

Abstract
The role of protein motions in enzymatic C-H→C transfer is an area of great contemporary debate. An effective tool in probing such a role is the temperature dependence of the intrinsic kinetic isotope effects for the enzyme-catalyzed reaction. The outcome of those experiments is interpreted within the context of phenomenological Marcus-like models of hydrogen tunneling. The current review focuses on recent studies of dihydrofolate reductase (DHFR) and how the role of protein motions in the catalyzed reaction has been demonstrated. The motions in DHFR are controlled by local effects of active site residues, global effects involving remote residues across the enzyme and appear to be preserved during the evolution of the enzyme from bacteria to human.
Introduction
Dihydrofolate reductase (DHFR) has emerged as a model system in many investigations of the role of protein motions in enzymatic C-H→C transfer, and enzyme catalysis in general. DHFR catalyzes the reduction of 7,8-dihydrofolate (DHF) to S-5,6,7,8-tetrahydrofolate (THF) through a hydride transfer of pro-R hydrogen from the C4 atom of NADPH to the C6 position of DHF. DHFR is critical in maintaining the intercellular pool of THF, which is required for the biosynthesis of many essential molecules including thymine. Due to its pharmacological importance, small size (18 kDa), and relative ease of study the enzyme has been the focus of many experimental and theoretical studies [3–6]. The current review focuses on the enzyme from Escherichia coli and outlines the recent advances made in elucidating the nature of the catalyzed hydride transfer. A particular focus is placed on the role of protein motions, how both active site and remote amino acid residues modulate these motions and the evolutionary preservation of protein dynamics to optimize the DHFR catalyzed reaction.
Temperature Dependence of Intrinsic Kinetic Isotope Effects (KIEs) and the Marcus-Like Model
A wealth of information concerning the nature of the hydride transfer reaction catalyzed by DHFR has been gained through measurements of the temperature dependence of the KIEs for wild-type (WT) and site-directed mutant forms of the enzyme [1,7–11]. Such results are best interpreted through full tunneling Marcus-like models of C-H transfer (also referred to as vibrationally enhanced tunneling, environmentally coupled tunneling and other terms found in literature) [4,6,12–19]. The Marcus-like model, adapted from Ruldoph Marcus’ landmark theory of electron tunneling [20], is based on three principles. First, the hydrogen should be treated quantum mechanically throughout the reaction coordinate and therefore it tunnels under the barrier once the needed conditions are there. Second, the model states that the rate of hydrogen transfer is determined by fluctuations of the electronic potential surface of the reaction. Finally, these fluctuations comprise two orthogonal coordinates: a rearrangement coordinate that adjusts the energy levels of the reactant and product while reaching the tunneling ready state (TRS), and a ‘gating’ coordinate that represents the fluctuations of the hydrogen donor and acceptor distance (DAD) at the TRS. These principles are depicted in Figure 1.
Figure 1. Marcus-Like models of hydrogen tunneling.

Three slices of the potential energy surface (PES) along components of the collective reaction coordinate showing the effect of heavy-atom motions on the zero point energy (ZPE) in the reactant (blue) and product (red) potential well. Panel A shows the heavy atom coordinate, and Panel B shows the H-atom position, which is orthogonal to the heavy atom coordinate. In the top panels the hydrogen is localized in the reactant well, and the ZPE of the product state is higher than that of the reactant state. Heavy atom reorganization brings the system to the tunneling ready state (TRS) (middle panels A and B), where the ZPEs in the reactant and product wells are degenerate and the hydrogen can tunnel between the wells. Further heavy atom reorganization breaks the transient degeneracy and traps the hydrogen in the product state (bottom panels). The rate of reaching the TRS depends on the reorganization energy (λ) and driving force (ΔG°). Panel C shows the effect of donor and acceptor distance (DAD) sampling on the wavefunction overlap at the TRS. Reprinted with permission from Annual Reviews [6].
The electronic potential surface of the reaction is not altered upon isotopic substitution of the reactants, so KIEs afford the opportunity to probe a minimally perturbed reaction coordinate. More importantly, it has been demonstrated by several theoretical studies that the temperature dependence of the KIE is highly sensitive to changes in DAD fluctuations at the TRS of the reaction [6,12,16,18,21–23]. At optimal DADs, there is a sufficient wave function overlap between donor and acceptor at the TRS so that all isotopes of H can tunnel (middle panels of A and B in Figure 1). If this is also a narrow and accurate distribution of DADs, the KIEs will be temperature independent, as most often observed for WT enzymes with their physiological substrates [6,12,16,22–26]. Under non-optimal reaction conditions, which can often be induced through site directed mutagenesis of the enzyme, the average DAD at the TRS is too long for efficient tunneling of heavier isotopes and the DADs’ distribution is broader (poorly organized TRS), which result in inflated and temperature dependent KIEs. This larger temperature dependency is due to thermally activated motions of the protein scaffold that at high temperature lead to a shorter DAD, from which all isotopes can tunnel (small KIE), but at low temperature are confined to a DAD that is too long for heavy isotope tunneling (large KIE). The temperature dependence of the KIE therefore provides an indirect, but very powerful probe of the involvement of enzyme motions in the catalyzed H transfer.
KIEs as a Probe of Local Effects on DHFR Catalyzed Chemistry
The KIEs for WT DHFR have been shown to be temperature independent, which suggests that the enzyme has well defined active site dynamics that maintain a short and narrowly distributed DAD for hydride tunneling [8]. To gain insights into how the dynamics of the active site of DHFR are maintained, I14 was studied trough a series of mutations that were chosen to decrease the side chain while not significantly perturbing the electrostatics of the active site [7,9]. As shown in Figure 2, I14 is positioned behind the nicotinamide ring of NADPH and assists in maintaining an optimal DAD between the cofactor (H donor) and the DHF substrate (H acceptor). Such a role of a hydrophobic residue has also been seen in morphonone reductase [27], horse liver alcohol dehydrodrogenase [28,29] and TauD [30]. I14A was mutated to V, A and G and the temperature dependence of the intrinsic H/T KIEs were measured for each mutant, along with other kinetic parameters. The study afforded both an explanation as to how DHFR maintains an optimal DAD for H tunneling and an important test to Marcus-like models. As seen in Figure 2, the isotope effects on the activation energy for catalyzed reaction (i.e., the temperature dependence of the intrinsic KIEs given by the slopes in the Arrhenius plots) progressively increased as the size of the amino acid at position 14 decreased. Molecular dynamics (MD) simulations of the WT and mutants reveled that the increased flexibility induced upon mutation resulted in populations of the enzyme active complexes whose average DAD is longer and has a broader distribution. Importantly, MD simulations confirmed the predictions of Marcus-like models, which state that longer and more broadly distributed DADs result in steeper temperature dependencies of the KIE.
Figure 2. Role of I14 on Local Motions in DHFR.
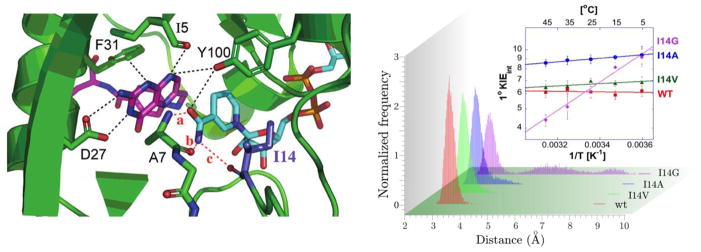
Left Panel: Active Site of E.coli DHFR (PDB ID 1RX2) where the nicotinamide is in blue, the folate in magenta and I14 highlighted as metallic blue. Right Panel. Histogram showing the normalized frequency versus DAD for WT (red), I14V (green), I14A (blue) and I14G (purple) [7]. The inset shows Arrhenius plots for the intrinsic H/T KIEs for the WT and mutants. Reprinted with permission from the American Chemical Society.
KIEs as a Probe of Global Effects on DHFR Catalyzed Chemistry
The studies of I14 serve as a framework for understanding the roles of remote residues whose functions are not as easily explained from 3D structures of the enzyme alone. Several remote residues were proposed to participate in a ‘global dynamic network’ based on quantum mechanical/molecular mechanical (QM/MM) calculations, MD simulations and bioinformatic analysis looking at co-evolution of the enzyme (Figure 3) [4,31–35]. Initial kinetic studies on G121 and M42 mutants implicated a synergy between the two residues in the catalyzed chemistry [36]. This was shown by measuring single turnover rates of G121V and M42W DHFRs and comparing the results to a G121V/M42W double mutant. The effects of the single mutants on single-turnover rates were smaller than the combined effects of both mutations on G121V/M42W DHFR [36]. The temperature dependence of the intrinsic H/T KIEs were measured for each of the mutants and the double mutant, in order to establish a possible link between these remote residues and hydride tunneling occurring at the active site [10,11]. The G121V and M42W single mutants exhibited only a modest increase in the temperature dependence of the KIE, whereas the G121V/M42W double mutant was steeply temperature dependent (Figure 3). Recently, another residue predicted to be part of that network [4,31–35], F125, has been also tested, and F125M and its double mutants with G121V and M42W indicated that this residue is also part of the same network [2]. By contrast, W133, which was predicted to comprise the global network of coupled motions based solely on bioinformatic analysis [34], was found to have no effect on the hydride transfer reaction. This confirms that results obtained with G121, M42 and F125 are associated with a functional role of a dynamic network and not a general role of all residues in the enzyme. These results suggest that motions across the entire protein provide the environmental reorganization needed to maintain the short and narrowly distributed DAD needed for efficient tunneling. The outcome confirmed the predictions of high level QM/MM simulations [4,31,33,35], suggesting those residues are part of the same dynamic network coupled to the chemical step. Importantly, the examination of the temperature dependence of intrinsic KIEs with the double mutants accords with the synergy between the remote residues that was suggested by single-turnover measurements [2,36,37], although those rates may involve more microscopic rate constants than just the C-H activation step.
Figure 3. Roles of Distal Residues on the DHFR catalyzed reaction.
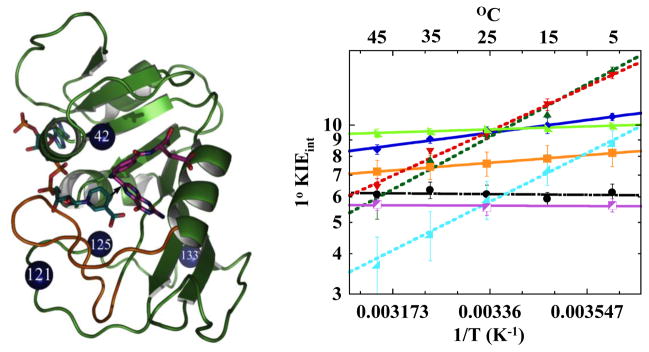
Left Panel. Structure of WT-DHFR (PDB Code 1RX2), with folate in magenta and NADP in light blue. A black arrow marks the hydride’s path from C4 of the nicotinamide to C6 of the folate, and the residues reviewed here are marked as blue spheres. Right Panel. Arrhenius plots of intrinsic H/T KIEs of WT (black) and distal DHFR mutants: W133F (magenta), M42W (orange), G121V (light green), F125M (dark blue), M42W-F125M (dark green), G121V-F125M (red), M42W-G121V (light blue) [2]. Reprinted with permission from the American Chemical Society.
KIEs as a Probe of Conservation of Functional Motions in Enzyme Evolution
A recent analysis of over 200 DHFR sequences from organisms ranging from bacteria to humans have identified phylogenetic coherent events (PCE) in the course of evolution from bacteria to human [38]. In particular the genomic analysis identified N23 and G51 (the numbering corresponds to the E. coli enzyme) as two such PCEs. Residue N23 was previously studied through a diproline insertion to create a N23PP E. coli DHFR (with or without S148A, which was found to have no added effect). NMR relaxation experiments indicated a restriction of msec timescale motions in this mutant as compared to the WT and both steady-state and single turnover rates were significantly decreased [39]. Based on the PCEs identified in ref [38], where N23PP was only present after another insertion at G51, a more ‘humanized’ form of E. coli DHFR (N23PP/G51PEKN) was created. The kinetic parameters of N23PP/G51PEKN were restored to that of the WT and QM/MM simulations showed that the mutant had as short and narrow distribution of DADs as WT E. coli DHFR. This is in contrast to the results of N23PP DHFR, which exhibits a long and broad distribution of DADs as shown by two different QM/MM calculations [38,40]. The Marcus-like model therefore predicts that N23PP should have steeply dependent KIEs, while the N23PP/G51PEKN mutant should exhibit the temperature independent KIEs of the WT. As shown in Figure 4, this is exactly what was observed [1]. The results demonstrate that the dynamics of DHFR are evolutionary preserved, which is interesting given that the chemical step is far from being rate limiting in the DHFR reaction [41].
Figure 4. Evolutionary Preservation of DHFR Dynamics.
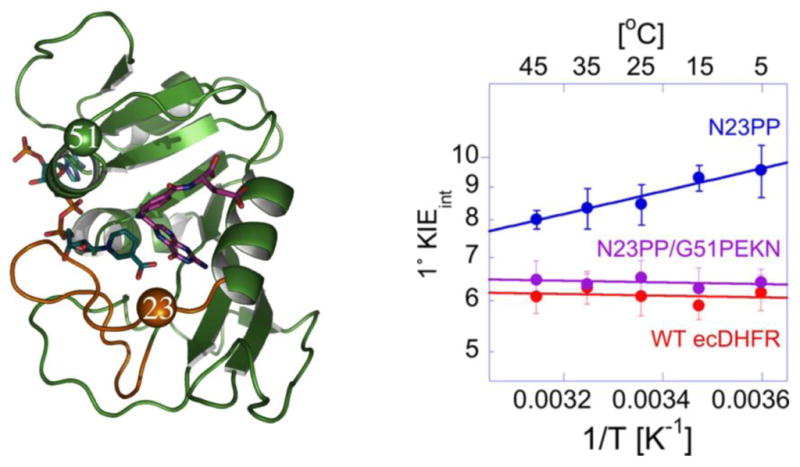
Left Panel: Structure of WT DHFR showing the positions of N23 and G51. The Met-20 loop is shown in brown, folate is shown in purple, and the nicotinamide ring of NADPH is in blue. Residues N23 and G51 are shown as brown and green spheres, respectively. Right Panel: Arrhenius plots for the intrinsic H/T KIEs for the WT, N23PP, N23PP/G51PEKN [1]. Reprinted with permission from the American Society of Biochemistry and Molecular Biology.
It should be noted that the studies of N23PP/S148A was not met without controversy. Empirical valence bond (EVB) calculations suggested that the impaired rates of the mutant are due to a increased reorganization energy for the reaction [42]. Furthermore, studies of the temperature dependence of the KIEs on single turnover rates [43] and a study on an isotopically labeled ‘heavy N23PP/S148A DHFR’ [40] failed to detect any effect of the altered dynamics on the single turnover rates. However, single-turnover rates reflect not only hydride transfer, but a combination of microscopic rate constants, which results in what is known as kinetic complexity [1]. This kinetic complexity masks effects on the chemical step (C-H→C transfer in this case), a problem that can be alleviated by studying intrinsic KIEs as in Figure 4 above [44,45]). Thus, while the mutation may have an effect on the reorganization energy as suggested by the EVB calculations [42], it also influences the dynamics at the TRS and the chemical step of the DHFR catalyzed reaction.
Conclusions
The temperature dependence of intrinsic KIEs, analyzed using Marcus-like models, has been applied to elucidate various aspects of the hydride transfer reaction catalyzed by DHFR. In the framework of these models, the observed temperature independent KIEs indicate that the WT enzyme activates and reorganize the reactants to an optimized TRS with well defined and narrow distribution of DADs, most suitable for H-tunneling. This orientation is achieved through motions of active site residues, solvent molecules, and residues across the protein. Those remote and active site residues constitute a dynamic network of functional correlated motions, directly affecting the formation of the TRS. Phylogenetic analysis of DHFR sequences and functional studies of mutants generated based on those studies suggest that the functional motions under study are evolutionarily conserved. The studies outlined here demonstrate the structural and dynamic role of residues across the protein in enzyme catalyzed C-H→C transfer. The findings may apply not only to other enzymes catalyzing hydride transfer, but may indicate a general role of the whole protein in enzyme catalysis.
Highlights.
The role of protein motions in catalysis by dihydrofolate reductase is reviewed.
Marcus-like models explain the temperature dependence of kinetic isotope effects.
Both local and global motions contribute to dihydrofolate reductase catalysis.
The functional motions of dihydrofolate reductase are evolutionarily conserved.
Acknowledgments
This work was supported by NSF CHE 1149023 and NIH R01 GM65368.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of the review have been highlighted as:
• of special interest
•• of outstanding interest
- 1••.Francis K, Stojkovic V, Kohen A. Preservation of Protein Dymanics in Dihydrofolate Reductase Evolution. J Biol Chem. 2013;288:35961–35968. doi: 10.1074/jbc.M113.507632. Intrinsic H/T kinetic isotope effects for a humanized form of E. coli DHFR (N23PP/G51PEKN) were temperature independent as are that of the wild-type indicating that the dynamics of the enzyme are evolutionary preserved. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh P, Sen A, Francis K, Kohen A. Extension and limits of the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. J Am Chem Soc. 2014 doi: 10.1021/ja411998h. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allemann RK, Evans RM, Loveridge EJ. Probing coupled motions in enzymatic hydrogen tunnelling reactions. Biochem Soc Trans. 2009;37:349–353. doi: 10.1042/BST0370349. [DOI] [PubMed] [Google Scholar]
- 4.Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu Rev Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- 5.Hammes GG, Benkovic SJ, Hammes-Schiffer S. Flexibility, diversity, and cooperativity: pillars of enzyme catalysis. Biochemistry. 2011;50:10422–10430. doi: 10.1021/bi201486f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6••.Klinman JP, Kohen A. Hydrogen Tunneling Links Protein Dynamics to Enzyme Catalysis. Ann Rev Biochem. 2013;82:471–496. doi: 10.1146/annurev-biochem-051710-133623. A comprehensive and up to date review on the development of Marcus-like models of hydrogen tunneling starting with semi-classical views of kinetic isotope effects, continuing with Bell-tunneling correction models and ending with modern full tunneling models and their application to select enzyme systems to elucidate the role of dynamics in catalysis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7••.Stojkovic V, Perissinotti LL, Willmer D, Benkovic SJ, Kohen A. Effects of the donor-acceptor distance and dynamics on hydride tunneling in the dihydrofolate reductase catalyzed reaction. J Am Chem Soc. 2011;134:1738–1745. doi: 10.1021/ja209425w. The temperature dependence of the KIEs of I14 mutants were analyzed to show that the residue modulates the donor and acceptor distance at the tunneling ready state of the wild-type and MD simulations confirmed the predictions of Marcus-like models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sikorski RS, Wang L, Markham KA, Rajagopalan PT, Benkovic SJ, Kohen A. Tunneling and coupled motion in the Escherichia coli dihydrofolate reductase catalysis. J Am Chem Soc. 2004;126:4778–4779. doi: 10.1021/ja031683w. [DOI] [PubMed] [Google Scholar]
- 9.Stojkovic V, Perissinotti LL, Lee J, Benkovic SJ, Kohen A. The effect of active-site isoleucine to alanine mutation on the DHFR catalyzed hydride-transfer. Chem Commun (Camb) 2010;46:8974–8976. doi: 10.1039/c0cc02988b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10•.Wang L, Goodey NM, Benkovic SJ, Kohen A. Coordinated effects of distal mutations on environmentally coupled tunneling in dihydrofolate reductase. Proc Natl Acad Sci U S A. 2006;103:15753–15758. doi: 10.1073/pnas.0606976103. Experimental measurements of single turnover rates and the temperature dependence of intrinsic KIEs for G121V, M42W and G121V/M42W mutants of E. coli DHFR confirmed theoretical simulations proposing a global network of couple motions across the enzyme to promote hydride tunneling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, Goodey NM, Benkovic SJ, Kohen A. The role of enzyme dynamics and tunnelling in catalysing hydride transfer: studies of distal mutants of dihydrofolate reductase. Philos Trans R Soc Lond B Biol Sci. 2006;361:1307–1315. doi: 10.1098/rstb.2006.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hammes-Schiffer S. Hydrogen tunneling and protein motion in enzyme reactions. Acc Chem Res. 2006;39:93–100. doi: 10.1021/ar040199a. [DOI] [PubMed] [Google Scholar]
- 13.Kohen A. Kinetic isotope Effects as Probes for hydrogen Tunneling in Enzyme Catalysis. In: Kohen A, Limbach HH, editors. Isotopes Effects in Chemistry and Biology. Taylor and Francis; 2006. pp. 743–764. [Google Scholar]
- 14.Kuznetsov AM, Ulstrup J. Proton and Hydrogen Tunneling in Hydrolytic and Redox Enzyme Catalysis. Can J Chem. 1999;77:1085–1096. [Google Scholar]
- 15.Marcus RA. H and other transfers in enzymes and in solution: theory and computations, a unified view. 2. Applications to experiment and computations. J Phys Chem B. 2007;111:6643–6654. doi: 10.1021/jp071589s. [DOI] [PubMed] [Google Scholar]
- 16.Nagel ZD, Klinman JP. Update 1 of: Tunneling and dynamics in enzymatic hydride transfer. Chem Rev. 2010;110:PR41–67. doi: 10.1021/cr1001035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwartz SD. Vibrationally Enhanced Tunneling and kinetic isotope effects in Enzymatic Reactions. In: Kohen A, Limbach HH, editors. Isotope effects in chemistry and biology. CRC Press: Taylor and Francis; 2006. pp. 475–498. [Google Scholar]
- 18.Wang Z, Roston D, Kohen A. Experimental and theoretical studies of enzyme-catalyzed hydrogen transfer reactions. In: Christov CZaK-C T., editor. Structural and Mechanistic Enzymology: Bringing together Experiments and Computing. Vol. 87. Elsevier Inc; 2012. pp. 155–180. [DOI] [PubMed] [Google Scholar]
- 19.Hay S, Sutcliffe MJ, Scrutton N. Probing Coupled Motions in Enzymatic Hydrogen Tunnelling Reactions: Beyond Temperature-Dependence Studies of Kinetic Isotope Effects. In: Allemann R, Scrutton N, editors. Quantum tunnelling in enzyme-catalyzed reactions. Royal Society of Chemistry; 2009. pp. 199–218. [Google Scholar]
- 20.Marcus RA, NS Electron transfers in chemistry and biology. BBA-Bioenergetics. 1985;811:265–322. [Google Scholar]
- 21.Antoniou D, Caratzoulas S, Kalyanaraman C, Mincer JS, Schwartz SD. Barrier passage and protein dynamics in enzymatically catalyzed reactions. Eur J Biochem. 2002;269:3103–3112. doi: 10.1046/j.1432-1033.2002.03021.x. [DOI] [PubMed] [Google Scholar]
- 22.Roston D, Cheatum CM, Kohen A. Hydrogen donor-acceptor fluctuations from kinetic isotope effects: a phenomenological model. Biochemistry. 2012;51:6860–6870. doi: 10.1021/bi300613e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutcliffe MJ, Masgrau L, Roujeinikova A, Johannissen LO, Hothi P, Basran J, Ranaghan KE, Mulholland AJ, Leys D, Scrutton NS. Hydrogen tunnelling in enzyme-catalysed H-transfer reactions: flavoprotein and quinoprotein systems. Philos Trans R Soc Lond B Biol Sci. 2006;361:1375–1386. doi: 10.1098/rstb.2006.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hay S, Scrutton NS. Good vibrations in enzyme-catalysed reactions. Nat Chem. 2012;4:161–168. doi: 10.1038/nchem.1223. [DOI] [PubMed] [Google Scholar]
- 25.Kanaan N, Ferrer S, Marti S, Garcia-Viloca M, Kohen A, Moliner V. Temperature dependence of the kinetic isotope effects in thymidylate synthase. A theoretical study. J Am Chem Soc. 2012;133:6692–6702. doi: 10.1021/ja1114369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan F, Gadda G. Oxygen- and temperature-dependent kinetic isotope effects in choline oxidase: correlating reversible hydride transfer with environmentally enhanced tunneling. J Am Chem Soc. 2005;127:17954–17961. doi: 10.1021/ja0560377. [DOI] [PubMed] [Google Scholar]
- 27.Pudney CR, Johannissen LO, Sutcliffe MJ, Hay S, Scrutton NS. Direct analysis of donor-acceptor distance and relationship to isotope effects and the force constant for barrier compression in enzymatic H-tunneling reactions. J Am Chem Soc. 2010;132:11329–11335. doi: 10.1021/ja1048048. [DOI] [PubMed] [Google Scholar]
- 28.Bahnson BJ, Colby TD, Chin JK, Goldstein BM, Klinman JP. A link between protein structure and enzyme catalyzed hydrogen tunneling. Proc Natl Acad Sci U S A. 1997;94:12797–12802. doi: 10.1073/pnas.94.24.12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colby TD, Bahnson BJ, Chin JK, Klinman JP, Goldstein BM. Active site modifications in a double mutant of liver alcohol dehydrogenase: structural studies of two enzyme-ligand complexes. Biochemistry. 1998;37:9295–9304. doi: 10.1021/bi973184b. [DOI] [PubMed] [Google Scholar]
- 30.McCusker KP, Klinman JP. An active-site phenylalanine directs substrate binding and C-H cleavage in the alpha-ketoglutarate-dependent dioxygenase TauD. J Am Chem Soc. 2010;132:5114–5120. doi: 10.1021/ja909416z. [DOI] [PubMed] [Google Scholar]
- 31.Agarwal PK, Billeter SR, Rajagopalan PT, Benkovic SJ, Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proc Natl Acad Sci U S A. 2002;99:2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radkiewicz JL, Brooks CL. Protein Dynamics in Enzymatic Catalysis: Exploration of Dihydrofolate Reductase. J Am Chem Soc. 2000;122:255–231. [Google Scholar]
- 33.Rod TH, Brooks CL., 3rd How dihydrofolate reductase facilitates protonation of dihydrofolate. J Am Chem Soc. 2003;125:8718–8719. doi: 10.1021/ja035272r. [DOI] [PubMed] [Google Scholar]
- 34.Suel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Mol Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 35••.Wong KF, Selzer T, Benkovic SJ, Hammes-Schiffer S. Impact of distal mutations on the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. Proc Natl Acad Sci U S A. 2005;102:6807–6812. doi: 10.1073/pnas.0408343102. QM/MM calculations on DHFR suggest that a network of coupled motions across the enzyme promote the hydride transfer reaction and allowed for the prediction of relevant distal residues that comprise that network. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajagopalan PTR, Stefan L, Benkovic SJ. Coupling Interactions of Distal Residues Enhance Dihydrofolate Reductase Catalysis: Mutaional Effects on Hydride Transfer Rates. Biochemistry. 2002;41:12618–12628. doi: 10.1021/bi026369d. [DOI] [PubMed] [Google Scholar]
- 37.Huang Z, Wagner CR, Benkovic SJ. Nonadditivity of mutational effects at the folate binding site of Escherichia coli dihydrofolate reductase. Biochemistry. 1994;33:11576–11585. doi: 10.1021/bi00204a020. [DOI] [PubMed] [Google Scholar]
- 38•.Liu CT, Hanoian P, French JP, Pringle TH, Hammes-Schiffer S, Benkovic SJ. Functional Significance of Evolving Protein Sequence in Dihydrofolate Reductase from Bacteria to Human. Proc Natl Acad Sci U S A. 2013;110:10159–10164. doi: 10.1073/pnas.1307130110. An extensive phylogentic analysis of 233 DHFR sequences reveal phylogenetically coherent events which were used to generate a humanized form of E. coli DHFR in which the effects of N23PP on the single turnover rates were overcome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhabha G, Lee J, Ekiert DC, Gam J, Wilson IA, Dyson HJ, Benkovic SJ, Wright PE. A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science. 2011;332:234–238. doi: 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruiz-Pernia JJ, Luk LY, Garcia-Meseguer R, Marti S, Loveridge EJ, Tunon I, Moliner V, Allemann RK. Increased Dynamic Effects in a Catalytically Compromised Variant of Escherichia coli Dihydrofolate Reductase. J Am Chem Soc. 2013;135:18689–18696. doi: 10.1021/ja410519h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fierke CA, Johnson KA, Benkovic SJ. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry. 1987;26:4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- 42.Adamczyk AJ, Cao J, Kamerlin SC, Warshel A. Catalysis by dihydrofolate reductase and other enzymes arises from electrostatic preorganization, not conformational motions. Proc Natl Acad Sci U S A. 2011;108:14115–14120. doi: 10.1073/pnas.1111252108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loveridge EJ, Behiry EM, Guo J, Allemann RK. Evidence that a ‘dynamic knockout’ in Escherichia coli dihydrofolate reductase does not affect the chemical step of catalysis. Nat Chem. 2012;4:292–297. doi: 10.1038/nchem.1296. [DOI] [PubMed] [Google Scholar]
- 44.Cook PF, Cleland WW. In: Enzyme Kinetics and Mechanism. Cook PF, Cleland WW, editors. NewYork, NY: Garland Publishing Inc; 2007. [Google Scholar]
- 45.Sen A, Yahashiri A, Kohen A. Triple isotopic labeling and kinetic isotope effects: exposing H-transfer steps in enzymatic systems. Biochemistry. 2011;50:6462–6468. doi: 10.1021/bi2003873. [DOI] [PMC free article] [PubMed] [Google Scholar]