

Abstract
Objective:
We report novel defects of mitochondrial translation elongation factor Ts (EFTs), with high carrier frequency in Finland and expand the manifestations of this disease group from infantile cardiomyopathy to juvenile neuropathy/encephalopathy disorders.
Methods:
DNA analysis, whole-exome analysis, protein biochemistry, and protein modeling.
Results:
We used whole-exome sequencing to find the genetic cause of infantile-onset mitochondrial cardiomyopathy, progressing to juvenile-onset Leigh syndrome, neuropathy, and optic atrophy in 2 siblings. We found novel compound heterozygous mutations, c.944G>A [p.C315Y] and c.856C>T [p.Q286X], in the TSFM gene encoding mitochondrial EFTs. The same p.Q286X variant was found as compound heterozygous with a splice site change in a patient from a second family, with juvenile-onset optic atrophy, peripheral neuropathy, and ataxia. Our molecular modeling predicted the coding-region mutations to cause protein instability, which was experimentally confirmed in cultured patient cells, with mitochondrial translation defect and lacking EFTs. Only a single TSFM mutation has been previously described in different populations, leading to an infantile fatal multisystem disorder with cardiomyopathy. Sequence data from 35,000 Finnish population controls indicated that the heterozygous carrier frequency of p.Q286X change was exceptionally high in Finland, 1:80, but no homozygotes were found in the population, in our mitochondrial disease patient collection, or in an intrauterine fetal death material, suggesting early developmental lethality of the homozygotes.
Conclusions:
We show that in addition to early-onset cardiomyopathy, TSFM mutations should be considered in childhood and juvenile encephalopathies with optic and/or peripheral neuropathy, ataxia, or Leigh disease.
Mitochondrial respiratory chain (RC) dysfunction is a major cause of metabolic disorders in adults and children. Mutations in mitochondrial and nuclear genes encoding mitochondrial protein synthesis machinery have been reported to cause an exceptionally variable spectrum of manifestations, despite all resulting in RC defects.1 The vast majority of identified mitochondrial translation defects originate from mutations in mitochondrial DNA–encoded transfer RNAs (tRNAs), but a growing number of nuclear genes are being identified. Mutations have been found in genes encoding elongation factors mtEFG1, EFTs, and elongation factor Tu (EFTu),2–6 mitoribosomal proteins (MRPL44, MRPS16, MRPS22, MRPL3, MRPL12),7–11 aminoacyl-tRNA synthetases,12–20 tRNA-modifying enzyme TRMU,21 pseudouridine synthase 1 PUS1,22 peptide release factor C12orf65,23 and RNase ELAC2.24 The complexity of the mitochondrial translation system suggests that the identified factors are only the tip of the iceberg.25,26
Next-generation sequencing methodology, in particular whole-exome sequencing, has shown to be an efficient way to find new disease-causing mutations in mitochondrial patients with variable clinical manifestations.3 Here, we applied exome sequencing to find the genetic cause of infantile-onset cardiomyopathy and Leigh disease, with slowly progressive, multiorgan RC deficiency, and report novel pathogenic mutations in the TSFM gene encoding for mitochondrial elongation factor Ts (EFTs).
METHODS
Standard protocol approvals, registrations, and patient consents.
All samples were taken according to the Declaration of Helsinki, with informed consent. The study was approved by the ethical review board of the Helsinki University Central Hospital and The Hospital District of Southwest Finland.
Exome sequencing.
The patients' genomic DNA was isolated from cultured fibroblasts (P1) or from muscle biopsy tissue (P3) and used for NimbleGen Sequence Capture 2.1M Human Exome v1.0 Array (Roche NimbleGen, Madison, WI) and sequenced with the Illumina Genome Analyzer-IIx platform (Illumina Inc., San Diego, CA) as previously described in reference 27. The identified single nucleotide polymorphisms (SNPs) were then filtered using dbSNP133, 1000 Genomes, and MitoCarta.28
Determination of the frequency of identified sequence variants.
Carrier frequencies were determined with 1000 Genomes, NHLBI (National Heart, Lung and Blood Institute Exome Variant Server) SNP, or dbSNP133 (National Center for Biotechnology Information) databases. Furthermore, because the patients originated from Finland, we utilized the exome data collection of more than 3,000 Finnish individuals from the Sequencing Initiative Suomi project (SISu; described in http://sisu.fimm.fi) and further characterized the carrier frequency of the p.Q286X variant by genotyping in a total of 32,497 subjects from the Finnish population (Finnish cohorts, http://sisu.fimm.fi).
The presence of the identified TSFM mutations in patients with mitochondrial disease was determined utilizing the data collection of 76 exomes in the laboratory of A.S.
Intrauterine fetal death samples were collected in Turku University Central Hospital between the years 2001 and 2011, excluding all early neonatal deaths, multiple pregnancies, and intrapartum deaths. The relevant conditions leading to intrauterine fetal death were classified according to the newly developed pathophysiologic classification system (ReCoDe, Relevant Condition of Death).29 DNA was extracted from paraffin blocks of 32 tissue samples by using NucleoSpin FFPE DNA kit (Macherey-Nagel, Düren, Germany) following the manufacturer's instructions. A 120–base pair amplification product was sequenced using the primers 5′-ctggacgatgagcctggg-3′ and 5′-ccctgaggctgcacatact-3′.
RNA analysis.
RNA was extracted with a standard Trizol and chloroform method from cultured fibroblasts of P1, P2, and one control, and from cultured myoblast of P3 and 2 controls with nonmitochondrial disease. Complementary DNA (cDNA) was synthesized using manufacturer's instructions (Maxima; Fermentas/Thermo Scientific, Pittsburgh, PA). The TSFM transcript levels were analyzed in 3 experimental replicates by real-time quantitative PCR with SYBR Green (Thermo Scientific) using the following primers: 5′-ggtgtttatcgcggctagag-3′ and 5′-ccacaagtctccagagctttc-3′ for TSFM and 5′-cgctctctgctcctcctgtt-3′ and 5′-ccatggtgtctgagcgatgt-3′ for GAPDH. The complete TSFM cDNA was PCR-amplified and sequenced with primers 5′-ggtgtttatcgcggctagag-3′ and 5′-ctcggtctgaagaggtttgg-3′. For solid-phase minisequencing, primers 5′-ccttaaggcacttttggaaat-3′ and 5′-B-ctcggtctgaagaggtttgg-3′ (B, biotinylated) and 5′-ggcagagactaagatgctgtcc-3′ (for Q286X mutation) were used with 3H-dCTP or 3H-dTTP, and radioactivity counts were measured by using the 2450 MicroBeta2 microplate counter (PerkinElmer, Waltham, MA).30
Protein modeling.
Structure modeling for human elongation factors EFTu and EFTs was based on resolved structures of bovine homologs (PDB id 1XB2, chain A for modeling EFTu and chain B for EFTs).31 Human and bovine EFTs sequences share 83.8% identity and 88.2% similarity, while EFTu sequences share 94.5% and 96.5%, respectively (identity matrix BLOSUM62). The following resources were used: structure modeling, SWISS-Model Server (http://swissmodel.expasy.org/), alignment mode; protein sequences of eukaryotic and prokaryotic EFTu and EFTs, UniProt knowledgebase (http://www.uniprot.org/); multiple sequence alignments, PROMALS3D (http://prodata.swmed.edu/promals3d/promals3d.php); assembly of EFTu-EFTs complex, structure analysis of the attained model and figure preparation, Discovery Studio V.2.5.5 (Accelrys).
Protein amount and translation analyses.
For Westerns, 15 µg of mitochondrial protein extracts were separated in 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. We used antibodies against SDH-70 kDa (MitoSciences, Eugene, OR) and EFTs/Tu (polyclonal, kindly provided by Linda Spremulli, Chapel Hill, NC). Experiments were done 3 times. Pulse translation assay was performed with 3 replicates as described previously.32 Total cellular proteins (30 µg) were separated in 12% to 20% polyacrylamide-gradient gel, which was imaged on a PhosphorImager plate (GE Healthcare, Waukesha, WI), analyzed with a Typhoon analyzer and ImageQuant software (Amersham Biosciences, Piscataway, NJ). As a control for fibroblast translation defect, we used patient fibroblasts with homozygous, previously published p.R312W mutation in EFTs (a kind gift from Professor Thorburn).3
RESULTS
Clinical findings.
Table 1 summarizes the clinical findings of the patients. Patient 1 (P1, II-3, family 1) (figure 1A), a female, is the third child of nonconsanguineous Finnish parents. Her brother (II-2) died suddenly at the age of 1 year during respiratory syncytial virus infection and was diagnosed postmortem to have cardiomyopathy. The early milestones of P1 were normal. At the age of 10 months, she was diagnosed with dilated cardiomyopathy. She had a severe episode of cardiac insufficiency during a respiratory infection at 1 year of age and was scheduled for cardiac transplantation, but this plan was canceled a few years later because the cardiomyopathy was no longer progressive. Her cardiac situation has remained stable on medication.
Table 1.
Clinical presentation of the patients
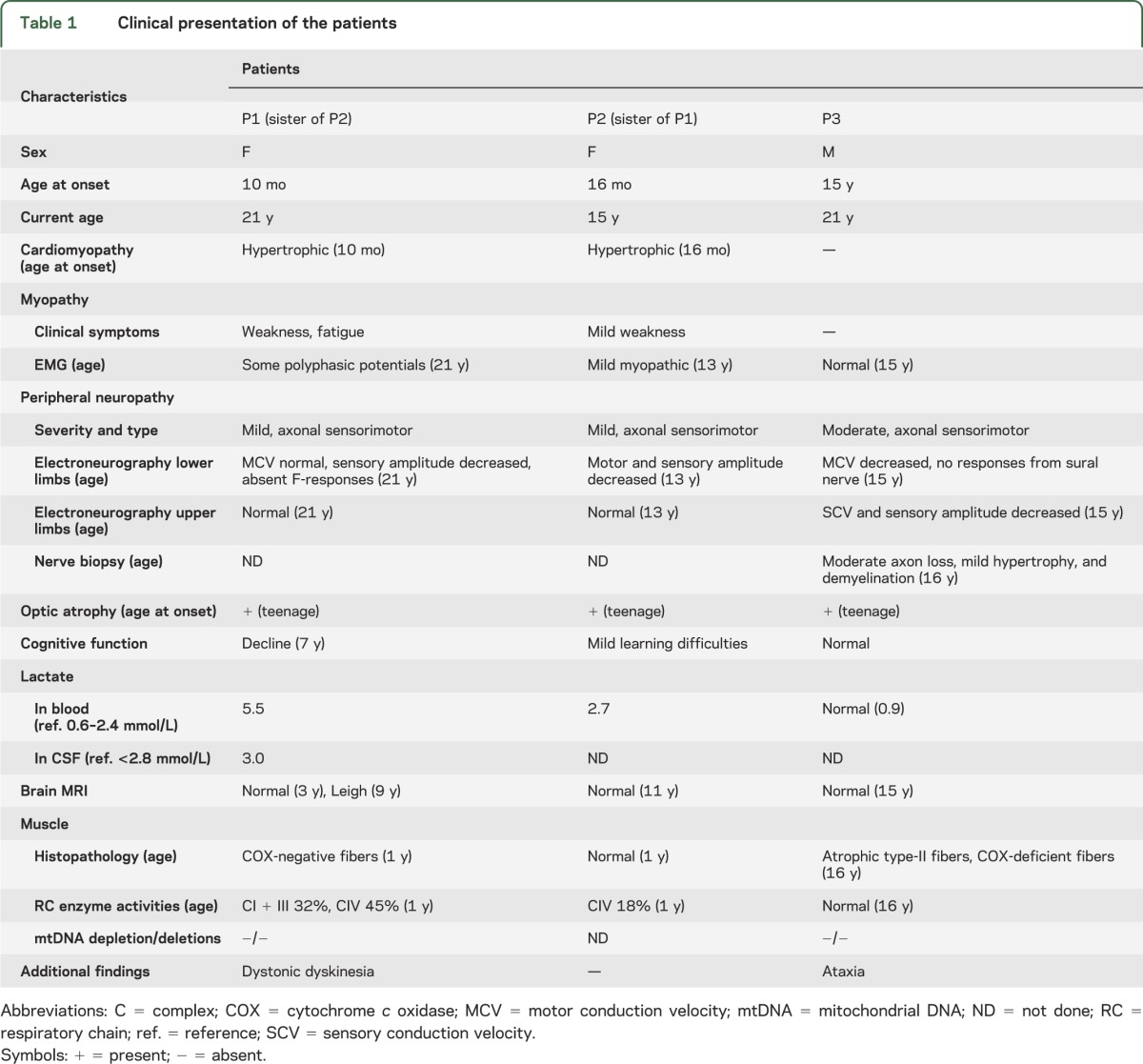
Figure 1. Exome sequencing revealed 3 novel mutations in the TSFM gene in 2 families.

(A) The pedigrees. Black symbols indicate affected individuals; strike through indicates deceased subject. (B) Exome data analysis and filtering steps of the single nucleotide variants of patient 1 (P1, family 1, II-3). TSFM, AGXT2L2 (PHYKPL, 5-phosphohydroxy-l-lysine phospho-lyase) and ACO2 (aconitase 2) indicate the genes in which potentially pathogenic variants were identified. TSFM gave best variant calling quality and was thus selected for further analysis. (C) TSFM gene structure with all 3 identified mutations marked in red. (D) Conservation of mutant elongation factor Ts amino acids Q286 and C315 in species. (E) Chromatograms of the 3 identified mutations (arrows). All mutations were confirmed to be heterozygous.
On examination at the age of 1 year, she had elevated serum and CSF lactate values. The skeletal muscle sample revealed combined deficiency of RC. Her brain MRI and early cognitive and motor development were normal. A cognitive decline manifested during the first school years, and brain MRI at the age of 9 years showed bilateral signal intensity in putamen and globus pallidus (in T2-weighted images), consistent with Leigh syndrome. Optic atrophy manifested at teenage and progressed to severe visual impairment. At present, at 21 years of age, she has frequent dystonic movements in her face, neck, and shoulder region, and mild peripheral axonal sensorimotor neuropathy.
Patient 2 (P2, II-4, family 1) (figure 1A), sister of P1, is a 15-year-old girl with hypertrophic cardiomyopathy diagnosed at the age of 16 months. Her cardiomyopathy has been stable on medication. She has mild learning difficulties, mild optic atrophy, and mild peripheral axonal sensorimotor neuropathy. Brain MRI at the age of 11 years was normal.
Patient 3 (P3, II-1, family 2) (figure 1A), a male, was born at full term to healthy Finnish parents. His development was normal until the age of 15 years when he experienced subacute deterioration of vision and was diagnosed with optic neuropathy. MRI of the brain was normal. He had mild tremor in the trunk and limbs, difficulties in keeping balance, and absent Achilles tendon reflexes. Electroneuromyography showed peripheral axonal neuropathy of mixed type. Histologic analysis of a sural nerve sample revealed a moderate loss of myelinated axons with a mild tendency to hypertrophy and demyelination. At the age of 21 years, the patient has ataxia and optical and peripheral neuropathy, but no cardiac symptoms.
Identification of TSFM mutations by whole-exome sequencing.
We determined the sequence of the whole exome of P1 by next-generation sequencing technology. Sequencing coverage was at 96.47% of reads mapped to the reference sequence with 20-fold coverage for 49.8% of the target bases. Figure 1B summarizes the filtering steps. P1 was found to carry compound heterozygous mutations in the TSFM gene that have been previously linked to mitochondrial cardiomyopathies: exon 6, missense mutation c.944G>A, Genbank cDNA NM_005726 [p.C315Y] and nonsense mutation c.856C>T [p.Q286X] (figure 1C). p.C315 is highly conserved in species, down to Drosophila melanogaster (figure 1D). The p.Q286X deletes the C-terminal 39 amino acids, leading to a truncated protein. Sanger sequencing confirmed the findings in P1 and in her affected sister P2 (figure 1E), and their parents were heterozygous carriers, indicating that the mutations cosegregated with the disease in the family. Sequencing the cDNAs from P1 and P2 fibroblast revealed that both alleles were present in cells, indicating that TSFM p.Q286X mRNA is not immediately degraded by nonsense-mediated decay.
A search for other TSFM variants in our mitochondrial disease patient cohort of 76 subjects revealed patient 3 (P3) to carry p.Q286X and a heterozygous intronic change c.106+4A>G. Sanger sequencing confirmed the findings. The mother of P3 carried the c.106+4A>G (figure 1E). This variant changed a conserved nucleotide within a splice site donor region, directly after the first exon (figure 1C), predicting aberrant splicing and a premature stop codon after 165 nucleotides. However, in cultured myoblasts of P3, c.106+4A>G did not affect TSFM transcript size, and when the alleles were quantified by solid-phase minisequencing, both were equally present in proportion. No other material was available from P3, and therefore we could not address a potential tissue-specific splicing defect. The total amount of TSFM transcript was reduced to 50% in P3 myoblasts, when compared with control myoblast lines (figure 2A), suggesting that the intronic mutation affected the general TSFM expression or the stability of the transcripts, possibly reducing the amount of TSFM available for translation.
Figure 2. TSFM mutations lead to protein degradation and mild reduction in translation activity.

(A) Relative messenger RNA expressions of EFTs in cultured cells of P1, P2, and P3. (B) Structural modeling of EFTu-EFTs complex showing the 2 amino acid changes p.Q286X (deleted sequence in yellow) and p.C315Y. Color coding: gray = EFTu; magenta = residues forming the GDP binding site in EFTu; pink = EFTs amino terminal domain (residues 1–99); green = the core domain (residues 100–210); blue = subdomain C (residues 211–318); and red = EFTs interaction site with EFTu and nucleotide exchange. p.C315 fits tightly into an uncharged pocket predicted to be formed by the residues from the opposite side of the β-sandwich. Substitution of cysteine 315 for tyrosine introduces a bulky side chain into the pocket designed for cysteine, resulting in van der Waals repulsion between the halves of the β-sandwich oriented toward the EFTu contact surface. This change in the local architecture affects stability of EFTs. Structural rearrangements caused by the mutation will affect the position of p.E104 locating in a distance of 3.8 Å from p.D126, which forms part of the α-turn in the subdomain N (marked in red) critical for interaction with EFTu and nucleotide exchange. (C) Immunoblot analysis showed almost complete loss of EFTs protein in P1 and P2 fibroblasts and substantial reduction in P3 myoblasts. Mild EFTu reduction was also seen in P1 and P2 but not in P3 samples. (D) Mitochondrial translation was partially reduced in P2 fibroblasts, but suggestively P1. An evident reduction of translation was seen in P3 myoblasts. R312W = patient fibroblast cell line with previously published EFTs mutation as a positive control for translation defect. ATP = ATP synthase; CO = cytochrome c oxidase; cytb = cytochrome b; EFTs = elongation factor Ts; EFTu = elongation factor Tu; GDP = guanosine diphosphate; ND = NADH dehydrogenase; SDH = succinate dehydrogenase.
Enrichment of p.Q286X mutation in the Finnish population.
TSFM p.Q286X variant was identified as a potentially pathogenic change with an independent, bioinformatics strategy. Exome data of more than 3,000 Finns (SISu) were compared with similar data from other populations to find which mutations were present in the Finnish population with high heterozygous carrier frequency, but lacking homozygotes. The p.Q286X was found to be enriched in Finland, with a heterozygous carrier frequency of 1:90. This frequency was verified by separate genotyping of a total of 32,497 Finnish subjects indicating even more common carrier frequency, 1:80. Based on Hardy-Weinberg equilibrium, 1 to 2 homozygotes would be expected in this population, but none existed. Such a carrier frequency predicted that, of 60,000 children born in Finland every year, 2 to 3 should be homozygous for this TSFM mutation. However, such patients were not identified in our mitochondrial patient cohort, suggesting that homozygosity is lethal, potentially leading to prenatal death. We therefore sequenced this TSFM mutation site from a sample collection from 32 intrauterine fetal deaths. However, all of the fetuses screened were carrying the wild-type allele.
No carriers for p.C315Y and c.106+4A>G variants were found among the 3,000 subjects from the Finnish population (SISu). We also searched this database for other TSFM variants, which would change amino acids, and found 2 such variants with maximal frequency of 1:1,500 (table e-1 on the Neurology® Web site at Neurology.org). These results show that variants in EFTs are rare, with the exception of the allele carrying the p.Q286X change.
TSFM mutations result in an unstable EFTs protein and reduced mitochondrial translation.
TSFM encodes for mitochondrial EFTs, which together with EFTu is essential for translation of mitochondrial DNA–encoded proteins. EFTu is a G protein, assisting in binding of charged aminoacyl-tRNA to the A-site of the mitochondrial ribosome in a guanosine triphosphate–dependent manner, and the EFTs functions as a nucleotide exchange factor. We used the previously resolved structure of the bovine EFTu-EFTs31 complex to model the human proteins (figure 2B). Human and bovine EFTs sequences show 83.8% identity and 88.2% similarity, while EFTu sequences show 94.5% and 96.5%, respectively. Our modeling indicated that C315 locates in the EFTs subdomain C of the core domain and was predicted to have an entirely structural role, as explained in detail in figure 2B. Change in local architecture would affect stability of the protein because of a less densely packed core and also would affect the positions of both p.D126 and p.F127, shown to be essential for interaction within the Tu-Ts complex.31 Therefore, p.C315Y change is likely to result in a largely unstable inactive protein and together with p.Q286X mutation to severely decreased amounts of EFTs.
As predicted by our structural analysis, EFTs was almost completely absent in P1 and P2 fibroblast mitochondria, and showed substantial reduction also in P3 myoblasts, in Western blot analysis, supporting the role of EFTs in the disease pathogenesis of all 3 patients (figure 2C). EFTu levels are known to follow EFTs levels,2,33 and consistent with this, the EFTu amount was clearly reduced in P1 and P2 fibroblasts, but not in P3 myoblasts, compared with controls (figure 2C). We then performed pulse labeling of mitochondrial translation products on all 3 cell lines and detected a partial translation defect in P2 fibroblasts, a suggestive defect in P1, and a clear reduction of overall mitochondrial translation in P3 myoblasts, especially in the cytochrome c oxidase subunits (figure 2D).
DISCUSSION
Herein, we report that TSFM mutations, leading to defects of mitochondrial translation elongation factor EFTs, can underlie childhood or juvenile-onset nervous system phenotypes, with or without cardiomyopathy. The infantile cardiac phenotype was severe and progressive, but later stabilized, whereas the nervous system phenotypes, including optic atrophy, peripheral axonal neuropathy, Leigh disease, and/or ataxia, manifested in school age. Previously, TSFM mutations have been described to always be lethal in infancy, with cardiomyopathy, encephalomyopathy, or both,3,5 or liver failure,34 and all the reported patients carried the same homozygous change, p.R312W. Our results support the previous findings that mitochondrial translation defects manifest in early infancy as cardiomyopathy.11,18,35 However, stabilization of the heart condition suggests that the translation defect may be compensated, if the patient survived the early stage. Indeed, similar early-onset severe disorders, followed by stabilization of cardiomyopathy, have been previously reported in single patients with mitochondrial translation defects. MRPL4411 and ELAC224 mutations cause mitochondrial cardiomyopathies with infantile lethality, but in the few surviving patients, cardiomyopathy transforms to slowly progressive in childhood. The lack of cardiomyopathy in patient 3 suggested that the residual EFTs was enough to rescue his heart, but not the nervous system, as he developed optic atrophy, neuropathy, and ataxia in juvenile age, mimicking the neurologic symptoms of the other 2 patients. Accumulation of data from the natural history of diseases, and their correlation with genotype information, may aid in treatment decisions, e.g., prioritization for heart transplantation.
We had the privilege of accessing an excessive genetic data collection of SISu. These data revealed that the p.Q286X mutation is highly enriched in the Finnish population, with 1:80 carrier frequency, but no homozygotes were found, strongly supporting a pathogenic role of this mutation. Very low frequency of other TSFM mutations in the population explained why defects in this gene are not causing a large proportion of mitochondrial diseases in Finland. Furthermore, the absence of p.Q286X allele homozygotes in mitochondrial disease, intrauterine death, and population cohorts suggested early developmental lethality. The frequency of the allele suggests it to potentially explain 2 to 3 early miscarriages per year in the country. These data emphasize the importance and power of large population-specific genomic data collections for identification of genetic causes for disease.
The pathogenic role of EFTs in all of our patients was strongly supported by the considerable reduction of EFTs protein in their samples, as well as consequent reduction in mitochondrial translation. This translation defect in fibroblasts was mild to moderate, despite very little residual EFTs, indicating that mitochondrial translation can be maintained with trace amounts of an essential factor. The overall reduction of TSFM transcript in P3, without an apparent splicing problem, leaves the pathogenic mechanism in his case still somewhat open. Cell-type–dependent splicing has been proposed to occur in nervous system manifesting disorders of mitochondrial translation, with increased probability to exon skipping especially in neural cell lines.14,36 Whether such a tissue-type–specific sensitivity to splicing errors could contribute to EFTs consequences in our patients remains to be verified.
EFTs binds the EFTu–guanosine diphosphate (GDP) complex, mediates GDP release, and forms a stable EFTu-EFTs complex.37,38 GDP can slowly dissociate from the protein, but in that transition state, EFTu becomes unstable39 unless stabilized by bound EFTs. EFTs and Tu protein amounts closely follow each other, the levels reflected in the amount of mitochondrial translation products,2,5,40 indicating the importance of their stoichiometric balance for mitochondrial translation. The β-sandwich structure of EFTs is important for interaction with Tu and stability of the complex.39 Mutation p.C315Y in human Ts destabilizes the central part of the β-sandwich core, especially affecting the contact surface for interaction with Tu. This structural change occurs at a site adjacent to a motif essential for binding to Ts and the nucleotide exchange. This protein-modeling prediction is in line with our findings of reduction of EFTs levels, which was accompanied by reduction in EFTu levels and a slight decrease in translation.
Our findings show that EFTs disorders manifest with wide variability in phenotypes, extending from early-lethal multisystem disorders with cardiomyopathy to childhood-onset Leigh disease, optic atrophy, neuropathy, and juvenile-onset ataxia neuropathies. Our findings support the notion that EFTs and mitochondrial translation are especially critical for cardiac function in early childhood, and if the patient survived the critical years, the cardiac status may stabilize. This finding emphasizes aggressive treatment to support cardiac function at the early stage, because the patient may stabilize without a cardiac transplant. The p.Q286X mutation was found to be enriched in the Finnish population, however, without homozygotes even in the mitochondrial patient population. This finding indicates that this severe mutation can be causing intrauterine deaths, most likely due to early developmental cardiac dysfunction.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all the contributors of the Sequencing Initiative Suomi collaboration for the possibility to utilize their population sequence material at its early stages. The authors acknowledge Professor David Thorburn (Murdoch Childrens Research Institute, University of Melbourne, Australia) for providing the p.R312W mutant EFTs fibroblast cells and Professor Linda Spremulli (Department of Chemistry, The University of North Carolina) for EFTu-EFTs antibody. The authors thank Anu Harju for technical help, and Brendan Battersby and Uwe Richter (Research Program Unit of Molecular Neurology, University of Helsinki, Finland) for discussion and critical comments.
GLOSSARY
- cDNA
complementary DNA
- EFTs
elongation factor Ts
- EFTu
elongation factor Tu
- GDP
guanosine diphosphate
- RC
respiratory chain
- SISu
Sequencing Initiative Suomi project
- SNP
single nucleotide polymorphism
- tRNA
transfer RNA
- TSFM
Ts translation elongation factor, mitochondrial
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
S. Ahola contributed to conception and design of the study, acquisition, analysis and interpretation of data, and drafted, revised, and approved the article. P. Isohanni, L. Euro, V. Brilhante, A. Palotie, H. Pihko, T. Lönnqvist, T. Lehtonen, J. Laine, and H. Tyynismaa contributed to acquisition and interpretation of data, and revised and approved the article. A. Suomalainen contributed to conception and design of the study, interpretation of data, and drafted, revised, and approved the article.
STUDY FUNDING
Supported by the Jane and Aatos Erkko Foundation, Academy of Finland, Sigrid Juselius Foundation, the University of Helsinki (to A.S.), the Helsinki Biomedical Graduate Program and the Biomedicum Helsinki Foundation (to S.A.), and the Finnish Medical Foundation (to P.I.). A.P. is funded by European Commission EU FP7-242167 and by Wellcome Trust Sanger Institute 091310/D/10/Z. None of the funding sources were involved in conception or design of the study, acquisition, or analysis or interpretation of the data; neither did any of the funding sources have insight in the statistical analysis or drafting or revising the article.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Ylikallio E, Suomalainen A. Mechanisms of mitochondrial diseases. Ann Med 2012;44:41–59 [DOI] [PubMed] [Google Scholar]
- 2.Antonicka H, Sasarman F, Kennaway NG, Shoubridge EA. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum Mol Genet 2006;15:1835–1846 [DOI] [PubMed] [Google Scholar]
- 3.Calvo SE, Compton AG, Hershman SG, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med 2012;4:118ra110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coenen MJ, Antonicka H, Ugalde C, et al. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med 2004;351:2080–2086 [DOI] [PubMed] [Google Scholar]
- 5.Smeitink JA, Elpeleg O, Antonicka H, et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am J Hum Genet 2006;79:869–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valente L, Tiranti V, Marsano RM, et al. Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet 2007;80:44–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller C, Saada A, Shaul N, et al. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann Neurol 2004;56:734–738 [DOI] [PubMed] [Google Scholar]
- 8.Saada A, Shaag A, Arnon S, et al. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet 2007;44:784–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galmiche L, Serre V, Beinat M, et al. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum Mutat 2011;32:1225–1231 [DOI] [PubMed] [Google Scholar]
- 10.Serre V, Rozanska A, Beinat M, et al. Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency. Biochim Biophys Acta 2013;1832:1304–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carroll CJ, Isohanni P, Poyhonen R, et al. Whole-exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J Med Genet 2013;50:151–159 [DOI] [PubMed] [Google Scholar]
- 12.Bayat V, Thiffault I, Jaiswal M, et al. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol 2012;10:e1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steenweg ME, Ghezzi D, Haack T, et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate “LTBL” caused by EARS2 mutations. Brain 2012;135:1387–1394 [DOI] [PubMed] [Google Scholar]
- 14.Edvardson S, Shaag A, Kolesnikova O, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet 2007;81:857–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 2007;39:534–539 [DOI] [PubMed] [Google Scholar]
- 16.Riley LG, Cooper S, Hickey P, et al. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am J Hum Genet 2010;87:52–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belostotsky R, Ben-Shalom E, Rinat C, et al. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet 2011;88:193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gotz A, Tyynismaa H, Euro L, et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet 2011;88:635–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pierce SB, Chisholm KM, Lynch ED, et al. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci USA 2011;108:6543–6548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elo JM, Yadavalli SS, Euro L, et al. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum Mol Genet 2012;21:4521–4529 [DOI] [PubMed] [Google Scholar]
- 21.Zeharia A, Shaag A, Pappo O, et al. Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet 2009;85:401–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet 2004;74:1303–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antonicka H, Ostergaard E, Sasarman F, et al. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet 2010;87:115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haack TB, Kopajtich R, Freisinger P, et al. ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am J Hum Genet 2013;93:211–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smits P, Smeitink J, van den Heuvel L. Mitochondrial translation and beyond: processes implicated in combined oxidative phosphorylation deficiencies. J Biomed Biotechnol 2010;2010:737385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Battersby BJ, Richter U. Why translation counts for mitochondria: retrograde signalling links mitochondrial protein synthesis to mitochondrial biogenesis and cell proliferation. J Cell Sci 2013;126:4331–4338 [DOI] [PubMed] [Google Scholar]
- 27.Sulonen AM, Ellonen P, Almusa H, et al. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol 2011;12:R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pagliarini DJ, Calvo SE, Chang B, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008;134:112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gardosi J, Kady SM, McGeown P, Francis A, Tonks A. Classification of stillbirth by relevant condition at death (ReCoDe): population based cohort study. BMJ 2005;331:1113–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suomalainen A, Syvanen AC. Analysis of nucleotide sequence variations by solid-phase minisequencing. Methods Mol Biol 1996;65:73–79 [DOI] [PubMed] [Google Scholar]
- 31.Jeppesen MG, Navratil T, Spremulli LL, Nyborg J. Crystal structure of the bovine mitochondrial elongation factor Tu.Ts complex. J Biol Chem 2005;280:5071–5081 [DOI] [PubMed] [Google Scholar]
- 32.Chomyn A. In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol 1996;264:197–211 [DOI] [PubMed] [Google Scholar]
- 33.Woriax VL, Bullard JM, Ma L, Yokogawa T, Spremulli LL. Mechanistic studies of the translational elongation cycle in mammalian mitochondria. Biochim Biophys Acta 1997;1352:91–101 [DOI] [PubMed] [Google Scholar]
- 34.Vedrenne V, Galmiche L, Chretien D, de Lonlay P, Munnich A, Rotig A. Mutation in the mitochondrial translation elongation factor EFTs results in severe infantile liver failure. J Hepatol 2012;56:294–297 [DOI] [PubMed] [Google Scholar]
- 35.Gotz A, Isohanni P, Liljestrom B, et al. Fatal neonatal lactic acidosis caused by a novel de novo mitochondrial G7453A tRNA-serine (UCN) mutation. Pediatr Res 2012;72:90–94 [DOI] [PubMed] [Google Scholar]
- 36.van Berge L, Dooves S, van Berkel CG, Polder E, van der Knaap MS, Scheper GC. Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation is associated with cell-type-dependent splicing of mtAspRS mRNA. Biochem J 2012;441:955–962 [DOI] [PubMed] [Google Scholar]
- 37.Clark BF, Nyborg J. The ternary complex of EF-Tu and its role in protein biosynthesis. Curr Opin Struct Biol 1997;7:110–116 [DOI] [PubMed] [Google Scholar]
- 38.Czworkowski J, Moore PB. The elongation phase of protein synthesis. Prog Nucleic Acid Res Mol Biol 1996;54:293–332 [DOI] [PubMed] [Google Scholar]
- 39.Jiang Y, Nock S, Nesper M, Sprinzl M, Sigler PB. Structure and importance of the dimerization domain in elongation factor Ts from Thermus thermophilus. Biochemistry 1996;35:10269–10278 [DOI] [PubMed] [Google Scholar]
- 40.Sasarman F, Antonicka H, Shoubridge EA. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum Mol Genet 2008;17:3697–3707 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.