

Abstract
Objective:
To clarify the relationship between depressive symptoms and the clinical and neuropathologic manifestations of dementia.
Methods:
In a clinical-pathologic cohort study, 1,764 older persons without cognitive impairment at enrollment completed annual clinical evaluations for a mean of 7.8 years. The evaluations included assessment of depressive symptoms (10-item Center for Epidemiological Studies Depression Scale) and cognitive function (battery of 17 performance tests). A total of 582 individuals died during follow-up and underwent a uniform neuropathologic examination to quantify β-amyloid plaques and tau tangle density in multiple brain regions and identify neocortical Lewy bodies, hippocampal sclerosis, and gross and microscopic cerebral infarcts.
Results:
Level of depressive symptoms slightly increased during follow-up. Incident mild cognitive impairment (52.2%) was associated with higher level of depressive symptoms before the diagnosis but not with change in symptoms after the diagnosis; incident dementia (17.9%) was associated with higher symptom level before dementia onset and with more rapid decline in symptoms after dementia onset. None of the neuropathologic markers was related to level of depressive symptoms or change in symptoms over time. In a mixed-effects model adjusted for the neuropathologic markers, higher level of depressive symptoms averaged over evaluations was associated with more rapid global cognitive decline, accounting for 4.4% of the variability in decline not attributable to the neuropathologic markers. Depressive symptoms did not modify the association of the neuropathologic markers with cognitive decline.
Conclusion:
In old age, depressive symptoms have an association with cognitive decline that is independent of the neuropathologic hallmarks of dementia.
Late-life depressive symptoms are associated with an increased rate of cognitive decline1,2 and risk of mild cognitive impairment (MCI)3,4 and dementia.1,5,6 The main hypotheses about the association, which are not mutually exclusive, are that depressive symptoms are a consequence of cognitive decline; depressive symptoms and cognitive decline are manifestations of the same underlying neuropathology; depressive symptoms alter the association of pathology with declining cognition; and depressive symptoms have an independent association with cognitive decline.1–7 The first hypothesis has been examined in several studies but only sporadically supported. In contrast, little research has been done on the latter hypotheses because testing them requires longitudinal clinical data on depressive symptoms and cognitive function plus postmortem data on the neurodegenerative and cerebrovascular lesions traditionally associated with cognitive decline and dementia.
In the present study, we used data from 2 clinical-pathologic cohort studies to examine the associations among depressive symptoms, cognitive decline, and neuropathologic lesions. Participants from the Religious Orders Study and Rush Memory and Aging Project completed annual assessments of depressive symptoms and cognitive function. Those who died during follow-up underwent a neuropathologic examination to quantify 6 common dementia-related lesions. In analyses, we modeled the relationship of incident MCI and dementia with depressive symptoms during follow-up. We then tested whether depressive symptoms were related to the neuropathologic lesions, modified the association of the neuropathologic lesions with cognitive decline, or had an association with residual cognitive decline not linked to neuropathologic burden.
METHODS
Participants.
Data are from individuals in 2 ongoing longitudinal clinical-pathologic studies begun in the 1990s, one involving older Catholic clergy recruited from multiple sites in the United States (Religious Orders Study)8 and the other involving older lay individuals from the Chicago area (Rush Memory and Aging Project).9 Eligibility for each study required baseline age older than 50 years and agreement to yearly clinical assessments and postmortem examination of the brain.
When these analyses were conducted, 2,792 persons had enrolled and finished the initial clinical assessment. As shown in figure 1, those with MCI or dementia at initial assessment were excluded, and some individuals were not eligible for follow-up because of death in the first year or enrollment in the past year. This left 1,832 people and 1,764 (96.3%) had follow-up data. They began the study at an average age of 76.6 years (SD = 7.5) with an average of 16.2 years of schooling (SD = 3.8); 26.6% were men. After an average of 7.8 years of annual observation (SD = 4.8), 52.2% had developed MCI and 17.9% had developed dementia. Of the 680 individuals who died during follow-up, a brain autopsy was performed on 612 (90.0%) (figure 1). At the time of these analyses, a uniform neuropathologic examination had been completed on the first consecutive 582 participants. They died at an average age of 87.6 years (SD = 6.7) with an average of 16.6 years of schooling (SD = 3.7); 33.7% were men. They completed an average of 8.4 years of follow-up (SD = 4.1). Compared with the 1,182 individuals without neuropathologic data, they were older at baseline (t1,291.7 = 11.0, p < 0.001), more likely to be men (χ21 = 22.4, p < 0.001), more educated (t1,762 = 2.8, p = 0.005), had more depressive symptoms at baseline (χ21 = 22.5, p < 0.001), and were more likely to develop MCI (χ21 = 62.2, p < 0.001) and dementia (χ21 = 74.0, p < 0.001).
Figure 1. Composition of the study groups.
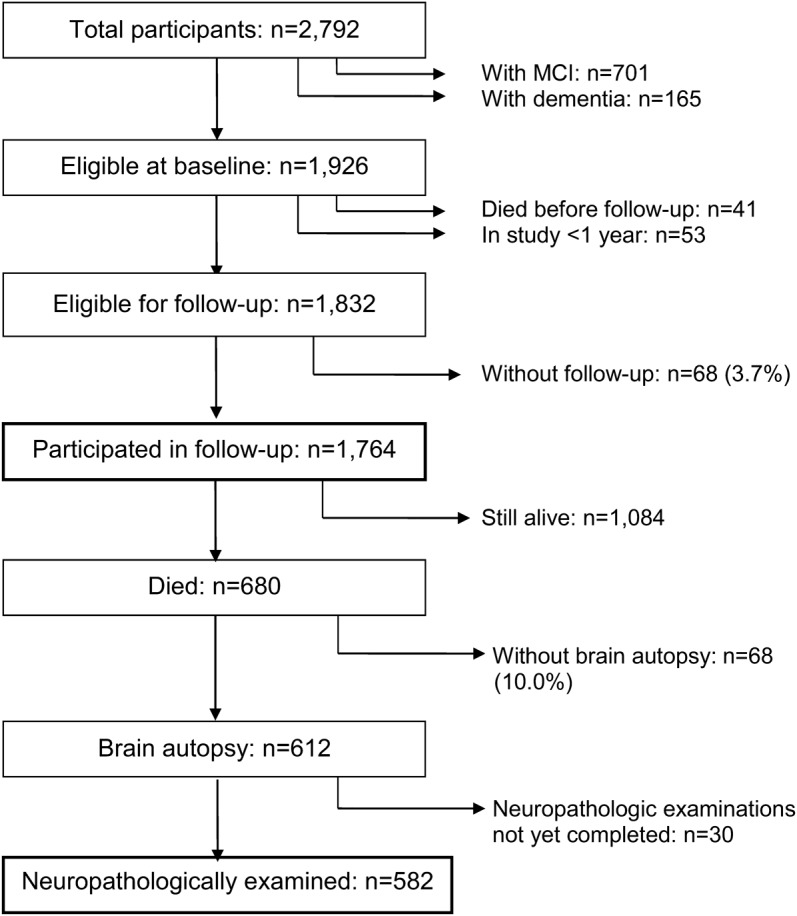
Flow diagram showing the composition of the full group and the neuropathologically examined subgroup. MCI = mild cognitive impairment.
Standard protocol approvals, registrations, and patient consents.
In each study, individuals signed informed consent forms after a detailed conversation with staff. Each project was approved by Rush University Medical Center's institutional review board.
Assessment of depressive symptoms.
At each annual visit, a short form10 of the Center for Epidemiological Studies Depression Scale11 was administered. On each of 10 items, the interviewer read a brief statement (e.g., “I felt lonely”) and asked the participant: “Have you felt this way much of the past week?” The score is the number of item responses indicative of depression. This 10-item measure predicts morbidity1 and mortality.12 It is psychometrically similar to the original version of the scale, including the ability to capture change in symptoms over time,13,14 but differs in 2 important respects: it does not include cognitive symptoms (i.e., “I had trouble keeping my mind on what I was doing,” “I talked less than usual”) and reduces participant burden (i.e., fewer items, yes/no response format).
Assessment of cognitive function.
Each study included annual administration of 18 cognitive tests.15–17 The Mini-Mental State Examination was included to facilitate comparisons with other cohorts. The other 17 tests consisted of performance measures of multiple cognitive domains, including episodic memory (n = 7), semantic memory (n = 3), working memory (n = 3), perceptual speed (n = 2), and visuospatial ability (n = 2). To construct a global cognitive outcome measure that capitalized on all cognitive data, we transformed the 17 raw test scores to z scores (based on the mean and SD from the initial evaluation of the combined cohorts) and computed the mean of these z scores.
Clinical classification.
Clinical diagnoses were rendered as part of each annual assessment. Dementia was based on historical evidence of declining cognition plus evidence on examination of impairment in at least 2 cognitive functions.18 MCI was diagnosed in individuals who had cognitive impairment but not dementia.19
Neuropathologic examination.
Death occurred a median of 7.3 months after the last clinical evaluation (interquartile range: 3.7–10.9) and the brain was removed a median of 6.3 hours after death (interquartile range: 4.8–9.8). A standard protocol for tissue preservation, tissue sectioning, and quantification of pathologic findings was implemented by examiners kept unaware of all clinical data.20,21 The cerebral hemispheres were coronally cut into 1-cm slabs, the cerebellar hemispheres were sagittally cut into 1-cm slabs, and the brainstem was removed at the level of the mamillary bodies and bisected at the mid-pons level.
We visually inspected slabs for gross infarcts. Slabs from one cerebral hemisphere and one cerebellar hemisphere and all slabs with suspected infarcts were fixed for at least 3 days in 4% paraformaldehyde. In addition, suspected infarcts were processed for histologic confirmation and the age was classified as acute, subacute, or chronic. One hemisphere was examined for microinfarcts in 6 cortical regions, 2 subcortical regions, and the midbrain using 6-μm paraffin-embedded sections stained with hematoxylin & eosin. Chronic microinfarcts included cavitated lesions with few macrophages and fibrillary gliosis or incomplete infarcts.22 In analyses, gross infarcts and microinfarcts were treated as binary variables.
β-Amyloid–immunoreactive plaques and tau-immunoreactive tangles were assessed in 8 areas: anterior cingulate cortex, dorsal lateral prefrontal cortex, superior frontal cortex, inferior temporal cortex, hippocampus (cornus ammonis subfield 1/subculum), entorhinal cortex, angular/supramarginal gyrus, and primary visual cortex. In the cortical regions, tissue blocks from adjacent slabs were paraffin-embedded and cut in 20-μm sections. In the hippocampus, we dissected 0.5-cm blocks from consecutive 1-cm slabs, embedded them in paraffin, and cut them in 20-μm sections.
An N-terminus–directed monoclonal antibody (1:1,000, 10D5; Elan Pharmaceuticals, Dublin, Ireland) was used to label β-amyloid. Immunohistochemistry was done with diaminobenzidine as the reporter, 2.5% nickel sulfate to sharpen immunoreaction product contrast, and a computer-assisted sampling procedure. We computed the proportion of pixels in each region that was β-amyloid–immunoreactive and used the mean of the regional values as an overall measure of β-amyloid burden.23
The density of tau-immunoreactive tangles was quantified with an anti-paired helical filament-tau antibody clone AT8 (1:2,000; ThermoScientific, Rockford, IL) and computer-assisted sampling. Tangle density/mm2 in each region was standardized and the mean of the standard scores was used as a composite measure of tangle density.23
The substantia nigra, 2 limbic regions (anterior cingulate cortex, entorhinal cortex), and 3 neocortical regions (midfrontal cortex, superior or middle temporal cortex, inferior parietal cortex) were assessed for Lewy bodies using a monoclonal antibody to phosphorylated α-synuclein (1:20,000; Wako Chemical USA Inc., Richmond, VA).20,24 Analyses are based on the presence or absence of neocortical disease, and affected individuals usually also had nigral and limbic Lewy bodies.24,25
A hemisection of the mid-hippocampus at the level of the lateral geniculate body was stained with hematoxylin & eosin to assess neuronal loss in the hippocampal subfields CA1–4 and subiculum. Severe diffuse or segmental neuronal loss in the pyramidal cell layer of any of the CA subfields or subiculum was classified as hippocampal sclerosis.
Statistical analysis.
Each analysis was adjusted for age, sex, and education. The distributions of depressive symptom scores at each evaluation were skewed. Therefore, to characterize change in symptoms over time and test whether clinical diagnoses were related to prediagnostic symptom level or postdiagnostic rate of change in symptoms, we constructed a series of proportional odds models26 with depressive symptoms treated as an ordinal variable with 6 levels (0, 1, 2, 3, 4, ≥5). The proportionality assumption, assessed with the score test for proportional odds and the trend odds model,27 was found to be adequately met. The first model included a term for time (from baseline in years), which indicates the log odds of the outcome shifting 1 level per year. We repeated the model with a time-varying term for MCI onset and its interaction with time and then again with a time-varying indicator for dementia onset and its interaction with time.
In the neuropathologically examined subgroup, we repeated the first proportional odds model and then added terms for each neuropathologic marker and its interaction with time. We estimated the associations of depressive symptoms (averaged across evaluations) and neuropathologic markers with cognitive decline in mixed-effects models.28 The initial model had terms for time (from death in years), mean depressive symptoms, and their interaction. A second model had terms for time, each neuropathologic marker, and the interaction of each marker with time. We repeated this analysis with terms added for mean depressive symptoms and its interaction with time. We repeated the latter analysis with terms added for the 2-way interactions of mean depressive symptoms with each neuropathologic marker and the 3-way interactions of mean depressive symptoms by each marker by time.
RESULTS
Depressive symptoms during development of dementia and MCI.
At baseline, the 1,764 participants (figure 1) reported a mean of 1.0 symptom (SD = 1.6, skewness = 2.0) on the Center for Epidemiological Studies Depression Scale (963 had no symptoms; 371 had 1; 171 had 2; 113 had 3; 64 had 4; and 82 had 5 or more). Over time, the within-person range of scores had a median of 2 (interquartile range: 1–4), suggesting substantial variability. We assessed change in depressive symptoms during a mean of 7.8 years of annual follow-up (SD = 4.8) in a proportional odds model adjusted for baseline age, sex, and education. In this analysis, depressive symptoms increased over time (estimate = 0.043, SE = 0.006, p < 0.001). This was mainly attributable to a slight decline in the likelihood of having no symptoms (figure 2A).
Figure 2. Change in depressive symptoms over time.
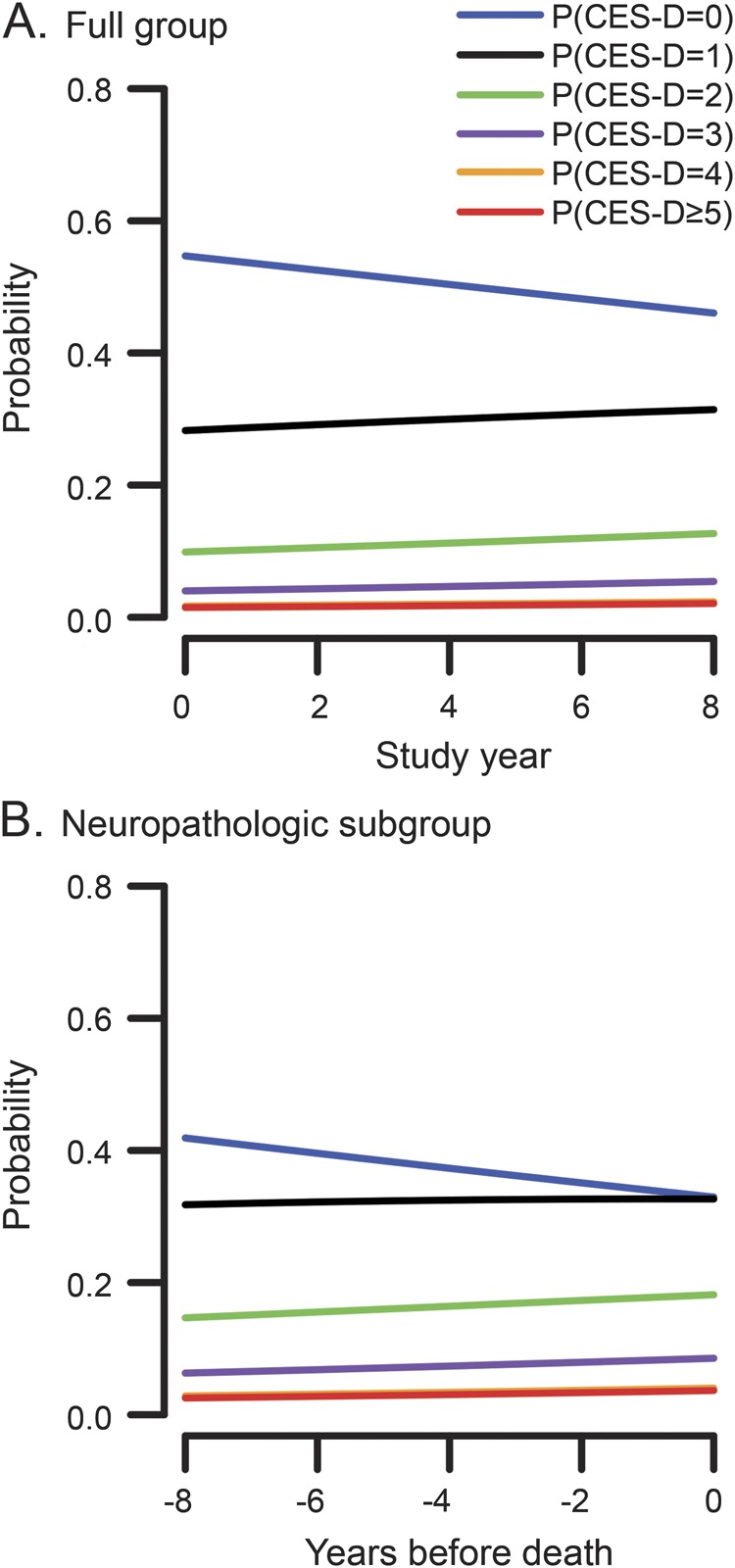
Probability of having different levels of depressive symptoms over time in the full group (A) and the neuropathologically examined subgroup (B), from proportional odds models adjusted for age, sex, and education. CES-D = Center for Epidemiological Studies Depression Scale.
During follow-up, 52.2% of the cohort (922/1,764) developed MCI, widely recognized as a precursor to dementia in old age. At baseline, they were older and more educated than those who remained cognitively intact (table e-1 on the Neurology® Web site at Neurology.org). To determine the relation of MCI to depressive symptoms, we repeated the analysis with the addition of a time-varying indicator of MCI onset and its interaction with time. Incident MCI was associated with a higher level of depressive symptoms before MCI onset but not with rate of change in symptoms after MCI onset (table 1, model A).
Table 1.
Relation of incident mild cognitive impairment and dementia to change in depressive symptoms over timea
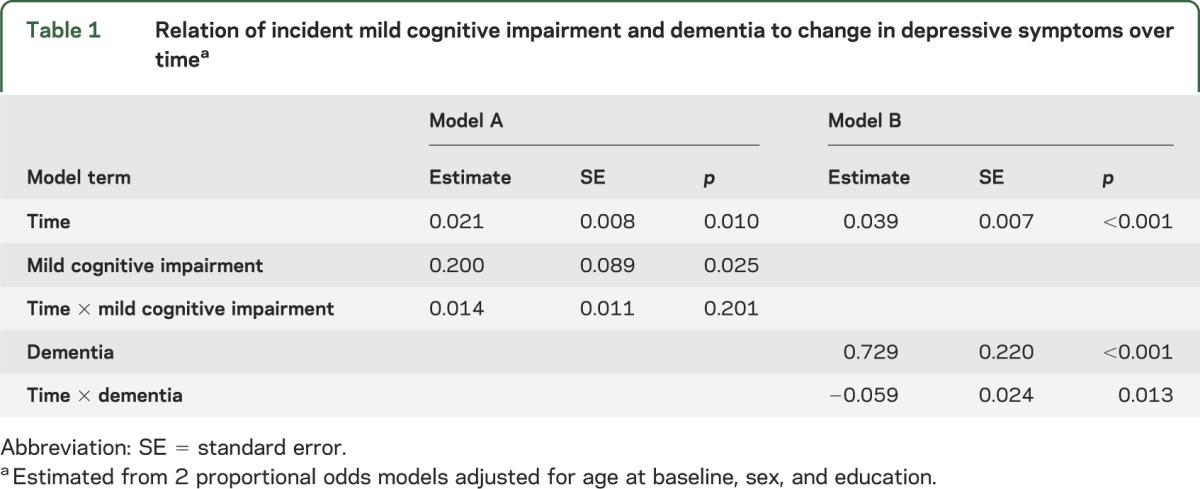
A total of 315 individuals (17.9%) developed dementia during follow-up. At baseline, they were older and more depressed than those who did not develop dementia (table e-1). Incident dementia was associated with a higher level of depressive symptoms before dementia onset, but a slight decrease in symptoms thereafter (table 1, model B).
Depressive symptoms and neuropathologic burden.
The remaining analyses were conducted on the 582 neuropathologically examined individuals (figure 1). At baseline, they reported a mean of 1.2 depressive symptoms (SD = 1.6, median = 1, skewness = 1.6). In a proportional odds model adjusted for age at death, sex, and education, depressive symptoms slightly increased during the mean of 8.4 years of observation (estimate = 0.048, SE = 0.013, p < 0.001; figure 2B).
On the postmortem neuropathologic examination, the composite measure of β-amyloid plaque burden ranged from 0 to 22.9 (mean = 3.9, SD = 4.3, median = 2.4, skewness = 1.3) and the composite measure of tau tangle density ranged from 0 to 32.2 (mean = 4.4, SD = 5.2, median = 2.8, skewness = 2.2). In addition, 32.1% had one or more chronic gross infarcts, 27.9% had one or more chronic microinfarcts, 12.1% had neocortical Lewy bodies, and 5.9% had hippocampal sclerosis.
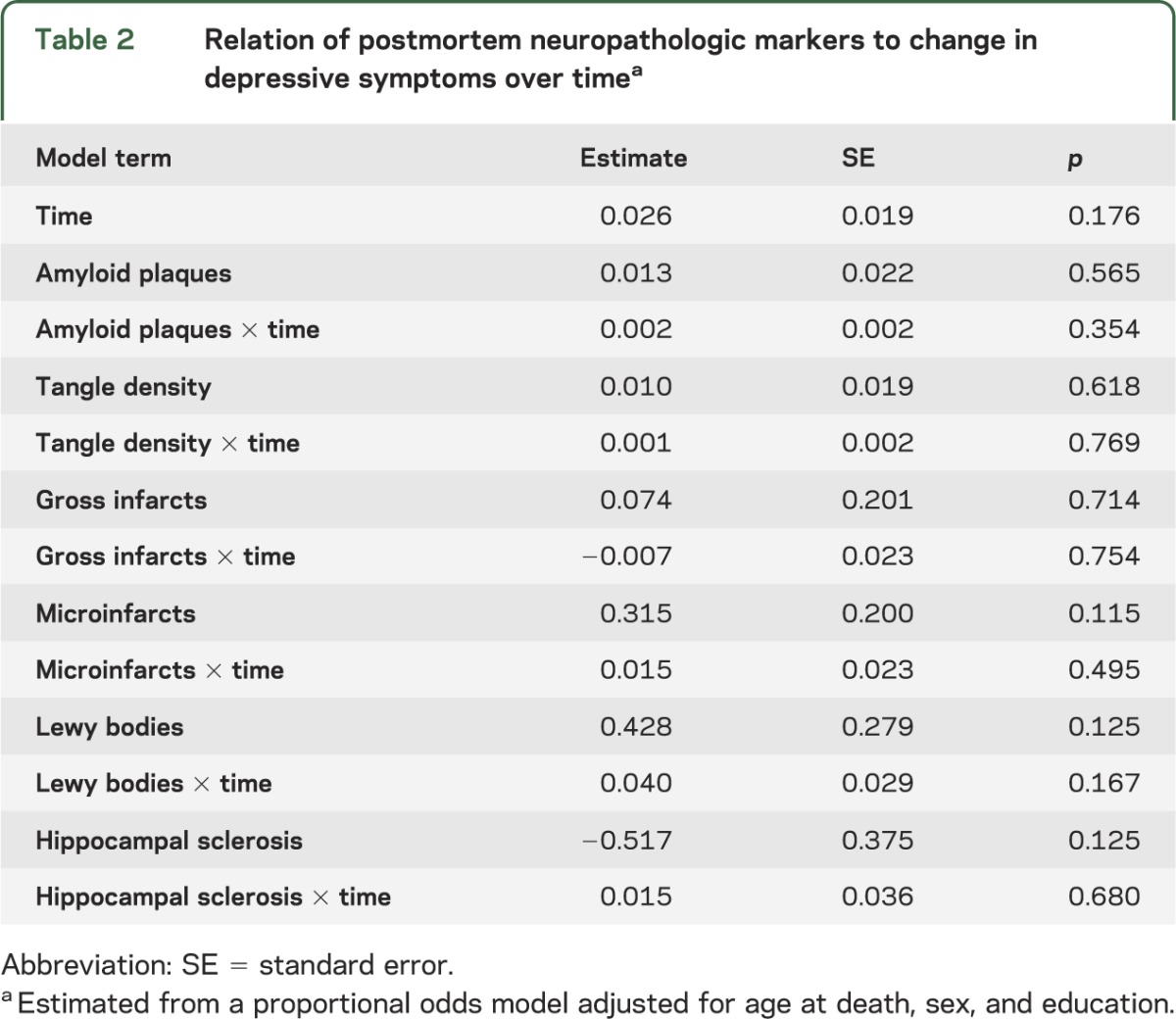
We constructed a proportional odds model adjusted for age at death, sex, and education to test whether the neuropathologic markers were related to level of depressive symptoms or to change in depressive symptoms over time. The neuropathologic markers were not related to level of depressive symptoms proximate to death or change in depressive symptoms over time (table 2).
Table 2.
Relation of postmortem neuropathologic markers to change in depressive symptoms over timea
Depressive symptoms, neuropathologic burden, and cognitive decline.
We examined the associations among depressive symptoms, neuropathologic burden, and change in cognitive function in mixed-effects models adjusted for age at death, sex, and education. Because dementia develops by minute degrees,13,29 we focused on a continuous outcome (i.e., rate of change in cognitive function) rather than a binary one (e.g., diagnosis of dementia) to better capture this process and maximize statistical power. In the initial model, higher level of depressive symptoms averaged across all clinical evaluations (mean = 1.3, SD = 1.3, median = 0.9, skewness = 1.6) was associated with more rapid cognitive decline (table 3, model A). We then constructed a model with terms for each neuropathologic marker and its interaction with time. Each neuropathologic marker except amyloid and microinfarcts was associated with more rapid cognitive decline (table 3, model B) and together the markers accounted for 31.8% of the variability in rates of cognitive decline. When depressive symptoms and the neuropathologic markers were included in the same analysis, higher level of depressive symptoms continued to be associated with faster rate of cognitive decline (table 3, model C), accounting for 4.4% of the residual variability in cognitive decline not attributable to neuropathologic burden. Figure e-1, which is based on this analysis, shows a dose-response relationship, with higher levels of depressive symptoms associated with increasingly rapid cognitive decline.
Table 3.
Relation of depressive symptoms and neuropathologic markers to global cognitive declinea
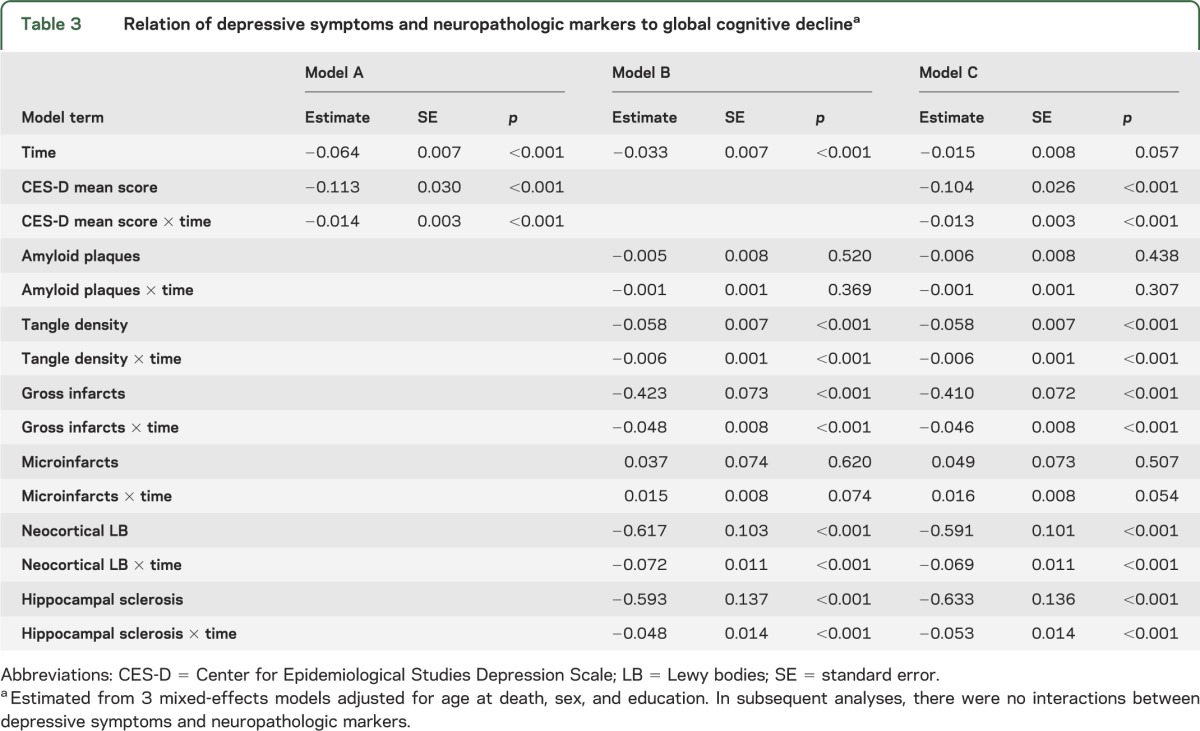
To determine whether depressive symptoms modified the relationship of neuropathologic burden to cognitive decline, we repeated the previous model with terms added for the 2-way interactions of depressive symptoms with each neuropathologic marker, and the 3-way interactions of depressive symptoms, time, and each neuropathologic marker. None of the 12 interactions was significant.
DISCUSSION
A group of more than 1,750 older persons without cognitive impairment at study onset completed annual clinical evaluations for a mean of about 8 years during which nearly 600 died and underwent a neuropathologic examination. Depressive symptoms were associated with rate of cognitive decline, and after adjustment for postmortem markers of 6 types of dementia-related pathology, higher level of depressive symptoms during the study period remained associated with faster rate of cognitive decline, accounting for 4.4% of the variability in rates of cognitive decline not attributable to neuropathologic burden.
The observed association of depressive symptoms with cognitive decline is consistent with most prior research,1,2 including an earlier analysis of data from the Religious Orders Study.1 The present results add to that literature by showing that depressive symptoms have an association with cognitive decline that is independent of the neuropathologic conditions most strongly linked to late-life cognitive decline and dementia.
How depressive symptoms contribute to cognitive impairment is not known. Prior studies have associated late-life depression, whether defined diagnostically or psychometrically, with abnormalities in frontal-subcortical and limbic networks involved in emotional regulation. These abnormalities include lower brain volume30,31 and white matter integrity32,33 in antemortem neuroimaging studies and lower density of neurons34 and dendritic spines per neuron35 in postmortem neuropathologic studies. It is not clear, however, whether these or other factors are contributing to the association of depressive symptoms with late-life cognitive decline and dementia.
We found no support for other hypotheses about the association between depression and late-life cognition. First, depressive symptoms did not increase with the onset of MCI and slightly decreased after dementia onset. Although this is not consistent with prior reports of a very slight increase in depressive symptoms before dementia onset,13,14 it is consistent with reports of stable14 or declining36 depressive symptoms after dementia onset and the lack of an association of cognitive level37–39 and cognitive change40 with change in depressive symptoms. Second, neither level of depressive symptoms nor change in symptoms over time was related to common neuropathologic lesions associated with cognitive decline and dementia. Third, depressive symptoms did not alter the association of neuropathology with cognitive decline. Overall, that depressive symptoms were related to cognitive decline after adjusting for pathology but neither dementia nor pathology was associated with increasing depressive symptoms suggests that depressive symptoms are a risk factor for late-life cognitive decline. Although the neurobiological basis of the association is uncertain, the present results suggest that interventions targeting depressive symptoms and psychological distress have the potential to help maintain cognitive health in old age.
This study has several strengths and limitations. The clinical diagnoses of MCI and dementia were based on a uniform evaluation and widely accepted criteria applied by experienced clinicians, minimizing diagnostic misclassification. Participation in clinical follow-up and brain autopsy was high, reducing the likelihood that selective attrition biased results. The availability of psychometrically sound measures of depressive symptoms and cognitive function administered annually for a mean of about 8 years maximized our ability to characterize change. An important limitation is that the volunteer participants were self-selected and so the generalizability of the results remains to be determined. In addition, our measures of some disease processes were incomplete (e.g., cerebrovascular disease), some disease processes were not measured (e.g., transactive response DNA-binding protein 43), and some markers were infrequently observed (e.g., hippocampal sclerosis). Use of a brief self-report scale may have led us to underestimate depressive symptoms especially as cognition became impaired, although informant report does not suggest that depressive symptoms increase in MCI or dementia.14
Supplementary Material
ACKNOWLEDGMENT
The authors thank the Illinois residents who participated in the Rush Memory and Aging Project and the many Catholic nuns, priests, and brothers who have participated in the Religious Orders Study; Traci Colvin, MPH, and Karen Skish, MS, for study coordination; John Gibbons, MS, and Greg Klein, MS, for data management; and Wenqing Fan, MS, for statistical programming.
GLOSSARY
- MCI
mild cognitive impairment
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Drafting/revising manuscript for content: Wilson, Capuano, Boyle, Hoganson, Hizel, Shah, Nag, Schneider, Arnold, Bennett. Study concept or design: Wilson, Capuano, Boyle, Hoganson, Hizel, Shah, Nag, Schneider, Arnold, Bennett. Analyses or interpretation of the data: Wilson, Capuano, Boyle, Shah, Nag, Schneider, Arnold, Bennett. Acquisition of data: Bennett, Nag, Schneider. Statistical analysis: Capuano. Study supervision or coordination: Bennett. Obtaining funding: Boyle, Bennett.
STUDY FUNDING
This research was supported by the National Institute on Aging (R01AG17917, P30AG10161, R01AG15819, R01AG34374) and the Illinois Department of Public Health. The funding organizations had no role in the design or conduct of the study; collection, management, analysis, or interpretation of the data; or preparation, review, or approval of the manuscript.
DISCLOSURE
R. Wilson serves as a consulting editor for Aging, Neuropsychology, and Cognition, Psychology and Aging, and Neuropsychology; has served as a consultant for Pain Therapeutics, Inc.; and receives research support from NIH (P30AG010161 [coinvestigator], R01AG015819 [coinvestigator], R01AG017917 [coinvestigator], R01AG034374 [coinvestigator], R01AG039478 [coinvestigator], R01AG036042 [coinvestigator], R01AG036836 [coinvestigator], R01AG041797 [coinvestigator], R01AG042210 [coinvestigator], and R01NR013151 [coinvestigator]) and Alzheimer's Association (NIRGD-11-205469). A. Capuano receives research support from NIH (R01AG022018 [coinvestigator], R01AG017917 [coinvestigator], R01AG015819 [coinvestigator], P30AG010161 [coinvestigator], R01AG043379 [coinvestigator], R01HL096944 [coinvestigator], and R01AG040039 [coinvestigator]). P. Boyle receives research support from the NIH (R01AG034374 [principal investigator], R01AG034119 [coinvestigator], R01AG033678 [principal investigator], and R01AG040039 [coinvestigator]). G. Hoganson and L. Hizel report no disclosures. R. Shah receives research support from NIH (P30AG010161 [coinvestigator], P20MD006886 [coinvestigator], U01AG024904 [coinvestigator], U01AG029824 [coinvestigator], U01AG046152 [coinvestigator], U01AG010483 [coinvestigator], R01AG036042 [coinvestigator], and R01AG017917 [coinvestigator]), Merck & Company (clinical trial site principal investigator), Genentech, Inc. (clinical trial site principal investigator), Eli Lilly & Company (clinical trial site principal investigator), and Navidea Biopharmaceuticals (clinical trial site principal investigator). S. Nag receives research support from NIH (R01AG017917 [coinvestigator], P30AG010161 [coinvestigator], R01AG043379 [coinvestigator], R01NS078009 [coinvestigator], and R01NS082416 [coinvestigator]). J. Schneider receives research support from NIH (R01AG042210 [principal investigator], P30AG010161 [coinvestigator], R01HL096944 [coinvestigator], R01AG039478 [coinvestigator], R01AG017917 [coinvestigator], R01AG015819 [coinvestigator], R01AG022018 [coinvestigator], R01AG036042, [coinvestigator], R01AG040039 [coinvestigator], R01AG036836 [coinvestigator], R01AG034374 [coinvestigator], U01AG046152 [coinvestigator], R01AG043379 [coinvestigator], R01NS078009 [coinvestigator], R01AG043975 [coinvestigator], R01AG033678 [coinvestigator], and R21ES021290 [coinvestigator]). S. Arnold serves as an associate editor for Translational Neuroscience and Journal of Alzheimer's Disease, has been a consultant for Teva Pharmaceuticals, Inc., and receives research support from the NIH (R01AG039478 [principal investigator], P30AG10124 [clinical core principal investigator], R01DA025201 [coinvestigator], U01DK057135 [coinvestigator], U01AG024904 [site principal investigator]), the BrightFocus Foundation (principal investigator), the Marian S. Ware Charitable Giving Fund (project principal investigator), Johnson & Johnson (coinvestigator), Merck (clinical trial site principal investigator), Bristol-Myers Squibb (clinical trial site principal investigator), and Pain Therapeutics, Inc. (site principal investigator). D. Bennett serves on the editorial board of Neurology®; has served as a consultant to Schering-Plough Corp., Mediation, Inc., and the Gerson Lehrman Group; and receives research support from Danone Inc., the NIH (R01AG017917 [principal investigator], R01AG015819 [principal investigator], R01AG036042 [principal investigator], U01AG032984 [co-principal investigator, leader of epidemiologic cohort studies], R01HL096944 [coinvestigator], R01AG033678 [coinvestigator], R01AG034374 [coinvestigator], R01AG022018 [coinvestigator], R01AG034119 [coinvestigator], P30AG010161 [principal investigator—administrative core leader, Religious Orders Study core leader], U01AG046152 [coinvestigator], R01AG040039 [coinvestigator], R01AG039478 [coinvestigator], R01AG038651 [coinvestigator], R01AG036836 [coinvestigator], R01AG041797 [coinvestigator], R01AG042210 [coinvestigator], P20MD006886 [coinvestigator], R01NS078009 [coinvestigator], R01AG043617 [coinvestigator], R01AG043975 [coinvestigator], U18NS082140 [coinvestigator], and R01NS082416 [coinvestigator]), and the Illinois Department of Public Health. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Wilson RS, Barnes LL, Mendes de Leon CF, et al. Depressive symptoms, cognitive decline, and risk of AD in older persons. Neurology 2002;59:364–370 [DOI] [PubMed] [Google Scholar]
- 2.Köhler S, van Boxtel MPJ, van Os J, et al. Depressive symptoms and cognitive decline in community-dwelling older adults. J Am Geriatr Soc 2010;58:873–879 [DOI] [PubMed] [Google Scholar]
- 3.Barnes DE, Alexopoulos GS, Lopez OL, Williamson JD, Yaffe K. Depressive symptoms, vascular disease, and mild cognitive impairment: findings from the Cardiovascular Health Study. Arch Gen Psychiatry 2006;63:273–279 [DOI] [PubMed] [Google Scholar]
- 4.Geda YE, Knopman DS, Mrazek DA, Jicha GA, Smith GE, Negash S. Depression, apolipoprotein E genotype, and the incidence of mild cognitive impairment: a prospective cohort study. Arch Neurol 2006;63:435–440 [DOI] [PubMed] [Google Scholar]
- 5.Saczynski JS, Beiser A, Seshadri S, Auerbach S, Wolf PA, Au R. Depressive symptoms and risk of dementia: the Framingham Heart Study. Neurology 2010;75:35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnes DE, Yaffe K, Byers AL, McCormick M, Schafer C, Whitmer RA. Midlife vs late-life depressive symptoms and risk of dementia. Arch Gen Psychiatry 2012;69:493–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panza F, Frisardi V, Capurso C, et al. Late-life depression, mild cognitive impairment, and dementia: possible continuum? Am J Geriatr Psychiatry 2010;18:98–116 [DOI] [PubMed] [Google Scholar]
- 8.Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Overview and findings from the Religious Orders Study. Curr Alzheimer Res 2012;9:628–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the Rush Memory and Aging Project. Curr Alzheimer Res 2012;9:646–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohout FJ, Berkman LF, Evans DA, Cornoni-Huntley J. Two shorter forms of the CES-D depression symptoms index. J Aging Health 1993;5:79–193 [DOI] [PubMed] [Google Scholar]
- 11.Radloff LS. The CES-D scale: a self-report depression scale for research in the general population. Appl Psychol Meas 1977;1:385–401 [Google Scholar]
- 12.Wilson RS, Bienias JL, Mendes de Leon CF, Evans DA, Bennett DA. Negative affect and mortality in older persons. Am J Epidemiol 2003;158:827–835 [DOI] [PubMed] [Google Scholar]
- 13.Amieva H, Goff ML, Millett X, et al. Prodromal Alzheimer's disease: successive emergence of the clinical symptoms. Ann Neurol 2008;64:492–498 [DOI] [PubMed] [Google Scholar]
- 14.Wilson RS, Hoganson GM, Rajan KB, Barnes LL, Mendes de Leon CF, Evans DA. Temporal course of depressive symptoms during the development of Alzheimer's disease. Neurology 2010;75:21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson RS, Beckett LA, Barnes LL, et al. Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging 2002;17:179–193 [PubMed] [Google Scholar]
- 16.Wilson RS, Barnes LL, Bennett DA. Assessment of lifetime participation in cognitively stimulating activities. J Clin Exp Neuropsychol 2003;25:634–642 [DOI] [PubMed] [Google Scholar]
- 17.Wilson RS, Barnes LL, Krueger KR, Hoganson G, Bienias JL, Bennett DA. Early and late life cognitive activity and cognitive systems in old age. J Int Neuropsychol Soc 2005;11:400–407 [PubMed] [Google Scholar]
- 18.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan E. Clinical diagnosis of Alzheimer's disease: report of the NINCDS/ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984;34:939–944 [DOI] [PubMed] [Google Scholar]
- 19.Bennett DA, Wilson RS, Schneider JA, et al. Natural history of mild cognitive impairment in older persons. Neurology 2002;59:198–205 [DOI] [PubMed] [Google Scholar]
- 20.Schneider JA, Li JL, Li Y, Wilson RS, Kordower JH, Bennett DA. Neurofibrillary tangles in the substantia nigra are related to gait impairment in older persons. Ann Neurol 2006;59:166–173 [DOI] [PubMed] [Google Scholar]
- 21.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006;66:1837–1844 [DOI] [PubMed] [Google Scholar]
- 22.Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA. Microinfarct pathology, dementia, and cognitive systems. Stroke 2011;42:722–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid with clinical AD and level of cognitive function. Arch Neurol 2004;61:378–384 [DOI] [PubMed] [Google Scholar]
- 24.Wilson RS, Yu L, Schneider JA, Arnold SE, Buchman AS, Bennett DA. Lewy bodies and olfactory dysfunction in old age. Chem Senses 2011;36:367–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. Neurology 1996;47:1113–1124 [DOI] [PubMed] [Google Scholar]
- 26.Min Y, Agresti A. Random effect models for repeated measures of zero-inflated count data. Stat Model 2005;5:1–19 [Google Scholar]
- 27.Capuano AW, Dawson JD. The trend odds model for ordinal data. Stat Med 2013;32:2250–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diggle PJ, Liang KY, Zeger SL. Analysis of Longitudinal Data. New York: Oxford University Press; 2002 [Google Scholar]
- 29.Wilson RS, Leurgans SE, Boyle PA, Bennett DA. Cognitive decline in prodromal Alzheimer disease and mild cognitive impairment. Arch Neurol 2011;68:351–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koolschijn PCMP, van Haren NEM, Lensvelt-Mulders GJLM, Hulshoff Pol HE, Kahn RS. Brain volume abnormalities in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Hum Brain Mapp 2009;30:3719–3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dotson VM, Davatzikos C, Kraut MA, Resnick SM. Depressive symptoms and brain volumes in older adults: a longitudinal magnetic resonance imaging study. J Psychiatry Neurosci 2009;34:367–375 [PMC free article] [PubMed] [Google Scholar]
- 32.Sexton CE, Mackay CE, Ebmeier KP. A systematic review of diffusion tensor imaging studies in affective disorders. Biol Psychiatry 2009;66:814–823 [DOI] [PubMed] [Google Scholar]
- 33.Sexton CE, Allan CL, Masurier ML, et al. Magnetic resonance imaging in late-life depression: multimodal examination of network disruption. Arch Gen Psychiatry 2012;69:680–689 [DOI] [PubMed] [Google Scholar]
- 34.Wilson RS, Nag S, Boyle PA, et al. Brainstem aminergic nuclei and late-life depressive symptoms. JAMA Psychiatry 2013;70:1320–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soetanto A, Wilson RS, Talbot K, et al. Anxiety and depression are associated with MAP2 and synaptopodin immunolabeled dendrite and spine densities in hippocampal CA3 of older humans. Arch Gen Psychiatry 2010;67:448–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holtzer R, Scarmeas N, Wegesin DJ, et al. Depressive symptoms in Alzheimer's disease: natural course and temporal relation to function and cognitive status. J Am Geriatr Soc 2005;53:2083–2089 [DOI] [PubMed] [Google Scholar]
- 37.Panza F, D'Introno A, Colacicco AM, et al. Temporal relationship between depressive symptoms and cognitive impairment: the Italian Longitudinal Study of Aging. J Alzheimers Dis 2009;17:899–911 [DOI] [PubMed] [Google Scholar]
- 38.Gale CR, Allerhand M, Deary IJ; HALCyon Study Team. Is there a bidirectional relationship between depressive symptoms and cognitive ability in older people? A prospective study using the English Longitudinal Study of Ageing. Psychol Med 2012;42:2057–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jajodia A, Borders A. Memory predicts changes in depressive symptoms in older adults: a bidirectional longitudinal analysis. J Gerontol B Psychol Sci Soc Sci 2011;66:571–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van den Kommer TN, Comijs HC, Aartsen MJ, Huisman M, Deeg DJH, Beekman ATF. Depression and cognition: how do they interrelate in old age? Am J Geriatr Psychiatry 2013;21:398–410 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.