgaki H. Amplification and overexpression of MDM2 in primary (de novo) glioblastomas. J Neuropathol Exp Neurol. 1997;56:180–185. doi: 10.1097/00005072-199702000-00009. [DOI] [PubMed] [Google Scholar]
- 28.Burton EC, Lamborn KR, Forsyth P, Scott J, O'Campo J, Uyehara-Lock J, Prados M, Berger M, Passe S, Uhm J, et al. Aberrant p53, mdm2, and proliferation differ in glioblastomas from long-term compared with typical survivors. Clin Cancer Res. 2002;8:180–187. [PubMed] [Google Scholar]
- 29.Matsumoto R, Tada M, Nozaki M, Zhang CL, Sawamura Y, Abe H. Short alternative splice transcripts of the mdm2 oncogene correlate to malignancy in human astrocytic neoplasms. Cancer Res. 1998;58:609–613. [PubMed] [Google Scholar]
- 30.Masciullo V, Khalili K, Giordano A. The Rb family of cell cycle regulatory factors: clinical implications. Int J Oncol. 2000;17:897–902. doi: 10.3892/ijo.17.5.897. [DOI] [PubMed] [Google Scholar]
- 31.Huang Z, Wu Q, Guryanova OA, Cheng L, Shou W, Rich JN, Bao S. Deubiquitylase HAUSP stabilizes REST and promotes maintenance of neural progenitor cells. Nat Cell Biol. 2011;13:142–152. doi: 10.1038/ncb2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Everett RD, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 1997;16:566–577. doi: 10.1093/emboj/16.3.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648–653. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- 34.Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 35.Huang HJ, Yee JK, Shew JY, Chen PL, Bookstein R, Friedmann T, Lee EY, Lee WH. Suppression of the neoplastic phenotype by replacement of the RB gene in human cancer cells. Science. 1988;242:1563–1566. doi: 10.1126/science.3201247. [DOI] [PubMed] [Google Scholar]
- 36.Becker K, Marchenko ND, Palacios G, Moll UM. A role of HAUSP in tumor suppression in a human colon carcinoma xenograft model. Cell Cycle. 2008;7:1205–1213. doi: 10.4161/cc.7.9.5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell. 2004;13:879–886. doi: 10.1016/s1097-2765(04)00157-1. [DOI] [PubMed] [Google Scholar]
- 38.Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, Pandolfi PP. The deubiquitinylation and localization of PTEN are regulated by a HAUSP–PML network. Nature. 2008;455:813–817. doi: 10.1038/nature07290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van der Horst A, de Vries-Smits AMM, Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM, Burgering BMT. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol. 2006;8:1064–1073. doi: 10.1038/ncb1469. [DOI] [PubMed] [Google Scholar]
- 40.Ronai Z. Balancing Mdm2 – a Daxx-HAUSP matter. Nat Cell Biol. 2006;8:790–791. doi: 10.1038/ncb0806-790. [DOI] [PubMed] [Google Scholar]
- 41.Yap DB, Hsieh JK, Chan FS, Lu X. mdm2: a bridge over the two tumour suppressors, p53 and Rb. Oncogene. 1999;18:7681–7689. doi: 10.1038/sj.onc.1202954. [DOI] [PubMed] [Google Scholar]
- 42.Blanchette P, Gilchrist CA, Baker RT, Gray DA. Association of UNP, a ubiquitin-specific protease, with the pocket proteins pRb, p107 and p130. Oncogene. 2001;20:5533–5537. doi: 10.1038/sj.onc.1204823. [DOI] [PubMed] [Google Scholar]
- 43.Dick FA. Structure–function analysis of the retinoblastoma tumor suppressor protein – is the whole a sum of its parts? Cell Div. 2007;2:26. doi: 10.1186/1747-1028-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kulathu Y, Komander D. Atypical ubiquitylation – the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13:508–523. doi: 10.1038/nrm3394. [DOI] [PubMed] [Google Scholar]
- 45.Sdek P, Ying H, Zheng H, Margulis A, Tang X, Tian K, Xiao Z-XJ. The central acidic domain of MDM2 is critical in inhibition of retinoblastoma-mediated suppression of E2F and cell growth. J Biol Chem. 2004;279:53317–53322. doi: 10.1074/jbc.M406062200. [DOI] [PubMed] [Google Scholar]
- 46.Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 47.Cheng C, Niu C, Yang Y, Wang Y, Lu M. Expression of HAUSP in gliomas correlates with disease progression and survival of patients. Oncol Rep. 2013;29:1730–1736. doi: 10.3892/or.2013.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colland F, Formstecher E, Jacq X, Reverdy C, Planquette C, Conrath S, Trouplin V, Bianchi J, Aushev VN, Camonis J, et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther. 2009;8:2286–2295. doi: 10.1158/1535-7163.MCT-09-0097. [DOI] [PubMed] [Google Scholar]
- 49.Reverdy C, Conrath S, Lopez R, Planquette C, Atmanene C, Collura V, Harpon J, Battaglia V, Vivat V, Sippl W, et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem Biol. 2012;19:467–477. doi: 10.1016/j.chembiol.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 50.Yamaguchi M, Miyazaki M, Kodrasov MP, Rotinsulu H, Losung F, Mangindaan REP, de Voogd NJ, Yokosawa H, Nicholson B, Tsukamoto S. Spongiacidin C, a pyrrole alkaloid from the marine sponge Stylissa massa, functions as a USP7 inhibitor. Bioorg Med Chem Lett. 2013;23:3884–3886. doi: 10.1016/j.bmcl.2013.04.066. [DOI] [PubMed] [Google Scholar]
- 51.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–682. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paul I, Ahmed SF, Bhowmik A, Deb S, Ghosh MK. The ubiquitin ligase CHIP regulates c-Myc stability and transcriptional activity. Oncogene. 2013;32:1284–1295. doi: 10.1038/onc.2012.144. [DOI] [PubMed] [Google Scholar]
- 53.Ahmed SF, Deb S, Paul I, Chatterjee A, Mandal T, Chatterjee U, Ghosh MK. The chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. J Biol Chem. 2012;287:15996–16006. doi: 10.1074/jbc.M111.321083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guturi KKN, Mandal T, Chatterjee A, Sarkar M, Bhattacharya S, Chatterjee U, Ghosh MK. Mechanism of β-catenin-mediated transcriptional regulation of epidermal growth factor receptor expression in glycogen synthase kinase 3 β-inactivated prostate cancer cells. J Biol Chem. 2012;287:18287–18296. doi: 10.1074/jbc.M111.324798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mahul-Mellier A-L, Pazarentzos E, Datler C, Iwasawa R, AbuAli G, Lin B, Grimm S. De-ubiquitinating protease USP2a targets RIP1 and TRAF2 to mediate cell death by TNF. Cell Death Differ. 2012;19:891–899. doi: 10.1038/cdd.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strayhorn WD, Wadzinski BE. A novel in vitro assay for deubiquitination of I kappa B alpha. Arch Biochem Biophys. 2002;400:76–84. doi: 10.1006/abbi.2002.2760. [DOI] [PubMed] [Google Scholar]
- 57.Chatterjee A, Chatterjee U, Ghosh MK. Activation of protein kinase CK2 attenuates FOXO3a functioning in a PML-dependent manner: implications in human prostate cancer. Cell Death Dis. 2013;4:e543. doi: 10.1038/cddis.2013.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhowmik A, Das N, Pal U, Mandal M, Bhattacharya S, Sarkar M, Jaisankar P, Maiti NC, Ghosh MK. 2,2′-diphenyl-3,3′-diindolylmethane: a potent compound induces apoptosis in breast cancer cells by inhibiting EGFR pathway. PLoS One. 2013;8:e59798. doi: 10.1371/journal.pone.0059798. [DOI] [PMC free article] [PubMed] [Google Scholar]