

Abstract
The 7-deazapurine nucleoside antibiotic tubercidin was converted into its 4-N-benzyl and 4-N-(4-nitrobenzyl) derivatives by alkylation at N3 followed by Dimroth rearrangement to the 4-N- isomer or by fluoro-diazotization followed by SNAr displacement of the 4-fluoro group by a benzylamine. The 4-N-(4-nitrobenzyl) derivatives of sangivamycin and toyocamycin antibiotics were prepared by the alkylation approach. Cross-membrane transport of labeled uridine by hENT1 was inhibited to a weaker extent by the 4-nitrobenzylated tubercidin and sangivamycin analogues than was observed with 6-N-(4-nitrobenzyl)adenosine. Type-specific inhibition of cancer cell proliferation was observed at μM concentrations with the 4-N-(4-nitrobenzyl) derivatives of sangivamycin and toyocamycin, and also with 4-N-benzyltubercidin. Treatment of 2′,3′,5′-O-acetyladenosine with aryl isocyanates gave the 6-ureido derivatives but none of them exhibited inhibitory activity against cancer cell proliferation or hENT1.
Keywords: Antiproliferation, 7-deazapurine nucleosides, nitrogen heterocycles, nucleosides, nucleoside transporter inhibition
Introduction
Analogues and derivatives of 7-deazapurine nucleoside antibiotics have been synthesized and subjected to extensive biological testing.[1] A sizeable number of active nucleoside compounds with such a pyrrolo[2,3-d]pyrimidine base has been prepared by chemical modifications of the parent antibiotics as well as by coupling base derivatives with protected sugars.[2] Noteworthy examples of such molecules include: (a) 5-iodotubercidin,[3] an up-field activator of the p53 pathway; (b) sangivamycin analogues such as 6-hydrazinosangivamycin[4] and xylocidine,[5] in vitro down-field inhibitors of PKC and CDK in cancer cell lines; (c) a methyl-substituted tubercidin,[6] which acts against the replication of polio and dengue viruses; (d) the anti-HSV agent xylotubercidin;[7] (e) substituted toyocamycin analogues[8] and 2′-β-C-methyl derivative of toyocamycin,[9] sangivamycin,[9] and tubercidin[10] that have activity against HCV; (f) 2′-deoxy-2′-fluoroarabinotubercidin[11] and 2-amino-2′-deoxy-2′-fluoroarabinotubercidin,[12] which exhibit antiviral activity; (g) 4N,5-diaryltubercidin derivatives, which act as adenosine kinase inhibitors;[2l, 13] and (h) tubercidin derivatives such as 4-(het)aryl,[14] 5-(het)aryl[15] and some 4-substituted-5-(het)aryl[16] compounds with nanomolar cytostatic activity against several cancer cell lines.
Direct alkylation of exocyclic amino groups on 7-deazapurine nucleosides has not been noted. Treatment of tubercidin with methyl iodide followed by sodium hydroxide produced 4-N-methyltubercidin[2b] (6-N-methyl-7-deazaadenosine) via Dimroth rearrangement. Several other 4-N-substituted tubercidin derivatives have been prepared by SNAr displacement of chloride from the 4-chloro analogue[2b, 16] of tubercidin or by displacement of 1,2,4-triazole from 4-N-(1,2,4-triazol-4-yl) intermediates.[17] Such 4-N-substituted toyocamycin derivatives also were prepared from the corresponding 4-chloro compounds.[8] The 7-chloro derivative of the C-nucleoside antibiotic formycin (3-β-D-ribofuranosylpyrazolo[4,3-d]pyrimidine) was prepared and then converted into 7-N-benzylformycin.[18]
Purine and pyrimidine nucleosides and their derivatives play crucial roles in human physiology and pharmacology.[19] These “salvage” metabolites can be converted into nucleotides, which in turn serve as the energy-rich currency of intermediary metabolism and as precursors of nucleic acid biosynthesis. Adenosine functions as a key signalling molecule and modulates diverse physiological processes through interactions with cell surface P1 purinergic receptors.
Many types of cancer and viral infections are treated with nucleoside drugs,[20] but drug availabilities within cells and in the extracellular environment are determined primarily by specific nucleoside transporter (NT) proteins that facilitate their movement across plasma membranes and some organellar membranes. Because of the hydrophilic nature of nucleosides, NTs play key roles in the physiology, pathophysiology, and therapeutic actions of many nucleoside drugs.[19] The human equilibrative nucleoside transporter 1 (hENT1) is the prototypical NT present in the outer plasma membrane and some intracellular membranes of most, if not all, human cells.[21]
Nitrobenzylmercaptopurine ribonucleoside (NBMPR) and structurally related hENT1 probes such as 6-N-(4-nitrobenzyl)adenosine[22] and 5′-S-{2-(6-[3-(fluorescein-5-yl)thioureido-1-yl]hexanamido)}ethyl-6-N-(4-nitrobenzyl)-5′-thioadenosine (FITC-SAHENTA)[23] bind with nM affinity within, or closely associated with, the outward-facing permeant-binding site of hENT1. Development of such probes has greatly aided investigations on the structure, function, and cell surface abundances of hENT1.[19, 23–24] We now report synthesis of analogues of these transport inhibitors with purine and 7-deazapurine bases that exhibit pronounced cytotoxicity in several cancer cell lines as well as moderate inhibition of hENT1 transport activity.
Results and Discussion
Chemistry
The N-benzylated 7-deazaadenosine analogues (2a–d) (Scheme 1) were prepared by two procedures. One involved alkylation of the 7-deazaadenosine antibiotics (1a–c) at N3 with a benzyl bromide followed by base-promoted Dimroth rearrangement.[18, 25] The second route employed diazotization-fluorodediazoniation followed by SNAr displacement of fluoride with a benzyl amine.[26] Alkylation of adenosine occurs at N1, and the resulting cation undergoes rearrangement in basic solution to give the N6-alkylated product.[27] Alkylation of tubercidin (1a) with benzyl bromide in DMF occurred at N3 (same as ring nitrogen N1 in adenosine), and treatment of the 3-benzyl intermediate with Me2NH/THF in MeOH resulted in rearrangement to give 4-N-benzyltubercidin (2a, 67%).
Scheme 1.
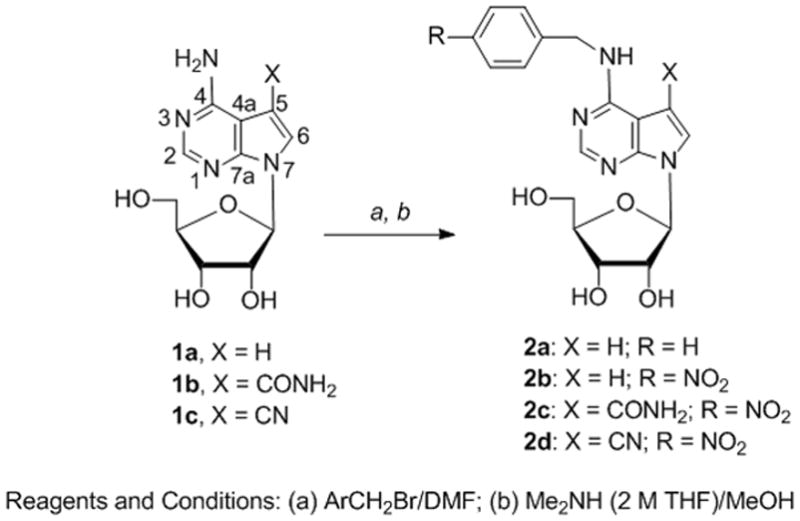
Synthesis of 4-N-benzylated nucleosides by alkylation
Reagents and Conditions: (a) ArCH2Br/DMF; (b) Me2NH (2 M THF)/MeOH
Because the 6-N-(4-nitrobenzyl) derivatives of adenosine and other adenine nucleosides are more potent inhibitors of nucleoside transport,[22] we also synthesized 4-nitrobenzyl derivatives of the 7-deaza antibiotics. Alkylation of 1a with 4-nitrobenzyl bromide followed by treatment with Me2NH/MeOH produced 4-N-(4-nitrobenzyl)tubercidin (2b, 56%). Analogous treatment of sangivamycin (1b) with 4-nitrobenzyl bromide and then Me2NH/MeOH gave 4-N-(4-nitrobenzyl)sangivamycin (2c, 46%).
Treatment of toyocamycin (1c) with 4-nitrobenzyl bromide unexpectedly gave 4-N-(4-nitrobenzyl)toyocamycin (2d) directly. Formation of an intermediate alkylated at N3 apparently did not occur because TLC of the reaction mixture showed only a more rapidly migrating material corresponding to 2d without indication of a more polar (cationic) product. Therefore, successive treatment with Me2NH/MeOH was not required. Although direct alkylation on the exocyclic amino group of adenine and guanine with quinone methides is known,[28] the observed direct alkylation on the exocyclic amine group of toyocamycin with 4-nitrobenzyl bromide has not been previously noted. This might have resulted from alteration of the nucleophilicity of the endocyclic N3 of toyocamycin relative to that of the exocyclic 4-amino nitrogen by the electron withdrawing pull of the cyano group at C5. A possible accompanying stereoelectronic effect might result from interference of the cylindrical cyano group with a synperiplanar orientation of the exocyclic amino group relative to the planar heterocyclic ring that would diminish lone-pair donation from the amino nitrogen toward N3 of that amidine system.[29]
Alternatively, tubercidin (1a, Scheme 2) was acetylated and then subjected to diazotive-fluorodeamination[23, 30] followed by SNAr displacement of fluoride with benzylamines.[23, 26] Thus, 2′,3′,5′-tri-O-acetyltubercidin (3) was treated with NaNO2 in freshly prepared ~55% HF-pyridine at −10 °C[31] for 15 min to yield the protected 4-fluorotubercidin 4 (82%). The concentration of HF-pyridine as well as the temperature are crucial.[31b] Treatment of 4 with 4-nitrobenzylamine hydrochloride/Et3N/MeOH[23] gave 2′,3′,5′-tri-O-acetyl-4-N-(4-nitrobenzyl)tubercidin (5, 47%), which was deacetylated (NH3/MeOH) to give 2b (85%).
Scheme 2.
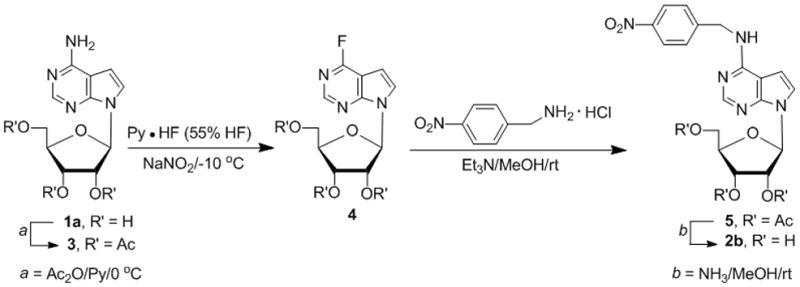
SNAr-based synthesis of 4-N-(4-nitrobenzyl)tubercidin (2b)
Some 2′,3′-bis-O-silylated adenosine derivatives with ureido substituents at the 5′- and 6-positions exhibit anti-proliferative activity.[32] We prepared the 6-N-[N-arylcarbamoyl]adenosine derivatives (9a–c) (Scheme 3) with phenyl, 4-methoxyphenyl, and 4-nitrophenyl moieties to probe for binding to hENT1 and inhibition of nucleoside transport as observed with 6-N-(4-nitrobenzyl)adenosine.[22] The 2′,3′,5′-tri-O-acetyl derivative 7 of adenosine (6) was treated with phenyl isocyanate to give 8a. Deacetylation gave 6-N-[N-phenylcarbamoyl]adenosine (9a), and the analogous sequential treatment of 7 with the respective isocyanates produced the 4-methoxyphenyl and 4-nitrophenyl analogues 9b and 9c.
Scheme 3.
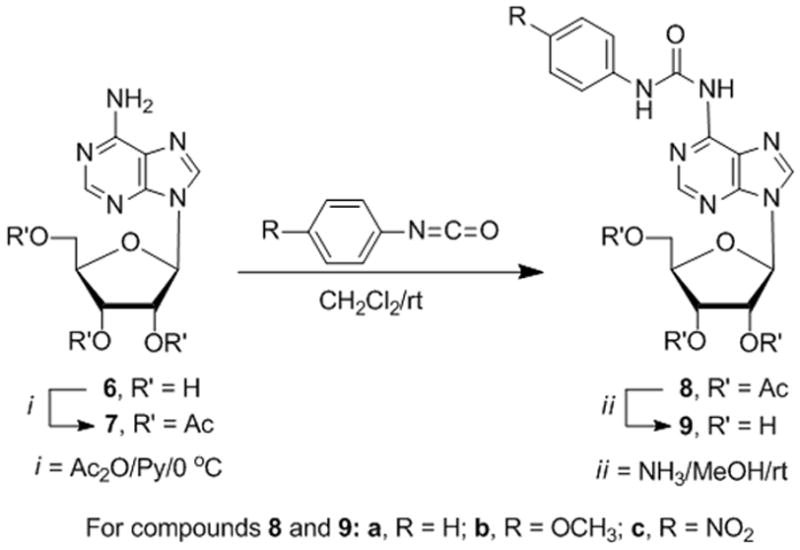
Synthesis of 6-ureidoadenosine analogues
For compounds 8 and 9: a, R = H; b, R = OCH3; c, R = NO2
Nucleoside Transport
Inhibition of the initial rate of 3H-uridine (20 μM) uptake by recombinant hENT1 produced in the Xenopus oocyte heterologous expression system was determined as described previously.[21] Minimal inhibition (italic>25%) of labelled uridine uptake was observed at a 100-μM concentration of each of the 6-ureidoadenosine compounds 9a, 9b, and 9c. The 4-N-benzyltubercidin (2a) and 4-N-(4-nitrobenzyl)toyocamycin (2d) analogues also exhibited weak inhibition (bold>45%) but both 4-N-(4-nitrobenzyl)tubercidin (2b) and 4-N-(4-nitrobenzyl)sangivamycin (2c) fully inhibited labelled uridine uptake at that concentration. Dose-response evaluations indicated that 2c (IC50 = 123 ± 31 nM) was a better inhibitor of uridine transport by hENT1 than 2b (IC50 > 1000 nM). However, the 7-deaza analogue 2c was a weaker inhibitor of hENT1-mediated transport than 6-N-(4-nitrobenzyl)adenosine, NBMPR, and other derivatives[22] containing a nitrogen atom at the 7-position in the adenine ring.
Inhibition of Cell Proliferation
Inhibition of the proliferation of murine leukemia (L1210) cells and human T-lymphocyte (CEM), cervix carcinoma (HeLa), prostate cancer (PC-3), and kidney carcinoma (Caki-1) cells by the adenine and 7-deazaadenine nucleoside analogues was evaluated. No pronounced inhibitory effect was observed with the 6-ureido compounds 9a, 9b, and 9c in any of the tumor cell lines (Table 1). Marked inhibition of the proliferation of PC-3 cells (IC50: 0.92 μM) and HeLa cells (IC50: 7.4 μM) by 4-N-benzyltubercidin (2a) was observed but no significant activity was found with its nitrobenzyl analogue 2b (IC50: >50 μM). The 4-N-(4-nitrobenzyl)sangivamycin (2c) and 4-N-(4-nitrobenzyl)toyocamycin (2d) analogues inhibited proliferation of L1210, HeLa, and PC-3 cells at ~0.9–9.4 μM with 2c showing more potent effects (0.92–3.4 μM). It is intriguing that such striking differences in cytostatic activity were dependent on the nature of the tumor cell lines. HeLa and PC-3 tumor cells were highly susceptible to the cytostatic activity of 2a, 2c, and 2d whereas the L1210 cells were sensitive only to 2c and 2d, and the CEM and Caki-1 cells were weakly sensitive to the antiproliferative effects of any of the 2a–2d compounds.
Table 1.
Inhibitory effects on the proliferation of cells in culture
| Compd | IC50 (μM)* | ||||
|---|---|---|---|---|---|
| L1210 | CEM | HeLa | PC-3 | Caki-1 | |
| 2a | 172 ± 47 | 182 ± 20 | 7.4 ± 2.2 | 0.92 ± 0.67 | 116 ± 23 |
| 2b | 125 ± 28 | ≥250 | 143 ± 4 | 76 ± 10 | 132 ± 30 |
| 2c | 0.92 ± 0.04 | 115 ± 28 | 1.8 ± 0.2 | 3.4 ± 0.9 | 116 ± 23 |
| 2d | 5.5 ± 1.5 | 109 ± 16 | 9.4 ± 3.0 | 5.6 ± 3.3 | 98 ± 10 |
| 9a | >250 | 221 ± 0 | 201 ± 44 | 115 ± 20 | 150 ± 33 |
| 9b | >250 | >250 | 161 ± 10 | 110 ± 31 | ≥250 |
| 9c | >250 | >250 | >250 | 45 ± 26 | >250 |
50% inhibitory concentration
Conclusions
Alkylation at N3 of tubercidin with 4-nitrobenzyl bromide and Dimroth rearrangement gave 4-N-(4-nitrobenzyl)tubercidin, which also was prepared by fluoro-diazotization of tubercidin and SNAr displacement of the 4-fluoro group by 4-nitrobenzylamine. The alkylation-rearrangement sequence was employed to convert sangivamycin (the 5-carboxamidotubercidin antibiotic) into its 4-N-(4-nitrobenzyl) derivative. However, no evidence of formation of an initial N3-alkylated intermediate was observed upon treatment of toyocamycin (the 5-cyanotubercidin antibiotic) with 4-nitrobenzyl bromide. Alkylation occurred directly on the amino group to give 4-N-(4-nitrobenzyl)toyocamycin. The 4-N-(4-nitrobenzyl) derivatives of tubercidin and sangivamycin inhibited cross-membrane transport of labelled uridine by the human equilibrative nucleoside transporter hENT1 at >1 μM and ~120 nM, respectively. Inhibition of the proliferation of L1210, HeLa, and PC-3 tumor cells in culture was observed with 4-N-benzyltubercidin and the 4-N-(4-nitrobenzyl) derivatives of sangivamycin and toyocamycin at 0.92–9.4 μM concentrations.
Experimental Section
1H (400 MHz), 13C (100.6 MHz), and 19F (376 MHz) NMR spectra were recorded at ambient temperature in solutions of CDCl3 or DMSO-d6. Reaction progress was monitored by TLC on Merck Kieselgel 60-F254 sheets with product detection by 254-nm light. Products were purified by column chromatography using Merck Kiselgel 60 (230–400 mesh) or by automated flash chromatography using a CombiFlash system. UV spectra were recorded with a Varian Cary 100 Bio UV-visible spectrophotometer. Reagent grade chemicals were used and solvents were dried by reflux and distillation from CaH2 under N2 unless otherwise specified, and an atmosphere of N2 was used for reactions. Purity of the synthesized compounds was determined to be ≥95% by elemental analysis (C, H, N) and/or HPLC.
4-Benzylamino-7-(β-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine (2a)
BnBr (345 μL, 496 mg, 2.9 mmol) was added to a stirred solution of tubercidin (1a; 266 mg, 1 mmol) in dried DMF (5 mL). After stirring for 48 hours at 40 °C, TLC showed almost complete conversion to a more polar product. Volatiles were evaporated to ~1 mL (< 40 °C, vacuum pump) and the resulting syrup was added dropwise to dried acetone (30 mL) with vigorous stirring. Et2O (60 mL) was added to the suspension, which was chilled for 20 min at 0 °C and vacuum filtered. The hygroscopic precipitate was quickly dissolved in MeOH (10 mL) and Me2NH/THF (2 M, 8 mL) was added. The resulting solution was stirred at reflux (65 °C, oil bath) for 20 h. TLC showed ~80% conversion to less polar spot, and volatiles were evaporated and coevaporated with MeOH (2 ×). The residue was dissolved in warm MeOH (10 mL), H2O (60 mL) was added, and the solution was extracted with EtOAc (5 × 20 mL). The combined organic phase was dried (Na2SO4), concentrated in vacuo, coevaporated (2 × EtOH) and flash chromatographed (EtOAc) to give 2a (238 mg, 67%) as a colorless oil: UV (MeOH) λmax 276 nm, λmin 244 nm; 1H NMR (DMSO-d6) δ 8.09–8.13 (m, 2 H, NH, H2), 7.38 (d, J = 3.7 Hz, 1 H, H6), 7.29–7.35 (m, 4 H, Ph), 7.22–7.25 (m, 1 H, Ph), 6.67 (d, J = 3.4 Hz, 1 H, H5), 6.01 (d, J = 5.9 Hz, 1 H, H1′), 5.27–5.32 (m, 2 H, 2′-OH, 5′-OH), 5.12 (d, J = 4.3 Hz, 1 H, 3′-OH), 4.70–4.78 (m, 2 H, CH2), 4.44 (q, J = 5.7 Hz, 1 H, H2′), 4.09 (“q”, J = 4.1 Hz, 1 H, H3′), 3.90 (q, J = 3.4 Hz, 1 H, H4′), 3.60–3.65 (m, 1 H, H5′), 3.50–3.56 (m, 1 H, H5″); 13C NMR (DMSO-d6) δ 156.1, 151.4, 149.5, 140.1, 128.2 (Ph), 127.1 (Ph), 126.6, 122.3 (C6), 103.4, 99.2 (C5), 87.6 (C1′), 85.1 (C4′), 73.7 (C2′), 70.7 (C3′), 61.8 (C5′), 43.1 (CH2); MS (ESI) m/z 357 (100%, MH+), HRMS (ESI) m/z 357.1581 (MH+), calcd for C18H29N4O4 357.1563.
4-(4-Nitrobenzylamino)-7-(β-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine (2b)
Method A
Tubercidin (1a; 266 mg, 1.0 mmol) was treated with 4-nitrobenzyl bromide (648 mg, 3.0 mmol) at 80 °C for 24 h as described above for 2a followed by addition of MeOH (15 mL) and Me2NH/THF (2 M, 8 mL). The reaction mixture was stirred at reflux for 20 h; TLC showed ~85% conversion to a less polar product 2b (244 mg, 56%, yellow oil): UV (MeOH) λmax 278 nm, λmin 240 nm; 1H NMR (DMSO-d6) δ 8.30 (t, J = 6.1 Hz, 1 H, NH), 8.19 (“d”, J = 8.8 Hz, 2 H, Ph), 8.12 (s, 1 H, H2), 7.58 (d, J = 8.8 Hz, 2 H, Ph), 7.43 (d, J = 3.7 Hz, 1 H, H6), 6.69 (d, J = 3.5 Hz, 1 H, H5), 6.04 (d, J = 6.3 Hz, 1 H, H1′), 5.28–5.30 (m, 2 H, 2 × OH), 5.14 (s, 1 H, OH), 4.80–4.91 (m, 2 H, CH2), 4.45 (“d”, J = 4.4 Hz, 1 H, H2′), 4.11–4.13 (m, 1 H, H3′), 3.92 (q, J = 3.5 Hz, 1 H, H4′), 3.64 (dt, J = 3.9, 11.8 Hz, 1 H, H5′), 3.52–3.57 (m, 1 H, H5″); 13C NMR (DMSO-d6-D2O) δ 155.8, 151.0, 148.8, 147.9, 146.3, 127.9 (Ph), 123.4 (Ph), 122.8 (C6), 103.6, 99.3 (C5), 87.5 (C1′), 84.9 (C4′), 73.5 (C2′), 70.4 (C3′), 61.6 (C5′), 42.7 (CH2); MS (ESI): m/z 402 (100%, MH+). Anal. Calcd for C18H19N5O6•1.25 H2O (423.89): C, 51.00; H, 5.11; N, 16.52. Found: C, 51.05; H, 5.06; N, 16.03.
Method B
Step a
Ac2O (377 μL, 408 mg, 4 mmol) was added to a stirred suspension of 1a (266 mg, 1 mmol) in dried pyridine (5 mL) at 0 °C (ice bath) and stirring was continued at 0 °C for 12 h and then at ambient temperature for 9 h (total reaction time: 21 h). MeOH was added, the reaction mixture was stirred at ambient temperature for 30 min, and volatiles were evaporated (vacuum pump, <25 °C). MeOH was added and evaporated, and the resulting gum was partitioned between CHCl3 (50 mL) and 2% AcOH/H2O (50 mL). The aqueous layer was extracted with CHCl3, and the combined organic phase was washed with NaHCO3/H2O, brine, and dried (MgSO4). Volatiles were evaporated in vacuo and the residue was column chromatographed (EtOAc) to give 2′,3′,5′-tri-O-acetyltubercidin (3; 337 mg, 86%) as a colorless foam with data as reported.[33]
Step b
NaNO2 (66 mg, 0.95 mmol) was added to a solution of 3 (150 mg, 0.38 mmol) in freshly prepared ~55% HF-pyridine[31b] (1.9 mL) at −10 °C in a sealed polypropylene vessel. The mixture was stirred at −10 °C for 15 min, and TLC showed almost complete conversion to a less polar spot. Ice/H2O was added and the mixture was extracted with CH2Cl2. The combined organic phase was washed with NaHCO3/H2O, brine, and dried (Na2SO4). Volatiles were removed in vacuo, and the resulting brown oil was column chromatographed (30% EtOAc in hexanes) to give 7-(2,3,5-tri-O-acetyl-β-D-ribofuranosyl)-4-fluoropyrrolo[2,3-d]pyrimidine (4; 124 mg, 82%) as a colorless oil: UV (MeOH) λmax 258 nm, λmin 233 nm; 1H NMR (CDCl3) δ 8.53 (d, 4JH–F = 0.5 Hz, 1 H, H2), 7.37 (d, J = 3.8 Hz, 1 H, H8), 6.66 (d, J = 3.8 Hz, 1 H, H7), 6.45 (d, J = 6.0 Hz, 1 H, H1′), 5.74 (t, J = 5.8 Hz, 1 H, H2′), 5.55 (dd or m, J = 4.1, 5.6 Hz, 1 H, H3′), 4.32–4.42 (m, 3 H, H4′, H5′, H5″), 2.12 (s, 3 H, CH3), 2.13 (s, 3 H, CH3), 2.02 (s, 3 H, CH3); 19F NMR (CDCl3): δ −64.81 ppm (s); 13C NMR (CDCl3) δ 170.2, 169.6, 169.4 (3 × C=O), 162.3 (d, 1JC–F = 253.8 Hz, C4), 155.1 (d, 3JC–F = 11.9 Hz, C7a), 151.1 (d, 3JC–F = 14.5 Hz, C2), 125.7 (d, 4JC–F = 2.6 Hz, C6), 105.4 (d, 2JC–F = 33.4 Hz, C4a), 99.6 (d, 3JC–F = 4.9 Hz, C5), 86.1 (C1′), 79.9 (C4′), 73.1 (C2′), 70.1 (C3′), 63.3 (C5′), 20.7, 20.5, 20.3 (3 × s, 3 × CH3); MS (ESI): m/z 396 (100%, MH+).
Step c
Freshly distilled Et3N (113 μL, 82 mg, 0.81 mmol) was added to a stirred suspension of 4 (93 mg, 0.23 mmol) and 4-nitrobenzylamine hydrochloride (67 mg, 0.35 mmol) in MeOH (3 mL) and stirring was continued for 5 h at ambient temperature. Volatiles were evaporated in vacuo and the residue was column chromatographed (50% EtOAc in hexanes) to give 2′,3′,5′-tri-O-acetyl-4-N-(4-nitrobenzyl)tubercidin (5; 58 mg, 47%) as a colorless oil: UV (MeOH) λmax 277 nm, λmin 238 nm. 1H NMR (CDCl3) δ 8.36 (s, 1 H, H2), 8.17 (d, J = 8.7 Hz, 2 H, Ph), 7.51 (d, J = 8.7 Hz, 2 H, Ph), 7.12 (d, J = 3.768 Hz, 1 H, H6), 6.42–6.46 (dd, 3.8, 6.0 Hz, 2 H, H1′, H5), 5.73 (t, J = 5.8 Hz, 1 H, H2′), 5.54–5.59 (m, 2 H, H3′, NH), 4.95 (d, J = 6.1 Hz, 2 H, CH2), 4.31–4.40 (m, 3 H, H4′, H5′, H5″), 2.14 (s, 6 H, 2 × CH3), 2.04 (s, 3 H, CH3); 13C NMR (CDCl3) δ 170.4, 169.7, 169.5 (3 × C=O), 156.0 (C4), 152.3 (C2), 150.8 (C7a), 147.3 (Ph), 146.7 (Ph), 128.0 (Ph), 123.9 (Ph), 121.5 (C6), 103.8 (C4a), 99.4 (C5), 85.4 (C1′), 79.5 (C4′), 73.1 (C2′), 70.8 (C3′), 63.5 (C5′), 44.2 (CH2), 20.8, 20.6, 20.4 (3 × s, 3 × CH3). MS (ESI): m/z 528 (100%, MH+).
Step d
NH3/MeOH (5 mL) was added to a stirred solution of 5 (54 mg, 0.1 mmol) in MeOH (1 mL) and stirring was continued at ambient temperature for 20 h. Volatiles were evaporated and the residue was column chromatographed with a 2 → 4% gradient of the upper phase of EtOAc/i-PrOH/H2O (4:1:2) in EtOAc to give 2b (35 mg, 85%) as a yellow oil.
5-Carboxamido-4-(4-nitrobenzylamino)-7-(β-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine (2c)
Sangivamycin (1b; 155 mg, 0.5 mmol) was treated with 4-nitrobenzyl bromide (162 mg, 1.5 mmol) for 26 h at 40 °C as described for 1a → 2a and the alkylated intermediate was then stirred with Me2NH/THF (2 M, 6 mL) and MeOH (12 mL) for 45 h at ambient temperature. Me2NH/THF (2 M, 2 mL) was added and the mixture was stirred at reflux for 32 h (total reaction time: 77 h). TLC showed ~80% conversion to the less polar 2c, which was obtained as off-white crystals (101 mg, 45%) after recrystallization from EtOH: mp 154–158 °C (dec.); UV (MeOH) 286 nm (ε 23 100), λmin 229 nm (ε 8700); 1H NMR (DMSO-d6) δ 10.29 (t, J = 6.0 Hz, 1 H, NH), 8.17–8.21 (m, 4 H, Ph, H2, H6), 8.11 and 7.47 (2 × s, 2 × 1 H, CONH2), 7.59 (brd, J = 8.8 Hz, 2 H, Ph), 6.06 (d, J = 6.0 Hz, 1 H, H1′), 5.43 (d, J = 6.3 Hz, 1 H, 2′-OH), 5.20 (d, J = 5.0 Hz, 1 H, 3′-OH), 5.09 (t, J = 5.8 Hz, 1 H, 5′-OH), 4.90 (“d”, J = 6.2 Hz, 2 H, CH2), 4.37 (q, J = 5.8 Hz, 1 H, H2′), 4.10 (“q”, J = 4.5 Hz, 1 H, H3′), 3.93 (q, J = 4.0 Hz, 1 H, H4′), 3.61–3.67 (m, 1 H, H5′), 3.53–3.58 (m, 1 H, H5″); 13C NMR (DMSO-d6) δ 166.5, 156.5, 152.6, 150.4, 148.0, 146.4, 128.0 (Ph), 125.7, 123.6, 110.9, 101.7, 87.2 (C1′), 85.3 (C4′), 73.9 (C2′), 70.5 (C3′), 61.9 (C5′), 42.9 (CH2); MS (ESI) m/z 445 (100%, MH+). Anal. Calcd for C19H20N6O7•1.5 H2O (471.42): C, 48.41; H, 4.92; N, 17.83. Found: C, 48.59; H, 4.66; N, 17.65.
5-Cyano-4-(4-nitrobenzylamino)-7-(β-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidine (2d)
Toyocamycin (1c; 146 mg, 0.5 mmol) was heated with 4-nitrobenzyl bromide (364 mg, 1.5 mmol) in dried DMF (3 mL) at 40 °C for 63 h (TLC showed ~85% conversion to a less polar product). Volatiles were evaporated to ~1 mL (< 40 °C, vacuum pump) and this material was added dropwise to 20 mL of vigorously stirred dried acetone. Et2O (40 mL) was added to the stirred suspension, which was chilled at 0 °C for 20 minutes and the precipitate was collected by vacuum filtration. Recrystallization from MeOH gave 2d (99 mg, 46%) as colorless crystals: mp 214–216 °C; UV (MeOH) λmax 274, 236 nm (ε 22 100, 18 900), λmin 250, 228 nm (ε 12 800, 18 000); 1H NMR (DMSO-d6) δ 8.35 (s, 1 H, H2), 8.19–8.23 (m, 3 H, H6, Ph), 7.59 (d, J = 8.6 Hz, 2 H, Ph), 6.87 (s, 1 H, NH), 5.93 (d, J = 5.6 Hz, 1 H, H1′), 5.47 (d, J = 6.0 Hz, 1 H, 2′-OH), 5.35 (“s”, 2 H, CH2), 5.21 (d, J = 5.0 Hz, 1 H, 3′-OH), 5.09 (t, J = 5.4 Hz, 1 H, 5′-OH), 4.31 (q, J = 5.5 Hz, 1 H, H2′), 4.08 (q, J = 4.6 Hz, 1 H, H3′), 3.92 (q, J = 3.7 Hz, 1 H, H4′), 3.62–3.68 (m, 1 H, H5′), 3.53–3.58 (m, 1 H, H5″); 13C NMR (DMSO-d6-D2O) δ 152.6, 149.4, 146.7, 144.9, 142.7, 129.4, 128.5 (Ph), 123.5 (Ph), 115.2, 105.0, 87.7 (C1′), 85.8 (C4′), 85.5, 74.6 (C2′), 70.1 (C3′), 61.1 (C5′), 48.8 (CH2); MS (ESI): m/z 427 (100%, MH+). Anal. Calcd for C19H18N6O6•0.5 H2O (435.39): C, 52.41; H, 4.40; N, 19.30. Found: C, 52.17; H, 4.29; N, 19.30.
6-N-[N-phenylcarbamoyl]adenosine (9a)
Step a
A solution of phenyl isocyanate (35 μL, 39 mg, 0.32 mmol) and 2′,3′,5′-tri-O-acetyladenosine (7; 106 mg, 0.27 mmol) in CH2Cl2 (5 mL) was stirred at ambient temperature under N2. An additional portion of phenyl isocyanate (35 μL, 39 mg, 0.324 mmol) was added after 48 h, and stirring was continued for 20 h (total reaction time: 68 h). The reaction mixture was partitioned (NaHCO3/H2O//CHCl3) and the organic layer was dried (Na2SO4), concentrated, and column chromatographed (70% EtOAc/hexanes) to give 2′,3′,5′-tri-O-acetyl-6-N-[N-phenylcarbamoyl]adenosine (8a; 111 mg, 80%): 1H NMR (CDCl3) δ 11.89 (s, 1 H, NH), 9.40 (s, 1 H, NH), 8.62 (s, 1 H, H2), 8.56 (s, 1 H, H8), 7.63 (“d”, J = 7.6 Hz, 2 H, Ph), 7.35 (“t”, J = 7.9 Hz, 2 H, Ph), 7.11 (“t”, J = 7.4 Hz, 1 H, Ph), 6.25 (d, J = 5.4 Hz, 1 H, H1′), 6.03 (t, J = 5.5 Hz, 1 H, H2′), 5.69 (“dd”, J = 4.4, 5.4 Hz, 1 H, H3′), 4.37–4.48 (m, 3 H, H4′, H5′, H5″), 2.15 (s, 3 H, 1 × CH3), 2.08 (s, 3 H, CH3), 2.07 (s, 3 H, CH3).
Step b
NH3/MeOH (6 mL) was added to 8a (103 mg, 0.2 mmol) in MeOH (3 mL) and the mixture was stirred at ambient temperature for 2 h. Volatiles were evaporated and the residual solid was recrystallized from MeOH to give 9a (66 mg, 85%): mp 193–195 °C; UV (MeOH) λmax 279 nm (ε 28 250), λmin 241 nm (ε 9200); 1H NMR (DMSO-d6) δ 11.76 (s, 1 H, NH), 10.17 (s, 1 H, NH), 8.72 (s, 1 H, H2), 8.69 (s, 1 H, H8), 7.63 (d, J = 7.7 Hz, 2 H, Ph), 7.36 (t, J = 7.9 Hz, 2 H, Ph), 7.10 (t, J = 7.4 Hz, 1 H, Ph), 6.01 (d, J = 5.6 Hz, 1 H, H1′), 5.54 (d, J = 6.0 Hz, 1 H, 2′-OH), 5.24 (d, J = 5.0 Hz, 1 H, 3′-OH), 5.14 (t, J = 5.5 Hz, 1 H, 5′-OH), 4.61 (q, J = 5.6 Hz, 1 H, H2′), 4.19 (q, J = 4.7 Hz, 1 H, H3′), 3.99 (q, J = 3.8 Hz, 1 H, H4′), 3.67–3.73 (m, 1 H, H5′), 3.55–3.61 (m, 1 H, H5″); 13C NMR (DMSO-d6) δ 150.83 (C=O), 150.81 (C2), 150.6, 150.0, 142.4 (C8), 138.4, 128.9 (Ph), 123.2 (Ph), 120.6, 119.4 (Ph), 87.7 (C1′), 85.7 (C4′), 73.8 (C2′), 70.3 (C3′), 61.3 (C5′); MS (ESI) m/z 387 (100%, MH+). Anal. Calcd for C17H18N6O5•0.5 H2O (395.37): C, 51.64; H, 4.84; N, 21.26. Found: C, 51.38; H, 4.77; N, 21.02.
6-N-[N-(4-Methoxyphenyl)carbamoyl]adenosine (9b)
Step a
A solution of 7 (197 mg, 0.5 mmol) and 4-methoxyphenyl isocyanate (78 μL, 90 mg, 0.6 mmol) in CH2Cl2 (5 mL) was stirred at ambient temperature under N2. Additional 4-methoxyphenyl isocyanate (78 μL, 90 mg, 0.6 mmol) was added after 12 h, and stirring was continued for 12 h (total reaction time: 24 h). The mixture was partitioned (NaHCO3/H2O//CHCl3) and the organic layer was dried (Na2SO4) and evaporated to give residual 2′,3′,5′-tri-O-acetyl-6-N-[N-(4-methoxyphenyl)carbamoyl]adenosine (8b; 260 mg, 96%) of sufficient purity for NMR characterization and use in the next step: 1H NMR (CDCl3) δ 11.45 (s, 1 H, NH), 8.62 (s, 1 H, H2), 8.59 (s, 1 H, H8), 8.12 (s, 1 H, NH), 7.53 (“d”, J = 8.9 Hz, 2 H, Ph), 6.91 (“d”, J = 9.0 Hz, 2 H, Ph), 6.21 (d, J = 5.3 Hz, 1 H, H1′), 5.97 (t, J = 5.4 Hz, 1 H, H2′), 5.66 (“t”, J = 4.8 Hz, 1 H, H3′), 4.38–4.48 (m, 3 H, H4′, H5′, H5″), 2.16 (s, 3 H, CH3), 2.14 (s, 3 H, CH3), 2.10 (s, 3 H, CH3).
Step b
NH3/MeOH (6 mL) was added to crude 8b (260 mg, 0.5 mmol) in MeOH (3 mL) and the mixture was stirred at ambient temperature for 2 h. Volatiles were evaporated and the solid residue was recrystallized from MeOH to give 9b (170 mg, 80%): mp 181–182 °C; UV (MeOH) λmax 280 nm (ε 21 300), λmin 247 nm (ε 11 100); 1H NMR (DMSO-d6) δ 11.59 (s, 1 H, NH), 10.07 (s, 1 H, NH), 8.70 (s, 1 H, H2), 8.67 (s, 1 H, H8), 7.53 (“d”, J = 4.5 Hz, 2 H, Ph), 6.94 (“d”, J = 4.5 Hz, 2 H, Ph), 6.01 (d, J = 5.7 Hz, 1 H, H1′), 5.54 (d, J = 6.0 Hz, 1 H, 2′-OH), 5.24 (d, J = 5.0 Hz, 1 H, 3′-OH), 5.14 (t, J = 5.6 Hz, 1 H, 5′-OH), 4.61 (q, J = 5.5 Hz, 1 H, H2′), 4.18 (q, J = 4.8 Hz, 1 H, H3′), 3.98 (q, J = 3.8 Hz, 1 H, H4′), 3.75 (s, 3 H, OCH3), 3.67–3.72 (m, 1 H, H5′), 3.55–3.61 (m, 1 H, H5″); 13C NMR (DMSO-d6) δ 155.4 (C=O), 150.9 (C2), 150.8, 150.4, 149.8, 142.3 (C8), 131.1, 121.2 (Ph), 120.3, 114.1 (Ph), 87.7 (C1′), 85.6 (C4′), 73.7 (C2′), 70.1 (C3′), 61.1 (C5′), 55.2 (OCH3); MS (ESI) m/z 417 (100%, MH+). Anal. Calcd for C18H20N6O6•1.5 H2O (443.41): C, 48.76; H, 5.23; N, 18.95. Found: C, 48.54; H, 4.97; N, 18.85.
6-N-[N-(4-Nitrophenyl)carbamoyl]adenosine (9c)
Step a
A solution of 4-nitrophenyl isocyanate (54 mg, 0.33 mmol) and 7 (117 mg, 0.30 mmol) in CH2Cl2 (20 mL) was stirred at ambient temperature under N2 for 17 h (TLC showed ~85 % conversion to a less polar product) and then was heated at reflux for 24 h (total reaction time: 41 h). The mixture was partitioned (NaHCO3/H2O//CHCl3) and the organic layer was dried (Na2SO4) and evaporated to give solid 2′,3′,5′-tri-O-acetyl-6-N-[N-(4-nitrophenyl)carbamoyl]adenosine (8c; 160 mg, 96%) of sufficient purity for NMR characterization and use in the next step: 1H NMR (DMSO-d6) δ 12.13 (s, 1 H, NH), 10.62 (s, 1 H, NH), 8.75 (s, 1 H, H2), 8.71 (s, 1 H, H8), 8.27 (d, J = 9.2 Hz, 2 H, Ph), 7.90 (d, J = 9.2 Hz, 2 H, Ph), 6.33 (d, J = 5.3 Hz, 1 H, H1′), 6.05 (d, J = 5.7 Hz, 1 H, H2′), 5.65 (t, J = 5.4 Hz, 1 H, H3′), 4.42–4.47 (m, 1 H, H4′, H5′), 4.25–4.3 (m, 1 H, H5″), 2.13 (s, 3 H, CH3), 2.05 (s, 3 H, CH3), 2.03 (s, 3 H, CH3).
Step b
NH3/MeOH (6 mL) was added to crude 8c (160 mg, 0.29 mmol) in MeOH (3 mL) and the mixture was stirred at ambient temperature for 2 h. Volatiles were evaporated and the residual solid was recrystallized from MeOH to give 9c (80 mg, 65%): mp 202–208 °C; UV (MeOH) λmax 317, 278 nm (ε 16 200, 11 300), λmin 286, 241 nm (ε 10 300, 5600). 1H NMR (DMSO-d6) δ 12.27 (s, 1 H, NH), 10.61 (s, 1 H, NH), 8.73 (s, 1 H, H2), 8.72 (s, 1 H, H8), 8.27 (“d”, J = 9.2 Hz, 2 H, Ph), 8.0 (“d”, J = 9.2 Hz, 2 H, Ph), 6.01 (d, J = 5.7 Hz, 1 H, H1′), 5.52 (“s”, J = 22.7 Hz, 1 H, 2′-OH), 5.24 (d, J = 14.1 Hz, 1 H, 3′-OH), 5.14 (“s”, 1 H, 5′-OH), 4.61 (“s’, 1 H, H2′), 4.18 (“s”, 1 H, H3′), 3.98 (q, J = 3.8 Hz, 1 H, H4′), 3.68–3.71 (m, 1 H, H5′), 3.55–3.61 (m, 1 H, H5″); 1H NMR (D2O/DMSO-d6) δ 8.71 (s, 1 H, H2), 8.68 (s, 1 H, H8 ), 8.24 (“d”, J = 9.2 Hz, 2 H, Ph), 7.88 (“d”, J = 9.2 Hz, 2 H, Ph), 6.00 (d, J = 5.7 Hz, 1 H, H1′), 4.58 (t, J = 5.4 Hz 1 H, H2′), 4.15 (“t”, J = 4.3 Hz 1 H, H3′), 3.99 (q, J = 3.7 Hz, 1 H, H4′), 3.68 (dd, J = 3.8, 12.1 Hz 1 H, H5′), 3.57 (dd, J = 3.9, 12.2 Hz 1 H, H5″); 13C NMR (DMSO-d6) δ 150.9, 150.72, 150.66, 149.4, 144.5, 142.6 (C8), 142.2, 125.0, 120.5, 119.1, 87.7 (C1′), 85.6 (C4′), 73.7 (C2′), 70.1 (C3′), 61.1 (C5′); MS (ESI) m/z 432 (100%, MH+). Anal. Calcd for C17H17N7O7•H2O (449.37): C, 45.44; H, 4.26; N, 21.82. Found: C, 45.10; H, 3.96; N, 21.59.
Cytostatic activity assays
All assays were performed in 96-well microtiter plates. To each well of 200 μL were added (5–7.5) × 104 tumor cells and a given amount of the test compound. The cells were allowed to proliferate for 48 h (murine leukemia L1210), 72 h (human lymphocytic CEM), 96 h (human cervix carcinoma HeLa), 144 h (human prostate cancer PC-3), or 168 h (human kidney Caki-1) at 37 °C in a humidified CO2-controlled atmosphere. At the end of the incubation period, the cells were counted in a Coulter counter. The IC50 (50% inhibitory concentration) was defined as the concentration of the compound that inhibited cell proliferation by 50%.
Acknowledgments
This work was partially supported by NIGMS/NCI (1SC1CA138176; SFW) and the KU Leuven (GOA 10/014; JB). We thank the NIH MARC U*STAR (GM083688-02) program for the fellowships supporting H.O. and P.T., and Mrs. Lizette van Berckelaer for technical assistance with the cytostatic evaluations.
References
- 1.a) Suhadolnik RJ. Nucleoside Antibiotics. Wiley-Interscience; 1970. [Google Scholar]; b) Suhadolnik RJ. Nucleosides as Biological Probes. Wiley; 1979. [Google Scholar]; c) Seela F, Peng X. Curr Top Med Chem. 2006;6:867–892. doi: 10.2174/156802606777303649. [DOI] [PubMed] [Google Scholar]; d) Seela F, Peng X, Budow S. Curr Org Chem. 2007;11:427–462. [Google Scholar]; e) Seela F, Budow S, Peng X. Curr Org Chem. 2012;16:161–223. [Google Scholar]
- 2.a) Gerster JF, Carpenter B, Robins RK, Townsend LB. J Med Chem. 1967;10:326–331. doi: 10.1021/jm00315a008. [DOI] [PubMed] [Google Scholar]; b) Tolman RL, Robins RK, Townsend LB. J Heterocycl Chem. 1967;4:230–238. [Google Scholar]; c) Tolman RL, Robins RK, Townsend LB. J Amer Chem Soc. 1969;91:2102–2108. doi: 10.1021/ja01036a040. [DOI] [PubMed] [Google Scholar]; d) Hinshaw BC, Gerster JF, Robins RK, Townsend LB. J Heterocycl Chem. 1969;6:215–221. [Google Scholar]; e) Uematsu T, Suhadolnik RJ. J Med Chem. 1973;16:1405–1407. doi: 10.1021/jm00270a021. [DOI] [PubMed] [Google Scholar]; f) Maruyama T, Wotring LL, Townsend LB. J Med Chem. 1983;26:25–29. doi: 10.1021/jm00355a006. [DOI] [PubMed] [Google Scholar]; g) Bergstrom DE, Brattesani AJ, Ogawa MK, Reddy PA, Schweickert MJ, Balzarini J, De Clercq E. J Med Chem. 1984;27:285–292. doi: 10.1021/jm00369a010. [DOI] [PubMed] [Google Scholar]; h) De Clercq E, Balzarini J, Madej D, Hansske F, Robins MJ. J Med Chem. 1987;30:481–486. doi: 10.1021/jm00386a007. [DOI] [PubMed] [Google Scholar]; i) Turk SR, Shipman C, Nassiri R, Genzlinger G, Krawczyk SH, Townsend LB, Drach JC. Antimicrob Agents Chemother. 1987;31:544–550. doi: 10.1128/aac.31.4.544. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Gupta PK, Daunert S, Nassiri MR, Wotring LL, Drach JC, Townsend LB. J Med Chem. 1989;32:402–408. doi: 10.1021/jm00122a019. [DOI] [PubMed] [Google Scholar]; k) Robins MJ, Wilson JS, Madej D, Low NH, Hansske F, Wnuk SF. J Org Chem. 1995;60:7902–7908. [Google Scholar]; l) Bookser BC, Ugarkar BG, Matelich MC, Lemus RH, Allan M, Tsuchiya M, Nakane M, Nagahisa A, Wiesner JB, Erion MD. J Med Chem. 2005;48:7808–7820. doi: 10.1021/jm050394a. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, Jia D, Liu H, Zhu N, Zhang W, Feng J, Yin J, Hao B, Cui D, Deng Y, Xie D, He L, Li B. PLoS ONE. 2013;8:1–11. doi: 10.1371/journal.pone.0062527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stockwin L, Yu S, Stotler H, Hollingshead M, Newton D. BMC Cancer. 2009;9:63–75. doi: 10.1186/1471-2407-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi BY, Lee CH. Bioorg Med Chem Lett. 2010;20:3880–3884. doi: 10.1016/j.bmcl.2010.05.037. [DOI] [PubMed] [Google Scholar]
- 6.Wu R, Smidansky ED, Oh HS, Takhampunya R, Padmanabhan R, Cameron CE, Peterson BR. J Med Chem. 2010;53:7958–7966. doi: 10.1021/jm100593s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Clercq E, Robins MJ. Antimicrob Agents Chemother. 1986;30:719–724. doi: 10.1128/aac.30.5.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varaprasad CVNS, Ramasamy KS, Girardet JL, Gunic E, Lai V, Zhong W, An H, Hong Z. Bioorg Chem. 2007;35:25–34. doi: 10.1016/j.bioorg.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Ding Y, An H, Hong Z, Girardet JL. Bioorg Med Chem Lett. 2005;15:725–727. doi: 10.1016/j.bmcl.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 10.a) Eldrup AB, Prhavc M, Brooks J, Bhat B, Prakash TP, Song Q, Bera S, Bhat N, Dande P, Cook PD, Bennett CF, Carroll SS, Ball RG, Bosserman M, Burlein C, Colwell LF, Fay JF, Flores OA, Getty K, LaFemina RL, Leone J, MacCoss M, McMasters DR, Tomassini JE, von Langen D, Wolanski B, Olsen DB. J Med Chem. 2004;47:5284–5297. doi: 10.1021/jm040068f. [DOI] [PubMed] [Google Scholar]; b) Olsen DB, Eldrup AB, Bartholomew L, Bhat B, Bosserman MR, Ceccacci A, Colwell LF, Fay JF, Flores OA, Getty KL, Grobler JA, LaFemina RL, Markel EJ, Migliaccio G, Prhavc M, Stahlhut MW, Tomassini JE, MacCoss M, Hazuda DJ, Carroll SS. Antimicrob Agents Chemother. 2004;48:3944–3953. doi: 10.1128/AAC.48.10.3944-3953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhattacharya BK, Rao TS, Revankar GR. J Chem Soc Perkin Trans. 1995;1:1543–1550. [Google Scholar]
- 12.a) Bhattacharya BK, Ojwang JO, Rando RF, Huffman JH, Revankar GR. J Med Chem. 1995;38:3957–3966. doi: 10.1021/jm00020a009. [DOI] [PubMed] [Google Scholar]; b) Ojwang JO, Bhattacharya BK, Marshall HB, Korba BE, Revankar GR, Rando RF. Antimicrob Agents Chemother. 1995;39:2570–2573. doi: 10.1128/aac.39.11.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Ugarkar BG, Castellino AJ, DaRe JM, Kopcho JJ, Wiesner JB, Schanzer JM, Erion MD. J Med Chem. 2000;43:2894–2905. doi: 10.1021/jm0000259. [DOI] [PubMed] [Google Scholar]; b) Boyer SH, Ugarkar BG, Solbach J, Kopcho J, Matelich MC, Ollis K, Gomez-Galeno JE, Mendonca R, Tsuchiya M, Nagahisa A, Nakane M, Wiesner JB, Erion MD. J Med Chem. 2005;48:6430–6441. doi: 10.1021/jm0503650. [DOI] [PubMed] [Google Scholar]
- 14.Naus P, Pohl R, Votruba I, Dzubak P, Hajduch M, Ameral R, Birkus G, Wang T, Ray AS, Mackman R, Cihlar T, Hocek M. J Med Chem. 2010;53:460–470. doi: 10.1021/jm901428k. [DOI] [PubMed] [Google Scholar]
- 15.Bourderioux A, Naus P, Perlikova P, Pohl R, Pichova I, Votruba I, Dzubak P, Konecny P, Hajduch M, Stray KM, Wang T, Ray AS, Feng JY, Birkus G, Cihlar T, Hocek M. J Med Chem. 2011;54:5498–5507. doi: 10.1021/jm2005173. [DOI] [PubMed] [Google Scholar]
- 16.Naus P, Caletkova O, Konecny P, Dzubak P, Bogdanova K, Kolar M, Vrbkova J, Slavetinska L, Tloust’ova E, Perlikova P, Hajduch M, Hocek M. J Med Chem. 2014;57:1097–1110. doi: 10.1021/jm4018948. [DOI] [PubMed] [Google Scholar]
- 17.Miles RW, Samano V, Robins MJ. J Am Chem Soc. 1995;117:5951–5957. [Google Scholar]
- 18.Robins MJ, Trip EM. Biochemistry. 1973;12:2179–2187. doi: 10.1021/bi00736a001. [DOI] [PubMed] [Google Scholar]
- 19.Young JD, Yao SYM, Baldwin JM, Cass CE, Baldwin SA. Mol Aspects Med. 2013;34:529–547. doi: 10.1016/j.mam.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Jordheim LP, Durantel D, Zoulim F, Dumontet C. Nat Rev Drug Discov. 2013;12:447–464. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- 21.Griffiths M, Beaumont N, Yao SYM, Sundaram M, Boumah CE, Davies A, Kwong FYP, Coe I, Cass CE, Young JD, Baldwin SA. Nat Med. 1997;3:89–93. doi: 10.1038/nm0197-89. [DOI] [PubMed] [Google Scholar]
- 22.Robins MJ, Asakura J-i, Kaneko M, Shibuya S, Jakobs ES, Agbanyo FR, Cass CE, Paterson ARP. Nucleosides and Nucleotides. 1994;13:1627–1646. [Google Scholar]
- 23.Robins MJ, Peng Y, Damaraju VL, Mowles D, Barron G, Tackaberry T, Young JD, Cass CE. J Med Chem. 2010;53:6040–6053. doi: 10.1021/jm100432w. [DOI] [PubMed] [Google Scholar]
- 24.Young JD, Yao SYM, Cass CE, Baldwin SA. In: Red Cell Membrane Transport in Health and Disease. Bernhardt I, Ellory JC, editors. Springer-Verlag; Berlin: 2003. pp. 321–337. [Google Scholar]
- 25.Dimroth O. Justus Liebigs Ann Chem. 1909;364:183–226. [Google Scholar]
- 26.Liu J, Robins MJ. J Am Chem Soc. 2007;129:5962–5968. doi: 10.1021/ja070021u. [DOI] [PubMed] [Google Scholar]
- 27.a) Engel JD. Biochem Biophys Res Commun. 1975;64:581–586. doi: 10.1016/0006-291x(75)90361-7. [DOI] [PubMed] [Google Scholar]; b) Fujii T, Itaya T. Heterocycles. 1998;48:359–390. [Google Scholar]
- 28.Pande P, Shearer J, Yang J, Greenberg WA, Rokita SE. J Am Chem Soc. 1999;121:6773–6779. [Google Scholar]
- 29.Zhong M, Robins MJ. J Org Chem. 2006;71:8901–8906. doi: 10.1021/jo061759h. [DOI] [PubMed] [Google Scholar]
- 30.a) Robins MJ, Uznański B. Can J Chem. 1981;59:2608–2611. [Google Scholar]; b) Liu J, Robins MJ. Org Lett. 2005;7:1149–1151. doi: 10.1021/ol050063s. [DOI] [PubMed] [Google Scholar]
- 31.Olah GA, Welch JT, Vankar YD, Nojima M, Kerekes I, Olah JA. J Org Chem. 1979;44:3872–3881.b) Typical procedure for the preparation of ~55% HF-pyridine: 1.8 mL of 70% HF-pyridine was added to 0.6 mL of anhydrous pyridine at −78 °C to generate 2.4 mL of ~55% HF-pyridine. The mixture was allowed to warm gradually to −10 °C and used directly.
- 32.a) Peterson MA, Oliveira M, Christiansen MA. Nucleosides Nucleotides Nucleic Acids. 2009;28:394–407. doi: 10.1080/15257770903044432. [DOI] [PubMed] [Google Scholar]; b) Shelton JR, Cutler CE, Browning MS, Balzarini J, Peterson MA. Bioorg Med Chem Lett. 2012;22:6067–6071. doi: 10.1016/j.bmcl.2012.08.050. [DOI] [PubMed] [Google Scholar]
- 33.Nowak I, Conda-Sheridan M, Robins MJ. J Org Chem. 2005;70:7455–7458. doi: 10.1021/jo051256w. [DOI] [PubMed] [Google Scholar]