

Abstract
The human immunodeficiency virus type-1 (HIV-1) genome contains multiple, highly conserved structural RNA domains that play key roles in essential viral processes. Interference with the function of these RNA domains either by disrupting their structures or by blocking their interaction with viral or cellular factors may seriously compromise HIV-1 viability. RNA aptamers are amongst the most promising synthetic molecules able to interact with structural domains of viral genomes. However, aptamer shortening up to their minimal active domain is usually necessary for scaling up production, what requires very time-consuming, trial-and-error approaches. Here we report on the in vitro selection of 64 nt-long specific aptamers against the complete 5′-untranslated region of HIV-1 genome, which inhibit more than 75% of HIV-1 production in a human cell line. The analysis of the selected sequences and structures allowed for the identification of a highly conserved 16 nt-long stem-loop motif containing a common 8 nt-long apical loop. Based on this result, an in silico designed 16 nt-long RNA aptamer, termed RNApt16, was synthesized, with sequence 5′-CCCCGGCAAGGAGGGG-3′. The HIV-1 inhibition efficiency of such an aptamer was close to 85%, thus constituting the shortest RNA molecule so far described that efficiently interferes with HIV-1 replication.
The human immunodeficiency virus type 1 (HIV-1) genome is a 9.2 kb long, single-stranded RNA (ssRNA) molecule of positive polarity with multiple open reading frames (ORFs), including those for structural and functional proteins gag, pol and env. The coding region is flanked at both ends by untranslated regions (UTR). HIV-1 infective particles contain two identical genomic RNA molecules. The genome is reverse transcribed into a cDNA copy that is integrated into the host genome. The transcription of the proviral cDNA generates capped, full-length genomic RNAs that must undergo alternative splicing to accommodate the different ORFs immediately downstream of the capped 5′-end. All the HIV-1 genomic and subgenomic RNAs share approximately the first 300 nt of their 5′-UTR. This sequence contains several well-characterized and conserved functional RNA domains1,2 including the trans-activator response (TAR) element3, the polyadenylation [poly(A)] region4, the primer-binding site (PBS)5, the dimerisation initiation site (DIS)6, the major splicing donor (SD)7 and part of the packaging signal (psi)8 (Fig. 1). These RNA domains are involved in alternative, functionally relevant RNA-RNA interactions that determine two mutually exclusive conformations of the overall 5′-UTR region, termed Branched Multiple Hairpins (BMH) and Long Distance Interaction (LDI)9.
Figure 1. Schematic representation of the BMH conformation of the HIV-1 UTR308.

Essential structural domains are indicated. Putative binding sites for RNApt16 (boxed) and the in vitro selected aptamers are shown in boldface, and labelled from (a) to (f). The alternative, partially overlapping binding region (a′) is shown.
The identification of highly conserved structural-functional elements within viral RNA genomes has attracted much attention due to their potential use as targets for novel antiviral drugs. Indeed, small RNA molecules have been proved to be efficient inhibitors targeting functional RNA genomic domains (including those at the 5′-UTR) as well as genetic products of HIV-110,11,12,13,14,15.Some of the inhibitory RNAs have been included in clinical trials for HIV-1 treatment16. This strategy has been also applied to other clinically relevant RNA viruses17,18,19,20.
Aptamers are short RNA or DNA oligonucleotides that, due to their three dimensional structure, can efficiently and specifically bind a molecular target21. Most of the developed aptamers have been artificially obtained using SELEX (Systematic Evolution of Ligands by EXponential enrichment) techniques22, and their potential as therapeutic agents has been widely established23,24,25. A SELEX procedure consists in the exposition of a random population of nucleic acid molecules to a desired target, in experimental conditions that enable the segregation of the molecules bound to the target for a subsequent amplification round. The successive selection-amplification cycles progressively enrich the population in molecules able to bind the target. Usually, the starting population for SELEX consists of molecules with a random sequence flanked by constant regions needed for primer binding. This fact imposes a minimal aptamer length, limiting in turn its usefulness as a therapeutic agent. Several deletion methods have been developed to experimentally obtain the minimal domain that allows efficient aptamer binding12,26,27. Bioinformatic strategies constitute an alternative, though less explored approach for this purpose28,29,30. An example of such a ‘rational truncation approach' has been successfully applied to the production of 40–50 nt long functional RNA aptamers31. We report here the in vitro selection of efficient anti-HIV-1 RNA aptamers targeting the viral 5′UTR, and their shortening to a 16 nt-long minimal aptamer by the application of computational strategies based on sequence and structure analysis to identify and improve core aptamer domains. The combination of an in vitro selection with the in silico approach has allowed us to design the shortest reported RNA molecule that efficiently inhibits HIV-1 production in a human cell line.
Results
In vitro selection of RNA aptamers against the 5′-UTR of HIV-1
An in vitro selection strategy was used to isolate anti-HIV-1 RNA aptamers targeting the first 308 nt of the 5′-UTR of the HIV-1 subtype B NL4.3 genome. This target corresponds to the sequence fragment of the 5′UTR that is common to all genomic and subgenomic HIV-1 RNAs2, including 8 nt from the unspliced SD domain that have been maintained in the molecule to prevent alternative foldings of the entire molecule due to SD partial deletion after splicing. The starting RNA population consisted of roughly 7 × 1015 variants of 64 nt long molecules. RNA molecules were binding-challenged against the target molecule fixed to a sepharose-streptavidin column. Fourteen cycles of selection-amplification were performed, and the selective pressure was stepwise modified by increasing the binding temperature from round IV on, and by reducing the target:aptamer ratio from round XI on. The yield of complex formation increased along the selection process, as shown by the evolution of Bmax32 (Fig. 2). A total of 299 individual clonal sequences derived from the populations selected at rounds 0 (initial random population, 30 seqs), I (30), III (31), V (24), VIII (28), IX (32), X (35), XI (52) and XIV (37) were analyzed (Fig. 3 and data not shown). The presence of intra- and/or inter-round repeated sequences supposed a reduction in the diversity of the analyzed collection to 216 different molecules (Table S1). The repeated sequences gained representation from round IX on, thus showing that the selection process was effective. Finally, 188 out of the 216 different sequences showed the expected length of 64 nt, while the others contained deletions of one or more nucleotides in their variable region. The clustering of those 188 different, 64 nt-long sequences, is shown in Figure S1.
Figure 2. Changes in relevant variables along the selection process.

Average distances were computed over all possible sequence/structure pairs at each round, and error bars show the mean square deviation of each sample. In Bmax, error bars represent the experimental error.
Figure 3. Aptamer sequences of rounds XI and XIV, grouped attending to putative common targets.

The variable region is shown in boldface. The single-stranded sequences predicted by RNAfold software59 are boxed. White text represents the sequences complementary to 5′-UTR loop regions (shown below each group). x N: sequence multiplicity in each round. Among these putative interaction sites, once the energy of the folded configuration is considered (Table S2) the only robust and energetically favoured interaction is that with the poly(A) apical loop.
Sequence analysis showed that three clonal sequences from round IX (9.4% of the population), 15 from round X (42.9%), 35 from round XI (67.3%), and 33 from XIV (89.2%) shared the consensus octamer 5′-GGCAAGGA-3′ (with a point mutation in sequence XIV32: 5′-GGCAGGGA-3′). Interestingly, all main groups of repeated sequences in rounds XI and XIV contained the consensus octamer. Two groups were particularly relevant at rounds XI and XIV: i) Group 1, formed by clone 21 and other 6 repeated sequences of round XI (termed XI21-7) identical to 6 repeated sequences of round XIV (termed XIV26-6); and ii) Group 2, constituted by 17 sequences of round XI (termed XI1-17) identical to 23 repeated sequences of round XIV (termed XIV22-23). The latter group was first observed in round IX (see Table S1). Thus, our results indicate that the selection procedure has been successful in reducing the initial variability to two major sequences that together represent the 51% and 80% of the total population in rounds XI and XIV, respectively. Sequence comparison showed that the hexanucleotide 5′-GGCAAG-3′ within the consensus octamer is complementary to the apical loop of the poly(A) domain within the repeated region (R) located at both 5′ and 3′-UTRs of HIV-1 (Fig. 1 and 3), thus suggesting the putative relevance of this recognition site for the selected aptamers.
The remaining sequences in rounds XI and XIV can be grouped into two additional groups of molecules, defined by common sequence motifs complementary to the TAR apical loop and the SD apical loop of HIV-1 5′-UTR, respectively (Fig. 3). Additionally, certain unique sequences were identified, which did not show a clear sequence complementarity to any domain within the target sequence (Fig. 3).
In silico sequence and structure analysis
To study the effect of selection along the process, we measured the similarity of the clonal sequences within each round. The distributions of the Hamming distance between pairs of sequences at rounds 0, I to IX, X to XIV, and I to XIV is shown in Figure S2. Since a different number of sequences were available for each round, the distributions are rescaled by the factor M(M-1)/2, with M being the number of sequences per round. For rounds 0-IX, the distributions are relatively symmetric, and peaked between 17 and 19. This value is close to the average Hamming distance between random sequences: 25*3/4 = 18.75, where 25 is the length of the variable region and 3/4 is the probability that a nucleotide is different from other randomly chosen nucleotide. In turn, for rounds XI-XIV the distribution showed several peaks at varying distances, and groups of equal (zero Hamming distance) and very similar (distances 1 and 2) sequences were present in the pool. Note that the height of the peak is proportional to the absolute number of repeated sequences within a group. The comparison of the similarities among secondary structures along the process yields qualitatively analogous results (Figure S3). A simultaneous comparison of the similarity between sequences and the corresponding structures is shown in Figure S4. That representation highlights the known fact that similar sequences can fold into significantly different minimum free energy (MFE) structures, and vice versa, identical structures may arise from significantly different sequences.
Regarding the secondary structures of the folded sequences, the evolution of two thermodynamic parameters, the ensemble diversity (ED) and the frequency of the MFE structure along the in vitro selection process (FME), is graphically depicted in Figure S5 for the 188 different, 64 nt-long sequences studied. Groups of sequences were first detected at round VIII, and the process subsequently led to the appearance of two major groups: i) Group 1, represented by the folded sequence XIV26-6 and characterized by a low ED and high FME; and ii) Group 2, represented by XIV22-23, showing a high ED and low FME.
Average values of the Hamming distance between sequences as well as the base-pair distance between structures along the process are depicted in Figure 2, in parallel to the evolution of the extension of the binding reaction Bmax. A trend towards lower average distance between all pairs of sequences and structures in the population becomes apparent from round IX to XIV. Despite the large dispersion of these measures, the increase in similarity among groups of molecules is genuine, as information on the full distributions of pair distances (Figs. S2, S3 and S4) show. Based on the base-pair distance values, a clustering of the secondary structures corresponding to the 188 sequences is shown in Figure S6.
Design of the RNApt16 and in silico analysis of the aptamers-target interaction
The in silico predicted MFE secondary structure model of the aptamers selected in round XIV is depicted in Figure 4A–C. Folding energies corresponding to each structure (in Kcal/mol) are ΔG[XIV22] = −10.80; ΔG[XIV1] = −11.80; ΔG[XIV32] = −10.80; ΔG[XIV12] = −10.90; ΔG[XIV26] = −15.70; ΔG[XIV5] = −11.10; ΔG[XIV48] = −9.80; ΔG[XIV37] = −7.32; ΔG[XIV25] = −11.50. The most abundant structures (belonging to the Groups 1 and 2 defined above) indicate that the consensus octamer 5′-GGCAAGGA-3′ is always placed in an apical loop flanked by complementary sequences that form a double stranded region of at least 4 bp in length. The RNAfold algorithm calculates the likelihood that any nucleotide in a sequence actually occupies the predicted structural position by analysing how often it appears in the structural ensemble of the analysed sequence. While some of the motifs present in the folded sequences have low reliability, we observed that the consensus octamer forming a loop plus its 4 nt flanking regions consistently shows a stem-loop configuration. Such an RNA motif is systematically found in most structures of the thermodynamic ensemble of sequences in round XIV (Figure 4A–B), and does not contribute significantly to their observed ED. The consensus sequence of that conserved motif, with structure 5′-((((........))))-3′, is 5′-NNDYGGCARGGARNNN-3′ (sequence alignment not shown). Based on this fact, a 16 nt-long stem-loop RNA molecule was in silico designed as a minimal aptamer potentially able to interact with the 5′-UTR of HIV-1. This aptamer was termed RNApt16, and included the consensus octamer in a loop closed by the 4 bp-long stem allowing the highest possible thermodynamic stability of the folded molecule, thus formed by four consecutive C-G base pairs: 5′-CCCCGGCAAGGAGGGG-3′. The secondary structure of RNApt16 is shown in Figure 4D, the folding energy associated to its MFE is −6, 50 kcal/mol, and the frequency of this MFE within the thermodynamic ensemble is 91,82%.
Figure 4. In silico prediction of the MFE secondary structure of the aptamers.
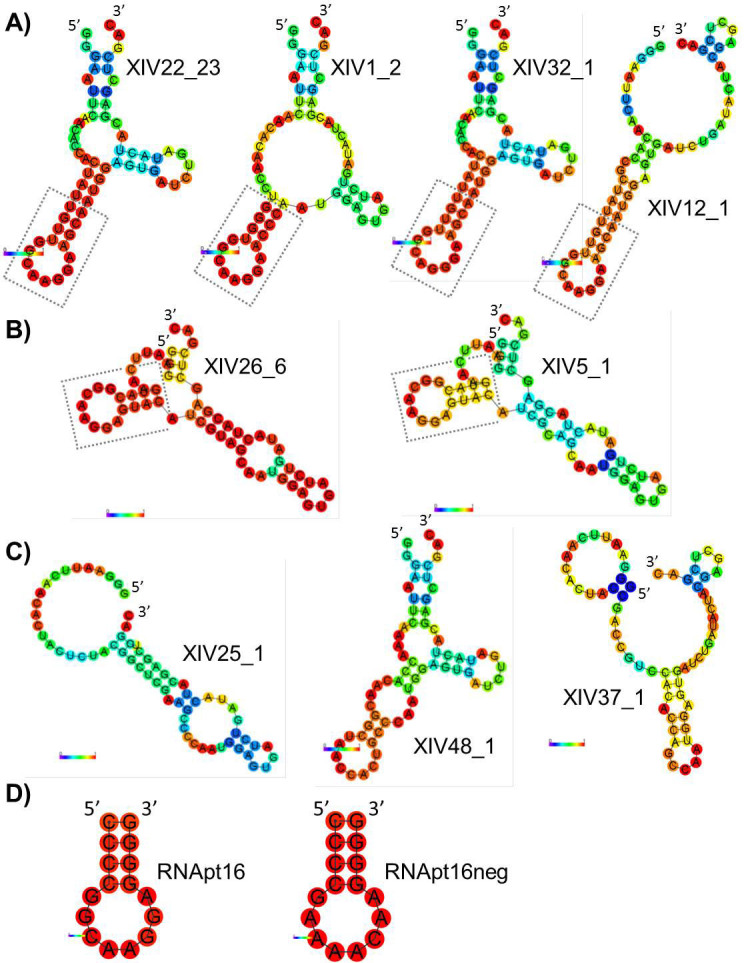
(A) Group 2. (B) Group 1. (C) Rest of structures. (D) RNApt16 and RNApt16neg. _N: number of repetitions of a particular sequence. The 16 nt-long stem-loop motif is boxed. Probabilities for every nucleotide to actually hold the structural position shown are represented by a colour code from deep blue (lowest) to red (highest).
We used RNAup to predict the preferred sites of interaction between either three selected aptamers (at round XIV) or the minimal engineered aptamer (RNApt16) with the target RNA. An example of the information yielded by RNAup can be seen in Figure S7. The regions of the target molecule with the highest probability of interaction with the aptamers are listed in Table S2. Among them, the binding site (a), placed at the poly(A) domain (Fig. 1), shows by far the most stable interaction.
Design of the molecule RNApt16neg as a negative control
The design of a negative control molecule to evaluate the activity of RNApt16 was computationally challenging. In order to do so, we extensively folded all sequences 5′-CCCCNNNNNNNNGGGG-3′ using RNAfold and kept those which had as their MFE the stem-loop structure 5′-((((........))))-3′. Out of 48 = 65,536 sequences of length 16 nt, 19,087 folded into the desired structure. For each of these sequences, we compared the frame of 8 unpaired, variable nucleotides in the trial molecules with a moving frame of 8 nucleotides along the target RNA. We investigated the possible antiparallel interactions (5′-3′ for the target UTR308 and 3′-5′ for negative control molecule). For each trial sequence, there are 302 possible interaction positions with the target. For each position of the moving frame, we counted: i) the total number of bases compatible for pairing; ii) the number of adjacent bases compatible for pairing. For each of the 19,087 sequences, we kept the maximum value of these two magnitudes over 302 frame positions as the overall pairing number.
Since we were interested in sequences with the structure of RNApt16, but binding the target with the lowest possible probability, we first selected those sequences with minimum overall pairing numbers. As a result, we obtained 112 sequences which had: i) a maximum of 5 possible pairing events in an 8 nt-frame along the target RNA; ii) a maximum of 4 possible adjacent pairs within the frame. Using RNAup, we obtained for all these sequences their total interaction energies, taking into account the two individual structures. The result showed interactions between 2 and 11 pairs of nucleotides and total free interaction energies between −0.40 and −7.20 kcal/mol.
After analyzing the 112 sequences we chose as the negative control, termed RNApt16neg, the molecule 5′-CCCCGAAAACAAGGGG-3′ (Fig. 4D) which has the following properties: i) it is the sequence of lowest total interaction energy with the target (−0.40 kcal/mol); ii) its interaction with the target is produced only in 2 nt (positions 5–6 of RNApt16neg and positions 22–23 of the target); iii) these interaction positions are in the loop of molecule RNApt16neg.
In vitro testing of the aptamer binding to the HIV-1 UTR308 molecule
Binding to the HIV-1 target UTR308 was analyzed for the two most abundant aptamers in populations XI and XIV (represented by sequences XIV22 and XIV26), and for RNApt16. Since further inhibition assays were performed using fused aptamer-U6 snRNA cassette molecules, we carried out binding assays with these chimeric molecules, termed LXIV22 and LXIV26. RNApt16 was assayed without any modification for both binding and inhibitory assays, as we were interested in studying the functional properties of the minimal aptamer. The interaction of the RNApt16neg molecule with the target was also assayed as a negative control. The binding efficiency was analyzed by gel electrophoresis mobility shift assays (Fig. 5). Multiple complex conformers were observed for the LXIV22 aptamer RNA, whereas a major complex conformer was obtained for LXIV26 (Fig. 5A) and RNApt16 aptamers (Fig. 5B). No interaction was detected for the control molecule RNApt16neg, thus demonstrating the usefulness of our in silico approach for designing such a negative control. Results obtained from three independent experiments were quantified and fitted to a hyperbolic one-site binding curve with R2 coefficient higher than 0.99 for aptamers LXIV26 and RNApt16, yielding a Kd of 82 ± 13 nM and 280 ± 60 nM, respectively. Binding of the aptamer LXIV22 responded to a one-site specific binding curve with Hill slope, also with R2 coefficient higher than 0.99 and with a Kd of 154 ± 5 nM.
Figure 5. Aptamers binding to the 5′-UTR of HIV-1.

Autoradiograms of representative native polyacrylamide gels resolving binding assays of LXIV22 and LXIV26 (A), RNApt16 and RNApt16neg (B), to the UTR308.  : unbound aptamer.
: unbound aptamer. 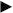 : aptamer-target complexes. Mean values of three independent experiments are represented on the right.
: aptamer-target complexes. Mean values of three independent experiments are represented on the right.
Efficient inhibition of viral particle production by the selected aptamers
The effect of the two most represented in vitro selected aptamers, XIV22 and XIV26, on HIV-1 production was assayed. Unmodified, pre-synthesized aptamers gave no satisfactory results (data not shown), probably due to their limited half-life or incorrect cellular location. Thus, we decided to flank them with the 5′ and 3′ stable hairpin-loop domains of the human U6 snRNA33 (Fig. 6A), thus giving rise to the modified aptamers termed LXIV22 and LXIV26. Aptamer sequences were cloned into vector pU614 and templates for in vitro transcription of aptamers LXIV22 or LXIV26 were prepared from the resulting plasmids. HEK 293T cells were co-transfected with 500 ng of in vitro transcribed LXIV22 or LXIV26 RNA molecules and 100 ng of plasmid pNL4.3 (containing the HIV-1 NL4.3 proviral DNA). Viral particle production was measured as p24 antigen abundance in the cellular supernatant, yielding inhibitions of 77 ± 7% and 80 ± 3%, respectively, in the HIV-1 viral particle production with respect to the control RNA termed ‘L-empty', that was transcribed from a pU6 plasmid without any cloned aptamer sequence, thus consisting in the two flanking U6 terminal 5′- and 3′-hairpins (Fig. 6B).
Figure 6. Ex vivo HIV-1 inhibition assays.

(A) Schematic representation of the U6-aptamer RNA constructs. (B) Inhibition of HIV-1 p24 antigen production by LXIV22 and LXIV26, using ‘L-empty' as control. (C) Inhibition by RNApt16 and RNApt16neg. Data represent the mean of three independent experiments. **: statistically significant differences as compared to the control (ANOVA, p < 0.01).
To validate the in silico shortening of the in vitro selected aptamers, inhibition of HIV-1 production by the designed RNApt16 was also tested. HEK 293T cells were co-transfected with 500 ng of chemically synthesized RNApt16 and 100 ng of pNL4.3. Inhibition of viral particles production of 85 ± 5% with respect to the synthetic RNApt16neg control molecule, which did not showed inhibitory effect, was observed (Fig. 6C). This result clearly points to the usefulness of the combined in vitro - in silico approach to design and optimize novel anti-HIV-1 agents.
Discussion
Anti-HIV-1 RNA aptamers have been described to inhibit viral replication by interacting with different viral or cellular targets34,35,36,37. In the present work, 64 nt-long RNA aptamers directed against the first 308 nt of the 5′-UTR of the HIV-1 genomic RNA have been selected in vitro. Based on our experimental results and on previous reports on the computational study of the SELEX outcome30,35, we have analysed in silico the RNA sequences and structures obtained along the process. Interestingly, a conserved, 16 nt-long sequence-structure motif present in most of the aptamers that target the viral RNA has been identified. The in silico designed RNApt16 aptamer showed a specific binding to the UTR308 RNA in vitro, and it efficiently inhibited the production of HIV-1 viral particles in transient transfection of HEK293T cell cultures. To our knowledge, this RNApt16 molecule is the shortest anti-HIV-1 efficient aptamer described to date. Actually, conventional SELEX procedures impose a minimal length to the selected molecules and to the minimal aptamers obtained by sequence trimming12,38,39, thus compromising the exploitation of aptamers as therapeutic agents. This limitation is here overcome by means of a combined in vitro – in silico approach.
The 16 nt-long, highly conserved stem-loop motif shared by most of the selected aptamers (in particular, the abundant sequences represented by XIV22 and XIV26) served as a guide for designing the RNApt16 molecule, together with RNApt16neg as a negative control. Binding of the RNApt16 molecule to the HIV-1 5′-UTR resembled the XIV26 binding behaviour with a single aptamer:target complex and fitting to a hyperbolic binding curve, while XIV22 showed different aptamer:target complexes and a sigmoidal binding curve. However, RNApt16 produced a less efficient binding with respect to that of aptamers XIV22 and XIV26, evidencing a contribution to the overall binding of the rest of the complete aptamer molecules apart from their 16 nt-long motif.
There are several quantitative properties that characterize the higher complexity of the selected, full-length aptamers in comparison to RNApt16. The analysis of the ensemble diversity (ED) and the frequency of the minimum free energy (FME) structure showed that two groups of aptamers were segregated from round IX on, with each of the two most represented molecules in round XIV belonging to one of these subpopulations (Figs. S1 and S5). The subpopulation represented by aptamer XIV26 (group 1) had low ED (3.12) and high FME (33.22%), while that represented by XIV22 (group 2) showed the opposite behaviour (ED = 11.65; FME = 6.07%). This observation eventually stems from the difference in stability of the two corresponding folded configurations (Table S1, Figs. 4A, 4B and S8). It is plausible that the structural plasticity of aptamer XIV22, with a fold of low stability outside the conserved, relevant interaction site, may promote multiple binding complexes, whereas aptamer XIV26 exhibits a robust secondary structure that likely entails a predominant binding complex. This possibility is in agreement with the in vitro results of the aptamer binding to the HIV-1 UTR308 molecule, where the participation of extra regions present in aptamer XIV22 might be responsible for the formation of multiple aptamer:target complexes and a binding curve with different topology with respect to that of XIV26 and RNApt16 (Fig. 5). Nonetheless, the dominant aptamer-target interaction is always that occurring at the consensus octamer 5′-GGCAAGGA-3′, located at the apical loop of a structural motif 5′-((((........))))-3′ within the aptamer. It is interesting that the observation of other potentially binding regions is conditional on the folded structure of the aptamer. Such is the case of region (a′) in Fig. 1 (see also Table S2), which interacts with a hexamer found in all assayed aptamers, since it belongs to the 3′ common flanking region (nucleotides 55–60): while in XIV22 and XIV1 that interaction is observed, and it occurs with a relatively high binding energy, we have not found it in XIV26. This observation is compatible with a dynamic (low folding energy) configuration in nucleotides 55–60 of the former two aptamers (Fig. 4A), while in the latter the breakage of the highly stable stem (Fig. 4B) followed by the binding to region (a′) is not favoured energetically and does not take place. In agreement with this result, we have checked that aptamer XIV5, characterized by a significantly lower folding energy in that domain, may indeed bind to (a′) (data not shown).
The HIV-1 inhibition efficiency of the aptamers seems to decrease when different competing interaction sites are present in the molecule, thus constituting an additional advantage of RNApt16. As a matter of fact, while RNApt16 alone inhibits the production of viral particles up to 85%, inhibition by the in vitro selected 64 nt-long aptamers was only produced (up to 75%) when fused to the U6 snRNA flanking hairpins (Fig. 6 and data not shown) that increase their molecular length in more than 60 nt. Indeed, we cannot discard that the addition of the flanking hairpins had a direct effect on the aptamer-target interaction.
The preferred target site of the selected aptamers and the RNApt16 molecule was the 5′-CUUGCC-3′ sequence (nts 81–86 of the HIV-1 genome) exposed in the apical loop of the essential poly(A) domain (Fig. 1). This hexamer is highly conserved among all the HIV-1 strains, subtypes, circulating intersubtype recombinant forms and groups, and even among the closely related simian immunodeficiency virus (SIV) from chimpanzee40,41 (Fig. S9 and data not shown). This sequence conservation makes interesting to analyze the inhibitory potential of the selected aptamers and RNApt16 against different clinical HIV-1 isolates belonging to distinct viral subtypes and groups. Although an identical poly(A) domain is present at both ends of all intracellular genomic and subgenomic HIV-1 RNAs, only the poly(A) at the 3′ end leads to polyadenylation of the HIV RNAs42. Therefore, we hypothesize that the achieved inhibitory effect of the in vitro selected aptamers and the in silico derived one (RNApt16) might be explained as a result of the interference with the proper 3′ end RNA polyadenylation (Fig. 7).
Figure 7. Model of putative functional effects of RNApt16 binding to poly(A) domain.

UTR308 region in genomic (A) and subgenomic (B) RNAs is shown in black. The RNApt16 is shown in red. Poly(A)-gag pseudoknot is represented by a gray line in (A). The arrowhead points the major splicing donor.
However, we cannot discard other inhibitory effects derived from aptamer binding to the 5′ poly(A) domain. The binding of the aptamers to the 5′ end poly(A) domain might additionally interfere with translation, reverse transcription, RNA dimerisation and encapsidation, by affecting the folding of neighbouring domains within the 5′UTR9,43,44,45. The interference with the later two processes may result in the promotion of aberrant genomic-subgenomic RNA heterodimers through 5′-UTR reshape, yielding viral particles that are incompetent for infection (Fig. 7). Additionally, aptamer binding might interfere with the previously described pseudoknot involving the 5′ poly(A)-targeted nucleotides and a downstream gag ORF region, whose function remains unknown46. In any case, we provide evidences that interfering with the poly(A) domain may effectively challenge the successful completion of the HIV-1 cycle.
Our results underline the usefulness of the engineered RNApt16 aptamer as an active inhibitor of HIV-1. RNApt16 includes a 4 bp-long stem entirely composed of G-C pairs that could protect the molecule against 5′-end degradation, as U6 snRNA hairpins may do in LXIV22 and LXIV26 aptamers. Indeed, uncapped and unpolyadenylated natural RNAs protect from exonucleases degradation by trapping their ends into G-C rich hairpins (e.g. naked RNA viruses, tRNAs and 5S rRNA38,39,47 and such a protection is usually essential to increase the half-life and, thus, the efficiency of inhibitory RNAs for therapeutic applications24,48,49. The remarkably small size of RNApt16 and its clear inhibitory effect without the need of further modifications makes of this poly(A) domain-binding molecule a very promising candidate for the development of anti-HIV strategies. A putative increase of the stability of either the in vitro selected aptamers or the in silico designed one will be explored by chemical modifications. Finally, the eventual selection of resistance mutations to the reported aptamers will be subject of further investigation. In any case, the combination of RNApt16 with other aptamers or with distinct inhibitory RNA molecules (e.g. siRNAs) of different specificity might help delaying the generation of escape HIV-1 mutant variants, as it has already been shown in mouse models50. Altogether, our results exemplify the applicability of an in vitro - in silico combined approach for the design and optimization of efficient anti-HIV-1 aptamers, useful as either drug candidates or diagnostic tools.
Methods
DNA templates and RNA synthesis
The HIV-1 5′-UTR RNA target molecule (termed UTR308 since it spans the first 308 nts of the genomic sequence) was synthesized by in vitro transcription of a PCR-amplified DNA template obtained from the pNL4.3 plasmid as previously described51. In turn, the pU6-based eukaryotic RNA expression vectors were obtained by cloning the aptamer coding sequences XIV22 and XIV26 within the KpnI and ApaI restriction sites of vector pU614. The aptamer-coding fragments were obtained by PCR amplification of the corresponding pGEM-T®easy (Promega, Madison, US-WI) aptamer-coding plasmid, using primers 5′KpnIC3 (5′-CGACTCGGTACCGGGAATTCAA-3′) and 3′ApaIC3 (5′-TCTGGGCCCGTCGAGCTCGTAGTATC-3′). The resulting plasmids were called pU6-LXIV22 and pU6-LXIV26, and led to the synthesis of the 128 nt-long LXIV22 and LXIV26 RNA molecules, composed of the corresponding RNA aptamer flanked by the 5′ and 3′ hairpins of the human U6 snRNA. RNA molecules LXIV22 and LXIV26 were obtained by in vitro transcription of the corresponding PCR templates as previously described51.
In vitro selection of aptamers
RNA aptamers were generated using a SELEX procedure against the HIV-1 molecule UTR308. The starting population of 84 nt-long DNA molecules consisted of 25 randomized nts flanked by constant sequences, and it was obtained by annealing and extension of 7.5 nmol of each of the oligonucleotides 5′EcoRIK (5′-GGATAATACGACTCACTATAGGGAATTCAA-3′) and 3′RANDOMK (5′-GTCGAGCTCGTAGTATCAGATCACTCCATN25TTGAATTCCCTATAGTG-3′), where the underlined sequence corresponds to the T7 RNA polymerase promoter and ‘N' stands for a randomized position. The amount of the oligonucleotide including the 25 nt-long random region was 4-fold higher than the theoretical number of different molecules (1.1 × 1015). The population of 64 nt-long RNA aptamers was in vitro transcribed as previously described52.
The target RNA molecule UTR308 was internally biotinylated during transcription by adding biotinylated-UTP (Roche, Indianapolis, US-IN) to the transcription mix at 0.106 mM final concentration (in the presence of 1 mM non-biotinylated UTP). This rendered the incorporation of, on average, one biotinylated-UTP residue per molecule. Two transcription reactions were performed in parallel at 37°C for 2 hours, and DNA templates were removed using RQ1 DNase (Promega, Madison, US-MA) at 1 U/μg DNA concentration, at 37°C for 30 min. All transcription products were gel-purified and ethanol-precipitated.
The target RNA was resuspended in 1 ml of binding buffer (150 mM sodium chloride, 20 mM sodium phosphate, pH 7.5) and renatured at 65°C for 10 min followed by an additional incubation at 37°C for 10 min. Then, biotinylated UTR308 was bound to a HiTrapTM Streptavidin HP column (GE Healthcare, Chalfont ST. Giles, UK) following manufacturer's instructions. The column was washed with 10 ml of binding buffer and equilibrated with 10 ml of TMN 1× buffer (20 mM TRIS-acetate; 10 mM magnesium acetate; 100 mM sodium chloride). The starting RNA aptamer population exceeded 40 μg weight, theoretically ensuring the presence of at least one copy of each of the possible sequence variants. Previous to the first selection round, a negative selection step was carried out to prevent selection of sepharose and/or streptavidin-binder molecules: for this purpose the initial RNA population was passed through an unloaded-sepharose column at 25°C and the unbound molecules were recovered. This RNA population was loaded into a target-containing column in 1 ml of TMN 1×, and incubated for binding at 25°C for 30 min. Unbound molecules were discarded by washing with 10 ml of TMN 1× at the binding temperature, and bound molecules were further recovered by elution with 10 ml of TMN 1× at 95°C. The first four 1 ml fractions recovered were concentrated using Centricon Ultracel YM-3 (Merck Millipore, Billerica, US-MA), and ethanol-precipitated.
One half of the recovered RNA pool was reverse transcribed and amplified by Tth DNA polymerase (Promega, Madison, US-WI) using the primers 3′XhoIK (5′-GTCGAGCTCGTAGTATCAGATCACTCCAT-3′) and 5′EcoRIK. The cDNA population was phenol-extracted, ethanol-precipitated and used as template for a new round of selection. Fourteen rounds of amplification-selection were performed, and the selection pressure was increased along the procedure as follows: i) binding temperature was 25°C for the rounds I to III, and 37°C for the rounds IV to XIV; ii) the aptamer:target ratio was 1:1 for the rounds I to X, and 1000:1 for the rounds XI to XIV. The second half of the cDNA population of each round was cloned in E. coli using pGEM-T®easy vector, and a minimum of 24 molecular clones were sequenced.
In silico methods for sequence analysis
For sequence comparison, Hamming distances between each pair of clonal sequences were calculated as the number of positions at which the corresponding nucleotides are different. Clustering of the non-repeated, 64 nt-long sequences was performed based on their mutual Hamming distances. An outgroup sequence was designed that contained the 10 nt and 29 nt-long constant sequences present at the 5′ and 3′ ends of all molecules, flanking an artificial 25 nt-long sequence 5′-(ACGT)6A-3′. Thus, the complete sequence of the outgroup was: 5′-GGGAATTCAAACGTACGTACGTACGTACGTACGTAATGGAGTGATCTGATACTACGAGCTCGAC-3′. The topology of the clustering was inferred by means of the neighbour-joining (NJ) method53 using the program NEIGHBOR from the PHYLIP v3.6 package54.
In silico methods for analyzing RNA structure
We used the Vienna RNA package55, version 1.5, to fold RNA sequences into their minimum free energy (MFE) secondary structure, as well as to compare the obtained structures (see below). The previously described standard parameter set56 was used, allowing for A-U, G-C, and wobble G-U base pairs, and disallowing the formation of isolated base pairs. Three relevant quantities were computed for every folded structure, in addition to its folding energy: i) its ensemble diversity (ED); ii) the frequency of the MFE structure (FME) in the thermodynamic ensemble; iii) the ensemble centroid structure (CE). The ED is the average distance between all the possible secondary structures present in the thermodynamic ensemble, which is in turn defined as the set of all different structures (each characterized by its corresponding folding energy) compatible with a given sequence. Structures with similar energy in the ensemble are found with comparable frequency. If the energy of the MFE structure is significantly lower than the rest of structures in the ensemble, then the corresponding sequence will fold most of the time in the MFE structure, and it will thus be much more frequent. The centroid of a set of structures is the structure that has the minimum total base-pair distance to the structures in the set, thus being the single structure that best represents the set as a whole57. For representation of RNA secondary structures the dot-bracket notation was used, where unpaired nucleotides are denoted by dots, and paired nucleotides by parentheses: ‘(' indicates that the partner is downstream, and ‘)' that the partner is upstream.
Structure comparison was performed by means of base-pair distances, which measure the number of base pairs that must be opened and closed in order to convert one structure into the other. We checked that structure comparison through Hamming or tree-edit distances yielded qualitatively equivalent results. The Hamming distance between two folded sequences (that must be of equal length) is defined as the number of positions at which the structural states [‘.', ‘(‘ or ‘)'] of the corresponding nucleotides differ. In turn, the tree–edit distance compares structural elements (e.g., hairpins, bulges or stems) and allows for a variable number of nucleotides involved in those motifs or separating them. It is slightly more computationally complex since it was devised to compare sequences of different length58.
A cluster analysis of the structures was performed, based on their base-pair distances. An artificial outgroup structure was designed, showing the maximum number of paired bases in their 64 nt-long sequence: 5′-((((((((((((((((((((((((((((((....))))))))))))))))))))))))))))))-3′. Clustering was performed as described above.
To predict those sites where interaction between aptamer and ligand is favoured, we used the RNAup algorithm from the Vienna RNA package, version 1.8.459. RNA-RNA binding is decomposed in two steps: first, the probability that a sequence interval (e.g. a binding site) remains unpaired is computed; in a second step, the binding energy provided that the binding site is unpaired is calculated as the optimum over all possible types of bindings60. Parameters are chosen as for RNAfold. In particular, isolated base pairs are not allowed and the length of the unstructured regions is 4 nts.
In vitro aptamer binding assays
The evaluation of the binding efficiency of the selected aptamers was essentially performed as previously described32, using trace amounts of renatured, 5′ end 32P-labelled aptamers and 0, 2, 20, 200 or 400 nM of non-labelled UTR308 target RNA.
Results obtained from three independent experiments were quantified and fitted to either a hyperbolic one-site binding curve (Equation 1) or a one-site specific binding curve with Hill slope (Equation 2):
 |
 |
where Bmax is the maximum value of Y (when X = ∞); Kd is the value of X when Y = Bmax/2; and h is the Hill slope and indicates the degree of cooperativity.
HIV-1 inhibition assays
The viral production inhibition assays were performed by transient transfection of human embryonic kidney (HEK) cells as previously described51, using 100 ng of plasmid pNL4.3 plus 500 ng of RNA aptamer in co-transfection experiments14.
Author Contributions
F.J.S.-L. performed all in vitro and cell culture experiments; M.S., S.M. and C.B. performed the computational analyses; A.B.-H. and C.B. designed the study and were responsible for the work; A.B.-H., C.B. and S.M. wrote the manuscript; all authors prepared figures, commented on the manuscript and approved the final version.
Supplementary Material
Supplementary data
Acknowledgments
We acknowledge Dr. J. Esté and C. Romero-López for critical reading of the manuscript, J.A. Reyes-Darias for his help with inhibition assays and HIV-1 detection by EIA, and V. Augustin-Vacas for his excellent technical assistance. We also thank Drs. M.C. López and M.C. Thomas for instrumental support. The support of Aptus Biotech is also acknowledged. This work was supported by the Spanish Ministerio de Ciencia e Innovación [BFU2009-08137 to A.B.-H., BIO2010-20696 to C.B. and FIS2011-27569 to S.M.]; by the Spanish Ministerio de Economía y Competitividad [BFU2012-31213 to A.B.-H.]; by the Junta de Andalucía [CVI-7430 to A.B.-H.]; by the Spanish National Research Council [201120E004 to A.B.-H.]; by Comunidad de Madrid [MODELICO, S2009/ESP-1691 to S.M.] and by FEDER funds from the EU to C.B. and A.B.-H. CIBERehd is funded by the Instituto de Salud Carlos III.
References
- Berkhout B. Structure and function of the human immunodeficiency virus leader RNA. Prog Nucleic Acid Res Mol Biol. 54, 1–34 (1996). [DOI] [PubMed] [Google Scholar]
- Berkhout B. HIV-1 as RNA evolution machine. RNA Biol. 8, 225–229 (2011). [DOI] [PubMed] [Google Scholar]
- Bannwarth S. & Gatignol A. HIV-1 TAR RNA: the target of molecular interactions between the virus and its host. Current HIV Res. 3, 61–71 (2005). [DOI] [PubMed] [Google Scholar]
- Zarudnaya M. I., Potyahaylo A. L., Kolomiets I. M. & Hovorun D. Structural model of the complete poly(A) region of HIV-1 pre-mRNA. J Biomol Struct & Dyn 31, 1044–1056 (2012). [DOI] [PubMed] [Google Scholar]
- Sleiman D. et al. Initiation of HIV-1 reverse transcription and functional role of nucleocapsid-mediated tRNA/viral genome interactions. Virus Res. 169, 324–339 (2012). [DOI] [PubMed] [Google Scholar]
- Ulyanov N. B. et al. NMR structure of the full-length linear dimer of stem-loop-1 RNA in the HIV-1 dimer initiation site. J Biol Chem 281, 16168–16177 (2006). [DOI] [PubMed] [Google Scholar]
- Harrison G. P. & Lever A. M. The human immunodeficiency virus type 1 packaging signal and major splice donor region have a conserved stable secondary structure. J Virol 66, 4144–4153 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clever J. L., Miranda D. Jr & Parslow T. G. RNA structure and packaging signals in the 5′ leader region of the human immunodeficiency virus type 1 genome. J Virol 76, 12381–12387 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huthoff H. & Berkhout B. Two alternating structures of the HIV-1 leader RNA. RNA (New York, N.Y 7, 143–157 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen P., Worland S. & Gold L. Isolation of high-affinity RNA ligands to HIV-1 integrase from a random pool. Virology 209, 327–336 (1995). [DOI] [PubMed] [Google Scholar]
- Barroso-DelJesus A. et al. Inhibition of HIV-1 replication by an improved hairpin ribozyme that includes an RNA decoy. RNA Biol. 2, 75–79 (2005). [DOI] [PubMed] [Google Scholar]
- Duconge F. & Toulme J. J. In vitro selection identifies key determinants for loop-loop interactions: RNA aptamers selective for the TAR RNA element of HIV-1. RNA (New York, N.Y 5, 1605–1614 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. J., Kim M. Y., Lee J. H., You J. C. & Jeong S. Selection and stabilization of the RNA aptamers against the human immunodeficiency virus type-1 nucleocapsid protein. Biochem Biophys Res Commun. 291, 925–931 (2002). [DOI] [PubMed] [Google Scholar]
- Puerta-Fernandez E. et al. Inhibition of HIV-1 replication by RNA targeted against the LTR region. AIDS (London, England) 19, 863–870 (2005). [DOI] [PubMed] [Google Scholar]
- Tuerk C., MacDougal S. & Gold L. RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc Natl Acad Sci USA 89, 6988–6992 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Darias J. A., Sanchez-Luque F. J. & Berzal-Herranz A. Inhibition of HIV-1 replication by RNA-based strategies. Current HIV Res. 6, 500–514 (2008). [DOI] [PubMed] [Google Scholar]
- Cheng C. et al. Potent inhibition of human influenza H5N1 virus by oligonucleotides derived by SELEX. Biochem Biophys Res Commun. 366, 670–674 (2008). [DOI] [PubMed] [Google Scholar]
- Kumar P. K. et al. Isolation of RNA aptamers specific to the NS3 protein of hepatitis C virus from a pool of completely random RNA. Virology 237, 270–282 (1997). [DOI] [PubMed] [Google Scholar]
- Romero-Lopez C., Berzal-Herranz B., Gomez J. & Berzal-Herranz A. An engineered inhibitor RNA that efficiently interferes with hepatitis C virus translation and replication. Antiviral Res 94, 131–138 (2012). [DOI] [PubMed] [Google Scholar]
- Romero-Lopez C., Diaz-Gonzalez R. & Berzal-Herranz A. Inhibition of hepatitis C virus internal ribosome entry site-mediated translation by an RNA targeting the conserved IIIf domain. Cell Mol Life Sci 64, 2994–3006 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington A. D. & Szostak J. W. In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822 (1990). [DOI] [PubMed] [Google Scholar]
- Tuerk C. & Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science (New York, N.Y 249, 505–510 (1990). [DOI] [PubMed] [Google Scholar]
- Cerchia L. & de Franciscis V. Targeting cancer cells with nucleic acid aptamers. Trends Biotech. 28, 517–525 (2010). [DOI] [PubMed] [Google Scholar]
- Marton S., Reyes-Darias J. A., Sanchez-Luque F. J., Romero-Lopez C. & Berzal-Herranz A. In vitro and ex vivo selection procedures for identifying potentially therapeutic DNA and RNA molecules. Molecules (Basel, Switzerland) 15, 4610–4638 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. et al. Aptamers as therapeutics in cardiovascular diseases. Current Med Chem 18, 4169–4174 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezhnoy A. et al. Isolation and optimization of murine IL-10 receptor blocking oligonucleotide aptamers using high-throughput sequencing. Mol Ther 20, 1242–1250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigdar S. et al. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett 330, 84–95 (2013). [DOI] [PubMed] [Google Scholar]
- Chushak Y. & Stone M. O. In silico selection of RNA aptamers. Nucleic Acids Res 37, e87 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoon S., Zhou B., Janda K. D., Brenner S. & Scolnick J. Aptamer selection by high-throughput sequencing and informatic analysis. BioTechniques 51, 413–416 (2011). [DOI] [PubMed] [Google Scholar]
- Thiel W. H. et al. Rapid identification of cell-specific, internalizing RNA aptamers with bioinformatics analyses of a cell-based aptamer selection. PloS One 7, e43836 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockey W. M. et al. Rational truncation of an RNA aptamer to prostate-specific membrane antigen using computational structural modeling. Nucleic Acid Ther 21, 299–314 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Lopez C., Diaz-Gonzalez R., Barroso-delJesus A. & Berzal-Herranz A. Inhibition of hepatitis C virus replication and internal ribosome entry site-dependent translation by an RNA molecule. J Gen Virol 90, 1659–1669 (2009). [DOI] [PubMed] [Google Scholar]
- Good P. D. et al. Expression of small, therapeutic RNAs in human cell nuclei. Gene Ther 4, 45–54 (1997). [DOI] [PubMed] [Google Scholar]
- Chaloin L., Lehmann M. J., Sczakiel G. & Restle T. Endogenous expression of a high-affinity pseudoknot RNA aptamer suppresses replication of HIV-1. Nucleic Acids Res 30, 4001–4008 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditzler M. A. et al. High-throughput sequence analysis reveals structural diversity and improved potency among RNA inhibitors of HIV reverse transcriptase. Nucleic Acids Res 41, 1873–1884 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange M. J. et al. Robust suppression of HIV replication by intracellularly expressed reverse transcriptase aptamers is independent of ribozyme processing. Mol Ther 20, 2304–2314 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whatley A. S. et al. Potent Inhibition of HIV-1 Reverse Transcriptase and Replication by Nonpseudoknot, “UCAA-motif” RNA Aptamers. Mol Ther 2, e71 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whipple J. M., Lane E. A., Chernyakov I., D'Silva S. & Phizicky E. M. The yeast rapid tRNA decay pathway primarily monitors the structural integrity of the acceptor and T-stems of mature tRNA. Genes & Dev 25, 1173–1184 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y. Y. & Worcel A. A 3′ exonuclease activity degrades the pseudogene 5S RNA transcript and processes the major oocyte 5S RNA transcript in Xenopus oocytes. Genes & Dev 3, 1008–1018 (1989). [DOI] [PubMed] [Google Scholar]
- Kuiken C. et al. HIV Sequence Compendium 2011. Los Alamos National Laboratory, Theoretical Biology and Biophysics, Los Alamos, New Mexico. LA-UR-11-11440 (2011). [Google Scholar]
- Pollom E. et al. Comparison of SIV and HIV-1 genomic RNA structures reveals impact of sequence evolution on conserved and non-conserved structural motifs. PLoS Pathogens 9, e1003294 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee A. H., Kasprzak W. & Shapiro B. A. Structural differentiation of the HIV-1 polyA signals. J Biomol Struct & Dyn 23, 417–428 (2006). [DOI] [PubMed] [Google Scholar]
- Abbink T. E. & Berkhout B. A novel long distance base-pairing interaction in human immunodeficiency virus type 1 RNA occludes the Gag start codon. J Biol Chem 278, 11601–11611 (2003). [DOI] [PubMed] [Google Scholar]
- Ashe M. P., Griffin P., James W. & Proudfoot N. J. Poly(A) site selection in the HIV-1 provirus: inhibition of promoter-proximal polyadenylation by the downstream major splice donor site. Genes & Dev 9, 3008–3025 (1995). [DOI] [PubMed] [Google Scholar]
- Paillart J. C. et al. First snapshots of the HIV-1 RNA structure in infected cells and in virions. J Biol Chem 279, 48397–48403 (2004). [DOI] [PubMed] [Google Scholar]
- Paillart J. C., Skripkin E., Ehresmann B., Ehresmann C. & Marquet R. In vitro evidence for a long range pseudoknot in the 5′-untranslated and matrix coding regions of HIV-1 genomic RNA. J Biol Chem 277, 5995–6004 (2002). [DOI] [PubMed] [Google Scholar]
- Esteban R., Vega L. & Fujimura T. 20S RNA narnavirus defies the antiviral activity of SKI1/XRN1 in Saccharomyces cerevisiae. J Biol Chem 283, 25812–25820 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behlke M. A. Chemical modification of siRNAs for in vivo use. Oligonucleotides 18, 305–319 (2008). [DOI] [PubMed] [Google Scholar]
- Thiel K. W. & Giangrande P. H. Therapeutic applications of DNA and RNA aptamers. Oligonucleotides 19, 209–222 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff C. P. et al. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice. Science Transl Med 3, 66ra6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Luque F. J., Reyes-Darias J. A., Puerta-Fernandez E. & Berzal-Herranz A. Inhibition of HIV-1 replication and dimerization interference by dual inhibitory RNAs. Molecules (Basel, Switzerland) 15, 4757–4772 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Lopez C., Barroso-delJesus A., Puerta-Fernandez E. & Berzal-Herranz A. Interfering with hepatitis C virus IRES activity using RNA molecules identified by a novel in vitro selection method. Biol Chem 386, 183–190 (2005). [DOI] [PubMed] [Google Scholar]
- Saitou N. & Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biology Evol 4, 406–425 (1987). [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP (Phylogeny Inference Package) version 3.6. Department of Genome Sciences, University of Washington, Seattle (2005). [Google Scholar]
- Hofacker I. L. et al. Fast Folding and Comparison of RNA Secondary Structures. Monatschefte F. Chemie 125, 911–940 (1994). [Google Scholar]
- Mathews D. H., Sabina J., Zuker M. & Turner D. H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol 288, 911–940 (1999). [DOI] [PubMed] [Google Scholar]
- Ding Y., Chan C. Y. & Lawrence C. E. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA (New York, N.Y 11, 1157–1166 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro B. A. An algorithm for comparing multiple RNA secondary structures. Comput Appl Biosci 4, 387–393 (1988). [DOI] [PubMed] [Google Scholar]
- Gruber A. R., Lorenz R., Bernhart S. H., Neubock R. & Hofacker I. L. The Vienna RNA websuite. Nucleic Acids Res 36, W70–74 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueckstein U. et al. Thermodynamics of RNA-RNA Binding. Bioinformatics 22, 1177–1182 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data