

Abstract
Background
IgG4-related disease (IgG4-RD) is a poorly understood, multi-organ, chronic inflammatory disease characterized by tumefactive lesions, storiform fibrosis, obliterative phlebitis and the accumulation of IgG4-expressing plasma cells at disease sites.
Objective
The role of B cells and IgG4 antibodies in IgG4-RD pathogenesis is not well defined. We evaluated patients with IgG4-RD for activated B cells in disease lesions as well as peripheral blood and investigated their role in disease pathogenesis.
Methods
B cell populations were analyzed from the peripheral blood of 84 patients with active IgG4-RD using flow cytometry. The repertoire of B cell populations was analyzed in a subset of patients by Next-generation Sequencing. Fourteen of these patients, were longitudinally followed for 9-15 months after Rituximab therapy.
Results
CD19+CD27+CD20-CD38hi plasmablasts, which are largely IgG4+, are elevated in patients with active IgG4-RD. These expanded plasmablasts are oligoclonal, exhibit extensive somatic hypermutation and their numbers decline following rituximab-mediated B-cell depletion therapy; this loss correlates with disease remission. A subset of patients relapse after rituximab therapy, and circulating plasmablasts that re-emerge in these subjects are clonally distinct and exhibit enhanced somatic hypermutation. Cloning and expression of Ig heavy and light chain genes from expanded plasmablasts at the peak of disease reveals that disease-associated IgG4 antibodies are self-reactive.
Conclusions
Clonally expanded CD19+CD27+CD20-CD38hi plasmablasts are a hallmark of active IgG4-RD. Enhanced somatic mutation in activated B cells and plasmablasts and emergence of distinct plasmablast clones upon relapse indicate that the disease pathogenesis is linked to de novo recruitment of naïve B cells into T-dependent responses by CD4+ T cells, likely driving a self-reactive disease process.
Keywords: IgG4-related disease, autoreactivity, rituximab, next-generation sequencing, somatic hypermutation, plasmablasts, IGHV repertoire, CDR3
Background
IgG4-related disease (IgG4-RD) is a multi-organ inflammatory condition that includes subjects previously diagnosed with other disorders that were defined earlier by the dominant pattern of organ involvement, e.g. type I autoimmune pancreatitis, Mikulicz's syndrome, Reidel's thyroiditis, retroperitoneal fibrosis, Küttner's tumor, tubulointerstitial nephritis and sclerosing cholangitis among others (1-4). IgG4 itself is generally considered to be a non-inflammatory immunoglobulin due to its limited ability to fix complement and bind activating Fc receptors (5, 6). Autoantibodies against antigens such as carbonic anhydrase II, pancreatic secretory trypsin inhibitor and lactoferrin have been described in IgG4-RD but they have poor specificity for this disease (7, 8). There is very limited evidence that the autoantibodies described so far are of the IgG4 subclass and it is unclear whether they are involved in disease pathogenesis (9).
The majority of patients with IgG4-RD have elevated levels of plasma IgG4 as well as increased infiltration of IgG4+ plasma cells in disease lesions (10, 11). The antigens inducing the plasma IgG4 and the immune processes leading to the infiltration of IgG4+ B cells and plasma cells into affected tissues remain largely unknown. It is likely that antigen-mediated processes, whether autoantigen or microbial driven, lead to expansion of specific B cells and, with the help of activated T follicular helper cells, facilitate their switching to IgG4, eventually resulting in clonal expansion of IgG4+ plasmablasts and plasma cells. Presumed oligoclonal IgG4 bands have also been observed in the cerebrospinal fluid of patients with IgG4-related pachymeningitis (12) and oligoclonal expansion of IgG4+ B cells has been inferred by Next generation sequencing of immunoglobulin (Ig) heavy (H) chain genes in subjects with IgG4-related sclerosing cholangitis (13).
Patients with IgG4-RD respond dramatically to the depletion of B cells with rituximab (an anti-CD20 monoclonal antibody), and this results in striking clinical improvement (14). In this study, we have determined that IgG4-RD patients with active disease exhibit large expansions of CD19+CD38+CD27+ plasmablasts that have undergone extensive somatic hypermutation. Rituximab-mediated B cell depletion, results in the reduction of plasmablasts and this loss coincides with disease remission. Subsequent relapse is linked to the re-emergence of clonally divergent and somatically hypermutated plasmablasts, suggesting that de novo reactivation of an underlying autoimmune disease process, likely driven by T cells, also drives the generation of somatically hypermutated IgG4 auto-antibodies.
Methods
Patients
This study was approved by the institutional review board and informed, written consent was obtained from all subjects with IgG4-RD referred to or presenting at the rheumatology clinic of the Massachusetts General Hospital. Samples from 84 patients with IgG4-RD were chosen for this study (organ involvement and patient demographics are listed in Table E1 in this article's online repository). The IgG4-RD patients were compared with 16 healthy controls (age 32-70 years). Twenty-three of these patients with active disease were treated with two 1000mg doses of rituximab, 15 days apart. 15 ml of peripheral blood was collected at initial presentation and each subsequent clinical visit. Fourteen of the rituximab-treated patients were longitudinally followed for 9-15 months at the rheumatology clinic.
IGHV Repertoire Analysis
Next-generation sequencing analysis of BCR IGH repertoire was undertaken using the ImmunoSeq® platform (Adaptive Biotechnologies Inc.) at the “survey” level of sequencing depth (15). Sorted cell numbers ranged from ∼5,000 to ∼40,000, assuring at least 5-fold depth of sequencing. Assembly of the V-D-J regions in the rearranged IGH sequences and analysis of somatic hypermutation was performed using the IMGT V-Quest server and NCBI VDJ solver (18). Single-cell PCR and sequencing of rearranged Ig genes from individually sorted plasmablasts was carried out with minor modifications to the method described by Tiller et al (19). Except for the IgG4-CH outer reverse primer (5′-AGGGCGCCAGGGGGAAGACG-3′), the primer sequences used were identical to those described by Tiller et al (19). Paired heavy and light chain PCR products from single plasmablasts were cloned and expressed as monoclonal antibodies in human embryonic kidney (HEK) 293T cells as previously described (19). Detailed methods are provided in this article's online repository.
Flow cytometry and microscopy
Detailed description of the methods used for PBMC isolation, flow cytometry, cell sorting, immunohistochemistry and immunofluorescence are provided in this article's Online Repository at www.jacionline.org.
Results
Expansion of plasmablasts in IgG4-RD
Immunohistochemical analysis of IgG4-RD lesions frequently reveals the presence of CD20+ B-cell follicles surrounded by CD19+CD20- cells with variable expression of IgG4. These latter cells could be either tissue plasmablasts or plasma cells (Fig 1, A). Flow cytometric analysis of the peripheral blood revealed large expansions of CD19+CD27+CD38hi plasmablasts in multiple IgG4-RD subjects (Fig 1, B). The plasmablasts are negative for CD20, largely surface IgG4+ and express high levels of SLAMF7, a CD2 family receptor that is expressed on activated human B cells (Fig 1, C) (20). These cells also show elevated expression of HLA-DR and BCMA, while IgM expression is not very prominent (Fig E1). The absolute numbers of circulating plasmablasts in the blood were significantly higher in subjects with disease (n = 84, p < 0.001) than in controls (n = 16) (Fig 1, D).
Fig 1. Expansion of CD19+CD20-IgG4+ plasmablasts in IgG4-related disease.
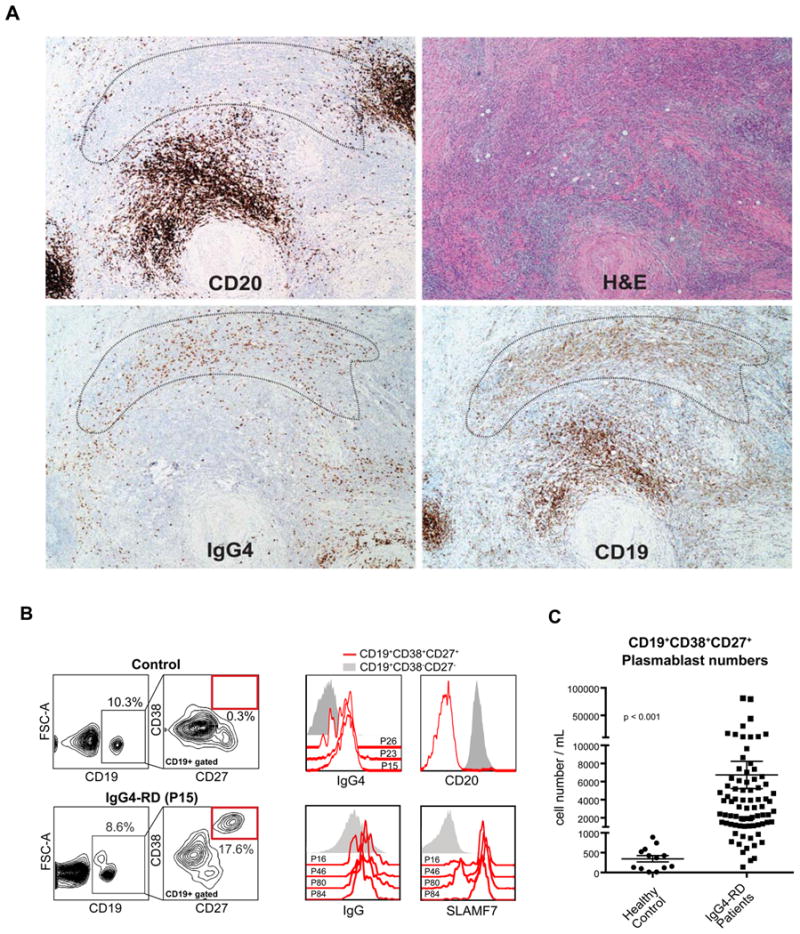
A, H&E and immunohistochemical stains for CD19, CD20, and IgG4 performed on serial sections of a submandibular gland biopsy are shown. An area peripheral to a CD19+CD20+B-cell follicle that is enriched for IgG4+CD19+CD20- cells is marked.
B, Flow cytometry gating scheme used to identify circulating CD19+CD27+CD38hi plasmablasts (red box).
C, Surface staining of CD19+CD27+CD38hi plasmablasts for IgG4, IgG, CD20 and SLAMF7 cells. Data from representative patients is shown as histograms overlaid on CD19+CD27-CD38- B cells as a negative control.
D, The numbers of plasmablasts in the peripheral blood of IgG4-RD subjects (n = 84) and healthy controls (n = 16) are shown (mean ± S.E.M., p < 0.001).
Plasmablasts share some surface markers with regulatory B cells (Bregs). We explored the possibility of CD19+CD38+CD24+IL-10+ Breg expansions in IgG4-RD subjects (21), but no difference in the proportion or counts of these cells was observed in PBMCs of 8 patients in comparison with healthy controls (Fig E2, A and B).
The expanded pool of plasmablasts is clonally restricted
Next-generation sequencing was used to analyze the IGHV gene repertoire of flow-cytometrically sorted plasmablasts from five patients with active IgG4-RD. Approximately 90% of the total sequences obtained exhibited productive rearrangements. Plasmablast expansions were found to be oligoclonal in all the subjects examined (Fig 2 A and Fig E3). The frequency of the expanded clones identified by single cell cloning and sequencing specifically of IgG4 genes of individual plasmablasts showed close consonance with the dominant clones determined by next-generation sequencing of plasmablast IGHV genes, confirming that the oligoclonal expansions detected by next-generation sequencing are unlikely to be affected by PCR bias (Fig 2, B and C), and that the major plasmablast clones in this disease make IgG4.
Fig 2. Restricted repertoire of expanded plasmablast populations.
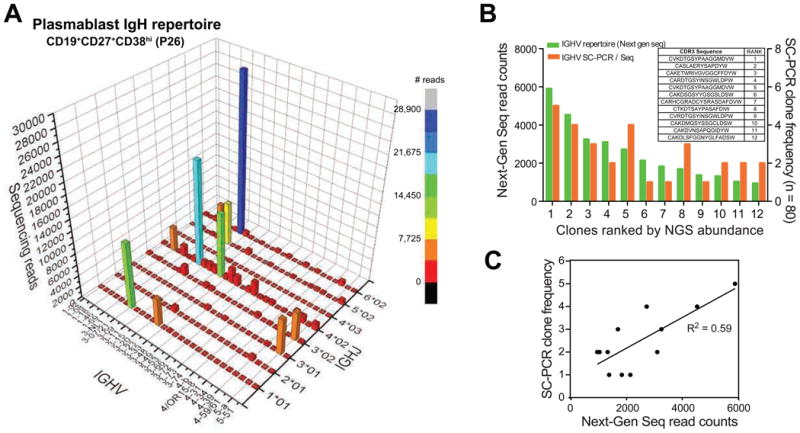
A, The immunoglobulin heavy chain (IGH) repertoire of CD19+CD27+CD38hi plasmablasts sorted from a representative patient (P26) is shown as a 3D histogram. The x- and y- axes represent IGH-V and IGH-J regions, respectively, and the z- axis indicates the number of productive IGH-rearrangements detected by next-generation sequencing.
B, Comparison of read counts of expanded clones from patient P26 detected by next-generation sequencing (green bars) and SC-PCR sequencing (orange bars). The corresponding CDR3 sequences ordered by frequency as seen on next-generation sequencing are shown in the table inlay.
C, Correlation between the absolute read counts of the most frequent IGH CDR3 sequences determined by next-generation sequencing and number of cells with the corresponding CDR3 sequence determined by SC-PCR (n = 80 cells) of flow-sorted IgG4+ plasmablasts from patient P26.
IgG4-RD relapse following rituximab treatment is associated with the re-emergence of circulating clonally divergent plasmablasts
Therapeutic B cell depletion with rituximab (an anti-CD20 monoclonal antibody) generally results in clinical improvement in IgG4-RD (22). The IgG4-RD Responder Index was used to quantify disease activity and to monitor the response to therapy (Fig 3, A)(23). An initial decline in serum IgG4 levels was observed in most of the treated patients but the residual levels of IgG4 were sustained at a steady level in a subset of patients (Fig 3, B), suggesting that a substantial fraction of the IgG4 is produced by long-lived plasma cells that do not express CD20 and are therefore resistant to Rituximab. These findings are also consistent with prior reports on the therapeutic use of rituximab in IgG4-RD (22). As expected, CD19+ B cells declined dramatically by day 30 after rituximab therapy (Fig 3, C). Although CD19+CD27+CD38hi plasmablasts do not express CD20, their counts also declined sharply following B cell depletion, likely due to depletion of their CD20+ precursors (Fig 3, D). These data suggest that most circulating plasmablasts are present transiently in the blood on their way to affected tissues or to the bone marrow, and their numbers fall rapidly after rituximab therapy because of a failure of repletion.
Fig 3. Rituximab-mediated depletion of plasmablasts improves clinical outcome.

A, Measurement of the therapeutic response to rituximab using the IgG4-RD Responder Index.
B, Decline in plasma-IgG4 concentrations after rituximab infusion. Colored lines represent individual patients followed upto 15 months after rituximab infusion. Dotted-horizontal line indicates the upper limit of normal (135 mg/dL). C and D, Rituximab-induced decline in the proportion of circulating B-cells (n =23, p < 0.001) (C) and plasmablasts (n=23, p < 0.001) (D) by day 30.
Following rituximab therapy, CD19+ B cell numbers remained very low typically for at least three months. The time of re-emergence of the CD19+CD27-CD38int naïve B cell pool after rituximab therapy varied between individuals, with the median duration for the restoration of B cell levels to 25% of the pre-rituximab value being 6.5 months (Fig 4, A). In a few cases, there was no B cell repopulation 6 months after rituximab therapy. The time for repopulation of the plasmablast pool was highly variable between patients, with the median time for newly generated plasmablast numbers to reach 10% of the pre-therapeutic levels being 8 months after rituximab infusion. Subjects displaying very limited or no repopulation of plasmablasts stayed in remission for up to 13 months post treatment and showed no signs of disease relapse until their most recent visit (Fig 4, B). However, some patients showed an early recurrence of plasmablasts within 3-5 months following therapy and some of them required a second infusion of rituximab to control the disease (Fig 4, C).
Fig 4. Repopulation of plasmablasts as a marker of disease relapse.

A, Longitudinal follow-up of the CD19+ B cell compartment in 14 IgG4-RD patients following rituximab therapy, plotted as a percentage of the baseline B cell count.
B and C, Longitudinal follow-up of CD19+CD27+CD38+ plasmablast counts following rituximab treatment in 6 IgG4-RD patients who experienced sustained remission (B) and 6 patients who relapsed (C).
In a longitudinal examination of a small number of subjects, we compared the clonal composition of the expanded plasmablast populations at disease presentation and at the time of relapse using single cell PCR sequencing for Ig heavy chain genes. Although specific populations of memory B cells such as IgM memory B cells have been shown to survive rituximab infusion and contribute to post-rituximab disease relapse in other conditions (24), B cells were almost completely depleted in our patients as seen in the peripheral blood, and in the 3 patients studied, the plasmablasts in circulation showed minimal overlap in the V-J segment usage before rituximab therapy and at the time of disease relapse (Fig 5, A & B and Fig E4). Plasmablast Ig repertoires were analyzed by next-generation sequencing before rituximab therapy and by single cell PCR sequencing after rituximab. The CDR3 sequences of re-emerging clones that shared a V-J combination before and after treatment exhibited <70% nucleotide sequence identity (Table E2). One possible interpretation of this data is that following the rituximab-mediated depletion of B cells, the plasmablast clones that appear in relapsing patients are derived from a new wave of naïve B cells emerging from the bone marrow with a distinct BCR repertoire. However, we cannot exclude the possibility that at least a subset of plasmablast clones at relapse arise from memory B cells that survive rituximab-mediated B cell depletion.
Fig 5. Clonal divergence in relapsing pool of plasmablasts.

The heat maps show the degree of expansion of plasmablast clones bearing specific IGH V and J combinations in two patients who relapsed, P11 (A) and P15 (B), using a blue-red color gradient with the most expanded clones shown in red. Unused V-J combinations are colored black. The tables show a comparison of the CDR3 amino acid sequences of the dominant clones sharing the same IGHV-J combinations before and after rituximab therapy. Highly expanded clones are colored red in the tables and the numbers in parentheses indicate CDR3 nucleotide sequence identity.
Ig genes of expanded plasmablasts before therapy and after relapse exhibit increased somatic hypermutation
Enhanced somatic hypermutation of the rearranged variable regions of the expanded plasmablasts at initial presentation was observed compared to naïve and memory B cells of healthy controls (Fig 6, A). This is consistent with the notion that IgG4-expressing plasmablasts are generated as part of a T-dependent immune response involving repeated rounds of mutation and selection by antigen. Interestingly, the hypermutation process was extensive enough to include both hypervariable (CDR3) and framework regions (FR3). Memory B cells and autoreactive B cells have been shown to have increased numbers of positively charged amino acids (25, 26). A significant increase in the number of positively charged residues was observed in the CDR3 regions of sequences from 5 IgG4-RD patients (Fig E5). The dominant clones of plasmablasts observed during relapse not only had V-D-J rearrangements that were distinct from the dominant clones prior to rituximab therapy in the same subject, but also had undergone extensive somatic hypermutation (Fig 6, B). The pathogenic immune processes driving the initial disease presentation as well as the processes causing relapse of disease are likely to be similar and presumably involve either persisting pathogenic T cells or the de novo generation of CD4+ T cell responses.
Fig 6. Expanded populations of plasmablasts exhibit enhanced somatic hypermutation and autoreactivity.
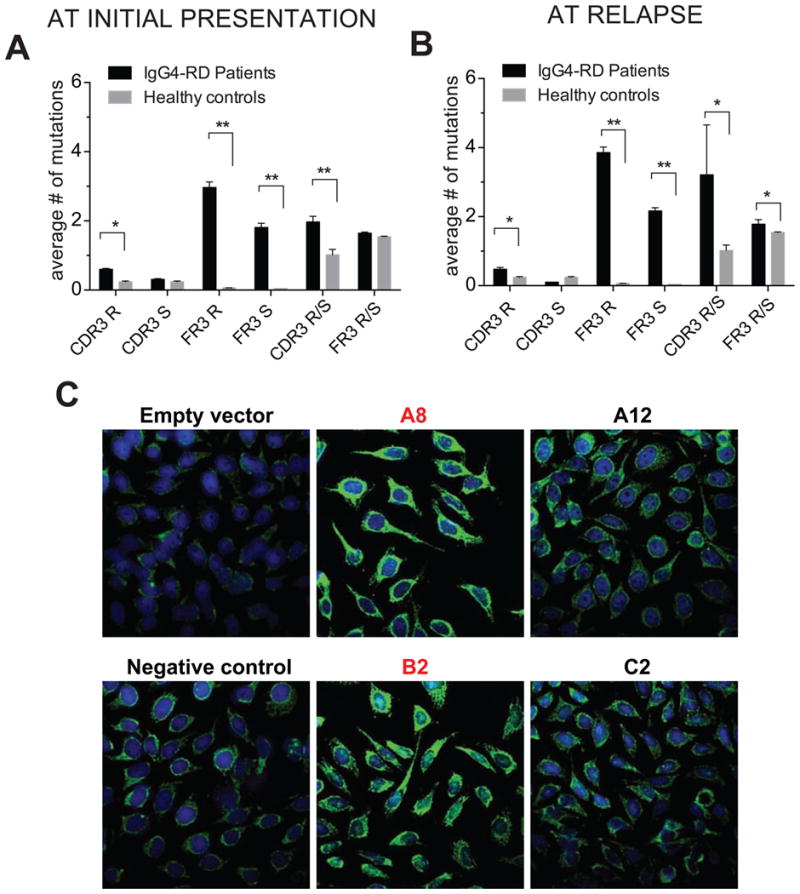
A & B, Comparison of replacement-R and silent-S mutations and R/S ratio in FR3 and CDR3 regions of IGHV-sequences of plasmablasts from (A) 5 untreated IgG4-RD subjects and (B) 3 relapsing IgG4-RD patients following rituximab therapy with CDR3 sequences from naïve B cells from 3-control samples as analyzed from NGS-reads (mean ± S.E.M., * p < 0.05, ** p < 0.005).
C, Immunofluorescence staining of Hep-2 coated slides with single plasmablast derived antibody clones. Clones with bright cytosolic staining are labeled in red.
Recombinant monoclonal immunoglobulins from active IgG-RD subjects are self-reactive
An obvious issue to address was whether these dominant plasmablast clones in patients with active disease are self-reactive. We cloned matched Ig heavy and light chain variable region genes of single cells from the most expanded IgG4 plasmablasts and expressed these in 293T cells and then examined whether the secreted monoclonal human antibodies were self-reactive using ELISA to detect binding to plate-adsorbed HeLa cell lysates and immunofluorescence to detect binding to fixed and permeabilized Hep2 cells. As seen in Fig 6, C and Fig E6, the recombinant monoclonal immunoglobulins were strongly self-reactive as assessed by both methods. The pattern of staining observed is predominantly cytosolic. These data suggest that the disease-associated oligoclonal plasmablast expansions and the T-dependent B cell activation events that likely drive disease progression represent responses to self-antigens.
Discussion
Expanded populations of oligoclonal CD19+CD20-CD27+CD38+ plasmablasts represent perhaps the best available indication of IgG4-RD disease activity, especially since serum IgG4 levels are not always elevated in this disease. The fact that IgG4 antibodies are generally considered to be non-inflammatory has made it difficult to cogently postulate a role for these antibodies in the disease process. The tight correlation between the loss of circulating IgG4+ plasmablasts and clinical improvement, and the re-emergence of circulating plasmablasts at the time of relapse, strongly suggest that IgG4+ plasmablasts in tissue sites may play a role in IgG4-RD pathogenesis and may be a potential therapeutic target. An antibody against SLAMF7, which is expressed on the disease-associated plasmablasts in IgG4-RD subjects, is currently being evaluated in clinical trials for multiple myeloma (27, 28) and may be worth evaluating as a potential therapy for IgG4-RD. However, the clinical utility of plasmablasts as a biomarker for tracking disease activity in IgG4-RD requires to be validated in a larger cohort of patients.
The high degree of somatic hypermutation seen in the IgG4+ plasmablasts suggests that T-dependent immune responses may be relevant in this disease and implies that at least some of these circulating plasmablasts may have the potential to home to the bone marrow and differentiate into long-lived plasma cells. This inference is supported by the observation that although there is a decline in serum IgG4 following rituximab therapy, the IgG4 levels stay well above the upper level of the reference range (135 mg/dL), especially in a subset of patients with multi-organ disease (4). In such patients, it is likely that a substantial pool of rituximab-resistant long-lived plasma cells has developed. However, most patients show a decline in IgG4 levels within the first few weeks following rituximab therapy. Since IgG4 molecules are estimated to persist in the circulation for about 3 weeks, it may be inferred that at least a portion of the circulating IgG4 is made by short-lived plasmablasts/plasma cells that are abundant in tissue lesions as well as in the circulation of patients with active disease, and these cells cannot be easily replenished after rituximab therapy due to the depletion of their precursor B cell pools.
There is essentially no clinical, histological or biochemical difference in the presentation of a subject seen with disease for the first time or when disease relapses. The same poorly understood pathogenic mechanisms likely drive the disease process initially and after B cell numbers recover after successful B-cell depletion therapy in some subjects. Given that the naive B cell repertoire has to be re-created from scratch after B cell depletion, it is not surprising that dominant IgG4 clones during peak disease activity at the time of the initial diagnosis are completely different from the dominant IgG4 plasmablast clones that re-emerge during relapse. However, in the setting of extensive somatic hypermutation, the finding of clonal divergence alone is not sufficient to exclude that the possibility that at least some of the dominant plasmablast clones at relapse arise from memory B cells that survived rituximab depletion (29, 30). The extensive degree of somatic hypermutation of IgG4 antibodies seen both initially and on relapse suggest that helper T cell dependent processes are likely important in this disease. Monoclonal immunoglobulins derived from the most abundant IgG4+ plasmablasts in patients with active disease are self-reactive. These findings, taken together, suggest that T cells and highly mutated self-reactive B lineage cells, perhaps activated B cells or plasmablasts themselves, may contribute in some way to the fibrotic disease process by mechanisms that are yet to be elucidated. Given the high levels of surface MHC Class II (HLA-DR) on plasmablasts, one could speculate that these cells serve as vital antigen-presenting cells to rogue CD4+ T cells in IgG4-RD, but it remains possible, although we consider this unlikely, that this disease is driven directly by B lineage cells with little or no role for T cells in pathogenesis. The identity of the antigens recognized by these monoclonal immunoglobulins remains to be determined, and it is not yet known whether the same antigens are recognized during the initial disease process and at the time of relapse. The specific contributions of both B and T cells to this process also remain to be identified.
The sequence of immunological events that occur within an IgG4-RD lesion is not well understood. The role of Th2 cells in IgG4-RD pathogenesis remains speculative given that other cell types like mast cells also secrete Th2 cytokines in IgG4-RD lesions (31) and the contribution of innate like lymphoid cells (ILCs) to the disease process has not yet been assessed. Large amounts of IL-10 produced by an exaggerated Treg response has also been postulated to contribute to the IgG4 class switch and to disease pathogenesis (32, 33). However the activation and expansion of B cells appears to be a necessary precursor of inflammatory T cell responses and our view, summarized in Fig 7, is that IgG4-RD may be caused by collaboration between CD4+ T cells and activated, somatically hypermutated IgG4+ B cells or plasmablasts. B-cell dependent activation of pathogenic CD4+ T cells presumably mediates the inflammation and fibrosis seen in this disease. Rituximab treatment causes a loss of these B cells and plasmablasts and leads to clinical improvement by breaking this vicious cycle of T-B collaboration. In a subset of treated patients, the disease relapses and newly generated B cells are recruited either by the same T cells that drove the initial disease manifestations or by a newly generated set of pathogenic CD4+ T cell clones. This recruitment, in response to either the same or distinct self-antigens, generates somatically mutated B cells and plasmablasts with a distinct repertoire from that of the clones that dominated at the time of initial presentation.
Fig 7. A speculative model for the role of activated B cells and plasmablasts in a presumed CD4+ T cell mediated pathogenic process causing IgG4-RD.

Activated somatically hypermutated B cells or plasmablasts may serve as effective antigen presenting cells for pathogenic effector CD4+ T cells in disease sites. In patients that relapse after rituximab-mediated B cell depletion therapy plasmablasts may be generated de novo from naïve B cells (solid arrow) or from CD20+ memory B cells that survive rituximab therapy (dashed arrow), facilitated by T-B cooperation in secondary lymphoid organs (not shown).
Plasmablast accumulations in the blood have been described in a small subset of patients with active systemic lupus erythematosus and rheumatoid arthritis (34-36). Interestingly it has been suggested that the approximately 20% of rheumatoid arthritis subjects who have elevated blood plasmablasts are likely to respond poorly to Rituximab therapy (35). In IgG4-RD however elevated plasmablasts are the norm and do not preclude a therapeutic response to anti-CD20 immunotherapy.
While we suspect that activated IgG4+ B cells participate in the process of pathogenesis, a demonstration of their direct role in disease pathogenesis is lacking and one could still argue that IgG4+ plasmablasts are secondarily induced bystanders in this disease. A clearer picture will likely emerge following the characterization of self-antigens and the clonal identification and characterization of disease-causing CD4+ T cells both at initial presentation and at the time of relapse.
Supplementary Material
Key Messages.
Oligoclonal expansions of IgG4 expressing plasmablasts are a hallmark of active and relapsing IgG4-RD.
Rituximab-mediated remission is associated with disappearance of plasmablasts while recurrence of clonally divergent plasmablasts is tightly correlated with disease relapse.
IgG4 antibodies expressed by clonally expanded plasmablasts in active disease are autoreactive.
Acknowledgments
This study was funded by grants AI 064930 and AI 076505 from the NIH and a pilot grant from the Harvard Institute of Translational Immunology supported by the Helmsley Foundation to SP.
Abbreviations
- IgG4-RD
IgG4-related disease
- Ig
Immunoglobulin
- Breg
Regulatory B cells
- BCR
B cell receptor
- SC-PCR
Single cell Polymerase chain reaction
- H & E
Hematoxylin and eosin
- IHC
Immunohistochemistry
- IGHV
Immunoglobulin heavy chain variable region
- V-D-J
Variable-Diversity-Joining
- PBMC
Peripheral blood mononuclear cell
- PMA
Phorbol myristate acetate
- R
Replacement
- S
Silent
- S.E.M
Standard error of mean
Footnotes
Conflict of Interest: The authors declare that they have no relevant conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mahajan VS, Mattoo H, Deshpande V, Pillai S, Stone JH. IgG4-Related Disease. Annual Review of Pathology: Mechanisms of Disease. 2014;9 doi: 10.1146/annurev-pathol-012513-104708. [DOI] [PubMed] [Google Scholar]
- 2.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539–551. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 3.Kamisawa T, Egawa N, Nakajima H. Autoimmune pancreatitis is a systemic autoimmune disease. Am J Gastroenterol. 2003;98:2811–2812. doi: 10.1111/j.1572-0241.2003.08758.x. [DOI] [PubMed] [Google Scholar]
- 4.Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, Fukushima M, Nikaido T, Nakayama K, Usuda N, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 5.Bindon CI, Hale G, Bruggemann M, Waldmann H. Human monoclonal IgG isotypes differ in complement activating function at the level of C4 as well as C1q. J Exp Med. 1988;168:127–142. doi: 10.1084/jem.168.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daeron M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–3725. doi: 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- 7.Asada M, Nishio A, Uchida K, Kido M, Ueno S, Uza N, Kiriya K, Inoue S, Kitamura H, Ohashi S, et al. Identification of a novel autoantibody against pancreatic secretory trypsin inhibitor in patients with autoimmune pancreatitis. Pancreas. 2006;33:20–26. doi: 10.1097/01.mpa.0000226881.48204.fd. [DOI] [PubMed] [Google Scholar]
- 8.Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut. 2005;54:274–281. doi: 10.1136/gut.2004.049064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aoki S, Nakazawa T, Ohara H, Sano H, Nakao H, Joh T, Murase T, Eimoto T, Itoh M. Immunohistochemical study of autoimmune pancreatitis using anti-IgG4 antibody and patients' sera. Histopathology. 2005;47:147–158. doi: 10.1111/j.1365-2559.2005.02204.x. [DOI] [PubMed] [Google Scholar]
- 10.Deshpande V. The pathology of IgG4-related disease: critical issues and challenges. Semin Diagn Pathol. 2012;29:191–196. doi: 10.1053/j.semdp.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Sah RP, Chari ST. Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol. 2011;23:108–113. doi: 10.1097/BOR.0b013e3283413469. [DOI] [PubMed] [Google Scholar]
- 12.Della-Torre E, Passerini G, Furlan R, Roveri L, Chieffo R, Anzalone N, Doglioni C, Zardini E, Sabbadini MG, Franciotta D. Cerebrospinal Fluid Analysis in Immunoglobulin G4-related Hypertrophic Pachymeningitis. J Rheumatol. 2013;40:1927–1929. doi: 10.3899/jrheum.130678. [DOI] [PubMed] [Google Scholar]
- 13.de Buy Wenniger LJ, Doorenspleet ME, Klarenbeek PL, Verheij J, Baas F, Elferink RP, Tak PP, de Vries N, Beuers U. IgG4+ clones identified by next-generation sequencing dominate the b-cell receptor repertoire in IgG4-associated cholangitis. Hepatology. 2013 doi: 10.1002/hep.26232. [DOI] [PubMed] [Google Scholar]
- 14.Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62:1755–1762. doi: 10.1002/art.27435. [DOI] [PubMed] [Google Scholar]
- 15.Larimore K, McCormick MW, Robins HS, Greenberg PD. Shaping of human germline IgH repertoires revealed by deep sequencing. J Immunol. 2012;189:3221–3230. doi: 10.4049/jimmunol.1201303. [DOI] [PubMed] [Google Scholar]
- 16.Brochet X, Lefranc MP, Giudicelli V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008;36:W503–508. doi: 10.1093/nar/gkn316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ehrenmann F, Lefranc MP. IMGT/3Dstructure-DB: querying the IMGT database for 3D structures in immunology and immunoinformatics (IG or antibodies, TR, MH, RPI, and FPIA) Cold Spring Harb Protoc. 2011;2011:750–761. doi: 10.1101/pdb.prot5637. [DOI] [PubMed] [Google Scholar]
- 18.Ohm-Laursen L, Nielsen M, Larsen SR, Barington T. No evidence for the use of DIR, D-D fusions, chromosome 15 open reading frames or VH replacement in the peripheral repertoire was found on application of an improved algorithm, JointML, to 6329 human immunoglobulin H rearrangements. Immunology. 2006;119:265–277. doi: 10.1111/j.1365-2567.2006.02431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods. 2008;329:112–124. doi: 10.1016/j.jim.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JK, Mathew SO, Vaidya SV, Kumaresan PR, Mathew PA. CS1 (CRACC, CD319) induces proliferation and autocrine cytokine expression on human B lymphocytes. J Immunol. 2007;179:4672–4678. doi: 10.4049/jimmunol.179.7.4672. [DOI] [PubMed] [Google Scholar]
- 21.Blair PA, Norena LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, Mauri C. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity. 2010;32:129–140. doi: 10.1016/j.immuni.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore) 2012;91:57–66. doi: 10.1097/MD.0b013e3182431ef6. [DOI] [PubMed] [Google Scholar]
- 23.Carruthers MN, Stone JH, Deshpande V, Khosroshahi A. Development of an IgG4-RD Responder Index. Int J Rheumatol. 2012;2012:259408. doi: 10.1155/2012/259408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maurer MA, Rakocevic G, Leung CS, Quast I, Lukacisin M, Goebels N, Munz C, Wardemann H, Dalakas M, Lunemann JD. Rituximab induces sustained reduction of pathogenic B cells in patients with peripheral nervous system autoimmunity. J Clin Invest. 2012;122:1393–1402. doi: 10.1172/JCI58743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu YC, Kipling D, Leong HS, Martin V, Ademokun AA, Dunn-Walters DK. High-throughput immunoglobulin repertoire analysis distinguishes between human IgM memory and switched memory B-cell populations. Blood. 2010;116:1070–1078. doi: 10.1182/blood-2010-03-275859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiller T, Kofer J, Kreschel C, Busse CE, Riebel S, Wickert S, Oden F, Mertes MM, Ehlers M, Wardemann H. Development of self-reactive germinal center B cells and plasma cells in autoimmune Fc gammaRIIB-deficient mice. J Exp Med. 2010;207:2767–2778. doi: 10.1084/jem.20100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsi ED, Steinle R, Balasa B, Szmania S, Draksharapu A, Shum BP, Huseni M, Powers D, Nanisetti A, Zhang Y, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res. 2008;14:2775–2784. doi: 10.1158/1078-0432.CCR-07-4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryan MC, Hering M, Peckham D, McDonagh CF, Brown L, Kim KM, Meyer DL, Zabinski RF, Grewal IS, Carter PJ. Antibody targeting of B-cell maturation antigen on malignant plasma cells. Mol Cancer Ther. 2007;6:3009–3018. doi: 10.1158/1535-7163.MCT-07-0464. [DOI] [PubMed] [Google Scholar]
- 29.Kamburova EG, Koenen HJ, Borgman KJ, ten Berge IJ, Joosten I, Hilbrands LB. A single dose of rituximab does not deplete B cells in secondary lymphoid organs but alters phenotype and function. Am J Transplant. 2013;13:1503–1511. doi: 10.1111/ajt.12220. [DOI] [PubMed] [Google Scholar]
- 30.Leandro MJ. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res Ther. 2013;15(Suppl 1):S3. doi: 10.1186/ar3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takeuchi M, Sato Y, Ohno K, Tanaka S, Takata K, Gion Y, Orita Y, Ito T, Tachibana T, Yoshino T. T helper 2 and regulatory T-cell cytokine production by mast cells: a key factor in the pathogenesis of IgG4-related disease. Mod Pathol. 2014 doi: 10.1038/modpathol.2013.236. [DOI] [PubMed] [Google Scholar]
- 32.Zen Y, Fujii T, Harada K, Kawano M, Yamada K, Takahira M, Nakanuma Y. Th2 and regulatory immune reactions are increased in immunoglobin G4-related sclerosing pancreatitis and cholangitis. Hepatology. 2007;45:1538–1546. doi: 10.1002/hep.21697. [DOI] [PubMed] [Google Scholar]
- 33.Kusuda T, Uchida K, Miyoshi H, Koyabu M, Satoi S, Takaoka M, Shikata N, Uemura Y, Okazaki K. Involvement of inducible costimulator- and interleukin 10-positive regulatory T cells in the development of IgG4-related autoimmune pancreatitis. Pancreas. 2011;40:1120–1130. doi: 10.1097/MPA.0b013e31821fc796. [DOI] [PubMed] [Google Scholar]
- 34.Nicholas MW, Dooley MA, Hogan SL, Anolik J, Looney J, Sanz I, Clarke SH. A novel subset of memory B cells is enriched in autoreactivity and correlates with adverse outcomes in SLE. Clin Immunol. 2008;126:189–201. doi: 10.1016/j.clim.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Owczarczyk K, Lal P, Abbas AR, Wolslegel K, Holweg CT, Dummer W, Kelman A, Brunetta P, Lewin-Koh N, Sorani M, et al. A plasmablast biomarker for nonresponse to antibody therapy to CD20 in rheumatoid arthritis. Sci Transl Med. 2011;3:101ra–192. doi: 10.1126/scitranslmed.3002432. [DOI] [PubMed] [Google Scholar]
- 36.Tsubaki T, Takegawa S, Hanamoto H, Arita N, Kamogawa J, Yamamoto H, Takubo N, Nakata S, Yamada K, Yamamoto S, et al. Accumulation of plasma cells expressing CXCR3 in the synovial sublining regions of early rheumatoid arthritis in association with production of Mig/CXCL9 by synovial fibroblasts. Clin Exp Immunol. 2005;141:363–371. doi: 10.1111/j.1365-2249.2005.02850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.