

Abstract
Transcription factors (TFs) influence cell fate by interpreting the regulatory DNA within a genome. TFs recognize DNA in a specific manner; the mechanisms underlying this specificity have been identified for many TFs, based on three-dimensional structures of protein-DNA complexes. More recently, structural views have been complemented with data from high-throughput in vitro and in vivo explorations of the DNA binding preferences of many TFs. Together, these approaches have greatly expanded our understanding of TF-DNA interactions. However, the mechanisms by which TFs select in vivo binding sites and alter gene expression remain unclear. Recent work has highlighted the many variables that influence TF-DNA binding, while demonstrating that a biophysical understanding of these many factors will be central to understanding TF function.
Keywords: transcription factor, TF-DNA binding specificity, high-throughput binding assays, quantitative modeling, cooperative TF binding, chromatin accessibility
Questions at the interface of genomics and structural biology
After decades of research, much is now understood about how transcription factors (TFs) recognize their cognate DNA binding sites in the genome to initiate gene regulatory functions. However, potential target sites of each TF occur many times in the genome. How proteins can very precisely identify their functional binding sites in a cellular environment has not been resolved. Although closely related proteins are known to bind to distinct target sites to execute different in vivo functions, the mechanisms by which paralogous TFs select very similar, but not identical, target sites are not understood. Current knowledge on the DNA binding specificities of TFs is largely derived from research in genomics and structural biology, two fields of research that have developed along parallel lines with limited interactions and have only just begun to become integrated.
Recent studies have focused on the question of how TFs recognize a subset of putative DNA binding sites (Figure 1A) by identifying features, beyond the sequence of the core binding site, which contribute to TF-DNA binding specificity [1–4]. Several features contribute to TF-DNA readout on multiple levels (Figure 1B), including the nucleotide sequence [5–11], three-dimensional structure and flexibility of TFs and their DNA binding sites [12–15], TF-DNA binding in the presence of cofactors [1, 16], cooperative DNA binding of TFs [12, 17–19], chromatin accessibility and nucleosome occupancy [20–25], indirect cooperativity via competition with nucleosomes [26, 27], pioneer TFs that bind nucleosomal DNA [28, 29] and DNA methylation [30]. Additionally, interactions exist among all of these factors, which might alter binding in a cell type-specific manner [29, 31]. Many comprehensive reviews [8, 32–48] have discussed these different aspects of TF-DNA binding specificity, often from either a genomics or structural biology perspective. This review attempts to integrate what has been learned at the various scales from studies by these two complementary approaches, and discusses the important progress that has been made in recent years.
Figure 1. Structure-based illustration of multiple levels of TF-DNA binding specificity.
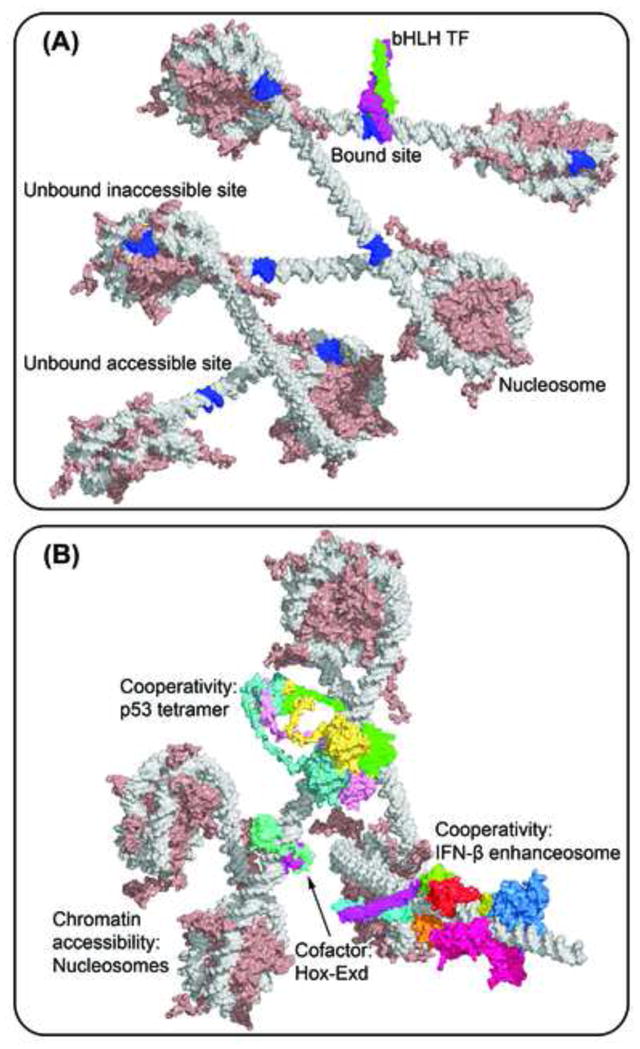
(A) The basic helix-loop-helix (bHLH) Mad-Max heterodimer (PDB ID 1nlw) binds to only a subset of putative binding sites (blue). Some TFBSs are inaccessible due to nucleosome formation (PDB ID 1kx5), while other accessible TFBSs are not selected by the TF.
(B) Higher-order determinants of TF binding include cooperativity with cofactors (e.g., Hox-Exd heterodimer; PDB ID 2r5z), multimeric binding (e.g., p53 tetramer; modeled based on PDB IDs 2ady and 1aie [228]), cooperativity through TF-TF interactions (e.g., IFN-β enhanceosome; modeled based on PDB IDs 1t2k, 2pi0, 2o6g and 2o61 [16]) and chromatin accessibility due to nucleosome formation (PDB ID 1kx5) [229].
Transcription factors recognize DNA through the interplay of base and shape readout
Structural biology has been at the forefront of the search for a protein-DNA recognition code. Cocrystal structures of protein-DNA complexes were first solved in the 1980s [49]. Since then, more than 1,600 entries of protein-DNA structures have been entered in the Protein Data Bank [50], including structures solved by nuclear magnetic resonance (NMR) spectroscopy. These structures have revealed why many TFs preferentially bind to a specific DNA sequence [39]. Namely, the preference for a given nucleotide at a specific position is mainly determined by physical interactions between the amino acid side chains of the TF and the accessible edges of the base pairs that are contacted. These contacts include direct hydrogen bonds, water-mediated hydrogen bonds and hydrophobic contacts. This form of protein-DNA recognition is known as base readout (Figure 2A). A prominent example for base readout is the formation of bidentate hydrogen bonds between arginine residues and guanine bases in the major groove of DNA [19].
Figure 2. Base and shape readout contribute to TF-DNA binding specificity.

(A) Base readout describes direct interactions between amino acids and the functional groups of the bases. Whereas the pattern of hydrogen bond acceptors (red) and donors (blue), heterocyclic hydrogen atoms (white) and the hydrophobic methyl group (yellow) is base pair-specific in the major groove, the pattern is degenerate in the minor groove.
(B) Shape readout includes any form of structural readout, based on global and local DNA shape features, including conformational flexibility and shape-dependent electrostatic potential. The IFN-β enhanceosome (PDB ID 1t2k; top) varies in minor groove shape. The human papillomavirus E2 protein (PDB ID 1jj4; bottom) binds to a binding site with intrinsic curvature.
(C) Most DNA binding proteins use interplay between the base- and shape-readout modes to recognize their DNA binding sites. However, the contribution of each mechanism to binding specificity might vary across TF families. Shape readout dominates for the minor groove-binding HMG box protein (PDB ID 2gzk; left). Base readout is a major contribution in DNA recognition by the bHLH protein Pho4 (PDB ID 1a0a; right). Both readout modes are more or less equally present in the DNA binding of a Hox-Exd heterodimer (PDB ID 2r5z; center).
TFs can also recognize the structural features of their DNA binding sites, such as sequence-dependent DNA bending [51, 52] and unwinding [53]. This phenomenon of recognizing sequence-dependent DNA structure is known as shape readout (Figure 2B). The DNA shape concept includes the static and dynamic properties of DNA structure, and the readout of enhanced negative electrostatic potential in narrow minor groove regions through arginine [13] or histidine [54] residues.
These two protein-DNA recognition mechanisms (i.e., base and shape readout, also known as direct and indirect readout [55]) were often historically presented as mutually exclusive driving forces for DNA recognition by a given protein. Only recently have structural studies [19, 56, 57] embraced the more realistic situation that most proteins use the interplay of base and shape readout to recognize their cognate binding sites. The contributions of base and shape readout, however, vary across protein families (Figure 2C; Figure 3). Recent structures of protein-DNA complexes accurately reflect the biologically correct architecture (which can affect cooperativity), revealing cofactors that bind (Figure 3A) [1] or do not contact [16] DNA, TF-DNA binding as dimers (Figure 3B–C) [58] or tetramers (Figure 3D) [19] and multiple TFs that bind DNA while forming protein-protein contacts (Figure 3E) [59].
Figure 3. Interplay of base and shape readout varies among TF families.

(A) A heterodimer of the homeodomain proteins (PDB ID 2r5z) Hox protein Sex combs reduced (Scr; cyan; top and center) and its cofactor Extradenticle (Exd; magenta; top and center) binds with its recognition helices through base readout to the major groove (blue box; bottom), whereas arginine residues of the N-terminal Scr linker read minor groove shape and electrostatic potential as a form of shape readout (beige box; bottom).
(B) A homodimer of the bHLH protein USF (PDB ID 1an4; green and pink; top and center) binds with its recognition helices through base readout to the E-box core-binding site (blue box; bottom) and recognizes flanking sequences (beige box; bottom) through extended linkers that connect the two α-helices of each USF monomer.
(C) The human papillomavirus (HPV) E2 homodimer (PDB ID 1jj4; purple and chartreuse; top and center) recognizes with its recognition helices the half-sites of its binding site through base readout (blue box; bottom), whereas the intrinsic curvature of the central spacer contributes to binding through shape readout (beige box; bottom).
(D) The four DBDs of the p53 tetramer (PDB ID 3kz8; cyan, yellow, pink, and green; top and center) bind to the major groove through base readout (blue box; bottom), whereas the Arg248 residues recognize the minor groove through shape readout (beige box; bottom).
(E) The c-Jun and ATF-2 TFs (cyan and magenta, respectively; top and center) of the IFN-β enhanceosome (PDB ID 1t2k) recognize the major groove through base readout (blue box; bottom), whereas the adjacent IRF-3 TFs (green and yellow; top and center) use their His40 residues to recognize the minor groove through shape readout (beige box; bottom).
Computational models for describing the DNA binding specificities of transcription factors
In parallel to structural biology approaches to studying protein-DNA binding specificity, sequence-based computational methods have been developed. These methods use a set of known protein-DNA binding sites to generate DNA motif models for predicting the binding specificity of any new site. Early DNA motif discovery methods [60–63] were trained and tested on: 1) small sets of aligned TF binding sites (TFBSs) collected from small-scale experiments, such as DNase I footprinting [64] or electrophoretic mobility shift assays [65], 2) simulated data, in which TFBSs were artificially inserted into background DNA [63], or 3) sets of promoter regions of coregulated genes [61]. The development of microarray- and sequencing-based assays for the high-throughput measurement of protein-DNA binding resulted in a burst of motif discovery methods; to date, hundreds of DNA motif discovery algorithms have been developed [9, 66, 67].
Most sequence-based DNA motif discovery methods use position weight matrices (PWMs) to represent the TF-DNA binding specificity [5, 8]. This type of model is simple, intuitive and can be learned from various data types: from small sets of known binding sites to high-throughput protein-DNA binding data. Traditional PWM models have the benefit of being easy to visualize as DNA motif logos [68]. However, these models are only able to describe the DNA base readout by a TF. Moreover, they implicitly assume that positions within a TFBS independently contribute to the binding affinity, an assumption that does not always hold [7, 10, 69–71]. Consequently, more complex sequence-based models of protein-DNA binding specificity have been developed (Figure 4; Table 1A) to account for positional dependencies within TFBSs, as well as other complexities in protein-DNA recognition [2, 9, 31, 72–74].
Figure 4. Timeline of genomic approaches for experimental and computational studies of TF-DNA binding specificity.
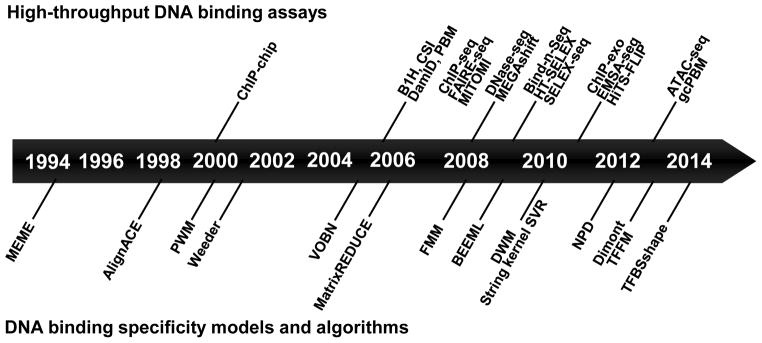
Development of experimental high-throughput DNA binding assays (above timeline axis) and computational DNA binding specificity models and algorithms (below timeline axis). Further examples of these experimental approaches and computational methods are provided in Table 1.
Table 1.
Computational models of protein-DNA binding specificity and high-throughput assays for generating the data used to train and test specificity models.
| (A) Computational models of protein-DNA binding specificity | ||
|---|---|---|
| Model type | Model Description | Refs. |
| Position weight matrices (PWMs) | Simple probabilistic models that assume independence between positions in TF binding sites (TFBSs) | [5] |
| Dinucleotide weight matrices (DWMs) | Generalization of PWM models that incorporates frequencies of dinucleotides | [73, 230] |
| Bayesian networks | Flexible probabilistic models that can incorporate dependencies between positions in TFBSs | [63] |
| Hidden Markov models | Probabilistic models that can incorporate dependencies between neighboring positions in TFBSs | [70, 231] |
| High-order Markov models | Flexible probabilistic models that can incorporate high order dependencies between neighboring positions in TFBSs | [232] |
| k-mer based regression models | Probabilistic models that predict the level of TF binding based on the frequencies of mono-, di-, and tri-nucleotides | [93, 233] |
| Markov networks | Flexible probabilistic models that can incorporate high-order dependencies within TFBSs | [72] |
| Neural networks | Flexible probabilistic models that represent TF binding specificities using a system of interconnected, artificial “neurons” | [75] |
| Random forest models | Flexible probabilistic models that represent TF specificity using a collection of decision trees | [92] |
| Support vector models | Probabilistic models that can incorporate complex patterns of similarities between TFBSs | [2, 31] |
| Variable-order Bayesian networks | Flexible probabilistic models that can incorporate high-order dependencies within TFBSs | [234] |
| Thermodynamic/Energy-based models | Models that infer DNA binding affinities by fitting thermodynamic equations to experimental data | [73, 81–83, 235–237] |
| Atomistic/Structure-based models | Models based on known structures of TFs bound to target DNA sites | [86, 90] |
| Probabilistic models that incorporate structural features | Models that incorporate structural features such as groove geometries and helical parameters | [2, 79, 91, 92] |
| Probabilistic models that incorporate in vivo data | Models that incorporate in vivo data such as DNA accessibility, histone modifications | [238, 239] |
| (B) In vivo high-throughput DNA binding assays | ||
|---|---|---|
| Assay name | Assay description | Refs |
| ChIP-chip | Chromatin immunoprecipitation followed by microarray hybridization | [240] |
| ChIP-seq | Chromatin immunoprecipitation followed by high-throughput sequencing | [241] |
| ChIP-exo | Chromatin immunoprecipitation with exonuclease digestion followed by high-throughput sequencing | [242] |
| DamID | DNA adenine methyltransferase identification | [243] |
| DNase-seq | DNase I cleavage followed by high-throughput sequencing | [151, 244] |
| FAIRE-seq | Formaldehyde-assisted isolation of regulatory elements, followed by high-throughput sequencing | [149] |
| ATAC-seq | Assay for transposase-accessible chromatin using high-throughput | [152] |
| (C) In vitro high-throughput DNA binding assays | ||
|---|---|---|
| Assay name | Assay description | Refs |
| B1H | Bacterial one-hybrid | [102, 245] |
| PBM | Protein binding microarray | [94, 246] |
| CSI | Cognate site identifier | [247] |
| MITOMI | Mechanically induced trapping of molecular interactions | [101, 248] |
| MEGAshift | Microarray evaluation of genomic aptamers by shift | [249] |
| TIRF-PBM | Total internal reflectance fluorescence protein-binding microarray | [103] |
| Bind-n-Seq | Analysis of in vitro protein-DNA interactions using massively parallel sequencing | [250] |
| SELEX-seq/HT-SELEX | Systematic evolution of ligands by exponential enrichment, followed by high-throughput sequencing | [1, 82, 110] |
| EMSA-seq | Electrophoretic mobility shift assay followed by deep sequencing | [95] |
| HiTS-FLIP | High-throughput sequencing - fluorescent ligand interaction profiling | [108] |
| gcPBM | Genomic-context protein binding microarray | [2] |
These complex models typically perform better than traditional PWMs [2, 63, 70, 73, 75], providing important insights into the DNA recognition mechanisms used by different TFs. For example, a dinucleotide-based model [73] revealed that including the nonindependent contributions between two specific positions in the DNA binding models of Hnf4a was critical for accurately predicting the genomic regions bound by Hnf4a in vivo. Another recent study [2] revealed that contributions from di- and tri-nucleotides in the DNA regions flanking TFBSs can influence TF binding specificity. Importantly, though, the flanking di- and tri-nucleotides in these models did not reflect base readout by the TFs; instead, the effect of the higher-order sequence features was exerted through local three-dimensional DNA structure (i.e., DNA shape) [13].
Interactions between adjacent base pairs are dominated by base stacking [76] and, to a lesser degree, by inter-base pair hydrogen bonds in the major groove [77]. These physical interactions give rise to DNA shape [78, 79] and explain the interdependencies between adjacent positions in a TFBS [73] and other, more complex situations. DNA shape features can be derived on a genomic scale by using a sliding pentamer window to mine Monte Carlo predictions [78]. This approach was the basis for generating a motif database of structural features of TFBSs [79], as well as for multiple studies in which hundreds of thousands of DNA sequences were analyzed in terms of DNA shape features [1, 2, 30, 79, 80].
A small but important class of sequence-based motif discovery methods represents approaches to infer DNA binding affinities by fitting thermodynamic energy-based models to experimental data (Table 1A). Similar to probabilistic models, some energy-based models assume independent contributions among positions in the TFBS [81–83], whereas others incorporate nonindependent contributions [73]. Structure-based atomistic models of DNA binding specificity have also been developed [84–90]. However, these models are not yet widely used, likely because they require knowledge of the structure of the protein (or one of its homologs) when bound to the target DNA site. Such data are not as easily available as DNA sequence data. Without having to model the complete structure of the TF-DNA complex, structural information on DNA alone can be incorporated into DNA motif discovery models. Recently, probabilistic models incorporating DNA structure-derived features [2, 79, 91, 92] were shown to perform better than models based on DNA sequence information alone. Thus, genomic and structural information are beginning to be integrated into protein-DNA binding models that account for both base- and shape-readout mechanisms.
Binding assays for probing the DNA binding specificities of transcription factors
With the emergence of new, high-throughput technologies for measuring protein-DNA binding (Figure 4; Table 1B, C), it has become more feasible to create complex models of DNA binding specificity through machine learning. However, all experimental datasets contain noise and (potentially substantial) biases, and complex models will fit the noise and biases more easily than simple PWM models. Thus, it is not surprising that in some recent studies of algorithms for training DNA binding specificity models from high-throughput data [9, 93], the models that performed best on certain in vitro datasets did not always generalize well to independent in vivo data. As more accurate datasets emerge (e.g., from genomic-context protein-binding microarrays [gcPBMs] [2, 74]), it is likely that more TFs will be better described by complex models of DNA binding specificity [43].
The rich datasets provided by high-throughput technologies have revolutionized our ability to characterize protein-DNA binding specificity. For example, the comprehensive nature of universal protein-binding microarray (PBM) data [94], which include measurements of TF binding specificity to all possible 8-bp sequences, has facilitated the characterization of low-affinity TF-DNA binding sites, which are often not captured by simple DNA binding models [95, 96]. Such sites, which are under widespread evolutionary selection [97, 98], are critical for interpreting the spatial and temporal TF gradients that arise during development [99, 100]. High-throughput datasets have revealed that closely related TFs, even when they exhibit a high degree of similarity in their DNA binding domains (DBDs; up to 67% amino acid identity), can have distinct DNA binding profiles [7, 95, 101–105]. Moreover, different TF family members can prefer different core binding sites [7, 102, 106, 107] or flanking DNA sequences [2, 108]. Thus, both base- and shape-readout mechanisms might play roles in the differential DNA binding specificity of paralogous TFs.
Perhaps the most striking finding suggested by high-throughput protein-DNA binding technologies is the large number of proteins that can bind DNA using two or more distinct modes [47]. A small number of such proteins were previously identified through structural studies [32, 39, 109]; however, recent high-throughput data suggest that this phenomenon is more common than anticipated. Variable binding modes can be classified into different categories: 1) variable spacing, in which TFs bind DNA motifs composed of two half-sites separated by different numbers of bases [7, 104]; 2) multiple DBDs, in which TFs contain multiple independent DBDs that allow them to recognize different DNA elements [7]; 3) multimeric binding, which might be more common than previously thought and can even occur in the case of TFs known to bind DNA primarily as monomers [10, 110]; and 4) alternate structural conformations, in which TFs with a single DBD can bind different DNA motifs, enabled by distinct conformations of the DBD (e.g., mouse TF SREBF1) or domains outside the DBD (e.g., yeast TF Hac1) [9, 111]. Importantly, the multiple modes of DNA binding observed in high-throughput in vitro studies are also enriched in the genomic regions bound by TFs in vivo [10, 104], suggesting that the different mechanisms of binding are biologically relevant. Further studies of TFs with multiple modes of binding are needed to understand the precise biochemical and biophysical mechanisms that allow such TFs to interact with diverse binding sites.
Studying the specificity of individual TFs via high-throughput in vitro technologies cannot provide a full picture of how these proteins achieve their diverse regulatory roles in the cell. Transcriptional regulation often involves the assembly of multiprotein complexes, which can modulate the DNA binding specificities of individual TFs [1, 16]. A complete understanding of the determinants of binding specificity in gene regulation requires the integration of all factors that affect protein-DNA binding in the cell, including cooperating or competing TFs and the local chromatin state.
From in vitro to in vivo transcription factor-DNA interactions
Transferring our knowledge of the in vitro biochemical and biophysical principles of protein-DNA interactions to an in vivo context is not straightforward. In contrast to the relatively well-defined components of a typical in vitro biochemical experiment, the cellular nucleus contains hundreds of millions of DNA base pairs (in metazoan genomes), as well as RNA, histones and countless nonhistone proteins. The overall concentration of macromolecules in the nucleus is estimated to be between 100 and 400 mg/ml [112, 113]. Within this crowded nucleoplasm [114], TFs somehow bind specific DNA sites and regulate gene expression. In addition, although the genome contains numerous potential binding sites for each TF, only some of them are actually bound in vivo, and only a fraction of the bound sites are functional. Consequently, predicting and interpreting in vivo TF-DNA binding are not trivial endeavors, even when the intrinsic sequence preferences of TFs are well characterized in vitro.
Regulatory genomic sequences targeted by TFs are primarily found in noncoding intergenic or intronic DNA, with a few exceptions [115]. The amount of noncoding genomic DNA varies from organism to organism, with metazoan genomes containing relatively large amounts of noncoding DNA (e.g., ~97% of the human genome is noncoding vs. <30% of the Saccharomyces cerevisiae genome [116]; Figure 5A). Although pioneering studies in S. cerevisiae have provided a tremendous foundation for our understanding of TF biology, the noncoding regulatory landscape in this organism is easier to parse than for metazoan eukaryotes.
Figure 5. Distinct cis-regulatory structure of unicellular and metazoan model organisms.

(A) Percentages of coding and noncoding DNA in select genomes, adapted from [116].
(B) Typical regulatory structure of a Saccharomyces cerevisiae gene, with most regulatory DNA binding sites falling within a few hundred bases of the gene’s TSS.
(C) Typical regulatory structure of a human gene, with several clusters of regulatory DNA sites (enhancers) distal to the TSS.
For (B) and (C), green dashed lines represent activating regulatory inputs, and red dashed lines represent repressive inputs.
For S. cerevisiae, most regulatory DNA sequences for a given gene fall within a few hundred base pairs of its transcription start site (TSS) (Figure 5B) [117]. In metazoans, by contrast, regulatory sequences often fall tens of kilobases or even megabases from the TSS of the target gene [118–120]. These distal elements can be upstream or downstream of the target gene, and they regularly bypass intervening genes (Figure 5C). The combination of a large search space (i.e., noncoding sequence) and the distal location of many enhancers complicates the search for regulatory DNA sequences in metazoans.
Making sense of regulatory DNA is further complicated by a lack of straightforward sequence grammar. Unlike genic coding regions, which are easily interpreted from the triplet code, noncoding regulatory elements are difficult to decode. Regulatory TFBSs are often clustered, with binding sites from different TFs in close proximity to one another. A group of TFBSs that function together to direct gene expression are referred to as a cis-regulatory module (CRM) or enhancer. The combinatorial nature of these groupings gives enhancers the ability to integrate inputs from multiple TFs, to direct the spatial and temporal patterns of gene expression. Although enhancers typically contain clusters of TFBSs and other common features (e.g., dinucleotide repeat sequences [121]), the patterns associated with these features are not sufficiently strong to permit easy discrimination between enhancers and nonregulatory DNA. In addition, sequence information is often an insufficient predictor of TF binding because in vivo TF binding preferences are influenced by additional variables, including interaction with cofactors and chromatin accessibility (discussed below). Ultimately, enhancers are difficult to decode and require further experimental work for their identification and functional characterization.
Chromatin and transcription factor-DNA binding
In the past decade, we have seen a dramatic expansion of the use of genome-wide technologies for studying in vivo TF-DNA binding and transcriptional regulation. These technologies include genome-wide chromatin immunoprecipitation (ChIP-seq) and related approaches (Figure 4; Table 1B), gene expression profiling and newer screening methods for the high-throughput identification of DNA regions with enhancer activity [122–130]. Collectively, these tools of the genomics era have facilitated the annotation of genomic regulatory regions and have served as a platform for understanding TF-DNA interactions on a global scale, informing models of how TFs achieve regulatory specificity in vivo.
One surprising finding from early genome-wide ChIP studies was that TF binding is widespread, with thousands to tens of thousands of binding events for many TFs. These numbers did not fit with existing ideas of the regulatory network structure, in which TFs were generally expected to regulate a few hundred genes, at most [131–133]. Binding is not necessarily equivalent to regulation, and it is likely that only a small fraction of all binding events will have an important impact on gene expression (Figure 6) (discussed below) [134, 135]. However, if we ignore preconceived notions regarding the expected number of direct target genes for a TF and, instead, focus only on DNA sequence, the genome-wide binding numbers begin to make sense.
Figure 6. In vitro versus in vivo transcription factor-DNA interactions.

(A) Standard and high-throughput in vitro DNA binding assays provide a motif or model representing a TF’s DNA binding preferences.
(B) Genomic DNA sequences matching a TF’s in vitro-derived motif represent potential TFBSs.
(C) Potential in vivo binding sites determined from a TF’s in vitro-derived motif far outnumber the actual number of in vivo binding sites as measured by ChIP-seq. In general, <5% of potential binding sites are identified as bound in vivo. In addition, in vivo binding strength does not always correlate with motif strength, and not all in vivo binding sites contain the expected motif. Non-DNA variables, such as nucleosomes and cofactor interactions, explain part of the difference between predicted and actual binding.
(D) Not all in vivo binding events have a regulatory impact on gene expression. Productive, functional binding must be validated experimentally using standard reporter assays or other measures of cis-regulatory function. In this hypothetical example, only Regions W and Y drive gene expression that is responsive to the TF being tested.
Considering the information content of a typical 6-bp human TF motif, one would expect matches to a motif to occur approximately once every 4 kb, with hundreds of thousands of potential binding sites genome-wide [136]. Thus, based on information theory alone, TFs actually bind far fewer regions than expected (Figure 6), due in large part to the restrictive nature of chromatinized DNA.
Nuclear DNA is associated with nucleosomes, which consist of two copies each of the histone proteins H2A, H2B, H3 and H4, or their variants. Nucleosome assembly facilitates DNA packaging in the nucleus, but also has major regulatory roles [22]. Histones are subject to extensive posttranslational modifications (PTMs) [137, 138], which can regulate chromatin compaction and affect the recruitment of certain transcriptional regulators [139, 140]. With more than 100 possible histone PTMs and a tremendous possibility for combinatorial PTM interactions, the burgeoning field of epigenomics is rapidly defining genome-wide chromatin states (i.e., distinct combinations of histone modifications and other chromatin-associated factors at a given locus) across many cellular contexts [137]. Findings from the integration of chromatin state data with TF binding data suggest that many TFs have specific histone PTM preferences that are consistent across multiple cell types [141]. Still, it is often unclear whether a specific chromatin state is simply permissive to TF binding, actively directs TF binding or is a result of TF binding. Further mechanistic elucidation of the relationships between TFs and histone PTMs will likely influence our models of TF-DNA targeting.
Aside from the regulatory potential of histone PTMs, nucleosome-DNA interactions can provide a steric impediment to TF binding and increase TF-DNA dissociation rates [142]. Consistent with this concept, most of the TFBSs identified by the Encyclopedia of DNA Elements (ENCODE) consortium fall within highly accessible (i.e., nucleosome-depleted) DNA regions [143]. Furthermore, for several TFs, simple thermodynamic models based on TF levels, DNA motif information and DNA accessibility [23, 24, 133, 144, 145] can largely explain genome-wide binding patterns. Carefully designed studies have suggested that the accessibility of TFBSs can explain most genome-wide binding patterns. However, recent studies indicate that some binding to accessible DNA regions may be a crosslinking-mediated ChIP artifact (discussed below) [146, 147], and there are factors whose binding patterns are not driven by DNA accessibility [148].
DNA accessibility in vivo is commonly measured through DNase-seq, FAIRE-seq, or, more recently, ATAC-seq (Figure 4; Table 1B) [149–152]. DNase-seq is based on the differential DNase I sensitivity of nucleosome-associated and nucleosome-free DNA. DNase I selectively cleaves DNA that is not protected by association with nucleosomes; therefore, accessible DNA regions manifest as DNase I-hypersensitive sites. TF binding to DNA protects DNA from cleavage by DNase I. Consequently, footprints of TF-DNA binding can be identified within hypersensitive regions [151]. These properties of DNase-seq data were recently exploited to characterize DNA accessibility profiles around TFBSs during a program to differentiate mouse embryonic stem cells (ESCs) into pancreatic and intestinal endoderm [153]. The data were used to quantify the impact of a given TF on DNA accessibility patterns. Ultimately, TFs were broken down into three categories: pioneers, settlers and migrants.
Pioneer TFs (Figure 7A) are characterized by their ability to bind DNA target sites, even in inaccessible regions, and, subsequently, to promote DNA accessibility. Although pioneer TF activity had been described previously [154, 155], the above DNase-seq based study expanded the catalog of TFs with pioneer activity [153]. Interestingly, TFBSs for the pioneer TF Pu.1 can be differentiated from nontargeted Pu.1 motif matches, based on DNA sequence and shape characteristics that favor nucleosome assembly [29]. True Pu.1 target sequences are highly associated with nucleosomes in cell types where Pu.1 is not expressed. This result suggests that selective pressures have favored sequences that are competent for both pioneer TF binding and nucleosome occupancy. It also highlights the importance of the interplay between these two forces in pioneer TF function.
Figure 7. Transcription factor-DNA binding strategies.

(A) Pioneer TFs can bind inaccessible, nucleosome-associated DNA sites. Pioneer factors then create an open chromatin environment that is permissive for the binding of nonpioneer factors (settler and migrant TFs).
(B) Settler TFs bind to essentially all accessible copies of their DNA target sites.
(C) Migrant TFs only bind a subset of their accessible target DNA sites.
(D) High- and low-affinity binding are driven by a TF’s specific DNA recognition properties. Nonspecific binding is driven by the electrostatic attraction between negatively charged DNA and positively charged DNA binding domains. Nonconsensus binding is driven by the attraction of TFs to repeated homo-oligomeric tracts. Indirect binding, or tethering, is driven by the interaction of TFs with another DNA binding factor (in this schematic, TF’).
In contrast, settler TFs (Figure 7B) almost always bind sites matching their DNA binding motif if these sites fall within accessible DNA; however, they do not bind inaccessible DNA sites [153]. The least defined group, migrant TFs (Figure 7C), are similar to settler TFs, although more selective [153]. Migrants only bind a subset of their target sites, even in accessible DNA; therefore, their selectivity is likely driven by interaction with additional cofactors. Although settler and migrant TFs do not evict nucleosomes like pioneer factors, TFs lacking pioneer activity can facilitate the binding of unrelated TFs by competing with nucleosomes for DNA binding; this process is termed collaborative competition or nucleosome-mediated cooperativity [26, 27]. Taken together, these data support the idea that DNA accessibility substantially contributes to the DNA binding selectivity of most TFs, with pioneer TFs being an important exception.
Functional and nonfunctional transcription factor-DNA binding
Regardless of whether one considers the widespread genomic binding of TFs to be expected or unexpected, most researchers acknowledge that a reasonable fraction of TF binding events are neutral or nonfunctional (i.e., they do not have a direct impact on target gene expression levels). ChIP-seq assays do not provide any information about regulatory function, just protein-DNA coassociation. In addition, similar to all biochemical purification assays, ChIP-seq assays must cope with false positives and false negatives (see [156] for what is necessary to confirm functional binding). Although functional binding events are certainly present within the thousands of genome-wide binding events for many TFs, neutral binding is likely to be common [135].
Thus, a major question in the TF genomics field regards how to identify functional TF binding events within the thousands of genome-wide TF-DNA interactions. What features distinguish functional from neutral binding? Can we use these distinctions to learn about TF-DNA binding strategies? The data suggest that functional binding can be identified on the basis of several distinguishing features, although these features will be influenced by the TF under study and the experimental design.
Developmentally dynamic or clustered TF peaks have been identified as enriched for functional binding events [157–162]. Functional analyses of TF targets in the Drosophila embryo suggested that the strongest ChIP peaks represent functional binding, whereas lower signal peaks do not [135]. Consistent with this model, strong ChIP peaks are more likely to be conserved across species [161, 163, 164]. However, ChIP peak strength is a less reliable indicator of function when monitoring binding in more heterogeneous tissues, likely because functional binding events only occur in a subset of cells within a tissue [162].
However, caution is needed when interpreting functionality or binding affinity from ChIP-seq signal strength. ChIP assays are usually based on the average signal across millions of cells. Thus, a medium peak might actually be a high-affinity TFBS that is only bound in 50% of cells, whereas a strong peak might be a medium-affinity TFBS that is bound in every cell. That is not to say that peak strength does not correlate with affinity or regulatory function for some TFs (as there clearly can be a strong correlation [135]); however, not all data follow this pattern. The experimental design must be considered when interpreting and building models from in vivo genome-wide TF binding data.
The implications of the many seemingly nonfunctional binding events identified by ChIP-seq should also be considered. As a point of clarification, discussions of ChIP-seq data often refer to regions of strong ChIP enrichment as TFBSs, which can be misleading. Immunopurification assays, especially those aided by crosslinking, can be rife with false positives. Indeed, recent carefully controlled ChIP-seq studies in yeast have indicated that many regions of the genome, especially those associated with highly expressed genes, are hyper-ChIPable. This situation makes it difficult to discern between functional and artifactual ChIP signals [146, 147]. The resulting high potential for artifact-based peaks in ChIP must be considered when interpreting ChIP-based studies.
The fact that potentially misleading ChIP signals are associated with highly expressed genes is interesting because highly occupied target (HOT) regions also exhibit this feature [165, 166]. HOT regions are often targeted by 10 or more unrelated TFs. They generally fall in nucleosome-depleted regions upstream of highly expressed genes. Although HOT regions can act as regulatory enhancers, many of the binding events within HOT regions are neutral (i.e., have no impact on gene expression patterns) and may result from nonspecific or indirect DNA binding [167, 168]. Interestingly, HOT region binding disappears when a modified, crosslinking-free ChIP protocol is used, suggesting that such binding could be an experimental artifact for certain TFs [169].
Nonfunctional ChIP signals may potentially be due to the capturing of transient nonspecific or indirect binding events in highly accessible DNA. Single-cell, single-molecule imaging studies of the TFs Sox2 and Oct4 demonstrated that nonspecific interactions with chromatin are central to the in vivo search for functional binding sites [170]. At physiological TF concentrations, at least for Sox2 and Oct4, these nonspecific interactions were sampled enough times to provide a measurable ChIP signal in a population of cells [170]. Nonregulatory protein-DNA interactions can be sequence-dependent, occurring via binding to spurious weak matches to the TF target sequence, as a result of the low-information motifs targeted by metazoan TFs (Figure 7D) [136]. Sequence-independent nonspecific interactions are also possible, through interactions with other chromatin-associated proteins or through the general electrostatic attraction between negatively charged DNA and positively charged DBDs (Figure 7D) [171, 172].
A recent theoretical model suggested that TFs are more attracted to repeated homo-oligomeric poly(dA:dT) and poly(dC:dG) tracts; the longer the segment, the greater the attraction (Figure 7D) [173–175]. This variation of nonspecific binding, termed nonconsensus binding, has also been observed in vitro [176]. It has the potential to shape nonfunctional and functional TF-DNA interactions [173]. Thus, although nonfunctional TF-DNA associations do not provide information about the regulatory targets of a TF, they may provide clues to the mechanisms by which TFs find their functional binding sites across the genome. To recognize their functional sites during this searching process, TFs are influenced by additional variables, including direct and indirect interactions with other TFs.
Transcription factor interactions at genomic regulatory regions
A clear theme from both classical enhancer-bashing studies and newer genomics data is that enhancers must integrate multiple TF inputs to direct precise patterns of gene expression. How, exactly, are multiple TFs assembled at enhancers? The answer to this question is likely to fall somewhere on a spectrum represented by two extremes: the enhanceosome model and the billboard model.
The enhanceosome model (Figure 8A) is based on pioneering work with the interferon-β (IFN-β) enhancer [177]. This model proposes that enhancer activity is dependent on the cooperative assembly of a set of TFs at the enhancer. Only once the cooperative unit is assembled on an enhancer will cofactor recruitment cause changes in gene expression. The cooperative assembly of an enhanceosome is dependent on protein-protein interactions and a highly constrained pattern of TF-DNA binding sites (or binding-site grammar). Enhancesome assembly does not tolerate shifts in the quality, spacing or orientation of the binding site, which can disrupt protein-protein interactions and cooperativity [59, 178].
Figure 8. Models of transcription factor assembly on enhancer DNA.

(A) Left: The enhanceosome model is characterized by cooperative TF binding and highly constrained binding site positioning. Right: Minor changes in enhancer sequence (i.e., inversion in this case, but insertions, deletions, mutations, etc., also apply) can lead to collapse of TF assembly and enhancer function.
(B) Left: The billboard model is characterized by highly flexible binding-site grammars. Although all TFs are important for enhancer function, TF binding and enhancer function are not affected by significant changes in binding site positioning or orientation.
The IFN-β enhanceosome probably represents an extreme example, as few enhancers are found under similarly stringent constraints. However, additional examples of organizationally constrained enhancers do exist [179–184]. Spatial constraints on select paired TF-TF coassociations and binding-site combinations are found in genome-wide ChIP data [148, 185, 186]. Interactions between TFs can lead to cooperative DNA binding, although this binding does not approach the extreme multifactorial cooperativity required for enhanceosome assembly. True enhanceosome and enhanceosome-like regulatory DNA elements are not common. It may be that they are only necessary under unique regulatory conditions, such as for the amplification of signals at enhancers regulated by low-abundance TFs [181] or to prevent unwanted TF synergy and ectopic enhancer activity [182].
The billboard model (Figure 8B), also known as the information display model [187, 188], hypothesizes that although individual TFBSs are essential for enhancer activity, binding-site grammar is very flexible. That is, the positioning of binding sites within an enhancer is not subject to strict spacing or orientation rules because, even though the TFs collaborate to regulate enhancer output, they do not target the enhancer as a cooperative unit. The TFs at a billboard enhancer work together in a combinatorial fashion to direct precise patterns of gene expression, but they do not depend on highly cooperative DNA binding to target the enhancer in an all-or-nothing manner. For example, the loss of a cell-specific repressive input to an enhancer will lead to ectopic target gene expression in that cell type, but will not cause the complete collapse of enhancer function. Billboard-like combinatorial binding is not uncommon in genome-wide ChIP data [189, 190]. Indeed, findings from the high-throughput dissection of mammalian enhancers suggest that the regulatory architecture of many enhancers is quite flexible [128, 191].
Another flexible enhancer architecture model—the TF collective model—was recently proposed on the basis of the genome-wide binding patterns of a panel of TFs that regulate heart development in Drosophila [192, 193]. Cardiac TFs were observed to bind their target regions in an all-or-nothing fashion, with binding driven by the collective action of many TFs, similar to cooperative binding. Similar all-or-nothing patterns of genome-wide binding have been seen in TFs that regulate mammalian hematopoiesis [194]. However, despite the similarity to cooperative binding, the binding-site grammar at targeted enhancers is flexible in the TF collective model [193].
Ultimately, the mechanisms by which multiple TFs assemble on enhancers probably fall on a continuum between the enhanceosome and billboard extremes. Distinct TF binding properties are better suited to different regulatory strategies. Noncooperative TF-DNA interactions are well suited for regulating graded gene expression, which is often necessary for homeostatic responses. Cooperative interactions are more appropriate for switch-like, on/off expression, which is often necessary in developmental cell-fate decisions [195–197]. The strategies employed by TFs and enhancers are subject to multiple evolutionary pressures. In the end, no single model can accurately describe all of the rules of transcriptional regulation.
Cellular context and transcription factor binding specificity
In multicellular organisms, gene regulatory networks are plastic, with spatial, temporal and environmental dynamics impacting gene expression patterns. Many TFs are reiteratively used in multiple cellular contexts, often directing the expression of distinct sets of genes. Characterizing the influence of cellular context on genome-wide TF-DNA binding is central to the understanding of binding specificity. Accordingly, there has recently been a dramatic increase in the number of ChIP-seq studies monitoring metazoan TF-DNA binding across multiple cell or tissue types [3, 162, 198–205] or across multiple environmental or signaling contexts [129, 206–208]. Although context-independent binding (i.e., binding events shared across multiple conditions) is common [162, 199, 204], context-specific binding is substantial in all cases, suggesting that regulatory specificity is often achieved at the level of TF-DNA binding. Importantly, DNA accessibility is dynamic, with important differences in accessibility across cell types or developmental stages within a cell type [143, 209–211]. Thus, the chromatin environment is modified by cellular context, likely through the pioneer TFs expressed in a given context, which, in turn, can impact the binding patterns of nonpioneer TFs.
Interestingly, context-independent and -dependent binding events for a given TF often represent distinct binding strategies. For example, estrogen receptor (ER) binding sites that are shared between breast and endometrial cancer cell lines are associated with high-affinity estrogen response elements (EREs), are not dependent on DNA accessibility, and tend not to colocalize with interacting TFs [204]. By contrast, cell type-specific ER binding sites are not associated with high-affinity EREs, fall within DNA that was accessible before ER activation, and colocalize with interacting TFs. Whether the colocalized TFs in the cell type-specific ER binding sites directly impact ER-DNA binding preferences, or whether they simply generate a permissive chromatin environment, remains to be tested. Nevertheless, it is clear that these binding sites represent a regulatory strategy that is distinct from that used at the cell type-independent ER binding sites.
Cell- and tissue-specific genomics data have clarified that precise patterns of gene expression result from collaboration between broadly expressed TFs and tissue-, cell-, or developmental stage-specific TFs [3, 129, 202, 212, 213]. This mechanism for refining the regulatory activity of a broadly expressed TF is not new to developmental biology. Indeed, the mechanism was evident from the findings of enhancer-bashing experiments that were performed before genomics experiments were common [214].
An interesting example of this refinement comes from two TF modules that direct the differentiation of mouse ESCs into spinal or cranial motor neurons (Figure 9A) [215]. The homeodomain TF Isl1 is an essential component of both modules. Homeodomain TFs Lhx3 and Phox2a determine whether a spinal or a cranial motor neuron, respectively, is generated. Inducible expression of these two ESC programming modules revealed that Isl1 binding is strongly influenced by context (i.e., the presence of Lhx3 or Phox2a is required for distinct Isl1-Lhx3 or Isl1-Phox2a composite binding sites, respectively). In this elegant experimental model, the programming TFs were induced concomitantly by using a polycistronic construct, in an identical cellular context (ESCs). Consequently, the observed binding differences were not due to basal differences in chromatin structure or expressed cofactors. The data suggested that Isl1 forms a complex with Lhx3 or Phox2a; the complex is then recruited to context-specific enhancers with distinct binding-site grammars to direct cranial or spinal motor neuron fate. Thus, Isl1 is necessary for both motor neuron fates, and its genome-wide DNA targeting is refined by interactions with additional cell type-specific TFs.
Figure 9. Cellular context and transcription factor-DNA binding.

(A) Isl1 is an essential factor in two separate embryonic stem cell (ESC) reprogramming modules, which generate spinal (left) and cranial (right) motor neurons, respectively. The genome-wide DNA targeting of Isl1 is markedly influenced by interaction with spinal- and cranial-specific TFs (Lhx3 and Phox2, respectively). DNA at different loci is represented in blue, red or black. DNA accessibility profiles of the reprogrammed stem cells resemble brain, not ESC, accessibility profiles, suggesting that the reprogramming TFs can induce DNA accessibility. However, this possibility remains to be functionally tested.
(B) Left column: GATA “switch” sites at the GATA2 locus remain continually bound by GATA factors through multiple stages of erythroid differentiation. GATA2 acts as an autoregulatory activator at these enhancers, and GATA1 is either repressive (red line) or neutral (gray dashed line). Right column: At the GATA1 locus, DNA methylation and, presumably, chromatin compaction prevent GATA2 from binding a “switch” enhancer in hematopoietic stem cells. As the epigenetic environment becomes permissive, GATA2 binds this enhancer and activates GATA1 expression. GATA1 then displaces GATA2 and acts as an autoregulatory activator at this enhancer.
Binding that is unaffected by a cellular context can be important and may represent the association of a TF with its canonical targets [199, 204]. For example, a variation in context-independent binding is central to the regulatory roles of the GATA transcription factors Gata1 and Gata2 (Figure 9B). These zinc finger proteins bind the DNA motif WGATAA (W = A or T) and are the primarily regulators of hematopoietic stem cell (HSC) maintenance and differentiation [216]. These factors were the subject of several recent ChIP-seq experiments covering multiple branches of hematopoietic lineage commitment. The studies identified substantial cell- and stage-specific GATA factor-DNA binding [213, 217, 218] and highlighted the key role that DNA-binding and non DNA-binding cofactors play in modifying GATA-DNA binding selectivity [194, 219–221].
GATA1 and GATA2 also act at binding sites that remain bound by GATA factors when HSCs differentiate into erythrocytes. In the GATA switch process, GATA2 (which maintains the HSC state) is displaced by GATA1 (which promotes erythroid commitment) [216]. This process is best characterized at autoregulatory enhancers targeting the GATA1 and GATA2 genes (Figure 9B), where the switch can have a neutral regulatory effect or can change the direction of an enhancer’s activity (e.g., activator to repressor) [216, 222–224]. Importantly, several ChIP-seq studies have demonstrated substantial overlap in the regions targeted by GATA1 and GATA2 at different stages, suggesting that the GATA switch might be part of a global mechanism during erythroid commitment [194, 218, 221, 225–227]. Thus, the potentially widespread GATA switch mechanism is dependent on highly similar GATA factors targeting the same DNA sequence at multiple stages of erythroid development.
Findings from the recent glut of context-specific ChIP-seq experiments demonstrate that the context-specific regulatory activity of a TF is often adjusted at the level of TF-DNA binding. A TF may bind and regulate the output of an enhancer in one cell, whereas it does not bind the same enhancer in another cell. Differential binding could be regulated via DNA accessibility or cofactor interactions; however, another mechanism is also prevalent. In many cases, a TF (or highly similar TFs, in the case of the GATA switch) targets the same enhancer across many cellular contexts. In these instances, changes in enhancer activity are likely to be regulated by changes in the coactivators or corepressors that are recruited by the bound TF, or by the action of collaborating TFs that target the same enhancer.
Selective pressures on regulatory DNA have resulted in finely tuned systems for increasing/decreasing the transcription of a given gene, although there clearly are many routes towards regulating enhancer output. It seems that the only common thread in the world of TF-DNA interactions and transcriptional regulation is that no single model is sufficient to explain all of the mechanisms used to achieve regulatory specificity.
Concluding remarks
TFs select their genomic target sites through multiple mechanisms at various levels. Some of these mechanisms are well understood; for instance, the determinants of base and shape readout are known because of the many high-resolution structures that are currently available. Models of TF-DNA binding specificity using PWMs or interdependencies between nucleotide positions in a binding site can quantitatively describe in vitro binding. Higher-order determinants of TF-DNA binding in vivo include cofactors, TF cooperativity and chromatin accessibility. However, an accurate model that integrates all of the known contributions to TF-DNA binding specificity is not yet available, because the interactions between the various factors of in vivo binding are highly complex, dynamic and dependent on many unknown parameters.
Thus, a simple recognition code does not exist between the amino acids of a TF’s DBD and the nucleotides in the TFBS. It is possible that some complex code, comprising rules from each of the different layers, contributes to TF-DNA binding; however, determining the precise rules of TF binding to the genome will require further high-quality structural and high-throughput binding data. Questions that remain to be determined include whether such a multi-rule system will ever be condensed into a single code and, if so, whether such a potential code represents the overarching principles of protein-DNA recognition or is highly specific for TF families and the cellular conditions of their activity.
Box 1. Outstanding questions.
Will it be possible to condense the different rules that determine TF-DNA binding specificity (e.g., base and shape readout, cofactors, cooperativity and chromatin accessibility) into a simple code?
Would such a code describe overarching principles that are valid for protein-DNA interactions in general, or would it be highly specific to a TF or a TF family?
If a single code cannot be defined, can a set of rules that describes binding specificity at multiple levels be integrated into a complex, but unified, model?
What kind of experimental data will be required to derive more accurate binding specificity models?
What kind of computational methods need to be developed to derive accurate models from high-throughput genome-wide binding data?
To what extent can higher-quality in vitro TF-DNA binding data be used to derive more accurate binding specificity models and explain in vivo TF-DNA binding?
Beyond using cofactors to alter DNA binding preferences, how much impact do variables, such as PTMs, have on TF-DNA binding specificity?
Considering the diverse, context-specific roles of many TFs, can a single motif ever capture a TF’s in vivo DNA binding preferences?
Within the same cell type, how important is cell-to-cell variation in TF-DNA interactions?
Will single-cell genomics reinforce or rewrite current models of in vivo TF-DNA binding?
Beyond DNA accessibility, are there any instances in which the chromatin state (e.g., presence of histone modifications) acts as an epigenetic specificity determinant, or is this state primarily an effect of TF binding?
Highlights.
TFs recognize their genomic target sites by using mechanisms at multiple levels
Models of DNA sequence and shape can capture the in vitro TF binding specificity
Cofactors, cooperativity, chromatin and other factors affect the in vivo TF binding
No simple code combines all of the various determinants of TF binding specificity
Acknowledgments
The authors thank the reviewers and the editor for their very constructive comments and suggestions. This work was supported by the National Institutes of Health (grants R01GM106056, U01GM103804 and, in part, R01HG003008 to R.R.). R.G. and R.R. are Alfred P. Sloan Research Fellows.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Slattery M, et al. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell. 2011;147:1270–1282. doi: 10.1016/j.cell.2011.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gordân R, et al. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 2013;3:1093–1104. doi: 10.1016/j.celrep.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yanez-Cuna JO, et al. Uncovering cis-regulatory sequence requirements for context-specific transcription factor binding. Genome Res. 2012;22:2018–2030. doi: 10.1101/gr.132811.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- 6.Bussemaker HJ, et al. Predictive modeling of genome-wide mRNA expression: from modules to molecules. Annu Rev Biophys Biomol Struct. 2007;36:329–347. doi: 10.1146/annurev.biophys.36.040306.132725. [DOI] [PubMed] [Google Scholar]
- 7.Badis G, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–1723. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stormo GD, Zhao Y. Determining the specificity of protein-DNA interactions. Nat Rev Genet. 2010;11:751–760. doi: 10.1038/nrg2845. [DOI] [PubMed] [Google Scholar]
- 9.Weirauch MT, et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat Biotechnol. 2013;31:126–134. doi: 10.1038/nbt.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jolma A, et al. DNA-binding specificities of human transcription factors. Cell. 2013;152:327–339. doi: 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 11.White MA, et al. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc Natl Acad Sci U S A. 2013;110:11952–11957. doi: 10.1073/pnas.1307449110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meijsing SH, et al. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohs R, et al. The role of DNA shape in protein-DNA recognition. Nature. 2009;461:1248–1253. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim S, et al. Probing allostery through DNA. Science. 2013;339:816–819. doi: 10.1126/science.1229223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watson LC, et al. The glucocorticoid receptor dimer interface allosterically transmits sequence-specific DNA signals. Nat Struct Mol Biol. 2013;20:876–883. doi: 10.1038/nsmb.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siggers T, et al. Non-DNA-binding cofactors enhance DNA-binding specificity of a transcriptional regulatory complex. Mol Syst Biol. 2011;7:555. doi: 10.1038/msb.2011.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panne D. The enhanceosome. Curr Opin Struct Biol. 2008;18:236–242. doi: 10.1016/j.sbi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Wasson T, Hartemink AJ. An ensemble model of competitive multi-factor binding of the genome. Genome Res. 2009;19:2101–2112. doi: 10.1101/gr.093450.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitayner M, et al. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat Struct Mol Biol. 2010;17:423–429. doi: 10.1038/nsmb.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, et al. Whole-genome comparison of Leu3 binding in vitro and in vivo reveals the importance of nucleosome occupancy in target site selection. Genome Res. 2006;16:1517–1528. doi: 10.1101/gr.5655606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaplan N, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai L, Morozov AV. Gene regulation by nucleosome positioning. Trends Genet. 2010;26:476–483. doi: 10.1016/j.tig.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan T, et al. Quantitative models of the mechanisms that control genome-wide patterns of transcription factor binding during early Drosophila development. PLoS Genet. 2011;7:e1001290. doi: 10.1371/journal.pgen.1001290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pique-Regi R, et al. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 2011;21:447–455. doi: 10.1101/gr.112623.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller JA, Widom J. Collaborative competition mechanism for gene activation in vivo. Mol Cell Biol. 2003;23:1623–1632. doi: 10.1128/MCB.23.5.1623-1632.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mirny LA. Nucleosome-mediated cooperativity between transcription factors. Proc Natl Acad Sci U S A. 2010;107:22534–22539. doi: 10.1073/pnas.0913805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glatt S, et al. Recognizing and remodeling the nucleosome. Curr Opin Struct Biol. 2011;21:335–341. doi: 10.1016/j.sbi.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 29.Barozzi I, et al. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol Cell. 2014;54:844–857. doi: 10.1016/j.molcel.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lazarovici A, et al. Probing DNA shape and methylation state on a genomic scale with DNase I. Proc Natl Acad Sci U S A. 2013;110:6376–6381. doi: 10.1073/pnas.1216822110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agius P, et al. High resolution models of transcription factor-DNA affinities improve in vitro and in vivo binding predictions. PLoS Comput Biol. 2010;6:e1000916. doi: 10.1371/journal.pcbi.1000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garvie CW, Wolberger C. Recognition of specific DNA sequences. Mol Cell. 2001;8:937–946. doi: 10.1016/s1097-2765(01)00392-6. [DOI] [PubMed] [Google Scholar]
- 33.von Hippel PH. From “simple” DNA-protein interactions to the macromolecular machines of gene expression. Annu Rev Biophys Biomol Struct. 2007;36:79–105. doi: 10.1146/annurev.biophys.34.040204.144521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong M, Marmorstein R. Structural Basis for Sequence-Specific DNA Recognition by Transcription Factors and their Complexes. In: Rice PA, Correll CC, editors. Protein-Nucleic Acid Interactions: Structural Biology. Royal Society of Chemistry; 2008. pp. 47–65. [Google Scholar]
- 35.Lawson CL, Berman HM. Indirect Readout of DNA Sequence by Proteins. In: Rice PA, Correll CC, editors. Protein-Nucleic Acid Interactions: Structural Biology. Royal Society of Chemistry; 2008. pp. 66–90. [Google Scholar]
- 36.Gorman J, Greene EC. Visualizing one-dimensional diffusion of proteins along DNA. Nat Struct Mol Biol. 2008;15:768–774. doi: 10.1038/nsmb.1441. [DOI] [PubMed] [Google Scholar]
- 37.Mann RS, et al. Hox specificity unique roles for cofactors and collaborators. Curr Top Dev Biol. 2009;88:63–101. doi: 10.1016/S0070-2153(09)88003-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan Y, et al. Mechanisms of transcription factor selectivity. Trends Genet. 2010;26:75–83. doi: 10.1016/j.tig.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rohs R, et al. Origins of specificity in protein-DNA recognition. Annu Rev Biochem. 2010;79:233–269. doi: 10.1146/annurev-biochem-060408-091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parker SC, Tullius TD. DNA shape, genetic codes, and evolution. Curr Opin Struct Biol. 2011;21:342–347. doi: 10.1016/j.sbi.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lelli KM, et al. Disentangling the many layers of eukaryotic transcriptional regulation. Annu Rev Genet. 2012;46:43–68. doi: 10.1146/annurev-genet-110711-155437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zakrzewska K, Lavery R. Towards a molecular view of transcriptional control. Curr Opin Struct Biol. 2012;22:160–167. doi: 10.1016/j.sbi.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Stormo GD. Quantitative Biology. Springer; 2013. Modeling the specificity of protein-DNA interactions; pp. 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ostuni R, Natoli G. Lineages, cell types and functional states: a genomic view. Curr Opin Cell Biol. 2013;25:759–764. doi: 10.1016/j.ceb.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 45.Weingarten-Gabbay S, Segal E. The grammar of transcriptional regulation. Hum Genet. 2014;133:701–711. doi: 10.1007/s00439-013-1413-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shlyueva D, et al. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15:272–286. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- 47.Siggers T, Gordan R. Protein-DNA binding: complexities and multi-protein codes. Nucleic Acids Res. 2014;42:2099–2111. doi: 10.1093/nar/gkt1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levo M, Segal E. In pursuit of design principles of regulatory sequences. Nat Rev Genet. 2014;15:453–468. doi: 10.1038/nrg3684. [DOI] [PubMed] [Google Scholar]
- 49.Rohs R, et al. Nuance in the double-helix and its role in protein-DNA recognition. Curr Opin Struct Biol. 2009;19:171–177. doi: 10.1016/j.sbi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berman HM, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stella S, et al. The shape of the DNA minor groove directs binding by the DNA-bending protein Fis. Genes Dev. 2010;24:814–826. doi: 10.1101/gad.1900610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hancock SP, et al. Control of DNA minor groove width and Fis protein binding by the purine 2-amino group. Nucleic Acids Res. 2013;41:6750–6760. doi: 10.1093/nar/gkt357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Y, et al. Structure of p53 binding to the BAX response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res. 2013;41:8368–8376. doi: 10.1093/nar/gkt584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang YP, et al. Mechanism of origin DNA recognition and assembly of an initiator-helicase complex by SV40 large tumor antigen. Cell Rep. 2013;3:1117–1127. doi: 10.1016/j.celrep.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dantas Machado AC, et al. Proteopedia: 3D visualization and annotation of transcription factor-DNA readout modes. Biochem Mol Biol Educ. 2012;40:400–401. doi: 10.1002/bmb.20650. [DOI] [PubMed] [Google Scholar]
- 56.Chen Y, et al. DNA binding by GATA transcription factor suggests mechanisms of DNA looping and long-range gene regulation. Cell Rep. 2012;2:1197–1206. doi: 10.1016/j.celrep.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang X, et al. Conformations of p53 response elements in solution deduced using site-directed spin labeling and Monte Carlo sampling. Nucleic Acids Res. 2014;42:2789–2797. doi: 10.1093/nar/gkt1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rohs R, et al. Structural and energetic origins of sequence-specific DNA bending: Monte Carlo simulations of papillomavirus E2-DNA binding sites. Structure. 2005;13:1499–1509. doi: 10.1016/j.str.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 59.Panne D, et al. An atomic model of the interferon-beta enhanceosome. Cell. 2007;129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Internat Conf Intell Syst Mol Biol (ISMB) 1994;2:28–36. [PubMed] [Google Scholar]
- 61.Roth FP, et al. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- 62.Pevzner PA, Sze SH. Combinatorial approaches to finding subtle signals in DNA sequences. Proc Internat Conf Intell Syst Mol Biol (ISMB) 2000;8:269–278. [PubMed] [Google Scholar]
- 63.Barash Y, et al. Modeling Dependencies in Protein-DNA Binding Sites. Proc Annu Internat Conf Res Comput Mol Biol (RECOMB) 2003 [Google Scholar]
- 64.Galas DJ, Schmitz A. DNAse footprinting: a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res. 1978;5:3157–3170. doi: 10.1093/nar/5.9.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garner MM, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 1981;9:3047–3060. doi: 10.1093/nar/9.13.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tompa M, et al. Assessing computational tools for the discovery of transcription factor binding sites. Nature Biotechnol. 2005;23:137–144. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- 67.Sandve GK, Drablos F. A survey of motif discovery methods in an integrated framework. Biol direct. 2006;1:11. doi: 10.1186/1745-6150-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Workman CT, et al. enoLOGOS: a versatile web tool for energy normalized sequence logos. Nucleic Acids Res. 2005;33:W389–392. doi: 10.1093/nar/gki439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Man TK, Stormo GD. Non-independence of Mnt repressor-operator interaction determined by a new quantitative multiple fluorescence relative affinity (QuMFRA) assay. Nucleic Acids Res. 2001;29:2471–2478. doi: 10.1093/nar/29.12.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bulyk ML, et al. Nucleotides of transcription factor binding sites exert interdependent effects on the binding affinities of transcription factors. Nucleic Acids Res. 2002;30:1255–1261. doi: 10.1093/nar/30.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tomovic A, Oakeley EJ. Position dependencies in transcription factor binding sites. Bioinformatics. 2007;23:933–941. doi: 10.1093/bioinformatics/btm055. [DOI] [PubMed] [Google Scholar]
- 72.Sharon E, et al. A feature-based approach to modeling protein-DNA interactions. PLoS Comput Biol. 2008;4:e1000154. doi: 10.1371/journal.pcbi.1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao Y, et al. Improved models for transcription factor binding site identification using nonindependent interactions. Genetics. 2012;191:781–790. doi: 10.1534/genetics.112.138685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mordelet F, et al. Stability selection for regression-based models of transcription factor-DNA binding specificity. Bioinformatics. 2013;29:i117–125. doi: 10.1093/bioinformatics/btt221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou Q, Liu JS. Extracting sequence features to predict protein-DNA interactions: a comparative study. Nucleic Acids Res. 2008;36:4137–4148. doi: 10.1093/nar/gkn361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Olson WK, et al. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci U S A. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Crothers DM, Shakked Z. DNA bending by adenine-thymine tracts. In: Neidle S, editor. Oxford Handbook of Nucleic Acid Structures. Oxford University Press; 1999. pp. 455–470. [Google Scholar]
- 78.Zhou T, et al. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 2013;41:W56–62. doi: 10.1093/nar/gkt437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang L, et al. TFBSshape: a motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2014;42:D148–155. doi: 10.1093/nar/gkt1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dror I, et al. Covariation between homeodomain transcription factors and the shape of their DNA binding sites. Nucleic Acids Res. 2014;42:430–441. doi: 10.1093/nar/gkt862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roider HG, et al. Predicting transcription factor affinities to DNA from a biophysical model. Bioinformatics. 2007;23:134–141. doi: 10.1093/bioinformatics/btl565. [DOI] [PubMed] [Google Scholar]
- 82.Zhao Y, et al. Inferring binding energies from selected binding sites. PLoS Comput Biol. 2009;5:e1000590. doi: 10.1371/journal.pcbi.1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun W, et al. TherMos: Estimating protein-DNA binding energies from in vivo binding profiles. Nucleic Acids Res. 2013;41:5555–5568. doi: 10.1093/nar/gkt250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mandel-Gutfreund Y, Margalit H. Quantitative parameters for amino acid-base interaction: implications for prediction of protein-DNA binding sites. Nucleic Acids Res. 1998;26:2306–2312. doi: 10.1093/nar/26.10.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Havranek JJ, et al. A simple physical model for the prediction and design of protein-DNA interactions. J Mol Biol. 2004;344:59–70. doi: 10.1016/j.jmb.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 86.Morozov AV, et al. Protein-DNA binding specificity predictions with structural models. Nucleic Acids Res. 2005;33:5781–5798. doi: 10.1093/nar/gki875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kaplan T, et al. Ab initio prediction of transcription factor targets using structural knowledge. PLoS Comput Biol. 2005;1:e1. doi: 10.1371/journal.pcbi.0010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Siggers TW, et al. Structural alignment of protein--DNA interfaces: insights into the determinants of binding specificity. J Mol Biol. 2005;345:1027–1045. doi: 10.1016/j.jmb.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 89.Siggers TW, Honig B. Structure-based prediction of C2H2 zinc-finger binding specificity: sensitivity to docking geometry. Nucleic Acids Res. 2007;35:1085–1097. doi: 10.1093/nar/gkl1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu LA, Bradley P. Atomistic modeling of protein-DNA interaction specificity: progress and applications. Curr Opin Struct Biol. 2012;22:397–405. doi: 10.1016/j.sbi.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maienschein-Cline M, et al. Improved predictions of transcription factor binding sites using physicochemical features of DNA. Nucleic Acids Res. 2012;40:e175. doi: 10.1093/nar/gks771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hooghe B, et al. A flexible integrative approach based on random forest improves prediction of transcription factor binding sites. Nucleic Acids Res. 2012;40:e106. doi: 10.1093/nar/gks283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kahara J, Lahdesmaki H. Evaluating a linear k-mer model for protein-DNA interactions using high-throughput SELEX data. BMC bioinformatics. 2013;14(Suppl 10):S2. doi: 10.1186/1471-2105-14-S10-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Berger MF, et al. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nature Biotechnol. 2006;24:1429–1435. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wong D, et al. Extensive characterization of NF-kappaB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. 2011;12:R70. doi: 10.1186/gb-2011-12-7-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Siggers T, et al. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-kappaB family DNA binding. Nature Immunol. 2012;13:95–102. doi: 10.1038/ni.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tanay A. Extensive low-affinity transcriptional interactions in the yeast genome. Genome Res. 2006;16:962–972. doi: 10.1101/gr.5113606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jaeger SA, et al. Conservation and regulatory associations of a wide affinity range of mouse transcription factor binding sites. Genomics. 2010;95:185–195. doi: 10.1016/j.ygeno.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rowan S, et al. Precise temporal control of the eye regulatory gene Pax6 via enhancer-binding site affinity. Genes Dev. 2010;24:980–985. doi: 10.1101/gad.1890410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.White MA, et al. A model of spatially restricted transcription in opposing gradients of activators and repressors. Mol Syst Biol. 2012;8:614. doi: 10.1038/msb.2012.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Maerkl SJ, Quake SR. A systems approach to measuring the binding energy landscapes of transcription factors. Science. 2007;315:233–237. doi: 10.1126/science.1131007. [DOI] [PubMed] [Google Scholar]
- 102.Noyes MB, et al. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell. 2008;133:1277–1289. doi: 10.1016/j.cell.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bonham AJ, et al. Tracking transcription factor complexes on DNA using total internal reflectance fluorescence protein binding microarrays. Nucleic acids research. 2009;37:e94. doi: 10.1093/nar/gkp424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gordân R, et al. Curated collection of yeast transcription factor DNA binding specificity data reveals novel structural and gene regulatory insights. Genome Biol. 2011;12:R125. doi: 10.1186/gb-2011-12-12-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nakagawa S, et al. DNA-binding specificity changes in the evolution of forkhead transcription factors. Proc Natl Acad Sci U S A. 2013;110:12349–12354. doi: 10.1073/pnas.1310430110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Berger MF, et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell. 2008;133:1266–1276. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chu SW, et al. Exploring the DNA-recognition potential of homeodomains. Genome Res. 2012;22:1889–1898. doi: 10.1101/gr.139014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nutiu R, et al. Direct measurement of DNA affinity landscapes on a high-throughput sequencing instrument. Nature Biotechnol. 2011;29:659–664. doi: 10.1038/nbt.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim J, Struhl K. Determinants of half-site spacing preferences that distinguish AP-1 and ATF/CREB bZIP domains. Nucleic Acids Res. 1995;23:2531–2537. doi: 10.1093/nar/23.13.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jolma A, et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010;20:861–873. doi: 10.1101/gr.100552.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fordyce PM, et al. Basic leucine zipper transcription factor Hac1 binds DNA in two distinct modes as revealed by microfluidic analyses. Proc Natl Acad Sci U S A. 2012;109:E3084–3093. doi: 10.1073/pnas.1212457109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hancock R. The crowded nucleus. Internat Rev Cell Mol Biol. 2014;307:15–26. doi: 10.1016/B978-0-12-800046-5.00002-3. [DOI] [PubMed] [Google Scholar]
- 113.Nolin F, et al. Changes to cellular water and element content induced by nucleolar stress: investigation by a cryo-correlative nano-imaging approach. Cell Mol Life Sci. 2013;70:2383–2394. doi: 10.1007/s00018-013-1267-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Goodsell DS. Miniseries: Illustrating the machinery of life: Eukaryotic cell panorama. Biochem Mol Biol Educ. 2011;39:91–101. doi: 10.1002/bmb.20494. [DOI] [PubMed] [Google Scholar]
- 115.Stergachis AB, et al. Exonic transcription factor binding directs codon choice and affects protein evolution. Science. 2013;342:1367–1372. doi: 10.1126/science.1243490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Alexander RP, et al. Annotating non-coding regions of the genome. Nature reviews Genetics. 2010;11:559–571. doi: 10.1038/nrg2814. [DOI] [PubMed] [Google Scholar]
- 117.Lin Z, et al. The spatial distribution of cis regulatory elements in yeast promoters and its implications for transcriptional regulation. BMC Genomics. 2010;11:581. doi: 10.1186/1471-2164-11-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.El-Kasti MM, et al. A novel long-range enhancer regulates postnatal expression of Zeb2: implications for Mowat-Wilson syndrome phenotypes. Hum Mol Genet. 2012;21:5429–5442. doi: 10.1093/hmg/dds389. [DOI] [PubMed] [Google Scholar]
- 119.Hosoya-Ohmura S, et al. An NK and T cell enhancer lies 280 kilobase pairs 3′ to the gata3 structural gene. Mol Cell Biol. 2011;31:1894–1904. doi: 10.1128/MCB.05065-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li L, et al. A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood. 2013;122:902–911. doi: 10.1182/blood-2012-08-447839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yanez-Cuna JO, et al. Dissection of thousands of cell type-specific enhancers identifies dinucleotide repeat motifs as general enhancer features. Genome Res. 2014 doi: 10.1101/gr.169243.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Slattery M, et al. Interpreting the regulatory genome: the genomics of transcription factor function in Drosophila melanogaster. Brief Funct Genomics. 2012;11:336–346. doi: 10.1093/bfgp/els034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Arnold CD, et al. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013;339:1074–1077. doi: 10.1126/science.1232542. [DOI] [PubMed] [Google Scholar]
- 124.Gisselbrecht SS, et al. Highly parallel assays of tissue-specific enhancers in whole Drosophila embryos. Nature Methods. 2013;10:774–780. doi: 10.1038/nmeth.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jory A, et al. A survey of 6,300 genomic fragments for cis-regulatory activity in the imaginal discs of Drosophila melanogaster. Cell Rep. 2012;2:1014–1024. doi: 10.1016/j.celrep.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Manning L, et al. A resource for manipulating gene expression and analyzing cis-regulatory modules in the Drosophila CNS. Cell Rep. 2012;2:1002–1013. doi: 10.1016/j.celrep.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jenett A, et al. A GAL4-driver line resource for Drosophila neurobiology. Cell Rep. 2012;2:991–1001. doi: 10.1016/j.celrep.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Patwardhan RP, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nature Biotechnol. 2012;30:265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shlyueva D, et al. Hormone-responsive enhancer-activity maps reveal predictive motifs, indirect repression, and targeting of closed chromatin. Mol Cell. 2014;54:180–192. doi: 10.1016/j.molcel.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 130.Kvon EZ, et al. Genome-scale functional characterization of Drosophila developmental enhancers in vivo. Nature. 2014 doi: 10.1038/nature13395. [DOI] [PubMed] [Google Scholar]
- 131.MacQuarrie KL, et al. Genome-wide transcription factor binding: beyond direct target regulation. Trends Genet. 2011;27:141–148. doi: 10.1016/j.tig.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Farnham PJ. Insights from genomic profiling of transcription factors. Nature Rev Genet. 2009;10:605–616. doi: 10.1038/nrg2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Biggin MD. Animal transcription networks as highly connected, quantitative continua. Dev Cell. 2011;21:611–626. doi: 10.1016/j.devcel.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 134.Li XY, et al. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol. 2008;6:e27. doi: 10.1371/journal.pbio.0060027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fisher WW, et al. DNA regions bound at low occupancy by transcription factors do not drive patterned reporter gene expression in Drosophila. Proc Natl Acad Sci U S A. 2012;109:21330–21335. doi: 10.1073/pnas.1209589110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wunderlich Z, Mirny LA. Different gene regulation strategies revealed by analysis of binding motifs. Trends Genet. 2009;25:434–440. doi: 10.1016/j.tig.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rivera CM, Ren B. Mapping human epigenomes. Cell. 2013;155:39–55. doi: 10.1016/j.cell.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tan M, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophysica Acta. 2014 doi: 10.1016/j.bbagrm.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rando OJ. Combinatorial complexity in chromatin structure and function: revisiting the histone code. Curr Opin Genet Dev. 2012;22:148–155. doi: 10.1016/j.gde.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ernst J, Kellis M. Interplay between chromatin state, regulator binding, and regulatory motifs in six human cell types. Genome Res. 2013;23:1142–1154. doi: 10.1101/gr.144840.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Luo Y, et al. Nucleosomes accelerate transcription factor dissociation. Nucleic Acids Res. 2014;42:3017–3027. doi: 10.1093/nar/gkt1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Li XY, et al. The role of chromatin accessibility in directing the widespread, overlapping patterns of Drosophila transcription factor binding. Genome Biol. 2011;12:R34. doi: 10.1186/gb-2011-12-4-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Simicevic J, et al. Absolute quantification of transcription factors during cellular differentiation using multiplexed targeted proteomics. Nature Methods. 2013;10:570–576. doi: 10.1038/nmeth.2441. [DOI] [PubMed] [Google Scholar]
- 146.Park D, et al. Widespread misinterpretable ChIP-seq bias in yeast. PloS ONE. 2013;8:e83506. doi: 10.1371/journal.pone.0083506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Teytelman L, et al. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc Natl Acad Sci U S A. 2013;110:18602–18607. doi: 10.1073/pnas.1316064110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cheng Q, et al. Computational identification of diverse mechanisms underlying transcription factor-DNA occupancy. PLoS Genet. 2013;9:e1003571. doi: 10.1371/journal.pgen.1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Giresi PG, et al. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17:877–885. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Song L, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21:1757–1767. doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hesselberth JR, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nature Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Buenrostro JD, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sherwood RI, et al. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nature Biotechnol. 2014;32:171–178. doi: 10.1038/nbt.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Magnani L, et al. Pioneer factors: directing transcriptional regulators within the chromatin environment. Trends Genet. 2011;27:465–474. doi: 10.1016/j.tig.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 155.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Carey MF, et al. Confirming the functional importance of a protein-DNA interaction. Cold Spring Harbor Protoc. 2012;2012:733–757. doi: 10.1101/pdb.top070060. [DOI] [PubMed] [Google Scholar]
- 157.Webber JL, et al. The relationship between long-range chromatin occupancy and polymerization of the Drosophila ETS family transcriptional repressor Yan. Genetics. 2013;193:633–649. doi: 10.1534/genetics.112.146647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hnisz D, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wilczynski B, Furlong EE. Dynamic CRM occupancy reflects a temporal map of developmental progression. Mol Syst Biol. 2010;6:383. doi: 10.1038/msb.2010.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.He Q, et al. High conservation of transcription factor binding and evidence for combinatorial regulation across six Drosophila species. Nature Genet. 2011;43:414–420. doi: 10.1038/ng.808. [DOI] [PubMed] [Google Scholar]
- 162.Slattery M, et al. Divergent transcriptional regulatory logic at the intersection of tissue growth and developmental patterning. PLoS Genet. 2013;9:e1003753. doi: 10.1371/journal.pgen.1003753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Paris M, et al. Extensive divergence of transcription factor binding in Drosophila embryos with highly conserved gene expression. PLoS Genet. 2013;9:e1003748. doi: 10.1371/journal.pgen.1003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bardet AF, et al. A computational pipeline for comparative ChIP-seq analyses. Nature Protoc. 2012;7:45–61. doi: 10.1038/nprot.2011.420. [DOI] [PubMed] [Google Scholar]
- 165.Negre N, et al. A cis-regulatory map of the Drosophila genome. Nature. 2011;471:527–531. doi: 10.1038/nature09990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yip KY, et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012;13:R48. doi: 10.1186/gb-2012-13-9-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kvon EZ, et al. HOT regions function as patterned developmental enhancers and have a distinct cis-regulatory signature. Genes Dev. 2012;26:908–913. doi: 10.1101/gad.188052.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Slattery M, et al. Diverse patterns of genomic targeting by transcriptional regulators in Drosophila melanogaster. Genome Res. 2014;24:1224–1235. doi: 10.1101/gr.168807.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kasinathan S, et al. High-resolution mapping of transcription factor binding sites on native chromatin. Nature Methods. 2014;11:203–209. doi: 10.1038/nmeth.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chen J, et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell. 2014;156:1274–1285. doi: 10.1016/j.cell.2014.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.von Hippel PH. Biochemistry. Completing the view of transcriptional regulation. Science. 2004;305:350–352. doi: 10.1126/science.1101270. [DOI] [PubMed] [Google Scholar]
- 172.Harris RC, et al. Opposites attract: shape and electrostatic complementarity in protein-DNA complexes. In: Schlick T, editor. Innovations in Biomolecular Modeling and Simulations. Royal Society of Chemistry; 2012. pp. 53–80. [Google Scholar]
- 173.Afek A, Lukatsky DB. Positive and negative design for nonconsensus protein-DNA binding affinity in the vicinity of functional binding sites. Biophys J. 2013;105:1653–1660. doi: 10.1016/j.bpj.2013.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Afek A, Lukatsky DB. Genome-wide organization of eukaryotic preinitiation complex is influenced by nonconsensus protein-DNA binding. Biophys J. 2013;104:1107–1115. doi: 10.1016/j.bpj.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sela I, Lukatsky DB. DNA sequence correlations shape nonspecific transcription factor-DNA binding affinity. Biophys J. 2011;101:160–166. doi: 10.1016/j.bpj.2011.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Orenstein Y, Shamir R. A comparative analysis of transcription factor binding models learned from PBM, HT-SELEX and ChIP data. Nucleic Acids Res. 2014;42:e63. doi: 10.1093/nar/gku117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Thanos D, Maniatis T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell. 1995;83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- 178.Escalante CR, et al. Structure of IRF-3 bound to the PRDIII-I regulatory element of the human interferon-beta enhancer. Mol Cell. 2007;26:703–716. doi: 10.1016/j.molcel.2007.04.022. [DOI] [PubMed] [Google Scholar]
- 179.Erives A, Levine M. Coordinate enhancers share common organizational features in the Drosophila genome. Proc Natl Acad Sci U S A. 2004;101:3851–3856. doi: 10.1073/pnas.0400611101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Crocker J, et al. Evolution acts on enhancer organization to fine-tune gradient threshold readouts. PLoS Biol. 2008;6:e263. doi: 10.1371/journal.pbio.0060263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Papatsenko D, Levine M. A rationale for the enhanceosome and other evolutionarily constrained enhancers. Curr Biol. 2007;17:R955–957. doi: 10.1016/j.cub.2007.09.035. [DOI] [PubMed] [Google Scholar]
- 182.Liu F, Posakony JW. Role of architecture in the function and specificity of two Notch-regulated transcriptional enhancer modules. PLoS Genet. 2012;8:e1002796. doi: 10.1371/journal.pgen.1002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Swanson CI, et al. Structural rules and complex regulatory circuitry constrain expression of a Notch- and EGFR-regulated eye enhancer. Dev Cell. 2010;18:359–370. doi: 10.1016/j.devcel.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Swanson CI, et al. Rapid evolutionary rewiring of a structurally constrained eye enhancer. Curr Biol. 2011;21:1186–1196. doi: 10.1016/j.cub.2011.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kazemian M, et al. Widespread evidence of cooperative DNA binding by transcription factors in Drosophila development. Nucleic Acids Res. 2013;41:8237–8252. doi: 10.1093/nar/gkt598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sorge S, et al. The cis-regulatory code of Hox function in Drosophila. The EMBO J. 2012;31:3323–3333. doi: 10.1038/emboj.2012.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Arnosti DN, Kulkarni MM. Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards? J Cell Biochem. 2005;94:890–898. doi: 10.1002/jcb.20352. [DOI] [PubMed] [Google Scholar]
- 188.Kulkarni MM, Arnosti DN. Information display by transcriptional enhancers. Development. 2003;130:6569–6575. doi: 10.1242/dev.00890. [DOI] [PubMed] [Google Scholar]
- 189.Jiang P, Singh M. CCAT: Combinatorial Code Analysis Tool for transcriptional regulation. Nucleic Acids Res. 2014;42:2833–2847. doi: 10.1093/nar/gkt1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Menoret D, et al. Genome-wide analyses of Shavenbaby target genes reveals distinct features of enhancer organization. Genome Biol. 2013;14:R86. doi: 10.1186/gb-2013-14-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Smith RP, et al. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nature Genet. 2013;45:1021–1028. doi: 10.1038/ng.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Erceg J, et al. Subtle changes in motif positioning cause tissue–specific effects on robustness of an enhancer’s activity. PLoS Genet. 2014;10:e1004060. doi: 10.1371/journal.pgen.1004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Junion G, et al. A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell. 2012;148:473–486. doi: 10.1016/j.cell.2012.01.030. [DOI] [PubMed] [Google Scholar]
- 194.Tijssen MR, et al. Genome-wide analysis of simultaneous GATA1/2, RUNX1, FLI1, and SCL binding in megakaryocytes identifies hematopoietic regulators. Dev Cell. 2011;20:597–609. doi: 10.1016/j.devcel.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Giorgetti L, et al. Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded transcriptional responses to environmental inputs. Mol Cell. 2010;37:418–428. doi: 10.1016/j.molcel.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 196.Lorberbaum DS, Barolo S. Gene regulation: when analog beats digital. Curr Biol. 2013;23:R1054–1056. doi: 10.1016/j.cub.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 197.Stewart-Ornstein J, et al. Msn2 coordinates a stoichiometric gene expression program. Curr Biol. 2013;23:2336–2345. doi: 10.1016/j.cub.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhang JA, et al. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell. 2012;149:467–482. doi: 10.1016/j.cell.2012.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kudron M, et al. Tissue-specific direct targets of Caenorhabditis elegans Rb/E2F dictate distinct somatic and germline programs. Genome Biol. 2013;14:R5. doi: 10.1186/gb-2013-14-1-r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Frietze S, et al. Cell type-specific binding patterns reveal that TCF7L2 can be tethered to the genome by association with GATA3. Genome Biol. 2012;13:R52. doi: 10.1186/gb-2012-13-9-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lodato MA, et al. SOX2 co-occupies distal enhancer elements with distinct POU factors in ESCs and NPCs to specify cell state. PLoS Genet. 2013;9:e1003288. doi: 10.1371/journal.pgen.1003288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Meireles-Filho AC, et al. cis-regulatory requirements for tissue-specific programs of the circadian clock. Curr Biol. 2014;24:1–10. doi: 10.1016/j.cub.2013.11.017. [DOI] [PubMed] [Google Scholar]
- 203.Gertz J, et al. Genistein and bisphenol A exposure cause estrogen receptor 1 to bind thousands of sites in a cell type-specific manner. Genome Res. 2012;22:2153–2162. doi: 10.1101/gr.135681.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gertz J, et al. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol Cell. 2013;52:25–36. doi: 10.1016/j.molcel.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zinzen RP, et al. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. 2009;462:65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- 206.Guertin MJ, Lis JT. Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet. 2010;6:e1001114. doi: 10.1371/journal.pgen.1001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.He HH, et al. Nucleosome dynamics define transcriptional enhancers. Nature Genet. 2010;42:343–347. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.John S, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nature Genet. 2011;43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Stergachis AB, et al. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell. 2013;154:888–903. doi: 10.1016/j.cell.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Thomas S, et al. Dynamic reprogramming of chromatin accessibility during Drosophila embryo development. Genome Biol. 2011;12:R43. doi: 10.1186/gb-2011-12-5-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gerstein MB, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Xu Z, et al. Impacts of the ubiquitous factor Zelda on Bicoid-dependent DNA binding and transcription in Drosophila. Genes Dev. 2014;28:608–621. doi: 10.1101/gad.234534.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Xu J, et al. Combinatorial assembly of developmental stage-specific enhancers controls gene expression programs during human erythropoiesis. Dev Cell. 2012;23:796–811. doi: 10.1016/j.devcel.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mann RS, Carroll SB. Molecular mechanisms of selector gene function and evolution. Curr Opin Genet Dev. 2002;12:592–600. doi: 10.1016/s0959-437x(02)00344-1. [DOI] [PubMed] [Google Scholar]
- 215.Mazzoni EO, et al. Synergistic binding of transcription factors to cell-specific enhancers programs motor neuron identity. Nat Neurosci. 2013;16:1219–1227. doi: 10.1038/nn.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bresnick EH, et al. Master regulatory GATA transcription factors: mechanistic principles and emerging links to hematologic malignancies. Nucleic Acids Res. 2012;40:5819–5831. doi: 10.1093/nar/gks281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Linnemann AK, et al. Genetic framework for GATA factor function in vascular biology. Proc Natl Acad Sci U S A. 2011;108:13641–13646. doi: 10.1073/pnas.1108440108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Dore LC, et al. Chromatin occupancy analysis reveals genome-wide GATA factor switching during hematopoiesis. Blood. 2012;119:3724–3733. doi: 10.1182/blood-2011-09-380634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Yu M, et al. Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol Cell. 2009;36:682–695. doi: 10.1016/j.molcel.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Chlon TM, et al. Cofactor-mediated restriction of GATA-1 chromatin occupancy coordinates lineage-specific gene expression. Mol Cell. 2012;47:608–621. doi: 10.1016/j.molcel.2012.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wilson NK, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7:532–544. doi: 10.1016/j.stem.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 222.Kaneko H, et al. GATA factor switching during erythroid differentiation. Curr Opin Hematol. 2010;17:163–168. doi: 10.1097/MOH.0b013e32833800b8. [DOI] [PubMed] [Google Scholar]
- 223.Snow JW, et al. Context-dependent function of “GATA switch” sites in vivo. Blood. 2011;117:4769–4772. doi: 10.1182/blood-2010-10-313031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Takai J, et al. The Gata1 5′ region harbors distinct cis-regulatory modules that direct gene activation in erythroid cells and gene inactivation in HSCs. Blood. 2013;122:3450–3460. doi: 10.1182/blood-2013-01-476911. [DOI] [PubMed] [Google Scholar]
- 225.Fujiwara T, et al. Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol Cell. 2009;36:667–681. doi: 10.1016/j.molcel.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wu W, et al. Dynamics of the epigenetic landscape during erythroid differentiation after GATA1 restoration. Genome Res. 2011;21:1659–1671. doi: 10.1101/gr.125088.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Suzuki M, et al. GATA factor switching from GATA2 to GATA1 contributes to erythroid differentiation. Genes to Cells: Mol Cell Mechan. 2013;18:921–933. doi: 10.1111/gtc.12086. [DOI] [PubMed] [Google Scholar]
- 228.Kitayner M, et al. Structural basis of DNA recognition by p53 tetramers. Mol Cell. 2006;22:741–753. doi: 10.1016/j.molcel.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 229.Davey CA, et al. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 230.Siddharthan R. Dinucleotide weight matrices for predicting transcription factor binding sites: generalizing the position weight matrix. PloS ONE. 2010;5:e9722. doi: 10.1371/journal.pone.0009722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Mathelier A, Wasserman WW. The next generation of transcription factor binding site prediction. PLoS Comput Biol. 2013;9:e1003214. doi: 10.1371/journal.pcbi.1003214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Grau J, et al. A general approach for discriminative de novo motif discovery from high-throughput data. Nucleic Acids Res. 2013;41:e197. doi: 10.1093/nar/gkt831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Annala M, et al. A linear model for transcription factor binding affinity prediction in protein binding microarrays. PloS ONE. 2011;6:e20059. doi: 10.1371/journal.pone.0020059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ben-Gal I, et al. Identification of transcription factor binding sites with variable-order Bayesian networks. Bioinformatics. 2005;21:2657–2666. doi: 10.1093/bioinformatics/bti410. [DOI] [PubMed] [Google Scholar]
- 235.Stormo GD, et al. Quantitative analysis of the relationship between nucleotide sequence and functional activity. Nucleic Acids Res. 1986;14:6661–6679. doi: 10.1093/nar/14.16.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Djordjevic M, et al. A biophysical approach to transcription factor binding site discovery. Genome Res. 2003;13:2381–2390. doi: 10.1101/gr.1271603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Foat BC, et al. Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Bioinformatics. 2006;22:e141–149. doi: 10.1093/bioinformatics/btl223. [DOI] [PubMed] [Google Scholar]
- 238.Narlikar L, et al. A nucleosome-guided map of transcription factor binding sites in yeast. PLoS Compu Biol. 2007;3:e215. doi: 10.1371/journal.pcbi.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Arvey A, et al. Sequence and chromatin determinants of cell-type-specific transcription factor binding. Genome Res. 2012;22:1723–1734. doi: 10.1101/gr.127712.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ren B, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 241.Johnson DS, et al. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 242.Rhee HS, Pugh BF. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011;147:1408–1419. doi: 10.1016/j.cell.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Greil F, et al. DamID: mapping of in vivo protein-genome interactions using tethered DNA adenine methyltransferase. Meth Enzymol. 2006;410:342–359. doi: 10.1016/S0076-6879(06)10016-6. [DOI] [PubMed] [Google Scholar]
- 244.Boyle AP, et al. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Meng X, et al. Counter-selectable marker for bacterial-based interaction trap systems. BioTechniques. 2006;40:179–184. doi: 10.2144/000112049. [DOI] [PubMed] [Google Scholar]
- 246.Berger MF, Bulyk ML. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nature Protoc. 2009;4:393–411. doi: 10.1038/nprot.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Warren CL, et al. Defining the sequence-recognition profile of DNA-binding molecules. Proc Natl Acad Sci U S A. 2006;103:867–872. doi: 10.1073/pnas.0509843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Fordyce PM, et al. De novo identification and biophysical characterization of transcription-factor binding sites with microfluidic affinity analysis. Nature Biotechnol. 2010;28:970–975. doi: 10.1038/nbt.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Tantin D, et al. High-throughput biochemical analysis of in vivo location data reveals novel distinct classes of POU5F1(Oct4)/DNA complexes. Genome Res. 2008;18:631–639. doi: 10.1101/gr.072942.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Zykovich A, et al. Bind-n-Seq: high-throughput analysis of in vitro protein-DNA interactions using massively parallel sequencing. Nucleic Acids Res. 2009;37:e151. doi: 10.1093/nar/gkp802. [DOI] [PMC free article] [PubMed] [Google Scholar]