

Abstract
The behavior of the Lewis-acid adducts of two copper-nitrene [Cu(NR)]+ complexes in nitrene-transfer and H atom abstraction reactions have been demonstrated to depend on the nature of the nitrene substituents. Two-state reactivity, in which a singlet ground state and a nearby triplet excited-state both contribute, provides a useful model for interpreting reactivity trends of the two compounds.
Copper-nitrene intermediates have attracted much interest in organometallic chemistry because they are implicated as key intermediates in the copper catalyzed alkane hydroxylation and alkene epoxidation reactions1–9. To engineer further improved catalysts, there is a substantial impetus to characterize fully the active species of existing catalysts, including the relevant higher oxidation states, intermediates and transition states so as to establish the mechanism(s) by which they affect C-N bond formation reactions. However, despite considerable interest, no copper nitrene intermediates have been isolated to allow for clear mechanistic studies9,10. In the absence of isolated examples of the proposed active intermediate, the mechanisms of copper-nitrene mediated aziridination and amination reactions have been elucidated by high-level quantum chemical calculations.1–3,7,8 In these calculations two alternative, limiting, electronic valence-bond structures have been characterized, namely, (closed-shell singlet) [CuIII(NR)]+ or (open-shell triplet or singlet) [CuII(NR•)]+ structures. While concerted aziridination reactions have been proposed to be favored on the closed-shell [CuIII(NR)]+ singlet surface,8 amination reactions were suggested to proceed via a stepwise H-atom abstraction/radical rebound (HAT/RR) pathway on the [CuII(NR•)]+ triplet surface.1 Moreover, in cases where the singlet and triplet states were very close in energy, a mechanistic picture involving both the singlet and triplet pathways for the aziridination7 and amination1 reactions has also been proposed.
What is required to test the theoretically predicted reactivity patterns is more direct experimental evidence that systematically shows the effect of singlet/triplet splitting energy on the reactivity of a copper nitrene species. Very recently, our group reported the spectroscopic characterization of a Lewis acid adduct of a cationic copper-tosylnitrene [CuII-(NTs•)Sc(OTf)3] species (1; Ts = tosyl; Scheme 1), the electronic structure of which could be best described in terms of a Cu(II) ion antiferromagnetically coupled to a nitrene radical thereby stabilizing an open-shell singlet ground state11. Reactivity studies revealed that this species aminates sp3 C–H bonds in substrates such as toluene and even cyclohexane in modest yields (21–35%). In this communication we now report the Lewis acid stabilization of a related copper nitrene species [CuII-(NMes•)Sc(OTf)3] (2; Mes=mesityl; Scheme 1), where the electron withdrawing heteroatom Ts nitrene substituent of 1 is replaced by the electron donating Mes group in 2. A comparative reactivity study between 1 and 2 shows that a two-state reactivity (TSR) model12,13 applies for the two compounds, in which a singlet ground state and a nearby triplet excited-state both contribute to the reactivity.
Scheme 1.
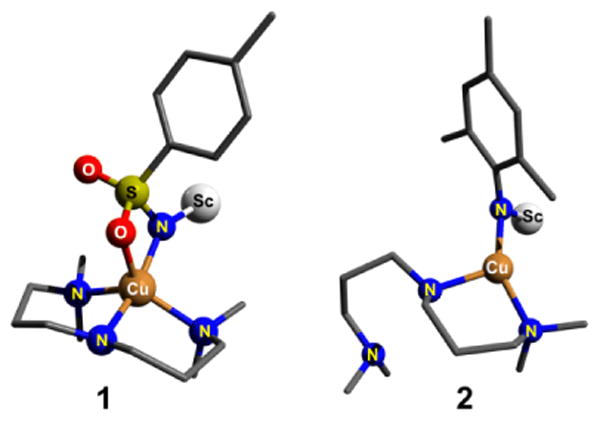
DFT optimized structures of 1 and 2 in their ground singlet states. Triflates bound to scandium and the hydrogen atoms are not shown for clarity.
Complex 2 is synthesized following the same procedure we reported previously for 1;11 the only difference is that we have now used an organic azide as a nitrene source in contrast to iminoiodane for the synthesis of 1. Thus the reaction of the previously synthesized [Cu(L1)](BF4)14 complex (L1 = 3,3′–iminobis(N,N–dimethylpropylamine)) in CH2Cl2 with one equivalent of mesityl azide at −90 °C in presence of 1.5 equivalent of scandium triflate for two hours generates the purple complex 2 with absorption maxima λmax(εmax) centered at 560 nm (2000 Lmol-1cm-1) and 670 nm (1227 Lmol-1cm-1) (Figure 1).15 Total consumption of the azide in the reaction was confirmed by the disappearance of the ν(N3) stretch at 2123 cm-1 in the IR spectrum of the resultant solution of 2 (Figure S1). Notably, the UV-Vis spectral changes associated with the formation of 2 are similar to that previously observed during the formation of 1;11 the corresponding bands for 1 are at 530 nm (3500 Lmol-1cm-1) and 750 nm (580 Lmol-1cm-1), respectively (Figure 1). 1H–NMR resonances of 2 are spread over a chemical shift range of 0 to +10 ppm, thereby, demonstrating a singlet ground state (S=0) similar to 1; the signals are, however, sharper compared to 1, which may point to a lesser contribution of the paramagnetic excited S=1 state in 2 (Figure S2). Notably, the 1H-NMR splitting pattern of 2 is similar to [Cu(L1)](BF4), which may also point to a similar geometry in the two cases.
Figure 1.
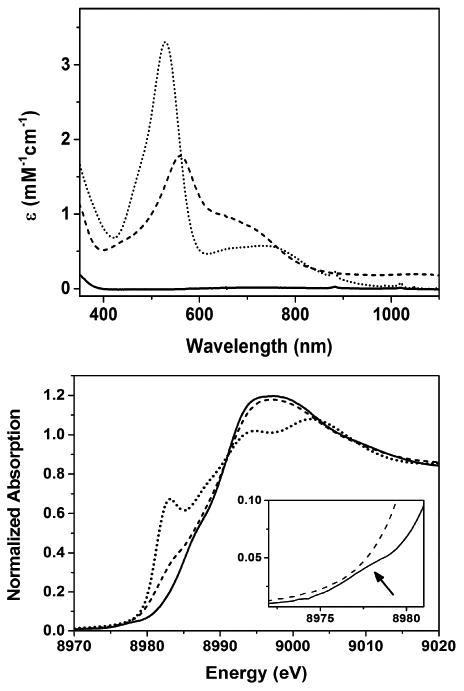
Top: Absorption Spectra of [Cu(L1)](BF4) (solid trace), 1 (dotted trace), and 2 (dashed trace) in CH2Cl2 at -90 °C; Bottom: Normalized Cu k-edge spectra for [Cu(L1)](BF4) (dotted line), 1 (solid line), and 2 (dashed line). The inset depicts an expansion of the pre-edge region; the pre-edge of 1 is depicted by the arrow
2 performed a two–electron reduction process (Figure S3) with a one electron reductant like decamethylferrocene (Me10Fc) at -90 °C with the corresponding formation of CuI (resultant solution is EPR silent) and decamethylferrocenium cation (Me10Fc+; 180 % yield); this confirmed that 2 is two oxidation levels (formal +3 oxidation state of copper) above [Cu(L1)](BF4) and its initial yield is at least 90%.
A comparison of the Cu K-edge X-ray absorption spectrum (XAS) of 2 with that of 1 and [Cu(L1)](BF4) is shown in Figure 2. Although the change in edge-shape and energies upon oxidation of [Cu(L1)](BF4) to 2 is similar to the earlier published result for 1, some interesting differences in the spectra of 1 and 2 are apparent. First of all, complex 1 exhibits a weak pre-edge at 8978 eV that is completely absent in 2. Moreover, in the spectrum of 2 a shoulder along the rising edge at 8981.5 eV, which is typically associated with a 1s-to-4p transition16–18, is present that is absent in 1. This may indicate a three coordinate trigonal planar (TP) geometry of copper in 216,19, in contrast to the previous report11 of a distorted square pyramidal (SP) geometry in 1. However, the lack of pre-edge in 2 removes the one sure way of assigning the physical oxidation state of copper (CuII vs CuIII); edge energies are known to be an ambiguous predictor for identifying whether a given complex is CuII or CuIII 16.
Complex 2 is very stable at -90 °C (< 5% decay in 2 hours) and could be further tested as oxidant for hydrogen atom transfer (HAT) and nitrene transfer (NT) reactions. This provided us an opportunity to perform a direct reactivity comparison of the two copper-nitrene complexes, 1 and 2, bearing a common supporting ligand (L1), but different nitrene substituents (Ts in 1 and Mes in 2) in HAT and NT reactions carried out under identical conditions. Rate constants for nitrene-transfer to PPh3 and thioanisole or C–H activation of 1,4-cyclohexadiene (CHD) and xanthene in CH2Cl2 at −90 °C were obtained from the pseudo-first order fit of the decay of the absorption band at 560 nm (Figures S4-S5). Analysis of the reaction mixture for CHD and PPh3 reactions show the formation of benzene and N-(mesityl)-iminotriphenylphosphorane products, respectively, in near-quantitative yields; the resultant solution is EPR silent, which may indicate the formation of Cu(I). Second order rate constants (k2; Table 1) for 2 were determined from the dependence of the first-order rate constants on substrate concentrations and then compared with the reported values11 for 1. A contrasting reactivity pattern is apparent (Table 1), in which complex 2 is a better NT oxidant, while complex 1 is a better HAT agent.
Table 1.
Second order rate constants of 1 and 2 determined for the different substrates by kinetic studies using UV-Vis spectroscopy.
| 1 k2 (-90°C; Lmol-1s-1) |
2 k2 (-90°C; Lmol-1s-1) |
|
|---|---|---|
| HAT | ||
| Xanthene | 0.3135 | 0.084 |
| CHD | 0.1554 | 0.030 |
| Toluene | 0.0051 | No reaction |
| NT | ||
| PPh3 | 0.196 | 2.89 |
| Thioanisole | No reaction | 0.5 |
DFT calculations (Figures S6-S9; Tables S1-S5) were carried out to understand the effect of the nitrene substituent on the reactivity of the [L1Cu(NR)Sc(OTf)3]+ (R= Ts for 1 and Mes for 2) complexes. Broken symmetry (BS) solutions20,21 were obtained for both 1 and 2. For 2 the calculated ground state is an open-shell [CuII(NMes•)]+ singlet with the excited triplet state lying 14.2 kcal/mol higher in energy (Table S1). For complex 1 the calculated ground state is also an open-shell singlet [CuII(NTs•)]+ state; the excited BS triplet state (Table S1) was, however, found to be only 2.5 kcal/mol higher in energy. The smaller singlet-triplet gap of 1 is consistent with a recent DFT study on Ni-nitrene complexes22–24, which predicted that singlet ground states are favored when aryl/alkyl groups are on the nitrene, while the triplet is favored with electron withdrawing heteroatom groups. The optimized geometry of 2 in the experimentally observed singlet ground state reveals a three coordinate TP geometry (Scheme 1; Figure S8; Table S4) fully consistent with the Cu K-edge XAS study; the ligand L1 acts as a bidentate ligand with one of the terminal tertiary amine nitrogen atoms remaining out of the coordination sphere of Cu. For 1 (Scheme 1; Figure S6; Table S2), in contrast, a SP geometry at copper is predicted for the BS singlet state, with a k2–N,O binding mode of the tosylnitrene ligand, consistent with the results of the previous calculations.8,11,25 Notably, the calculated Cu–N distance (Table S1) in 1 of 1.95 Å is significantly longer than the calculated distance for 2 of 1.84 Å, in their ground singlet states, thereby implying a more covalent Cu-N bond in 2.
Our DFT results provide a basis for interpreting the reactivity properties of 1 and 2 using a two-state reactivity model. In 1, the triplet state with a low activation barrier1 for HAT is low lying and easily accessible, which accounts for the higher HAT reactivity of 1 than 2. The NT reaction, in contrast, is thought to proceed predominantly on the singlet surface;8 the higher reactivity of 2 can then be possibly explained by the greater electrophilicity of the copper-nitrene unit in 2, owing to the larger contribution of the [CuIII(NR)]+ valence-bond resonance form compared to 1.
In summary, we have demonstrated that the behavior of [L1Cu(NR)Sc(OTf)3]+ (R= Ts for 1 and Mes for 2) complexes in both nitrene-transfer and H atom abstraction reactions depends on the nature of the nitrene substituent. Interestingly, whereas complex 2 is a better nitrene-transfer reagent than 1, an opposite reactivity trend is apparent for the H-atom abstraction reactions. DFT calculations provide a plausible framework within which to rationalize these reactivity patterns. It is postulated that the observed HAT reactivity reflects the availability of two closely lying spin states for 1 and 2, the so-called TSR hypothesis,12,13 wherein the excited triplet state has a much lower reaction barrier than the ground singlet state. Thus, the tosyl substrate of 1 results in a decrease in the singlet-triplet gap and an increased participation of the triplet state in determining the rate of reaction. Hence, in this study we provide evidence that the TSR theory, well established for iron-oxo complexes12,26, may also provide a framework for understanding reactivity of copper nitrenes. This study may therefore be of extreme importance in understanding the reactivity of mononuclear copper monooxygenases27–30, where copper-oxo intermediates, which are isoelectronic to the presently described copper-nitrene species, have often been implicated as the active species responsible for oxidations.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support of this work from the Cluster of Excellence “Unifying Concepts in Catalysis” (EXC 314/1), Berlin. XAS data were obtained on NSLS beamline X3B (Brookhaven National Laboratory), with support from NIH Grant P30-EB-009998 and the U.S. Department of Energy. I.M.P thanks BIG-NSE for a scholarship.
Footnotes
Electronic Supplementary Information (ESI) available: [Experimental and theoretical details]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Barman DN, Liu P, Houk KN, Nicholas KM. Organometallics. 2010;29:3404–3412. [Google Scholar]
- 2.Cundari TR, Dinescu A, Kazi AB. Inorg Chem. 2008;47:10067–10072. doi: 10.1021/ic801337f. [DOI] [PubMed] [Google Scholar]
- 3.Tekarli SM, Williams TG, Cundari TR. J Chem Theory Comput. 2009;5:2959–2966. doi: 10.1021/ct900277m. [DOI] [PubMed] [Google Scholar]
- 4.Aguila MJB, Badiei YM, Warren TH. J Am Chem Soc. 2013;135:9399–9406. doi: 10.1021/ja400879m. [DOI] [PubMed] [Google Scholar]
- 5.Badiei YM, Dinescu A, Dai X, Palomino RM, Heinemann FW, Cundari TR, Warren TH. Angew Chem Int Ed Engl. 2008;47:9961–9964. doi: 10.1002/anie.200804304. [DOI] [PubMed] [Google Scholar]
- 6.Badiei YM, Krishnaswamy A, Melzer MM, Warren TH. J Am Chem Soc. 2006;128:15056–7. doi: 10.1021/ja065299l. [DOI] [PubMed] [Google Scholar]
- 7.Maestre L, Sameera WMC, Díaz-Requejo MM, Maseras F, Pérez PJ. J Am Chem Soc. 2013;135:1338–1348. doi: 10.1021/ja307229e. [DOI] [PubMed] [Google Scholar]
- 8.Brandt P, So MJ, Andersson PG, Norrby PO. J Am Chem Soc. 2000;122:8013–8020. [Google Scholar]; b) Comba P, Lang C, Lopez de C, Laorden C, Muruganantham A, Rajaraman G, Wadepohl H, Zajaczkowski M. Chem Eur J. 2008;14:5313–5328. doi: 10.1002/chem.200701910. [DOI] [PubMed] [Google Scholar]
- 9.Gephart R, Warren TH. Organometallics. 2012;31:7728–7752. [Google Scholar]
- 10.A copper bis nitrene complex has been structurally characterized Dielmann F, Andrada DM, Frenking G, Bertrand G. J Am Chem Soc. 2014;136:3800–3802. doi: 10.1021/ja5007355. but no reactivity study is reported
- 11.Kundu S, Miceli E, Farquhar E, Pfaff FF, Kuhlmann U, Hildebrandt P, Braun B, Greco C, Ray K. J Am Chem Soc. 2012;134:14710–14713. doi: 10.1021/ja306674h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Usharani D, Janardanan D, Li C, Shaik S. Acc Chem Res. 2013;46:471–482. doi: 10.1021/ar300204y. [DOI] [PubMed] [Google Scholar]
- 13.Schröder D, Holthausen MC, Schwarz H. J Phys Chem B. 2004;108:14407–14416. [Google Scholar]
- 14.Liang HC, Zhang CX, Henson MJ, Sommer RD, Hatwell KR, Kaderli S, Zuberbühler AD, Rheingold AL, Solomon EI, Karlin KD. J Am Chem Soc. 2002;124:4170–1. doi: 10.1021/ja0125265. [DOI] [PubMed] [Google Scholar]
- 15.Complex 2 is EPR silent. However, when the reaction is performed in the absence of scandium a new species 3 is formed which shows a typical Cu(II) EPR signal (Figure S10). Although further characterization of 3 is not performed we assign it to the coper(II)-amidate complex [L1CuII(HNMes)]+ based on our previous study.
- 16.Kau LS, Spira-Solomon DJ, Penner-Hahn JE, Hodgson KO, Solomon EI. J Am Chem Soc. 1987;109:6433–6442. [Google Scholar]
- 17.Dubois JL, Mukherjee P, Stack TDP, Hedman B, Solomon EI, Hodgson KO. J Am Chem Soc. 2000;122:5775–5787. [Google Scholar]
- 18.Donoghue PJ, Tehranchi J, Cramer CJ, Sarangi R, Solomon EI, Tolman WB. J Am Chem Soc. 2011;133:17602–17605. doi: 10.1021/ja207882h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mankad NP, Antholine WE, Szilagyi RK, Peters JC. J Am Chem Soc. 2009;131:3878–3880. doi: 10.1021/ja809834k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noodleman L, Norman JG. J Chem Phys. 1979;70:4903–4906. [Google Scholar]
- 21.Noodleman L. J Chem Phys. 1981;74:5737–5743. [Google Scholar]
- 22.Cundari TR, Morello GR. J Org Chem. 2009;74:5711–5714. doi: 10.1021/jo900941u. [DOI] [PubMed] [Google Scholar]
- 23.Cundari TR, Vaddadi S. J Mol Struct THEOCHEM. 2006;801:47–53. [Google Scholar]
- 24.Cundari TR, Jimenez-Halla JOC, Morello GR, Vaddadi S. J Am Chem Soc. 2008;130:13051–13058. doi: 10.1021/ja803176j. [DOI] [PubMed] [Google Scholar]
- 25.Grove LE, Xie J, Yikilmaz E, Miller AF, Brunold TC. Inorg Chem. 2008;47:3978–92. doi: 10.1021/ic702412y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sastri CV, Lee J, Oh K, Lee J, a Jackson T, Ray K, Hirao H, Hirao W, a Halfen J, Kim J, Que L, Shaik S, Nam W. Proc Natl Acad Sci U S A. 2007;104:19181–19186. doi: 10.1073/pnas.0709471104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ray K, Heims F, Pfaff FF. Eur J Inorg Chem. 2013;2013:3784–3807. [Google Scholar]
- 28.Yoshizawa K, Kihara N, Kamachi T, Shiota Y. Inorg Chem. 2006;45:3034–3041. doi: 10.1021/ic0521168. [DOI] [PubMed] [Google Scholar]
- 29.Crespo A, Martí MA, Roitberg AE, Amzel LM, a Estrin D. J Am Chem Soc. 2006;128:12817–12828. doi: 10.1021/ja062876x. [DOI] [PubMed] [Google Scholar]
- 30.Hong S, Huber SM, Gagliardi L, Cramer CC, Tolman WB. J Am Chem Soc. 2007;129:14190–14192. doi: 10.1021/ja0760426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.