

Abstract
We present a patient with complete androgen insensitivity syndrome (CAIS) diagnosed with a serous papillary cystadenofibroma. A 41-year-old married female with a mass in the left inguinal region and a history of primary amenorrhea. A bulging mass of 13.7 cm × 8 cm × 12.4 cm in the left inguinal region extending from the hip joint to the level of labia majus, and a 3.2 cm × 2.8 cm mass in her right inguinal region were found by ultrasonography and magnetic resonance imaging. We performed bilateral gonadectomy. The pathology showed testicular tissue in the right inguinal mass and a serous papillary cystadenofibroma in the left one. CAIS is an infrequent clinical entity, occurrence of serous papillary cystadenofibroma is even rarer in this syndrome serous cystadenofibroma should come to mind in patients with a huge inguinal mass. Gonadectomy should be performed right after puberty to prevent the risk of malignancy development in the testes.
KEY WORDS: Complete androgen insensitivity syndrome, cystadenofibroma, gonadal tumor, inguinal mass, primary amenorrhea
INTRODUCTION
Complete androgen insensitivity syndrome (CAIS), formerly known as “testicular feminization syndrome,” was first described by Morris in 1953 in a review of 82 cases.[1] CAIS is the most common form of male pseudohermaphroditism and is a sex-linked recessive inherited disorder caused by a mutation in the androgen receptor gene located at Xq11-q12.[2] Subsequently, Wolffian duct development and male external genitalia differentiation do not occur correctly, and the Mullerian ducts regress due to the presence of anti-Mullerian hormone produced by the sertoli cells of normally developed gonads. In approximately one third of patients, residual Mullerian structures exist.[3] CAIS patients exhibit female phenotype because of the insensitivity to the androgen receptor. Differentiation of the gonads is normal, and serum androgen level is comparable with that of a normal male.
Patients with CAIS have undescended testes with solid immature tubules and markedly decreased or absent germ cells. It is well-known that testicular tumors develop in patients with CAIS, and the risk of gonadal malignancies increases with age. Fortunately, CAIS is usually diagnosed at puberty in phenotypically female patients with primary amenorrhea.[4] However, it may seldom be diagnosed in the older population. Many types of testicular tumors have been reported in patients with CAIS, however to the best of our knowledge this is the second case diagnosed with a gonadal serous papillary cystadenofibroma.[5]
CASE REPORT
A 41-year-old married female applied to our clinic with a left inguinal mass. The patient had been primary amenorrheic, but had not consulted a doctor. Clinical examination showed a bulging mass of 15 cm × 10 cm on the lateral side of the left inguinal canal and another 3 cm × 4 cm mass in the right inguinal region [Figure 1]. The vulva and perineum were normal in appearance, the vagina was 6 cm long but ended blind, pubic and axillary hair was sparse; however, the patient had normal breast development as well as a normal female phenotype. The abdominal ultrasonography (USG) and magnetic resonance imaging revealed the absence of the uterus and ovaries. They also showed a 13.7 cm × 8 cm × 12.4 cm polypoid cystic mass with a regular surface in the left inguinal region extending from the hip joint to the level of labia majus, and a solid 3.2 cm × 2.8 cm mass in the right inguinal region. In hormonal analysis, follicle stimulating hormone was found to be 9.68 mIU/mL, luteinizing hormone was 15.74 mIU/mL, estradiol was 56.07 pg/mL, and testosterone was 1.83 ng/mL. The tumor markers including carcinoembryonic antigen, CA125, alpha-fetoprotein, lactate dehydrogenase, and alkaline phosphatase were within normal ranges. The chromosome test revealed a 46, XY karyotype. The diagnosis of CAIS was made based on these findings.
Figure 1.

A bulging mass of 15 cm × 10 cm on the left side of inguinal canal
After the diagnosis, we performed an exploratory laparotomy. Internal female organs were absent. The right testis was of normal size, and the left one was swollen with a cyst. A gonadal tumor in the left inguinal region measuring 13 cm × 8 cm × 12 cm and a 3 cm × 4 cm one in the right inguinal region were extirpated. Vaginoplasty was not performed as the vagina was 6 cm long, and this was thought to be adequate for sexual intercourse.
Histopathologic examination revealed immature seminiferous tubules surrounded by immature germ cells and sertoli cells in the mass from the right inguinal region, and a serous papillary cystadenofibroma in the one from the left. No signs of spermatogenesis or leydig cell hyperplasia in the interstitial tissue were seen in both gonads [Figures 2-4]. Estrogen therapy was initiated postoperatively.
Figure 2.
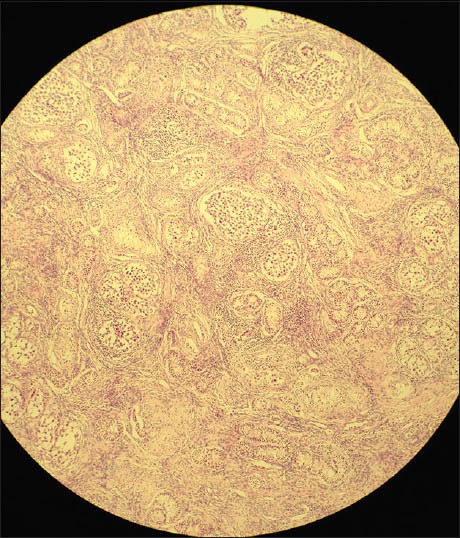
Microscopic appearance of the right gonad (H and E, ×10)
Figure 4.
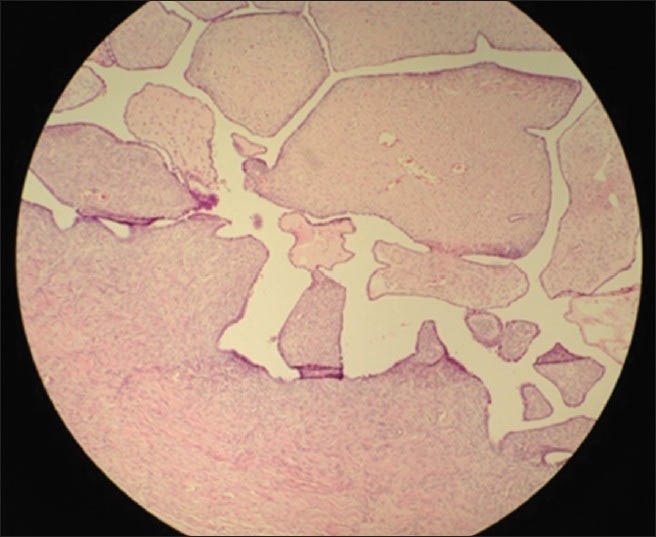
Microscopic appearance of the left gonadal serous papillary cystadenofibroma (H and E, ×10)
Figure 3.
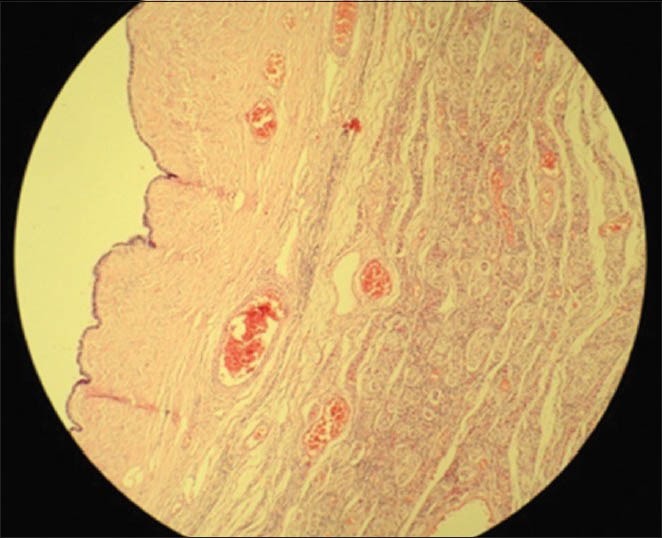
Microscopic appearance of the left gonad (H and E, ×10)
DISCUSSION
The typical presentation for CAIS is either primary amenorrhea in adolescence or inguinal swellings in an infant. A female adolescent with the disorder has normal breast development and a pubertal growth spurt at the appropriate age, but is primary amenorrheic. Development of estrogen-dependent secondary sexual characteristics occurs as the result of excess aromatization of androgens. Pubic and axillary hair is usually absent or can be present in sparse amounts.[4] This could be attributed to the mildly elevated levels of testosterone and levels of estradiol that were near the lowest limit of the normal range.
The risk of malignant transformation of the gonads in adult life is well documented. The best time for gonadectomyis soon after the completion of puberty. The point here is that estradiol produced from testes is necessary for a normal phenotypic female pubertal development. After gonadectomy, estrogen replacement therapy should be started. If gonadectomy was not done in puberty, it is generally recommended to be performed in early adulthood to avoid the risk of development of gonadal tumors. The procedure may be performed laparoscopically if the gonads are situated intraabdominal.
One mode of presentation includes a known family history of CAIS, and occasionally the discovery of a pelvic mass arising from a gonadal tumor.[6] Our patient had three sisters; one of them was 46, XX and married with children. The other two also had CAIS, however they refused to undergo surgery. The first case with CAIS and serous cystadenofibroma reported in the literature was a 56-year-old patient with a unilateral 10.6 cm mass in the inguinal region.[5] There is no circulating specific reliable tumor marker for monitoring tumor development, and USG of intra-abdominal gonads is not sufficiently sensitive.
In patients with CAIS, the incidence of gonadal tumor (e.g. seminoma) is 3.6% and 33% at the age of 25 years and 50 years, respectively.[7] Gonadectomy is recommended after puberty to eliminate the risk of gonadal malignancy and aid the development of feminine secondary sexual characteristics.[8] Studies have suggested an increased tumor risk <30% in late adulthood if gonadectomy is not performed.[9,10] Specific analyses in a large sample groups suggest a germ cell tumor risk as low as 0.8-2%, especially before puberty.[11,12] Benign tumors of nongerm cell origin include sertoli cell adenomas, hamartomas and cystadenofibromas.
Herein, we reported the second patient with CAIS with a large gonadal serous papillary cystadenofibroma in the literature. CAIS is an infrequent clinical entity occurrence of serous papillary cystadenofibroma is even rarer in this syndrome. Serous cystadenofibroma should come to mind in patients with a huge inguinal mass. Gonadectomy should be performed right after puberty to prevent the risk of tumor development in the testes.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Morris JM. The syndrome of testicular feminization in male pseudohermaphrodites. Am J Obstet Gynecol. 1953;65:1192–211. doi: 10.1016/0002-9378(53)90359-7. [DOI] [PubMed] [Google Scholar]
- 2.Brinkmann AO. Molecular basis of androgen insensitivity. Mol Cell Endocrinol. 2001;179:105–9. doi: 10.1016/s0303-7207(01)00466-x. [DOI] [PubMed] [Google Scholar]
- 3.Nichols JL, Bieber EJ, Gell JS. Case of sisters with complete androgen insensitivity syndrome and discordant Müllerian remnants. (e15-8).Fertil Steril. 2009;91:932. doi: 10.1016/j.fertnstert.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 4.Hughes IA, Davies JD, Bunch TI, Pasterski V, Mastroyannopoulou K, MacDougall J. Androgen insensitivity syndrome. Lancet. 2012;380:1419–28. doi: 10.1016/S0140-6736(12)60071-3. [DOI] [PubMed] [Google Scholar]
- 5.Müllauer KB, Obwegeser R, Ulm MR. Testicular feminization – sequelae of chronic neglect of a neoplastic process. Zentralbl Gynakol. 2000;122:287–90. [PubMed] [Google Scholar]
- 6.Stewart CJ, Baker E, Beaton C, Crook M, Peverall J, Wallace S. Detection of Y-chromosome in gonadal tumours using fluorescence in situ hybridization: Diagnostic value in intersex conditions including older patients with clinically unsuspected androgen insensitivity syndrome. Histopathology. 2008;52:175–82. doi: 10.1111/j.1365-2559.2007.02927.x. [DOI] [PubMed] [Google Scholar]
- 7.Yanai Y, Hiroi H, Osuga Y, Fujimoto A, Momoeda M, Yano T, et al. Androgen insensitivity syndrome with serous gonadal cyst. (e9-11).Fertil Steril. 2008;90:2018. doi: 10.1016/j.fertnstert.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Baksu A, Kabukcuoglu F, Baksu B, Goker N. Bilateral sertoli cell adenoma and serous cyst in a patient with androgen insensitivity syndrome. Eur J Obstet Gynecol Reprod Biol. 2004;114:104–7. doi: 10.1016/j.ejogrb.2003.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Rutgers JL, Scully RE. The androgen insensitivity syndrome (testicular feminization): A clinicopathologic study of 43 cases. Int J Gynecol Pathol. 1991;10:126–44. doi: 10.1097/00004347-199104000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Deans R, Creighton SM, Liao LM, Conway GS. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): Patient preferences and clinical evidence. Clin Endocrinol (Oxf) 2012;76:894–8. doi: 10.1111/j.1365-2265.2012.04330.x. [DOI] [PubMed] [Google Scholar]
- 11.Hannema SE, Scott IS, Rajpert-De Meyts E, Skakkebaek NE, Coleman N, Hughes IA. Testicular development in the complete androgen insensitivity syndrome. J Pathol. 2006;208:518–27. doi: 10.1002/path.1890. [DOI] [PubMed] [Google Scholar]
- 12.Looijenga LH, Hersmus R, Oosterhuis JW, Cools M, Drop SL, Wolffenbuttel KP. Tumor risk in disorders of sex development (DSD) Best Pract Res Clin Endocrinol Metab. 2007;21:480–95. doi: 10.1016/j.beem.2007.05.001. [DOI] [PubMed] [Google Scholar]