

ABSTRACT
The fibronectin matrix plays a crucial role in the regulation of angiogenesis during development, tissue repair and pathogenesis. Previous work has identified a fibronectin-derived homophilic binding peptide, anastellin, as an effective inhibitor of angiogenesis; however, its mechanism of action is not well understood. In the present study, we demonstrate that anastellin selectively inhibits microvessel cell signaling in response to the VEGF165 isoform, but not VEGF121, by preventing the assembly of the complex containing the VEGF receptor and neuropilin-1. Anastellin treatment resulted in the inactivation of α5β1 integrins but was not accompanied by a change in either adhesion complexes or adhesion-based signaling. Integrin inactivation was associated with a masking of the fibronectin synergy site within the extracellular matrix (ECM), indicating that α5β1 inactivation resulted from a decrease in available ligand. These data demonstrate that anastellin influences the microvessel cell response to growth factors by controlling the repertoire of ligated integrins and point to anastellin as an effective regulator of fibronectin matrix organization. These studies further suggest that homophilic fibronectin binding peptides might have novel applications in the field of tissue regeneration as tools to regulate neovascularization.
KEY WORDS: Fibronectin, Integrin, Angiogenesis, Neuropilin, VEGF, Anastellin, Extracellular matrix
INTRODUCTION
Fibronectin is a high-molecular-mass (450-kDa) protein found in plasma and also synthesized locally within tissues. Fibronectin is an integral matrix protein that is polymerized into an extracellular network of interacting fibers that function to support cell adhesion, migration, proliferation and survival. Genetic studies in mice have indicated that fibronectin and its integrin receptors are essential to the processes of both angiogenesis and vasculogenesis (reviewed in Argraves and Drake, 2005; Hynes, 2007). Structurally, fibronectin consists of repeating individually folded domains termed type I, II and III based on shared amino-acid homology. The type I and II domains are further stabilized by intra-domain disulfide bonds. Chemical and mechanical signals emanating from the fibronectin matrix are transduced through the integrin family of adhesion receptors. Matrix-based signals integrate with those from signaling pathways initiated through growth factors, cytokines and intercellular adhesion receptors (Schwarzbauer and DeSimone, 2011). It is through these matrix-based signaling networks that fibronectin exerts its influence over nearly all aspects of endothelial cell biology (Astrof and Hynes, 2009; Malinin et al., 2012). The fibronectin matrix is a dynamic structure that undergoes constant assembly and turnover. Changes in the synthesis of fibronectin isoforms coupled with contraction-mediated changes in protein tertiary and secondary structure result in changes in the availability and spacing of biologically active sites within the polymerized fibronectin fibers. Because they are not stabilized by disulfide bonds, the type III domains are particularly sensitive to mechanical unfolding. Therefore, alternative splicing and force-induced unfolding of fibronectin type III domains provide the cell with numerous opportunities to modulate the topographical display of bioactive sites within the matrix (Klotzsch et al., 2009; White and Muro, 2011).
Angiogenesis is a process of neovascularization that occurs during development and tissue repair to meet the nutrient and gas-exchange demands of remodeling tissue. Angiogenesis is also an essential step in pathological conditions such as cancer, where it provides the growing tumor with a blood supply. Angiogenesis is regulated by the vascular endothelial growth factor (VEGF) family and its receptors. There are five human genes for VEGF. VEGFA and its receptors, VEGFR2 and neuropilin-1 (NRP1), are known to be the major regulators of angiogenesis. VEGFR2 is the primary VEGFA receptor on endothelial cells and is essential for vascular development and angiogenesis, during which it regulates proliferation, migration and tube formation (reviewed in Koch and Claesson-Welsh, 2012). VEGFA can be processed into various isoforms through alternative splicing mechanisms, resulting in both pro- and anti-angiogenic subtypes (Nowak et al., 2008). NRP1 is a transmembrane protein that binds to certain isoforms of VEGFA and works in concert with VEGFR2 to promote VEGFA signaling. The proangiogenic isoforms, VEGF165 and VEGF121, are the primary isoforms of VEGFA. They have distinct but overlapping functions and differ in their requirement for NRP1 to mediate VEGFR2 activation and downstream signaling. Genetic studies of NRP1 in mice have demonstrated a role for NRP1 in both vascular development and angiogenesis (Kawasaki et al., 1999). The mechanism of action of NRP1 is not well understood but it appears to promote VEGF signaling by regulating VEGFR2 internalization and trafficking (Ballmer-Hofer et al., 2011).
The assembly of the fibronectin matrix is a cell-dependent stepwise process that first requires the binding of soluble protomeric fibronectin to the cell surface, followed by a series of homophilic binding events that promote the assembly of fibronectin monomers into the detergent-insoluble polymers that comprise the extracellular matrix (ECM) (reviewed in Xu and Mosher, 2011). Previous work has led to the identification of fibronectin-derived cryptic sequences that can promote fibronectin polymerization in the absence of cells. Cryptic self-polymerization activity has been identified in the III1 and III10 domains of fibronectin (Gee et al., 2013; Hocking et al., 1996; Morla et al., 1994). The cryptic polymerization activity present in the III1 domain of fibronectin can be recapitulated in a peptide termed anastellin (Morla et al., 1994; Morla and Ruoslahti, 1992). Subsequent studies showed that anastellin can bind directly to previously assembled fibronectin matrix and alter its conformation (Klein et al., 2003; Prabhakaran et al., 2009). Anastellin has been found to inhibit tumor angiogenesis in vivo as well as endothelial cell proliferation in vitro; however, its mechanism of action has remained elusive (Ambesi et al., 2005; Yi and Ruoslahti, 2001).
In the present study, we show that anastellin selectively inhibits VEGF165 signaling by preventing the formation of the VEGFR2–NRP1 complex. Consistent with this observation, anastellin had no effect on signaling from VEGF121, which does not require NRP1 for signaling. Loss of VEGFR2–NRP1 signaling was associated with the decreased availability of the synergy site within the fibronectin matrix and the inactivation of α5β1 integrin. The decrease in β1 activation state had no effect on cell adhesion or adhesion-based signaling. Our data suggest that the inhibitory effect on angiogenesis results from a conformational reorganization of the fibronectin matrix that alters the repertoire of ligated integrin receptors. Our data also implicate α5β1 integrin as a functional regulator of NRP1. These data point to a role for α5β1 in the regulation of VEGFR2 trafficking and further suggest that targeting ECM topography could provide a novel approach for the selective modification of the endothelial cell response to angiogenic growth factors.
RESULTS
Anastellin inhibits VEGF165-dependent signaling in microvessel cells
To determine the effects of anastellin on VEGF165-dependent microvessel cell proliferation, VEGF165 was incubated with human dermal microvessel cells that had been pretreated with increasing amounts of anastellin or a control fibronectin type III domain, FnIII13. The addition of anastellin to microvessel endothelial cells prevented cell proliferation in response to VEGF165 (Fig. 1A) while completely inhibiting the activation of ERK (also known as MAPK) (Fig. 1B,C), suggesting that anastellin was inhibiting proliferation by blocking signaling from growth factor receptors. The binding of VEGF165 to its receptor, VEGFR2, leads to the dimerization and activation of the VEGFR2 receptor kinase, which phosphorylates VEGFR2 at specific tyrosine residues. Phosphorylation of VEGFR2 at Tyr1175 is a crucial step in the downstream activation of ERK and is essential for VEGF-induced angiogenesis (Takahashi et al., 2001). As shown in Fig. 1D,E, the phosphorylation of VEGFR2 at Tyr1175 in response to VEGF165 remained near baseline in anastellin-treated cells. The inhibitory effect of anastellin on VEGFR2 Tyr1175 phosphorylation was dose dependent and occurred over the same dose range that blocked cell proliferation. Anastellin also inhibited the VEGF165-dependent phosphorylation of VEGFR2 on Tyr residues 1059 and 1054 (Fig. 1F,G).
Fig. 1.

Anastellin inhibits VEGF-dependent microvessel endothelial cell proliferation and receptor activation. (A) Human dermal microvessel cells were cultured for 3 days in medium supplemented with or without (‘C’) 10 ng/ml VEGF165 in the presence of increasing concentrations of anastellin. FnIII13 served as a negative control. Cells were fixed and proliferation was determined by Toluidine Blue staining. Anastellin significantly inhibited proliferation in response to VEGF165 at all doses. Data show the mean±s.e.m.; *P<0.001. (B,D,F) Microvessel cells were starved overnight in serum-free medium containing 0.5% BSA and pretreated for 60 minutes with increasing concentrations of anastellin prior to stimulation with 10 ng/ml VEGF165 for 6 minutes. Cell lysates were analyzed by western blotting for phospho-ERK1/2 and VEGFR2 phosphorylation. Total ERK2 and VEGFR2 staining served as loading controls. (C,E,G) Western blots from the experiments shown in panels B,D and F were quantified using ImageJ software. Data show the mean±s.e.m. (three independent experiments).
Anastellin inhibits the assembly of the VEGFR2–NRP1 receptor complex
In response to VEGF165, VEGFR2 forms a complex on the cell surface with its co-receptor, NRP1. VEGF165 binds to both VEGFR2 and NRP1, and the resulting complex promotes receptor activation and downstream signaling. To investigate a role for NRP1 in the regulation of VEGF165 signaling in microvessel cells, a blocking antibody against NRP1 was tested for its effect on VEGF165 signaling. As shown in Fig. 2A, the NRP1-blocking antibody completely inhibited the phosphorylation of both VEGFR2 and ERK in response to VEGF165. Similar findings were obtained under conditions of NRP1 knockdown, where VEGF165-dependent activation of both VEGFR2 and ERK were markedly reduced (Fig. 2B). The relative levels of phosphorylation of VEGFR and ERK in control and NRP1-knockdown cells were quantified by scanning of western blots (Fig. 2C,D). These data suggest that NRP1 plays a significant role in VEGF165-mediated signaling in microvessel endothelial cells. Consistent with its effect on cell signaling, NRP1 was also required for VEGF165-dependent cell growth. As shown in Fig. 2E, cell proliferation in response to VEGF165 was inhibited by ∼60% under conditions of NRP1 knockdown. To examine the effect of anastellin on the formation of the VEGFR2–NRP1 complex, co-immunoprecipitation experiments were performed in the presence and absence of anastellin. As shown in Fig. 2F, immunoprecipitation of NRP1 from VEGF165-treated cells resulted in the co-precipitation of VEGFR2. There was no VEGFR2 detected in the precipitates in the absence of VEGF165. Pretreatment of cells with anastellin caused a marked reduction in the amount of VEGFR2 that was co-precipitated with NRP1, consistent with anastellin disrupting the assembly of the VEGFR2–NRP1 complex in response to VEGF165. These findings confirm a role for NRP1 in VEGFR2 activation and downstream signaling in response to VEGF165 and are consistent with anastellin inhibiting VEGFR2 signaling by preventing the assembly of the VEGFR2–NRP1 complex.
Fig. 2.

Anastellin disrupts VEGFR2–NRP1 complex assembly and signaling. (A) Serum-starved microvessel cell monolayers were pretreated for 2 hours with increasing concentrations of an NRP1-function-blocking antibody and then stimulated with 5 ng/ml VEGF165 for 6 minutes. Cell lysates were analyzed by western blotting for phosphorylation of VEGFR2 and ERK. (B) Microvessel cells treated with either siRNA against NRP1 or control siRNA were stimulated with increasing concentrations of VEGF165 for 6 minutes and analyzed for the phosphorylation of VEGFR2 and ERK by western blotting. Total VEGFR2 and ERK2 served as loading controls. (C,D) Blots from B were quantified using ImageJ software. Data show the mean±s.e.m. (three independent experiments). (E) Microvessel cells were grown for 3 days in basal medium containing the indicated amount of VEGF165. Cells were fixed and cell counts were obtained by Toluidine Blue staining. Knockdown of NRP1 significantly inhibited cell growth. Data show the mean±s.e.m. (three independent experiments); *P<0.001. (F) Microvessel cells were treated for 60 minutes with 20 µM anastellin prior to stimulation with 10 ng/ml VEGF165 for 15 minutes. Cell monolayers were washed, and NRP1 was immunoprecipitated (IP) from cell lysates. Immunoprecipitates were analyzed by western blotting for co-precipitation of VEGFR2. Input amounts of VEGFR2 and NRP1 present in lysates prior to immunoprecipitation are shown on the left.
Anastellin inhibits signaling from VEGF165 but not VEGF121
Our data indicate that anastellin blocks VEGF signaling by preventing the formation of VEGFR2–NRP1 complexes. We therefore evaluated the response of anastellin-treated microvessel cells to an isoform of VEGF, VEGF121, which, unlike VEGF165, is not dependent on NRP1 for signaling through VEGFR2 (Pan et al., 2007b). As shown in Fig. 3A, pretreatment of microvessel cells with increasing doses of anastellin had no effect on the phosphorylation of VEGFR2 by VEGF121. A similar effect of anastellin was seen on ERK activation (Fig. 3A). By contrast, the phosphorylation of VEGFR2 in response to VEGF165 was nearly completely inhibited by anastellin. These data are quantified in Fig. 3B. Consistent with this observation, VEGF121 was less effective in mediating the formation of VEGFR2–NRP1 complexes. As shown in Fig. 3C, treatment of cells with VEGF165 resulted in the formation of a VEGFR2–NRP1 complex. In the presence of VEGF165, the immunoprecipitation of NRP1 resulted in the co-precipitation of VEGFR2. As expected, the formation of this complex was prevented when cells were pretreated with anastellin. By contrast, VEGF121-treated cells exhibited much less VEGFR2 in NRP1 immunoprecipitates, and this complex was unaffected by anastellin. These data indicate that anastellin selectively inhibits VEGF165 signaling by disrupting the association of VEGFR2 with NRP1.
Fig. 3.

Anastellin has no effect on VEGF121-induced VEGFR2 activation and signaling. (A) Serum-starved microvessel cell monolayers were pretreated for 60 minutes with increasing concentrations of anastellin, followed by stimulation with either 10 ng/ml VEGF121 or VEGF165 for 6 minutes. Cell lysates were analyzed by western blotting for changes in the phosphorylation of VEGFR2 and ERK. Total levels of VEGFR2 served as a loading control. (B) Blots were quantified using ImageJ software. Data show the mean±s.e.m. (three independent experiments). ‘C’, untreated control. (C) Microvessel cell monolayers were pretreated with 20 µM anastellin for 60 minutes prior to stimulation with 10 ng/ml VEGF165 or VEGF121 for 15 minutes. NRP1 was immunoprecipitated (IP) from all lysates, and precipitates were analyzed by western blotting for the presence of VEGFR2. Input amounts of VEGFR2 and NRP1 present in lysates prior to immunoprecipitation are shown on the left.
The matrix remodeling activity of anastellin is required for the inhibitory effects of anastellin on growth factor signaling
Earlier studies have shown that anastellin binds to soluble protomeric fibronectin and promotes its self polymerization (Morla et al., 1994). Anastellin also binds to polymerized matrix fibronectin, causing conformational changes within matrix fibrils that selectively impact on the availability of specific fibronectin epitopes (Klein et al., 2003). A mutant form of anastellin, L37A/Y40A, has been described, which retains fibronectin-binding activity but is significantly less effective at promoting conformational changes in fibronectin (Briknarová et al., 2003; You et al., 2009). To determine whether anastellin-mediated conformational changes in the fibronectin matrix were required for the effects of anastellin on VEGF165 signaling, microvessel cells pretreated with either anastellin or the anastellin mutant were compared for their ability to respond to VEGF165. As shown in Fig. 4A, anastellin caused a dose-dependent inhibition of both VEGFR2 and ERK phosphorylation in response to VEGF165 challenge. By contrast, the L37A/Y40A mutant form of anastellin had little effect on the phosphorylation of either VEGFR2 or ERK in response to VEGF165. These data are quantified in Fig. 4B. Mutant anastellin was also less effective than wild-type anastellin in blocking the VEGF165-induced complex formation between NRP1 and VEGFR2 (Fig. 4C). The negative control, FnIII13, had no effect on complex formation. These results indicate that the matrix conformation remodeling activity of anastellin is required for its inhibitory effect on VEGF165 signaling.
Fig. 4.

Anastellin inhibition of VEGF165 signaling requires fibronectin matrix remodeling. (A) Serum-starved microvessel cell monolayers were pretreated with increasing concentrations of anastellin or the anastellin mutant for 60 minutes prior to stimulation with 10 ng/ml VEGF165 for 6 minutes. Cell lysates were analyzed by western blotting for phosphorylated ERK and VEGFR2. Total VEGFR2 and ERK2 served as loading controls. (B) VEGF-dependent phosphorylation of Y1175VEGFR2 in the presence of 20 µm anastellin or anastellin mutant was quantified by scanning blots from three separate experiments. ‘C’, untreated control. Data show the mean±s.e.m. (C) Microvessel cells were treated with 20 µM anastellin, anastellin mutant or the control fibronectin module FnIII13 for 60 minutes prior to stimulation with 10 ng/ml VEGF165 for 15 minutes. NRP1 was immunoprecipitated (IP) from cell lysates. Immunoprecipitates were analyzed by western blotting for co-precipitation of VEGFR2. Input amounts of VEGFR2 and NRP1 present in lysates prior to immunoprecipitation are shown on the left.
Anastellin decreases the activation state of the α5β1 integrin
Earlier studies have shown that cell adhesion is a crucial regulator of growth factor signaling in endothelial cells. Adhesion increases the strength and duration of signaling in response to VEGF165 as well as other growth factors (Cabodi et al., 2010). To assess whether anastellin might be affecting the adhesion of microvessel cells to the matrix, microvessel cells were incubated with anastellin for 60 minutes and active β1 integrins were imaged by immunofluorescence using the 9EG7 antibody, which recognizes the high-affinity ligand-bound conformation of β1 integrin. As shown in Fig. 5A, in control cells, β1 integrin was localized in clusters typical of adhesion complexes. Following a 1-hour treatment with anastellin, 9EG7 staining was nearly completely absent, indicative of a change in the activation state of β1 integrin. The control fibronectin domain, FnIII13, had no effect on 9EG7 staining. Similar results were seen using another antibody (12G10) that also recognizes an active β1 integrin conformation (data not shown). Quantification of contacts recognized by 9EG7 (Fig. 5B) indicated that anastellin reduced the number of contacts containing active β1 integrin by ∼80%. Interestingly, anastellin had no effect on the organization of actin, suggesting that integrin inactivation was not affecting overall cell adhesion (Fig. 5C). By contrast, anastellin had no effect on the localization of β5 integrin, which remained associated with adhesion complexes (Fig. 5D,E). As shown in Fig. 5F, all the β1 adhesions co-stained for α5 integrin, suggesting that α5β1 integrin is the predominant fibronectin-binding integrin on endothelial microvessel cells. Both integrin subunits were lost from adhesion sites following treatment with anastellin, suggesting that the inactivation of β1 integrin was associated with a decrease in α5β1 binding to the fibronectin matrix. Loss of both α5 and β1 subunits from adhesion sites occurred between 20 and 60 minutes following the addition of anastellin (Fig. 6A). Similarly, western blot analysis (Fig. 6B,C) showed that the inhibitory effect of anastellin on VEGF165-mediated activation of ERK was seen after cells were pretreated with anastellin for 10–60 minutes. As a control, anastellin and VEGF165 were preincubated for 30 minutes prior to being added to cells. VEGF165 that had been preincubated with anastellin was still able to elicit a full activation of ERK, suggesting that anastellin was inhibiting VEGF165 signaling by acting on the cells and not on VEGF165 (Fig. 6C, last bar). The inhibitory effects of anastellin were reversible. Removal of anastellin from the medium resulted in the reappearance of α5β1 integrin at adhesion sites, and the amount of α5β1 integrin returned to control levels within 60 minutes (Fig. 6D). Similarly, VEGF165-mediated activation of ERK was also returned to control levels within 60 minutes following the removal of anastellin from the cells (Fig. 6E,F). These data suggest that the inhibitory effects of anastellin on both α5β1 integrin activation and VEGF165 signaling occur within 30–60 minutes and are completely reversible within 1 hour of anastellin removal.
Fig. 5.

Anastellin causes inactivation of β1 integrin. Microvessel cells were treated with vehicle (control), 20 µM anastellin or the control fibronectin module FnIII13 for 60 minutes. (A) Cells were washed, fixed and stained for active β1 integrin using the monoclonal antibody 9EG7. Nuclei were counterstained with Hoechst 33258. (B) NIH ImageJ software was used to quantify the number of β1-containing focal adhesions obtained from at least five fields of view. Data are shown normalized to control values. Data averaged over five independent experiments are represented as the mean±s.e.m.; *P<0.001. (C) Cells were stained for both actin (red) and active β1 integrin (green) using phalloidin and the 9EG7 antibody, respectively. Control cells show β1 integrin localized at actin termini in focal adhesions (arrowheads). (D,E) Microvessel cells were stained with a monoclonal antibody directed against integrin β5 and quantified as in B. (F) Microvessel cells were dual stained for β1 (9EG7, green) and α5 (AB1949, red) integrin subunits. Merged images are shown on the right. Scale bars: 25 µm.
Fig. 6.

Inactivation of α5β1 integrin and VEGFR2 by anastellin is both time dependent and reversible. (A) Microvessel cells were treated with 20 µM anastellin for the times indicated, washed, fixed and stained for β1 (9EG7, green) and α5 (AB1949, red) integrins. Nuclei were counterstained with Hoechst 33258. (B) Serum-starved microvessel cells were pretreated with 20 µM anastellin for increasing amounts of time up to 60 minutes prior to stimulation with 10 ng/ml VEGF165 for 6 minutes. Alternatively, VEGF165 and anastellin (VEGF/Anastellin) were mixed (10 ng/ml VEGF165, 20 µM anastellin) and incubated for 30 minutes prior to addition to cell monolayers for 6 minutes. ‘C’, vehicle (PBS) control. Phosphorylation of ERK was examined by western blotting. Blots were stripped and reprobed for ERK2, which served as a loading control. (C) ERK phosphorylation was quantified from three independent experiments using ImageJ software and was normalized to pERK levels obtained in the absence of anastellin. Data show the mean±s.e.m. (D) Microvessel cells were treated with 20 µM anastellin for 60 minutes and were incubated with serum-free medium (no anastellin) for the indicated times. Cells were then washed, fixed and dual stained for integrin β1 (9EG7, green) and α5 (AB1949, red) subunits. Nuclei were counterstained with Hoescht 33258. Control cells received no anastellin treatment. Scale bars: 10 µm. (E) Microvessel cells were pretreated with 20 µM anastellin for 60 minutes. The medium was replaced with serum-free medium. At the indicated times, cells were stimulated with 10 ng/ml VEGF165 for 6 minutes, and lysates were analyzed by western blotting for phosphorylated ERK. Blots were stripped and reprobed for ERK2, which served as loading control. (F) ERK phosphorylation was quantified from three independent experiments using ImageJ software and was normalized to VEGF-stimulated levels of pERK observed in the absence of anastellin (data not shown). Data show the mean±s.e.m.
Anastellin-dependent inactivation of β1 integrin is not accompanied by decreases in either adhesion sites or adhesion-based signaling
To determine whether the anastellin-mediated inactivation of β1 integrin resulted in changes in the activation of adhesion-based signaling molecules, cells were analyzed for the presence of activated paxillin and focal adhesion kinase (FAK, also known as PTK2). Paxillin and FAK are phosphorylated in response to integrin ligation and are localized to adhesion sites. As shown in Fig. 7A, microvessel cells contained clustered β1 integrins that colocalized with paxillin at adhesion sites. Following the addition of anastellin to microvessel cells, 9EG7 staining for β1 integrin was completely lost from adhesion sites but the number of paxillin-containing contacts was unchanged (Fig. 7B), suggesting that the inactivation of β1 integrin was not accompanied by a disassembly of the adhesion complex. Integrin adhesion results in the tyrosine phosphorylation of paxillin by adhesion-activated kinases, such as Src and FAK. Following loss of adhesion, paxillin is rapidly dephosphorylated by phosphatases (Retta et al., 1996). To evaluate the effect of anastellin on the phosphorylation status of paxillin, paxillin was immunoprecipitated from cell lysates and tyrosine phosphorylation evaluated by western blotting. As shown in Fig. 7C, phosphorylated paxillin was not seen in suspended microvessel cells but was seen in adherent cells. The amount of phosphorylated paxillin was unchanged in cells treated with anastellin or the control type III module, FnIII13, suggesting that anastellin was not affecting the level of phosphorylated paxillin. A similar effect was seen on the localization and phosphorylation of FAK. Microvessel cells treated with anastellin or the control module FnIII13 were analyzed for the presence of phosphorylated FAK (pY397FAK), an immediate downstream target of activated integrins. The addition of anastellin to cells resulted in a loss of active β1 integrins but no detectable change in the localization and number of pY397FAK-containing adhesions (Fig. 7D,E). Analysis of cell lysates by western blotting showed that anastellin had no effect on the level of pY397FAK within the cell (Fig. 7F). These data suggest that anastellin acts selectively on the activation state of the α5β1 integrin present in the adhesion site, decreasing its activation state without dissembling the adhesion complex and without impacting on the overall organization of actin.
Fig. 7.

Anastellin does not decrease the number of focal adhesion complexes. (A) Microvessel cell monolayers were treated with 20 µM anastellin for 60 minutes and stained for both active β1 integrin (green) and paxillin (red). (B) Microvessel cells were treated as in A, and paxillin-containing contacts were quantified by ImageJ software as described for Fig. 5. Data show the mean±s.e.m. (six fields of view, four independent experiments). (C) Paxillin was immunoprecipitated (IP) from microvessel cell lysates prepared from cells treated with 20 µM anastellin or the control fibronectin module, FnIII13. Lysates prepared from cells kept in suspension (Susp) served as a control for the loss of adhesion, whereas those prepared from cells treated with PBS served as adherent untreated control. Immunoprecipitates were analyzed by western blotting for phosphotyrosine (pTyr) using the monoclonal antibody 4G10. Blots were stripped and reprobed for paxillin, which served as the loading control. (D) Cells were treated as described in A and stained with antibody against phosphorylated FAK (pTyr397FAK, red). Scale bars: 10 µm. (E) pTyr397FAK-containing focal adhesions were quantified by ImageJ software. Data show the mean±s.e.m. (six fields of view, three independent experiments). (F) Lysates from cells treated as described in C were probed for pTyr397FAK by western blotting. Blots were stripped and reprobed for total FAK, which served as loading control.
Anastellin inactivates the synergy site of fibronectin
Both the αvβ5 and α5β1 integrins bind to the RGD-containing III10 domain of fibronectin; however, full adhesion of the α5β1 integrin also requires α5 binding to the synergy site within the adjacent III9 domain (Aota et al., 1994). Therefore, we evaluated whether the binding of anastellin to the fibronectin matrix was impacting on synergy site accessibility, thereby specifically targeting the α5β1 integrin. Fig. 8A shows that the fibronectin matrix could be visualized in control cells by immunofluorescence using monoclonal antibodies directed against the synergy site (HFN7.1) or the neighboring FnIII8 module (clone 568). Following a 1-hour treatment with anastellin, there was a marked decrease in staining using the synergy-site-specific antibody, consistent with this epitope being no longer accessible to the antibody. However, matrix fibrils were still visible in cells stained with the antibody directed against the FnIII8 domain. Total fluorescence was used as a quantitative measure of antibody binding, and it indicated that anastellin treatment resulted in a 70% loss of the synergy site epitope (Fig. 8B). There was also a small but significant decrease in the binding of the antibody directed against the FnIII8 domain. The data suggest that conformational changes in matrix fibronectin result in the masking of the synergy site epitope. The data are consistent with a model in which anastellin-mediated reorganization of the fibronectin matrix changes the topographical display of integrin binding sites, thus influencing VEGF signaling by dictating the pattern of ligated integrins.
Fig. 8.
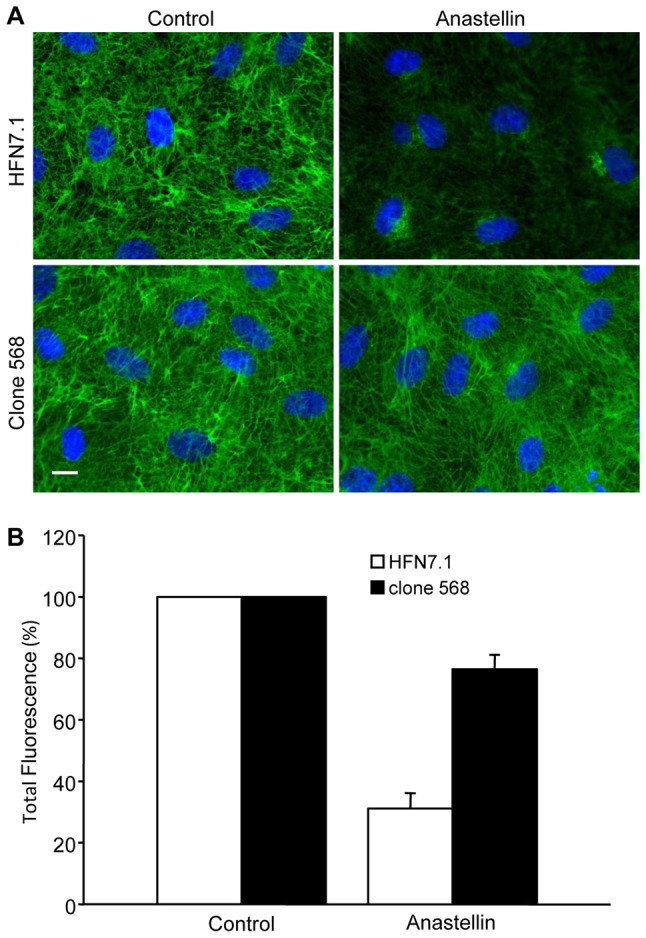
Anastellin decreases the availability of the synergy site epitope in matrix fibronectin. (A) Microvessel cell monolayers were treated with 5 µM anastellin for 60 minutes, fixed, permeabilized and stained using a fibronectin-blocking antibody (HFN7.1) against the synergy site in the FnIII9 module or an antibody (clone 568) that binds to the FnIII8 module. Scale bar: 10 µm. (B) Total fluorescence was quantified using NIH ImageJ software. Data show the mean±s.e.m. (at least six fields of view, two independent experiments).
DISCUSSION
The fibronectin matrix has a profound influence on endothelial cell morphology and differentiation. Early genetic studies demonstrated a requirement for fibronectin and the α5β1 integrin in vascularization (Kawasaki et al., 1999). Later studies showed that the ligation of integrins to fibronectin might not be sufficient to promote angiogenesis and that ongoing polymerization of fibronectin into ECM is also required (Zhou et al., 2008). The requirement for polymerized versus protomeric fibronectin in the angiogenic process is not understood, but the finding suggests that the polymerizing fibronectin fibrils provide important angiogenic cues that are not found in protomeric fibronectin. This concept is supported by accumulating evidence indicating that fibronectin, its isoforms, its polymerization and its secondary structure all impact on neovascularization, indicative of an essential role for topographical organization of the fibronectin matrix in endothelial cell biology (Cseh et al., 2010; Le Saux et al., 2011; Mitsi et al., 2006; Uemura et al., 2006). In the present study, we show that anastellin, a homophilic binding peptide of fibronectin, drives conformational changes within the assembled fibronectin matrix that influence the endothelial microvessel cell response to angiogenic growth factors. Incubation of microvessel cells with anastellin resulted in the selective inhibition of VEGF165 signaling. Inhibition of VEGF165 signaling was accompanied by anastellin-mediated conformational changes within the fibronectin matrix, leading to the inactivation of the α5β1 integrin and the loss of NRP1 function.
Anastellin prevented the formation of the VEGFR2–NRP1 complex in response to VEGF165, leading to the inhibition of downstream signaling. Several VEGF165-dependent activities in the vasculature are known to be influenced by NRP1, including endothelial cell migration, branching and survival (Kawamura et al., 2008; Kawasaki et al., 1999; Pan et al., 2007a). Our data indicate that NRP1 is also required for microvessel cell growth, as both anastellin and NRP1 knockdown inhibited VEGF165-mediated cell proliferation. VEGFR2 signaling in response to VEGF165 is regulated by clathrin-mediated endocytosis, and downstream signals are propagated from specific intracellular compartments (reviewed in Berger and Ballmer-Hofer, 2011). NRP1-regulated recycling of VEGFR2 back to the plasma membrane is known to influence the extent of ERK activation (Ballmer-Hofer et al., 2011; Lanahan et al., 2010). The strength of the ERK signal depends on the time for which VEGFR2 is exposed to PTP1b (also known as PTPN1), the endosomal phosphatase that controls the dephosphorylation of Tyr1175, the VEGFR2 residue required for ERK activation. In the absence of NRP1, trafficking through the endosomal compartments is delayed, thus prolonging the exposure of VEGFR2 to PTP1b, resulting in a loss of Tyr1175 phosphorylation and a decreased activation of ERK (Zhang et al., 2013). In our studies, anastellin had a more profound inhibitory effect on the phosphorylation of Tyr1175 as compared with either Tyr1059 or Tyr1054 (see Fig. 1). This observation was consistent with anastellin mediating its inhibitory effect on ERK signaling by preventing the formation of the VEGFR2–NRP1 complex. These data suggest a possible mechanism by which anastellin inhibits microvessel cell growth by disrupting NRP1-dependent VEGFR2 trafficking and prolonging the time for which VEGFR2 remains in the endosomal compartment. These data are also consistent with the impact of anastellin on VEGFR2 trafficking being initiated by conformational changes in the fibronectin matrix that decrease the activation state of α5β1 integrin. Taken together, these data point to a role for α5β1 integrin in regulating the functional activity of NRP1 in microvessel cells. Emerging evidence has implicated NRPs in the regulation of integrin function (reviewed in Goel et al., 2012; Goel and Mercurio, 2013). NRP1 has been shown to promote α5β1 activation and function in both matrix assembly (Yaqoob et al., 2012) and endothelial cell adhesion to fibronectin (Valdembri et al., 2009). Our studies now present evidence that the α5β1 integrin can regulate NRP1 function, thus demonstrating the bi-directionality of integrin–NRP1 crosstalk.
Our experiments demonstrate that the binding of anastellin to the fibronectin matrix results in the loss of the synergy site epitope. This epitope has been reported to be sensitive to changes in fibronectin conformation, and epitope loss is associated with decreased accessibility of the synergy site (PHSRN peptide) and decreased α5β1 integrin binding to fibronectin (García et al., 1999; Mao and Schwarzbauer, 2006). In previous studies, we used a panel of monoclonal anti-fibronectin antibodies to show that anastellin binds directly to the fibronectin matrix, causing conformational changes within the fibronectin extra domain A (FnEDA) while leaving several other epitopes unaffected (Klein et al., 2003). These studies also showed that anastellin does not bind directly to the EDA domain, suggesting that its effect on the EDA epitope is conformational rather than steric. The present study now identifies the synergy site epitope as a second conformationally regulated site within the fibronectin matrix that can be targeted by anastellin. The FnEDA domain is an alternatively spliced domain that is inserted between the FnIII11 and FnIII12 domains. The synergy site epitope lies between the FnIII9 and FnIII10 domains. As the FnIII11 domain has been identified as a major binding site on fibronectin for anastellin (Ohashi and Erickson, 2005), these data suggest that the binding of anastellin to the FnIII11 domain causes conformational changes in the neighboring FnEDA domain (Klein et al., 2003) and the FnIII9–10 domains. The FnEDA and FnIII9 domains of fibronectin represent major ligands for the α4β1 and α5β1 integrin receptors, respectively. Both of these integrins have been identified as important regulators of vascular function (reviewed in Astrof and Hynes, 2009). The ability of anastellin to impact on the secondary structure of these integrin-binding domains provides a possible molecular basis for the previously reported inhibitory effect of anastellin on in vivo angiogenesis (Yi and Ruoslahti, 2001). Anastellin inhibition of angiogenesis has also been reported to require plasma fibronectin (Yi et al., 2003). A previous study has shown that plasma fibronectin makes up ∼50% of the fibronectin in tissues (Moretti et al., 2007). This finding suggests that the requirement for plasma fibronectin in mediating the action of anastellin stems from the ability of plasma fibronectin to bind to anastellin and target it to tissues undergoing active remodeling.
Loss of the synergy site was accompanied by the inactivation of the α5β1 integrin. We used the term ‘inactivation’ to reflect the loss of specific antibody epitopes (9EG7 and 12G10) that report active ligand-bound conformations. In this instance, we are proposing that integrin inactivation is occurring secondary to a disengagement of the synergy site from the bound integrin. Whether this loss of ligand activates the inside-out signaling pathways that typically control integrin activation states is not known. Surprisingly, the inactivation of α5β1 integrin by anastellin was not accompanied by changes in either paxillin-containing adhesion sites or in the phosphorylation of FAK and paxillin. As both FAK and paxillin are rapidly dephosphorylated in response to loss of adhesion (Hartman et al., 2013; Mitola et al., 2006; Souza et al., 2012), this observation suggests that the disengagement of α5β1 from the matrix does not necessarily result in the activation of integrin-regulated phosphatases. In our study, loss of α5β1 from the focal adhesion is likely a response to the unavailability of the synergy site in fibronectin. The αvβ5 integrins, which bind to fibronectin but do not require the synergy site, remained associated with focal adhesions. The mechanism by which active integrins are released from focal adhesions is not well understood. The inactivation of α5β1 integrin by anastellin suggests that in the absence of a matrix ligand (i.e. synergy site) the integrin is uncoupled from the cytoplasmic molecules mediating high-affinity conformations (i.e. talin, kindlin) (reviewed in Bouvard et al., 2013). It is possible that, following the loss of the synergy site, integrins are actively transitioned into a closed inactive conformation and trafficked out of adhesion sites through the action of negative regulators of integrin activation, such as sharpin, filamin or ICAP1 (also known as ITGB1BP1). Interestingly, ICAP1-mediated regulation of β1 integrin activation has recently been linked to both aberrant vasculogenesis and ECM remodeling (Faurobert et al., 2013). Our data suggest that, following anastellin treatment, the αvβ5 integrins function to maintain adhesion as well as the activation of integrin-associated signaling proteins, whereas α5β1-specific functions are selectively inhibited. Our studies further suggest that in microvessel cells α5β1 functions to promote VEGF165 signaling by regulating the assembly of the VEGFR2–NRP1 complex and subsequent VEGFR2 trafficking.
The demonstration that anastellin regulates angiogenesis by targeting conformationally sensitive sites within the established fibronectin matrix suggests that homophilic binding peptides of fibronectin might be useful reagents for targeting conformationally regulated bioactive sites within the matrix. The ability of anastellin to affect signaling from one isoform of VEGF and not the other suggests that by targeting the topographical display of ligand binding sites within the fibronectin matrix it is possible to reprogram the cellular response to growth factors. This reprogramming might have important applications to the design of engineered tissue scaffolds used for tissue repair and regeneration. Additionally, pathologies characterized by extensive remodeling of the fibronectin matrix (i.e. tissue dysplasia and fibrosis) would be expected to respond to reagents designed to remodel the fibronectin matrix. A recent study has now shown that a single-chain variable-fragment monoclonal antibody directed at a cryptic homophilic binding site in fibronectin can be used to modulate the fibrotic response during vitreoretinopathy (Sharma et al., 2013). Understanding the contribution of conformationally regulated bioactive sites within the matrix to tissue repair and disease progression represent crucial steps towards the rational design of matrix-based therapeutics.
MATERIALS AND METHODS
Reagents
Unless indicated otherwise, reagents were obtained from Sigma-Aldrich (St Louis, MO). Fetal bovine serum (FBS) was from Hyclone (Logan, UT). Vitrogen-100 (type-I collagen) was from Cohesion Technologies (Palo Alto, CA). Recombinant human VEGF165 was obtained from R&D Systems (Minneapolis, MN) and VEGF121 from PeproTech (Rocky Hill, NJ). Recombinant fragments of human fibronectin, including FnIII1c (anastellin), FnIII 1cL37A/Y40A (anastellin mutant) and FnIII13, were generated and purified as described previously (Klein et al., 2003). Rabbit polyclonal antibodies against ERK2 (sc-154), FAK (sc-557) and NRP1 (sc-7239) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against phosphorylated (phospho)-p44/42 MAPK (Thr202/Tyr204; pERK1/2; 9106), phospho-VEGFR2 (Tyr1175; 2478) and VEGF receptor 2 (2479) were obtained from Cell Signaling Technology (Beverly, MA). Antibodies against phosphotyrosine (clone 4G10; 05-321), phospho-VEGFR2 (Tyr1054; 07-722), phospho-VEGFR2 (Tyr1059; 36-019), integrin αvβ5 (clone 15F11; MAB2019z), integrin β1 (clone 12G10; MAB2247), integrin α5 (AB1949) and the cell-binding domain of fibronectin (clone 568; MAB1937) were obtained from EMD Millipore (Billerica, MA). The fibronectin synergy site blocking antibody (HFN7.1; ab80923) was obtained from Abcam (Cambridge, MA). The function-blocking antibody against NRP1 (AF3870) was obtained from R&D Systems (Minneapolis, MN). Monoclonal antibodies directed against paxillin (610052) and the activated integrin chain beta 1 (clone 9EG7; 553715) were obtained from BD Biosciences (San Jose, CA). Phospho-FAK (Tyr397; 44-624G) polyclonal antibody, Alexa-Fluor-594-conjugated phalloidin (A12381) and all Alexa-Fluor-conjugated secondary antibodies were obtained from Life Technologies (Grand Island, NY).
Cell proliferation assay
Primary adult human dermal microvessel endothelial cells were obtained from VEC Technologies (Rensselaer, NY) and cultured in complete medium [MCDB-131, 20% defined FBS, 2 mM GlutaMAX (Gibco) and EGM-2MV (Cambrex, East Rutherford, NJ)] in a humidified incubator at 37°C under 5% CO2 on collagen-coated (20 µg/ml) tissue culture dishes. In order to address the effects of anastellin and NRP1 silencing on VEGF-dependent proliferation, microvessel cells were seeded onto collagen-coated tissue culture dishes at a density of 2×103 cells/cm2 in complete medium and allowed to attach and spread for 24 hours prior to treatment. Adherent cells were rinsed and incubated in basal medium (MCDB-131 containing 10% serum) supplemented with VEGF or preincubated with either anastellin or recombinant human FnIII13 for 1 hour prior to VEGF stimulation. Cells were maintained in culture for 3–5 days and then fixed for 30 minutes in 3% paraformaldehyde. Fixed cells were washed three times with distilled water and then stained with 0.05% Toluidine Blue for 1 hour. Following washing three times in distilled water, Toluidine Blue was extracted with 10% acetic acid and its absorbance was measured at 650 nm.
RNA interference experiments
ON-TARGETplus human NRP1 siRNA (SMART pool) was obtained from Dharmacon (Lafayette, CO). Human microvessel endothelial cells were seeded in complete medium onto collagen-coated tissue culture dishes at 2×103 cells/cm2 and allowed to attach and spread overnight. Cells were transfected in the presence of 50 nM siRNA (siNRP1) or under control conditions (control siRNA or nothing) in OptiMEM using Oligofectamine transfection reagent (Life Technologies) according to the manufacturer's instructions. Transfected cells were cultured in complete medium for 3 days and serum starved in MCDB-131, 0.5% bovine serum albumin (BSA) (starving medium) for 4–16 hours prior to treatment. To assess the effects of NRP1 knockdown on VEGF-dependent proliferation, MCDB-131 containing 10% FBS and supplemented with or without VEGF was added 4–6 hours after initial transfection. Cells were cultured for 3 days, fixed and stained with Toluidine Blue as described above.
Immunoblotting
To evaluate the activation of signaling intermediates, cells were placed in starving medium for the designated times prior to stimulation with growth factors or fibronectin fragments. Cell monolayers were washed twice with ice-cold PBS containing 1 mM Na3VO4 before solubilization in a solution of 20 mM Tris-HCl pH 7.4, 1% Triton X-100, 0.5% Nonidet P-40, 0.1 M NaCl, 40 mM NaF, 30 mM Na4P2O7, 2 mM EGTA, 1 mM Na3VO4 and 0.5 mM PMSF, containing Complete Mini protease inhibitors (Roche Biochemical, Indianapolis, IN). Cell lysates were cleared by centrifugation (20,000 g for 10 minutes at 4°C) and stored at −80°C until use. Protein concentrations were determined with a bicinchoninic acid (BCA) protein assay reagent kit (Pierce, Rockford, IL), using BSA as standard. Cell lysates were subjected to SDS-PAGE under reducing conditions and transferred onto nitrocellulose membranes (Schleicher and Schuell Bioscience, Keene, NH). Membranes were blocked overnight with 3% BSA (w/v) in Tris-buffered saline containing 1% Tween-20 (TBS-T) and incubated with primary antibodies overnight at 4°C. Blots were then rinsed with TBS-T containing 3% BSA and incubated with horseradish peroxidase (HRP)-linked secondary antibodies (1∶10,000) for 1 hour at room temperature. Immunoreactive bands were detected using Amersham ECL Western Blotting Detection Reagent (Amersham Biosciences; Piscataway, NJ). Blots were reprobed after stripping in 62.5 mM Tris-HCl pH 6.7, 2% SDS and 10 mM β-mercaptoethanol for 30 minutes at 60°C. Quantification of immunoblots was carried out using ImageJ analysis software. Values were normalized relative to VEGF-stimulated controls.
Immunoprecipitation
Endothelial cells seeded at 4×105 cells/cm2 onto collagen-coated dishes were allowed to attach and spread overnight in complete medium. Cells were then serum-starved for 4–16 hours in MCDB-131 containing 0.5% BSA prior to treatment with recombinant fibronectin fragments for 1 hour. For immunoprecipitation of VEGFR2–NRP1 complexes, treated cells were stimulated with 10 ng/ml VEGF165 or VEGF121 for 15 minutes prior to lysis in a solution of 50 mM Tris-HCl pH 7.8, 1% Nonidet P-40, 0.15 M NaCl, 10 mM NaF, 10 mM Na4P2O7, 2 mM EDTA, 1 mM Na3VO4 and 0.5 mM PMSF containing Complete Mini protease inhibitors (immunoprecipitation lysis buffer). Lysates were cleared by centrifugation (20,000 g for 10 minutes at 4°C), and equal amounts of protein (500 µg) were incubated with 2 µg of polyclonal anti-NRP1 antibodies and Protein A/G PLUS-Agarose (Santa Cruz) for 2 hours at 4°C with gentle rocking. Agarose beads were washed three times with immunoprecipitation lysis buffer by dilution and centrifugation (1000 g, 3 minutes at 4°C) and immune complexes were eluted with standard Laemmli reducing buffer. NRP1 and VEGFR2 were detected in complexes by immunoblotting.
Fluorescence microscopy
Microvascular endothelial cells were cultured in complete medium for 48–72 hours on collagen-coated (20 µg/ml) glass coverslips, rinsed once in serum-free medium and treated for 1 hour with recombinant fibronectin fragments. Treated cells were then washed once with serum-free medium, fixed for 20 minutes in 3% paraformaldehyde, permeabilized in 0.5% Triton X-100 for 10 minutes, blocked in 1% BSA and immunostained with antibodies as indicated. F-actin was visualized with Alexa-Fluor-594-conjugated phalloidin. After staining, coverslips were mounted with Prolong Antifade according to the manufacturer's instructions (Molecular Probes). Cell monolayers were examined using an Olympus BMX-60 microscope equipped with a cooled CCD sensi-camera (Cooke, Auburn Hills, MI), and images were acquired using Slidebook software (Intelligent Imaging Innovation, Denver, CO). Fluorescence images were processed with ImageJ analysis software for the quantification of focal complexes and fibronectin staining.
Statistical analysis
Data are presented as the mean±s.e.m. of at least three independent experiments performed in duplicate or triplicate. Proliferation and immunofluorescent staining results were analyzed using a one-way ANOVA with Tukey's multiple comparison test. Statistical analysis was performed with SigmaPlot, version 11.0 (Systat Software, Chicago, IL), with P<0.05 considered significant.
Acknowledgments
The authors would like to thank Carol Horzempa (Albany Medical College, Albany, NY) for technical assistance with the purification of recombinant fibronectin fragments.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
P.J.M.-L. designed the study and wrote the manuscript. A.A. performed all experiments. Both authors read, revised and approved the manuscript.
Funding
This study was supported by the National Institutes of Health [grant number CA58626 to P.J.M.-L.]. Deposited in PMC for release after 12 months.
References
- Ambesi A., Klein R. M., Pumiglia K. M., McKeown-Longo P. J. (2005). Anastellin, a fragment of the first type III repeat of fibronectin, inhibits extracellular signal-regulated kinase and causes G(1) arrest in human microvessel endothelial cells. Cancer Res. 65, 148–156 [PubMed] [Google Scholar]
- Aota S., Nomizu M., Yamada K. M. (1994). The short amino acid sequence Pro-His-Ser-Arg-Asn in human fibronectin enhances cell-adhesive function. J. Biol. Chem. 269, 24756–24761 [PubMed] [Google Scholar]
- Argraves W. S., Drake C. J. (2005). Genes critical to vasculogenesis as defined by systematic analysis of vascular defects in knockout mice. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 286A, 875–884 10.1002/ar.a.20232 [DOI] [PubMed] [Google Scholar]
- Astrof S., Hynes R. O. (2009). Fibronectins in vascular morphogenesis. Angiogenesis 12, 165–175 10.1007/s10456-009-9136-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballmer-Hofer K., Andersson A. E., Ratcliffe L. E., Berger P. (2011). Neuropilin-1 promotes VEGFR-2 trafficking through Rab11 vesicles thereby specifying signal output. Blood 118, 816–826 10.1182/blood-2011-01-328773 [DOI] [PubMed] [Google Scholar]
- Berger P., Ballmer-Hofer K. (2011). The reception and the party after: how vascular endothelial growth factor receptor 2 explores cytoplasmic space. Swiss Med. Wkly. 141, w13318. [DOI] [PubMed] [Google Scholar]
- Bouvard D., Pouwels J., De Franceschi N., Ivaska J. (2013). Integrin inactivators: balancing cellular functions in vitro and in vivo. Nat. Rev. Mol. Cell Biol. 14, 430–442 10.1038/nrm3599 [DOI] [PubMed] [Google Scholar]
- Briknarová K., Akerman M. E., Hoyt D. W., Ruoslahti E., Ely K. R. (2003). Anastellin, an FN3 fragment with fibronectin polymerization activity, resembles amyloid fibril precursors. J. Mol. Biol. 332, 205–215 10.1016/S0022-2836(03)00890-8 [DOI] [PubMed] [Google Scholar]
- Cabodi S., Di Stefano P., Leal M. P., Tinnirello A., Bisaro B., Morello V., Damiano L., Aramu S., Repetto D., Tornillo G. et al. (2010). Integrins and signal transduction. Adv. Exp. Med. Biol. 674, 43–54 10.1007/978-1-4419-6066-5_5 [DOI] [PubMed] [Google Scholar]
- Cseh B., Fernandez-Sauze S., Grall D., Schaub S., Doma E., Van Obberghen-Schilling E. (2010). Autocrine fibronectin directs matrix assembly and crosstalk between cell-matrix and cell-cell adhesion in vascular endothelial cells. J. Cell Sci. 123, 3989–3999 10.1242/jcs.073346 [DOI] [PubMed] [Google Scholar]
- Faurobert E., Rome C., Lisowska J., Manet-Dupé S., Boulday G., Malbouyres M., Balland M., Bouin A-P., Kéramidas M., Bouvard D. et al. (2013). CCM1-ICAP-1 complex controls β1 integrin-dependent endothelial contractility and fibronectin remodeling. J. Cell Biol. 202, 545–561 10.1083/jcb.201303044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García A. J., Vega M. D., Boettiger D. (1999). Modulation of cell proliferation and differentiation through substrate-dependent changes in fibronectin conformation. Mol. Biol. Cell 10, 785–798 10.1091/mbc.10.3.785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee E. P., Yüksel D., Stultz C. M., Ingber D. E. (2013). SLLISWD sequence in the 10FNIII domain initiates fibronectin fibrillogenesis. J. Biol. Chem. 288, 21329–21340 10.1074/jbc.M113.462077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel H. L., Mercurio A. M. (2013). VEGF targets the tumour cell. Nat. Rev. Cancer 13, 871–882 10.1038/nrc3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel H. L., Pursell B., Standley C., Fogarty K., Mercurio A. M. (2012). Neuropilin-2 regulates α6β1 integrin in the formation of focal adhesions and signaling. J. Cell Sci. 125, 497–506 10.1242/jcs.094433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman Z. R., Schaller M. D., Agazie Y. M. (2013). The tyrosine phosphatase SHP2 regulates focal adhesion kinase to promote EGF-induced lamellipodia persistence and cell migration. Mol. Cancer Res. 11, 651–664 10.1158/1541-7786.MCR-12-0578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking D. C., Smith R. K., McKeown-Longo P. J. (1996). A novel role for the integrin-binding III-10 module in fibronectin matrix assembly. J. Cell Biol. 133, 431–444 10.1083/jcb.133.2.431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes R. O. (2007). Cell-matrix adhesion in vascular development. J. Thromb. Haemost. 5, Suppl. 132–40 10.1111/j.1538-7836.2007.02569.x [DOI] [PubMed] [Google Scholar]
- Kawamura H., Li X., Harper S. J., Bates D. O., Claesson-Welsh L. (2008). Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res. 68, 4683–4692 10.1158/0008-5472.CAN-07-6577 [DOI] [PubMed] [Google Scholar]
- Kawasaki T., Kitsukawa T., Bekku Y., Matsuda Y., Sanbo M., Yagi T., Fujisawa H. (1999). A requirement for neuropilin-1 in embryonic vessel formation. Development 126, 4895–4902 [DOI] [PubMed] [Google Scholar]
- Klein R. M., Zheng M., Ambesi A., Van De Water L., McKeown-Longo P. J. (2003). Stimulation of extracellular matrix remodeling by the first type III repeat in fibronectin. J. Cell Sci. 116, 4663–4674 10.1242/jcs.00778 [DOI] [PubMed] [Google Scholar]
- Klotzsch E., Smith M. L., Kubow K. E., Muntwyler S., Little W. C., Beyeler F., Gourdon D., Nelson B. J., Vogel V. (2009). Fibronectin forms the most extensible biological fibers displaying switchable force-exposed cryptic binding sites. Proc. Natl. Acad. Sci. USA 106, 18267–18272 10.1073/pnas.0907518106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S., Claesson-Welsh L. (2012). Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2, a006502 10.1101/cshperspect.a006502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanahan A. A., Hermans K., Claes F., Kerley-Hamilton J. S., Zhuang Z. W., Giordano F. J., Carmeliet P., Simons M. (2010). VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev. Cell 18, 713–724 10.1016/j.devcel.2010.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Saux G., Magenau A., Gunaratnam K., Kilian K. A., Böcking T., Gooding J. J., Gaus K. (2011). Spacing of integrin ligands influences signal transduction in endothelial cells. Biophys. J. 101, 764–773 10.1016/j.bpj.2011.06.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinin N. L., Pluskota E., Byzova T. V. (2012). Integrin signaling in vascular function. Curr. Opin. Hematol. 19, 206–211 10.1097/MOH.0b013e3283523df0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y., Schwarzbauer J. E. (2006). Accessibility to the fibronectin synergy site in a 3D matrix regulates engagement of α5β1 versus alphavbeta3 integrin receptors. Cell Commun. Adhes. 13, 267–277 10.1080/15419060601072215 [DOI] [PubMed] [Google Scholar]
- Mitola S., Brenchio B., Piccinini M., Tertoolen L., Zammataro L., Breier G., Rinaudo M. T., den Hertog J., Arese M., Bussolino F. (2006). Type I collagen limits VEGFR-2 signaling by a SHP2 protein-tyrosine phosphatase-dependent mechanism 1. Circ. Res. 98, 45–54 10.1161/01.RES.0000199355.32422.7b [DOI] [PubMed] [Google Scholar]
- Mitsi M., Hong Z., Costello C. E., Nugent M. A. (2006). Heparin-mediated conformational changes in fibronectin expose vascular endothelial growth factor binding sites. Biochemistry 45, 10319–10328 10.1021/bi060974p [DOI] [PubMed] [Google Scholar]
- Moretti F. A., Chauhan A. K., Iaconcig A., Porro F., Baralle F. E., Muro A. F. (2007). A major fraction of fibronectin present in the extracellular matrix of tissues is plasma-derived. J. Biol. Chem. 282, 28057–28062 10.1074/jbc.M611315200 [DOI] [PubMed] [Google Scholar]
- Morla A., Ruoslahti E. (1992). A fibronectin self-assembly site involved in fibronectin matrix assembly: reconstruction in a synthetic peptide. J. Cell Biol. 118, 421–429 10.1083/jcb.118.2.421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morla A., Zhang Z., Ruoslahti E. (1994). Superfibronectin is a functionally distinct form of fibronectin. Nature 367, 193–196 10.1038/367193a0 [DOI] [PubMed] [Google Scholar]
- Nowak D. G., Woolard J., Amin E. M., Konopatskaya O., Saleem M. A., Churchill A. J., Ladomery M. R., Harper S. J., Bates D. O. (2008). Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 121, 3487–3495 10.1242/jcs.016410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi T., Erickson H. P. (2005). Domain unfolding plays a role in superfibronectin formation. J. Biol. Chem. 280, 39143–39151 10.1074/jbc.M509082200 [DOI] [PubMed] [Google Scholar]
- Pan Q., Chanthery Y., Liang W-C., Stawicki S., Mak J., Rathore N., Tong R. K., Kowalski J., Yee S. F., Pacheco G. et al. (2007a). Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell 11, 53–67 10.1016/j.ccr.2006.10.018 [DOI] [PubMed] [Google Scholar]
- Pan Q., Chathery Y., Wu Y., Rathore N., Tong R. K., Peale F., Bagri A., Tessier-Lavigne M., Koch A. W., Watts R. J. (2007b). Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J. Biol. Chem. 282, 24049–24056 10.1074/jbc.M703554200 [DOI] [PubMed] [Google Scholar]
- Prabhakaran S., Liang X., Skare J. T., Potts J. R., Höök M. (2009). A novel fibronectin binding motif in MSCRAMMs targets F3 modules. PLoS ONE 4, e5412 10.1371/journal.pone.0005412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retta S. F., Barry S. T., Critchley D. R., Defilippi P., Silengo L., Tarone G. (1996). Focal adhesion and stress fiber formation is regulated by tyrosine phosphatase activity. Exp. Cell Res. 229, 307–317 10.1006/excr.1996.0376 [DOI] [PubMed] [Google Scholar]
- Schwarzbauer J. E., DeSimone D. W. (2011). Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harb. Perspect. Biol. 3, a005041 10.1101/cshperspect.a005041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M., Tiwari A., Sharma S., Bhoria P., Gupta V., Gupta A., Luthra-Guptasarma M. (2013). Fibrotic remodeling of the extracellular matrix through a novel (engineered, dual-function) antibody reactive to a cryptic epitope on the N-terminal 30 kDa fragment of fibronectin. PLoS ONE 8, e69343 10.1371/journal.pone.0069343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza C. M., Davidson D., Rhee I., Gratton J. P., Davis E. C., Veillette A. (2012). The phosphatase PTP-PEST/PTPN12 regulates endothelial cell migration and adhesion, but not permeability, and controls vascular development and embryonic viability. J. Biol. Chem. 287, 43180–43190 10.1074/jbc.M112.387456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Yamaguchi S., Chida K., Shibuya M. (2001). A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-γ and DNA synthesis in vascular endothelial cells. EMBO J. 20, 2768–2778 10.1093/emboj/20.11.2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura A., Kusuhara S., Wiegand S. J., Yu R. T., Nishikawa S. (2006). Tlx acts as a proangiogenic switch by regulating extracellular assembly of fibronectin matrices in retinal astrocytes. J. Clin. Invest. 116, 369–377 10.1172/JCI25964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdembri D., Caswell P. T., Anderson K. I., Schwarz J. P., König I., Astanina E., Caccavari F., Norman J. C., Humphries M. J., Bussolino F. et al. (2009). Neuropilin-1/GIPC1 signaling regulates α5β1 integrin traffic and function in endothelial cells. PLoS Biol. 7, e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. S., Muro A. F. (2011). Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 63, 538–546 10.1002/iub.493 [DOI] [PubMed] [Google Scholar]
- Xu J., Mosher D. (2011). Fibronectin and other adhesive glycoproteins. The Extracellular Matrix: An Overview Mecham R P, ed43–55Berlin; Heidelberg: Springer-Verlag [Google Scholar]
- Yaqoob U., Cao S., Shergill U., Jagavelu K., Geng Z., Yin M., de Assuncao T. M., Cao Y., Szabolcs A., Thorgeirsson S. et al. (2012). Neuropilin-1 stimulates tumor growth by increasing fibronectin fibril assembly in the tumor microenvironment. Cancer Res. 72, 4047–4059 10.1158/0008-5472.CAN-11-3907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M., Ruoslahti E. (2001). A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc. Natl. Acad. Sci. USA 98, 620–624 10.1073/pnas.98.2.620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M., Sakai T., Fassler R., Ruoslahti E. (2003). Antiangiogenic proteins require plasma fibronectin or vitronectin for in vivo activity. Proc. Natl. Acad. Sci. USA 100, 11435–11438 10.1073/pnas.1635112100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You R., Klein R. M., Zheng M., McKeown-Longo P. J. (2009). Regulation of p38 MAP kinase by anastellin is independent of anastellin's effect on matrix fibronectin. Matrix Biol. 28, 101–109 10.1016/j.matbio.2009.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Lanahan A. A., Simons M. (2013). VEGFR2 trafficking: speed doesn't kill. Cell Cycle 12, 2163–2164 10.4161/cc.25536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Rowe R. G., Hiraoka N., George J. P., Wirtz D., Mosher D. F., Virtanen I., Chernousov M. A., Weiss S. J. (2008). Fibronectin fibrillogenesis regulates three-dimensional neovessel formation. Genes Dev. 22, 1231–1243 10.1101/gad.1643308 [DOI] [PMC free article] [PubMed] [Google Scholar]