

Abstract
Because mutations are mostly deleterious, mutation rates should be reduced by natural selection. However, mutations also provide the raw material for adaptation. Therefore, evolutionary theory suggests that the mutation rate must balance between adaptability—the ability to adapt—and adaptedness—the ability to remain adapted. We model an asexual population crossing a fitness valley and analyse the rate of complex adaptation with and without stress-induced mutagenesis (SIM)—the increase of mutation rates in response to stress or maladaptation. We show that SIM increases the rate of complex adaptation without reducing the population mean fitness, thus breaking the evolutionary trade-off between adaptability and adaptedness. Our theoretical results support the hypothesis that SIM promotes adaptation and provide quantitative predictions of the rate of complex adaptation with different mutational strategies.
Keywords: stress-induced variation, mutation rate, mathematical model, adaptive peak shifts, trade-off, evolvability
1. Introduction
There is experimental, clinical and theoretical evidence that high mutation rates increase the rate of adaptation and that during adaptive evolution, constitutive mutators—alleles that constitutively increase the mutation rate—can rise in frequency because of the beneficial mutations they generate [1–3]. However, during evolution in a stable environment, constitutive mutators become associated with poor genetic backgrounds due to increased accumulation of deleterious mutations; this was evidenced both in the laboratory [4] and in the clinic [5]. Classical models suggest the ‘reduction principle’, which states that natural selection reduces the mutation rate in a stable environment [6,7]. But many adaptations require new beneficial mutations, especially in asexual populations. This tension between the effects of beneficial and deleterious mutations leads to ‘the rise and fall of the mutator allele’ [8], where mutator alleles increase in frequency in a maladapted population, only to be eliminated by natural selection when the population is well adapted. This dynamic was studied using experimental evolution [9,10], mathematical analysis and simulations [11–13].
Thus, the mutation rate must balance between two evolutionary traits, as Leigh [14] suggested: adaptability—the capacity to adapt to new environmental conditions—and adaptedness—the capacity to remain adapted to existing conditions.
Stress-induced mutagenesis (SIM)—the increase of mutation rates in stressed or maladapted individuals—has been demonstrated in several species, including both prokaryotes and eukaryotes [15]. SIM has been observed in laboratory strains [16,17] and natural populations of Escherichia coli [18] (but see [19]), and in other species of bacteria such as pseudomonads [20], Helicobacter pylori [21], Vibrio cholera [22] and Streptococcus pneumonia [23]. SIM has also been observed in yeast [24,25], algae [26], nematodes [27], flies [28] and human cancer cells [29]. Several stress responses regulate the mutation rate in bacteria by shifting replication to error-prone DNA polymerases [30] and by inhibiting the mismatch repair system [31]. These stress responses include the SOS DNA-damage response, the RpoS-controlled general or starvation stress response, and the RpoE membrane protein stress response [32].
It is still not clear how SIM affects evolution and adaptation. Some authors have proposed that SIM has a significant impact on adaptability or evolvability [17,33,34], but there is no theoretical treatment of this impact. On the other hand, the effect of SIM on adaptedness was studied with deterministic [35] and stochastic [36] models. These models showed that without beneficial mutations SIM does not affect the mean fitness of asexual populations in stable environments, in contrast with constitutive mutagenesis (CM), which decreases the population mean fitness. More recently, we have shown that with rare beneficial mutations, if maladapted individuals increase their mutation rate then the population mean fitness of asexual populations increases [37].
Complex traits, coded by multiple genes, present an open evolutionary problem, first described by Sewall Wright in 1931 [38,39]: if different alleles are separately deleterious but jointly advantageous, how can a population evolve from one co-adapted gene complex to a fitter one, crossing a less fit ‘valley’? Wright suggested the ‘shifting balance theory of evolution’. His solution is valid [40–42], but possibly limited to specific parameter ranges [43–46]. As a result, other mechanisms have been proposed: increased phenotypic variance after population bottlenecks [47], environmental fluctuations [48], environmental heterogeneity [49], fitness-associated recombination [50], stochastic tunnelling in large asexual populations [51] and intermediate recombination rates [52].
Here, we analyse population genetics models of adaptive evolution to explore the rate of complex adaptation on rugged fitness landscapes, in which adaptations require two separately deleterious mutations. We develop analytic approximations and stochastic simulations, and compare normal, constitutive and stress-induced mutagenesis. We show that SIM can break the trade-off between adaptability and adaptedness by increasing the rate of complex adaptation without decreasing the population mean fitness.
2. Model
We model a population of N haploid asexual individuals with a large number of loci in full linkage. The model includes the effects of mutation, selection and genetic drift. Individuals are characterized by their genotype in two specific bi-allelic loci—ab, Ab, aB and AB—and by the number of deleterious mutations they carry in the rest of the non-specific loci. For example, aB/3 is the aB genotype with three additional deleterious mutations in non-specific loci.
We focus on adaptation to a new rugged fitness landscape. The fitness of the wild-type ab/0 is 1, the fitness of the single mutants Ab/0 and aB/0 is 1 − s, and the double mutant AB/0 has the highest fitness 1 + sH, where s is the selection coefficient and H is the relative advantage of the double mutant. This is the simplest case of a rugged fitness landscape: the single mutants Ab/0 and aB/0 are fitness valleys between the local and global fitness peaks ab/0 and AB/0 (figure 1). We do not consider smooth fitness landscapes in which single mutants have intermediate fitness (1 + sH > 1 − s > 1). We have already shown that SIM has higher mean fitness in changing environments on smooth landscapes [37]; however, analysis of the effect of SIM on the adaptation rate on smooth landscapes will be the subject of future efforts.
Figure 1.

Adaptation on a rugged fitness landscape. The figure shows the fitness of the possible genotypes, which are represented by the allele combination at the specific loci (ab, Ab, aB and AB) and the number of deleterious alleles across the genome following the forward-slash (‘/’). The y-axis represents fitness: the wild-type ab/0 has fitness 1; the fittest genotype AB/0 has fitness 1 + sH; deleterious alleles, either at the A/a and B/b loci, or at the non-specific loci, reduce fitness by 1 − s. The x-axis represents the number of accumulated mutations (genotypes are jittered to increase separation). Solid lines represent mutations at the a/A and b/B loci, occurring with probability μ. Dashed lines represent deleterious mutations in the rest of the genome, occurring with rate U. Mutagenesis is induced in stressed genotypes with fitness less than 1 (grey background). Fit genotypes, with fitness more than or equal to 1, do not hypermutate (white background). (a) In the analytic model, genotypes with deleterious alleles in non-specific loci are considered ‘evolutionary dead ends’ and do not contribute to adaptation. (b) In the simulations, individuals can accumulate up to 25 deleterious alleles (the figure only shows three). Multiple mutations can occur simultaneously but are not shown for simplicity of the illustration.
Deleterious mutations in the non-specific loci independently (multiplicatively) reduce the fitness of the individual by 1 − s. Mutations occur in the specific loci with probability μ. The number of new mutations per replication in the rest of the genome (the non-specific loci) is Poisson distributed with an average U. The model neglects back-mutations and compensatory mutations due to their insignificant short-term effects.
We consider three mutational strategies: normal mutagenesis (NM), where there is no increase in the mutation rate; CM, where all individuals always increase their mutation rate by τ, the mutation rate fold increase; and SIM, where only stressed or maladapted individuals increase their mutation rate by τ. Individuals are considered stressed if their fitness is below a specific threshold, so stress can be caused by a deleterious mutation (either in the specific A/a and B/b loci or in non-specific loci). Evidence shows that numerous stress responses induce mutagenesis in bacteria [16,32]. These responses can be activated due to deteriorating environmental conditions (see §3e) or due to mutations that impair important cell functions, thereby reducing fitness and inducing a stress response. For example, a frameshift mutation in the lac gene causes cells to starve on lactose, thus inducing mutagenesis via a stress response [17,53].
The main analysis assumes that SIM induces mutagenesis in individuals less fit than the wild-type; that is, the mutation rate U of individuals with fitness ω is
| 2.1 |
This equation models a scenario in which an environmental change (i.e. appearance of a new ecological niche or a new carbon source) provides an opportunity for adaptation without affecting the fitness of the wild-type (ab/0). We also study a different scenario in which the environmental change reduces the absolute fitness of the wild-type so that it is also stressed (see §3e). The assumption of a threshold relationship between fitness and the mutation rate in equation (2.1) is relaxed in the electronic supplementary material, appendix E, in which we explore continuous relationships between fitness and the mutation rate. The results are robust to this relaxation (see §3c).
We are interested in calculating the adaptation rate of a population homogeneous for each of the above mutational strategies (NM, CM or SIM). The adaptation process is separated into two distinct stages. In the first stage, a double mutant AB appears in the population, usually in a single copy. In the second stage, the double mutant either goes to extinction or avoids extinction, increases in frequency and goes to fixation.
We analysed this model with two methods. The first is analytic (figure 1a), in which we assume that: (i) genotypes with deleterious backgrounds (deleterious alleles in the non-specific loci) do not contribute to the adaptation process, and (ii) the number of deleterious alleles per individual before the appearance of a double mutant is at a mutation-selection balance (MSB) and is Poisson distributed with mean U/s [54]. The former assumption requires that mutation is weaker than selection (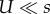 ); the later assumption only requires that mutation is not much stronger than selection. Specifically, the expected number of mutation-free individuals is at least one:
); the later assumption only requires that mutation is not much stronger than selection. Specifically, the expected number of mutation-free individuals is at least one: 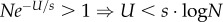 [55].
[55].
The second method is a stochastic Wright–Fisher simulation with selection, mutation and genetic drift (figure 1b), in which (i) individuals with a deleterious background can contribute to adaptation, and (ii) a mutation-free population evolves towards a MSB without assuming a Poisson distribution of the number of deleterious alleles.
(a). Wright–Fisher simulations
We track the number of individuals in each genotype class: ab/x, Ab/x, aB/x and AB/x, where x ≥ 0 is the number of deleterious alleles in non-specific loci. The simulations start with a single-peak smooth fitness landscape (the fitness of AB/x is (1 − s)2+x) and a mutation-free population (all individuals start in the optimal ab/0 genotype with fitness 1) that accumulates deleterious mutations over the first 5000 generations of the simulation. With s = 0.05 and 0.005, 180 and 1800 generations are enough for the average number of deleterious alleles per individual to reach 99.99% of its MSB value, U/s [56].
After 5000 generations, the fitness landscape changes to a rugged one, making AB the optimal genotype with fitness 1 + sH (figure 1b). The simulation then proceeds until an AB genotype appears and either fixates in the population or goes extinct (either all or no individuals are in the AB classes, respectively). Therefore, each simulation provides one sample of the waiting time for the appearance of a double mutant and one sample of the probability of fixation of a double mutant. At least 1000 simulations were performed for each parameter set.
Table 1 summarizes the model parameters with estimated values for E. coli.
Table 1.
Model parameters and estimated values for E. coli.
3. Results
(a). Appearance of a double mutant
We are interested in the waiting time for the appearance of a double mutant AB either by a double mutation in a wild-type individual ab, or via a single mutation in a single mutant Ab or aB (figure 1a). Denoting the population size by N, we note that (i) if Ne−U/s(μ/s)2 > 1, then double mutants are already expected at the MSB and adaptation will not require new mutations, and (ii) if Ne−U/sμ/s < 1, then no single mutants are expected at the MSB and double mutants must be generated by a double mutation in a wild-type individual. In this case, increasing the mutation rate of individuals with fitness below 1 will have no effect on the appearance of the double mutant and there is no point in analysing the effect of SIM.
Combining the two constraints, we get this constraint on the population size N: eU/ss/μ < N < eU/s(s/μ)2. This constraint is reasonable for bacterial populations (see table 1).
The frequencies of wild-type (ab) and single mutants (aB and Ab combined) that are mutation-free at the MSB are roughly e−U/s and 2μ/s · e−U/s, respectively. The probability that an offspring of a wild-type or single mutant parent is a double mutant AB is μ2 and μ, respectively. The probability that such an offspring is also mutation-free in the rest of its genome (the only mutations that occurred were at the specific loci) is e−U. Therefore, the probability q that a random offspring is a double mutant, given there are no double mutants in the current generation, is approximated by
| 3.1 |
The first expression assumes that individuals with a deleterious background do not contribute to adaptation and that the MSB distribution of deleterious alleles is Poisson. The second expression also assumes that mutation is much weaker than selection: 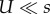 .
.
With SIM, the mutation rate of single mutants is increased τ-fold and the probability that a random offspring is a double mutant is
| 3.2 |
These expressions use the same assumptions as in equation 3.1. The second expression also assumes that τU < 1.
Appendix A in the electronic supplementary material includes full derivations of the above equations and figure S1 compares them with simulation results.
(b). Fixation probability of the double mutant
Assuming an advantage to the double mutant (H > 1) and a large population size (see the above constraint on N), a double mutant has two possible fates after its appearance: fixation or extinction. Following Eshel [64], the fixation probability ρ of the double mutant (see the electronic supplementary material, appendix B) is
| 3.3 |
That is, the fixation probability of the double mutant is roughly twice its adaptive advantage. This is a classic result of population genetics theory [65,66].
The fixation probability with SIM equals that of NM and CM because the mutation rate of the wild-type ab equals that of the double mutant AB (but see an exception in §3e).
(c). Adaptation rate
From the probability q that a random offspring is a double mutant, we can derive the probability that one or more double mutants appear in the next generation: 1 − (1 − q)N ≈ Nq. This is a good approximation because Nq is very small due to the constraint on N. Once a double mutant appears, it goes to fixation with probability ρ.
When fixation is much faster than appearance of the double mutant AB, the time for adaptation T can be approximated by the waiting time for a double mutant that goes to fixation. This waiting time follows a geometric distribution with rate Nqρ, and therefore the adaptation rate ν (the inverse of the waiting time for adaptation) is approximately
| 3.4 |
Plugging equations (3.1)–(3.3) in equation (3.4), we get these approximations:
| 3.5 |
| 3.6 |
| 3.7 |
The middle expression in each equation is the full approximation, which assumes a Poison distribution and no contribution of deleterious genotypes to adaptation. The right-hand sides are first-order approximations that assume mutation is much weaker than selection (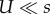 for NM and SIM,
for NM and SIM, 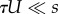 for CM) and that 1 < τ < 1/U. See table 1 for description of model parameters and an article by Weinreich & Chao [67] for a result similar to equation (3.5).
for CM) and that 1 < τ < 1/U. See table 1 for description of model parameters and an article by Weinreich & Chao [67] for a result similar to equation (3.5).
The main conclusions from equations (3.5)–(3.7): first, adaptation with CM is faster than with NM. Second, adaptation with SIM is also faster than with NM, but not as fast as with CM because the mutation-free wild-type (ab/0) does not hypermutate.
If mutation is weaker than selection (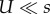 ), then the adaptation rate with CM increases with τ2 and the adaptation rate with SIM increases with τ. In addition, because the fixation probability is the same for NM, CM and SIM, the differences in the adaptation rate are due to differences in the appearance probability q (electronic supplementary material, figure S1); see §3e for a different scenario in which SIM also increases the fixation probability.
), then the adaptation rate with CM increases with τ2 and the adaptation rate with SIM increases with τ. In addition, because the fixation probability is the same for NM, CM and SIM, the differences in the adaptation rate are due to differences in the appearance probability q (electronic supplementary material, figure S1); see §3e for a different scenario in which SIM also increases the fixation probability.
Figure 2 compares the analytic approximations with simulation results for the weak mutation regime (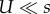 ; see the electronic supplementary material, appendix G, for results with strong mutation U = s/10). This regime is relevant for asexual microbes in which the deleterious mutation rate is generally 10−4 to 10−3 mutations per genome per generation and selection coefficients are estimated to be between 10−1 and 10−2 (see table 1). When the mutation rate fold increase τ is high (more than 10), the approximations slightly overestimate the adaptation rate because the double mutant AB is more likely to appear on a deleterious background (AB/1 instead of AB/0). Because the fitness of AB/1 is higher than that of the wild-type ab/0 (this happens because 1 + sH > (1 − s)−1 ≈ 1 + s), the double mutant can go to fixation even when it appears on a deleterious background, sweeping the deleterious alleles with it to fixation in a process called ‘genetic hitch-hiking’ [68]. However, these sweeps result in a lower fixation probability for the double mutant (electronic supplementary material, figure S2).
; see the electronic supplementary material, appendix G, for results with strong mutation U = s/10). This regime is relevant for asexual microbes in which the deleterious mutation rate is generally 10−4 to 10−3 mutations per genome per generation and selection coefficients are estimated to be between 10−1 and 10−2 (see table 1). When the mutation rate fold increase τ is high (more than 10), the approximations slightly overestimate the adaptation rate because the double mutant AB is more likely to appear on a deleterious background (AB/1 instead of AB/0). Because the fitness of AB/1 is higher than that of the wild-type ab/0 (this happens because 1 + sH > (1 − s)−1 ≈ 1 + s), the double mutant can go to fixation even when it appears on a deleterious background, sweeping the deleterious alleles with it to fixation in a process called ‘genetic hitch-hiking’ [68]. However, these sweeps result in a lower fixation probability for the double mutant (electronic supplementary material, figure S2).
Figure 2.

Complex adaptation with different mutational strategies. The figure shows the adaptation rate ν as a function of the mutation rate fold increase τ (both in log scale). The black circle is NM (τ = 1); solid line with circles is CM; solid line with squares is SIM; dashed lines with triangles is SIM with environmental stress (SIMe; see §3e). Lines are analytic approximations. Markers are the means of stochastic simulation results. Error bars represent 95% CI of the mean (at least 1000 simulations per point; computed with bootstrap with 1000 samples per point). Parameters (see table 1): U = 0.0004, s = 0.05, β = 0.0002, H = 2, N = 106. (Online version in colour.)
The adaptation rate of SIM with continuous relationships between fitness and mutation rate (electronic supplementary material, appendix E) is comparable to that of SIM with threshold relationships (electronic supplementary material, figure E2). This is because the main factors determining the adaptation rate are the mutation rates of the wild-type and the single mutants (ab, aB and Ab), as individuals with more than a single mutation do not have a significant contribution to adaptation. Therefore, our results are robust to the choice of the relationship between fitness and the mutation rate.
(d). The trade-off between adaptability and adaptedness
Next, we explore how different mutational strategies (NM, CM and SIM) balance between adaptability—the ability to adapt to new conditions—and adaptedness—the ability to remain adapted to current conditions. For this purpose, we define adaptedness as 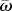 , the population mean fitness in a stable environment, and adaptability as ν, the rate of complex adaptation.
, the population mean fitness in a stable environment, and adaptability as ν, the rate of complex adaptation.
We used the above approximations (equations (3.5)–(3.7)) to calculate the rate of complex adaptation of populations with NM, CM and SIM. We also extended an existing model [37] to calculate the population mean fitness at the MSB. This extended model includes rare back or compensatory mutations (which have a stronger effect on MSB dynamics than on adaptive dynamics) and allows more than one mutation to occur in the same individual and generation. The details of this model and the calculation of population mean fitness with various mutational strategies are given in the electronic supplementary material, appendix D.
The mutation rate with CM is constant and uniform across the population, and the population mean fitness mainly depends on the fitness and mutation rate of the fittest individuals. Therefore, the population mean fitness decreases when the mutation rate increases; this decrease is due to generation of deleterious mutations in the fittest individuals. The adaptation rate, however, increases with the mutation rate (equation (3.6)). This trade-off between adaptability and adaptedness constrains the population: after a long period of environmental stability it can lose the potential for adaptation, and after a long period of environmental change the population can be susceptible to reduced fitness and mutational meltdowns [69].
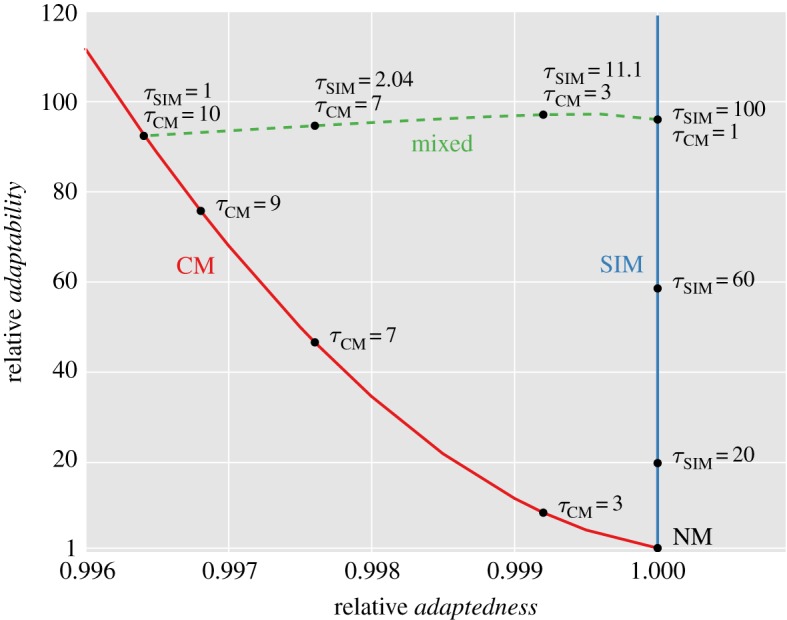
However, this trade-off between adaptability and adaptedness can be broken if mutation rates are not uniform across the population. Increased mutation rates in unfit individuals increase the population mean fitness, as long as beneficial (or compensatory) mutations can occur [37]. Figure D1 in the electronic supplementary material shows this advantage of SIM over NM in terms of the difference in population mean fitness (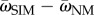 ). Moreover, increased mutation rates in unfit individuals also increase the adaptation rate (equation (3.7); figure 2). Therefore, SIM breaks the trade-off between adaptability and adaptedness.
). Moreover, increased mutation rates in unfit individuals also increase the adaptation rate (equation (3.7); figure 2). Therefore, SIM breaks the trade-off between adaptability and adaptedness.
Figure 3 shows the adaptation rate and population mean fitness of CM and SIM compared to NM for different values of τ, the mutation rate fold increase.
Figure 3.
The trade-off between adaptedness and adaptability. The figure shows the relative adaptedness and the relative adaptability of different mutational strategies in comparison to NM. Adaptedness is defined by the population mean fitness at MSB, 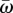 (see the electronic supplementary material, appendix D). Adaptability is defined by the rate of complex adaptation, ν (equations (3.5)–(3.7)). CM increases the mutation rate of all individuals τCM-fold; SIM increases the mutation rate of stressed individuals τSIM-fold; mixed strategies (dashed line) increase the mutation rate of all individuals τCM-fold and of stressed individuals an additional τSIM-fold. SIM breaks off the adaptability–adaptedness trade-off of CM, increasing the adaptability without compromising the adaptedness of the population. Parameters (see table 1): N = 106, U = 0.0004, β = 0.0002, s = 0.05, H = 2,
(see the electronic supplementary material, appendix D). Adaptability is defined by the rate of complex adaptation, ν (equations (3.5)–(3.7)). CM increases the mutation rate of all individuals τCM-fold; SIM increases the mutation rate of stressed individuals τSIM-fold; mixed strategies (dashed line) increase the mutation rate of all individuals τCM-fold and of stressed individuals an additional τSIM-fold. SIM breaks off the adaptability–adaptedness trade-off of CM, increasing the adaptability without compromising the adaptedness of the population. Parameters (see table 1): N = 106, U = 0.0004, β = 0.0002, s = 0.05, H = 2, 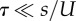 . (Online version in colour.)
. (Online version in colour.)
Any realistic rate of adaptation ν can be realized using both CM and SIM. The highest mean fitness will always be attained with SIM, which has a small advantage over NM (it cannot be seen in figure 3, but see the electronic supplementary material, figure D1) due to the increased generation of beneficial mutations in individuals with low fitness. If, for some rate of adaptation, the mutation rate fold increase τ required by SIM is too high (i.e. τU > s), the same adaptation rate can be realized by a mixed strategy (dashed line in figure 3). For example, a 96-fold increase in adaptation rate can be achieved with CM with τ = 10, with SIM with τ = 96 or with a mixed strategy with τCM = 7 and τSIM = 2 in which all individuals increase their mutation rate sevenfold and stressed individuals further increase their mutation rate twofold. However, these increases in adaptation rates have a price: the mutational load will decrease the population mean fitness from 0.9996 with NM to 0.996 with CM and 0.9972 with the mixed strategy. This price in not paid by populations with SIM because the mean fitness mainly depends on the mutation rate of fit individuals.
(e). Environmental stress
So far, we have considered the case in which the environmental change creates an opportunity for adaptation without affecting the absolute fitness of the population—for example, a new ecological niche can be favourable without affecting the well-being of the current population. In that scenario, the wild-type ab was not stressed and did not hypermutate.
Next, we consider a different scenario in which an environmental change affects the well-being of the entire population: for example, exposure to an antibiotic drug or a host immune response. In this case, the environmental change does not just create an opportunity for adaptation but also causes stress in the entire population. We use a subscript ‘e’ to denote quantities related with this scenario.
As before, the double mutant AB is resistant to the stress (i.e. the drug or immune response) and therefore has a higher fitness than either the wild-type or the non-resistant single mutants. However, in this scenario, the wild-type ab is also stressed and therefore hypermutates with SIM (compare with equation (2.1))
| 3.8 |
This scenario has an important biological relevance, as SIM has been implicated in the evolution of drug resistance in bacteria and yeast [34,70,71], and could be involved in the evolution of pathogen virulence and the evolution of drug resistance and progression in cancer cells [72].
We assume that after the environmental change, the SIMe population has reached a new MSB [56] with mutation rate τU, before the appearance of the double mutant (with s = 0.05 and U = 0.0004, for example, the average number of deleterious mutations is 0.99 · U/s after 90 generations, whereas the adaptation time is well over 1000 generations). Under this assumption, the adaptation rate with SIMe is (see the electronic supplementary material, appendix C, for full derivation)
| 3.9 |
That is, adaptation with SIMe is faster than with CM (figure 2a). The fixation probability of double mutants is higher with SIMe than with CM, because the mutation rate of double mutants is lower than that of the rest of the population. This difference in mutation rates confers an additional selective advantage to the double mutants (see the electronic supplementary material, appendix C), which increases their fixation probability
| 3.10 |
This additive advantage increases linearly with τ with a slope of U/sH and can be significant: for s = 0.05, H = 2 and U = 0.0004, increasing the mutation rate of stressed individuals 10-fold increases the fixation probability by 3.6%. The increased fixation probability was verified by simulations (electronic supplementary material, figure S2).
4. Discussion
We studied the effect of SIM on both adaptability (the capacity of populations to adapt to new complex conditions) and adaptedness (the ability of populations to stay adapted to existing conditions) [14]. We showed that SIM breaks the trade-off between adaptability and adaptedness, allowing rapid adaptation to complex environmental challenges without compromising the population mean fitness in a stable environment.
In addition to the pure strategies of CM and SIM, our model also considers a mixed mutational strategy. There are two examples of such a mixed strategy. First, if individuals have incomplete information regarding their condition (this is the case in most realistic biological scenarios), then we expect errors in the induction of mutagenesis: induction of mutagenesis without stress and failure to induce mutagenesis under stress. In this case, the population would, on average, use a mixed strategy. Second, a mutator allele can increase the mutation rate constitutively and further increase it under stress—for example, a recent study with Pseudomonas aeruginosa found that although the mutS, mutY and mutM mutator alleles always increase the mutation rate in comparison with the wild-type, the level of this increase depends on the level of stress the cell experiences [73].
We do not assume direct fitness costs for any of the mutational strategies. A ‘cost of DNA replication fidelity’ [74]—the energy and time expended in order to maintain a low mutation rate—could make both CM and SIM more successful. The ‘cost of fidelity’ may require further study, but empirical evidence suggests that it does not play an important role in the evolution of the mutation rate [75–78]. Another fitness cost might be associated with the regulation of the mutation rate: for individuals to determine whether their condition calls for the induction of mutagenesis, they must invest resources and energy in costly sensory mechanisms. However, such mechanisms already exist for various unrelated purposes, such as the maintenance of cell cycle and homeostasis. Therefore, we consider these mechanisms as ‘free’ in terms of fitness costs. For example, in E. coli mutagenesis is induced by several stress responses that serve other cellular functions [16,32], and this is probably the case in other organisms as well.
This article focuses on asexual populations, ignoring recombination, segregation and sexual reproduction. These mechanisms are important for adaptation on a rugged fitness landscape both because they help to cope with deleterious mutations and because they allow different single mutants to produce double mutants without an increased mutation rate. Recombination may reduce the advantage of SIM over NM in terms of population mean fitness [35], direct competitions [79] and adaptation rate (due to the Fisher–Muller effect).
Mean fitness and adaptation rate are both population-level traits. But simply because SIM has the most efficient balance between these traits does not mean it will necessarily evolve, because individual-level selection and population-level selection can act in opposing directions. In a previous article, we have demonstrated that second-order selection can lead to the evolution of SIM [37]: in an asexual population evolving on a smooth fitness landscape, selection favoured SIM over both NM and CM. In the current article, we show that selection also favours SIM on a rugged fitness landscape (electronic supplementary material, appendix F).
Several authors have suggested that the mutation rate must balance between adaptability and adaptedness: Kimura [6] found a mutation rate that balances between mutational and substitutional load; Johnson & Barton [80] found an optimal mutation rate that balances the generation of beneficial and deleterious mutations during adaptation; Leigh [81] found an optimal mutation rate that balances the generation of deleterious mutations and maintenance of standing variation in a fluctuating environment; Komarova & Wodarz [82] found an optimal rate of chromosome loss that balances the unmasking of recessive alleles and genetic load during carcinogenesis; Komarova et al. [83] and Agur et al. [84] found a time-dependent mutation rate strategy that optimizes carcinogenesis and adaptive immune response, respectively. By contrast, we find that SIM breaks, rather than balances, the trade-off between adaptability and adaptedness: it allows individuals to switch between rates optimized for stressful and benign conditions according to the circumstances.
Mutators have been suggested to play a role in cancer [85–87]. Furthermore, there is evidence that cancer cells increase their mutation rate in response to stresses such as hypoxia [88,89]. Our results suggest that such increases can have an important effect on the emergence of drug resistance, progression and metastasis of tumours [87,90].
Our model of complex adaptation on rugged fitness landscapes is similar to that of Weinreich & Chao [67], but our model includes various mutational strategies and the effects of stress and deleterious mutations. Our results (figure 2) suggest that SIM can help resolve the problem of fitness valley crossing by reducing the time required for a population to shift an adaptive peak.
Our results provide theoretical basis to the conjecture that SIM facilitates adaptation. This conjecture can be tested experimentally; for example, with E. coli, where it is possible to interfere with the regulation of mutagenesis [34]. The adaptation time with and without SIM can be measured in an experimental population adapting on a two-peak fitness landscape [91]. These measurements can then be compared to our analytic approximations to determine the relative advantage and disadvantage of the different mutational strategies.
5. Conclusion
SIM has been implicated as a driver of adaptive evolution for several decades. We provide theoretical treatment of this concept. Our results show that SIM increases the rate of complex adaptation and that in contrast to CM it does not jeopardize the fitness of populations under stable conditions. Because mutation is a fundamental force in every biological system, these results have important implications for many fields in the medical and life sciences, including epidemiology, oncology, ecology and evolutionary biology.
Supplementary Material
Supplementary Material
Acknowledgements
We thank U. Obolski for advice on statistical analysis. We are grateful to A. F. Agrawal, T. F. Cooper and two anonymous reviewers for helpful comments on an earlier version of the manuscript.
Data accessibility
The simulation data are deposited on Dryad (doi:10.5061/dryad.3066j). The IPython [92] code used to produce the figures is deposited as electronic supplementary material.
Funding statement
This research has been supported in part by the Israeli Science Foundation 1568/13 (L.H.) and by Marie Curie reintegration grant no. 2007–224866 (L.H.).
References
- 1.Sniegowski PD, Gerrish PJ, Johnson T, Shaver A. 2000. The evolution of mutation rates: separating causes from consequences. BioEssays 22, 1057–1066. () [DOI] [PubMed] [Google Scholar]
- 2.Denamur E, Matic I. 2006. Evolution of mutation rates in bacteria. Mol. Microbiol. 60, 820–827. ( 10.1111/j.1365-2958.2006.05150.x) [DOI] [PubMed] [Google Scholar]
- 3.De Visser JAGM. 2002. The fate of microbial mutators. Microbiology 148, 1247–1252. [DOI] [PubMed] [Google Scholar]
- 4.Funchain P, Yeung A, Stewart JL, Lin R, Slupska MM, Miller JH. 2000. The consequences of growth of a mutator strain of Escherichia coli as measured by loss of function among multiple gene targets and loss of fitness. Genetics 154, 959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montanari S, et al. 2007. Biological cost of hypermutation in Pseudomonas aeruginosa strains from patients with cystic fibrosis. Microbiology 153, 1445–1454. ( 10.1099/mic.0.2006/003400-0) [DOI] [PubMed] [Google Scholar]
- 6.Kimura M. 1967. On the evolutionary adjustment of spontaneous mutation rates. Genet. Res. 9, 23–34. ( 10.1017/S0016672300010284) [DOI] [Google Scholar]
- 7.Liberman U, Feldman MW. 1986. Modifiers of mutation rate: a general reduction principle. Theor. Popul. Biol. 30, 125–142. ( 10.1016/0040-5809(86)90028-6) [DOI] [PubMed] [Google Scholar]
- 8.Giraud A, Radman M, Matic I, Taddei F. 2001. The rise and fall of mutator bacteria. Curr. Opin. Microbiol. 4, 582–585. ( 10.1016/S1369-5274(00)00254-X) [DOI] [PubMed] [Google Scholar]
- 9.Sniegowski PD, Gerrish PJ, Lenski RE. 1997. Evolution of high mutation rates in experimental populations of E. coli. Nature 387, 703–705. ( 10.1038/42701) [DOI] [PubMed] [Google Scholar]
- 10.Wielgoss S, et al. 2012. Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load. Proc. Natl. Acad. Sci. USA 110, 222–227. ( 10.1073/pnas.1219574110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taddei F, Radman M, Maynard Smith J, Toupance B, Gouyon P-H, Godelle B. 1997. Role of mutator alleles in adaptive evolution. Nature 387, 700–702. ( 10.1038/42696) [DOI] [PubMed] [Google Scholar]
- 12.Kessler D, Levine H. 1998. Mutator dynamics on a smooth evolutionary landscape. Phys. Rev. Lett. 80, 2012–2015. ( 10.1103/PhysRevLett.80.2012) [DOI] [Google Scholar]
- 13.Tenaillon O, Toupance B, Le Nagard H, Taddei F, Godelle B. 1999. Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics 152, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leigh EGJ. 1970. Natural selection and mutability. Am. Nat. 104, 301–305. ( 10.1086/282663) [DOI] [Google Scholar]
- 15.Galhardo RS, Hastings PJ, Rosenberg SM. 2007. Mutation as a stress response and the regulation of evolvability. Crit. Rev. Biochem. Mol. Biol. 42, 399–435. ( 10.1080/10409230701648502) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster PL. 2007. Stress-induced mutagenesis in bacteria. Crit. Rev. Biochem. Mol. Biol. 42, 373–397. ( 10.1080/10409230701648494) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenberg SM, Shee C, Frisch RL, Hastings PJ. 2012. Stress-induced mutation via DNA breaks in Escherichia coli: a molecular mechanism with implications for evolution and medicine. BioEssays 34, 885–892. ( 10.1002/bies.201200050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bjedov I, Tenaillon O, Gérard B, Souza V, Denamur E, Radman M, Taddei F, Matic I. 2003. Stress-induced mutagenesis in bacteria. Science 300, 1404–1409. ( 10.1126/science.1082240) [DOI] [PubMed] [Google Scholar]
- 19.Katz S, Hershberg R. 2013. Elevated mutagenesis does not explain the increased frequency of antibiotic resistant mutants in starved aging colonies. PLoS Genet. 9, e1003968 ( 10.1371/journal.pgen.1003968) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kivisaar M. 2010. Mechanisms of stationary-phase mutagenesis in bacteria: mutational processes in pseudomonads. FEMS Microbiol. Lett. 312, 1–14. ( 10.1111/j.1574-6968.2010.02027.x) [DOI] [PubMed] [Google Scholar]
- 21.Kang JM, Iovine NM, Blaser MJ. 2006. A paradigm for direct stress-induced mutation in prokaryotes. FASEB J. 20, 2476–2485. ( 10.1096/fj.06-6209com) [DOI] [PubMed] [Google Scholar]
- 22.Baharoglu Z, Mazel D. 2011. Vibrio cholerae triggers SOS and mutagenesis in response to a wide range of antibiotics: a route towards multiresistance. Antimicrob. Agents Chemother. 55, 2438–2441. ( 10.1128/AAC.01549-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henderson-Begg SK, Livermore DM, Hall LMC. 2006. Effect of subinhibitory concentrations of antibiotics on mutation frequency in Streptococcus pneumoniae. J. Antimicrob. Chemother. 57, 849–854. ( 10.1093/jac/dkl064) [DOI] [PubMed] [Google Scholar]
- 24.Heidenreich E. 2007. Adaptive mutation in Saccharomyces cerevisiae. Crit. Rev. Biochem. Mol. Biol. 42, 285–311. ( 10.1080/10409230701507773) [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez GP, Romanova NV, Bao G, Rouf NC, Kow YW, Crouse GF. 2012. Mismatch repair-dependent mutagenesis in nondividing cells. Proc. Natl Acad. Sci. USA 109, 6153–6158. ( 10.1073/pnas.1115361109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goho S, Bell G. 2000. Mild environmental stress elicits mutations affecting fitness in Chlamydomonas. Proc. R. Soc. Lond. B 267, 123–129. ( 10.1098/rspb.2000.0976) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuba C, Ostrow DG, Salomon MP, Tolani A, Baer CF. 2012. Temperature, stress and spontaneous mutation in Caenorhabditis briggsae and Caenorhabditis elegans. Biol. Lett. 9, 20120334 ( 10.1098/rsbl.2012.0334) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharp NP, Agrawal AF. 2012. Evidence for elevated mutation rates in low-quality genotypes. Proc. Natl Acad. Sci. USA 109, 6142–6146. ( 10.1073/pnas.1118918109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bristow RG, Hill RP. 2008. Hypoxia and metabolism: hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 8, 180–192. ( 10.1038/nrc2344) [DOI] [PubMed] [Google Scholar]
- 30.Ponder RG, Fonville NC, Rosenberg SM. 2005. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol. Cell 19, 791–804. ( 10.1016/j.molcel.2005.07.025) [DOI] [PubMed] [Google Scholar]
- 31.Debora BN, Vidales LE, Ramírez R, Ramírez M, Robleto EA, Yasbin RE, Pedraza-Reyes M. 2010. Mismatch repair modulation of MutY activity drives Bacillus subtilis stationary-phase mutagenesis. J. Bacteriol. 193, 236–245. ( 10.1128/JB.00940-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Al Mamun AAM, et al. 2012. Identity and function of a large gene network underlying mutagenic repair of DNA breaks. Science 338, 1344–1348. ( 10.1126/science.1226683) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tenaillon O, Denamur E, Matic I. 2004. Evolutionary significance of stress-induced mutagenesis in bacteria. Trends Microbiol. 12, 264–270. ( 10.1016/j.tim.2004.04.002) [DOI] [PubMed] [Google Scholar]
- 34.Cirz RT, Romesberg FE. 2007. Controlling mutation: intervening in evolution as a therapeutic strategy. Crit. Rev. Biochem. Mol. Biol. 42, 341–354. ( 10.1080/10409230701597741) [DOI] [PubMed] [Google Scholar]
- 35.Agrawal AF. 2002. Genetic loads under fitness-dependent mutation rates. J. Evol. Biol. 15, 1004–1010. ( 10.1046/j.1420-9101.2002.00464.x) [DOI] [Google Scholar]
- 36.Shaw FH, Baer CF. 2011. Fitness-dependent mutation rates in finite populations. J. Evol. Biol. 24, 1677–1684. ( 10.1111/j.1420-9101.2011.02320.x) [DOI] [PubMed] [Google Scholar]
- 37.Ram Y, Hadany L. 2012. The evolution of stress-induced hypermutation in asexual populations. Evolution 66, 2315–2328. ( 10.1111/j.1558-5646.2012.01576.x) [DOI] [PubMed] [Google Scholar]
- 38.Wright S. 1931. Evolution in Mendelian populations. Genetics 16, 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wright S. 1988. Surfaces of selective value revisited. Am. Nat. 131, 115–123. ( 10.1086/284777) [DOI] [Google Scholar]
- 40.Crow JF, Engels WR, Denniston C. 1990. Phase three of Wright's shifting-balance theory. Evolution 44, 233 ( 10.2307/2409403) [DOI] [PubMed] [Google Scholar]
- 41.Wade MJ, Goodnight CJ. 1991. Wright's shifting balance theory: an experimental study. Science 253, 1015–1018. ( 10.1126/science.1887214) [DOI] [PubMed] [Google Scholar]
- 42.Peck SL, Ellner SP, Gould F. 2000. Varying migration and deme size and the feasibility of the shifting balance. Evolution 54, 324–327. ( 10.1111/j.0014-3820.2000.tb00035.x) [DOI] [PubMed] [Google Scholar]
- 43.Moore FB-G, Tonsor SJ. 1994. A simulation of Wright's shifting-balance process: migration and the three phases. Evolution 48, 69–80. ( 10.2307/2410004) [DOI] [PubMed] [Google Scholar]
- 44.Gavrilets S. 1996. On phase three of the shifting-balance theory. Evolution 50, 1034–1041. ( 10.2307/2410644) [DOI] [PubMed] [Google Scholar]
- 45.Coyne JA, Barton NH, Turelli M. 2000. Is Wright's shifting balance process important in evolution? Evolution 54, 306–317. ( 10.1111/j.0014-3820.2000.tb00033.x) [DOI] [PubMed] [Google Scholar]
- 46.Whitlock MC, Phillips PC. 2000. The exquisite corpse: a shifting view of the shifting balance. Trends Ecol. Evol. 15, 347–348. ( 10.1016/S0169-5347(00)01930-3) [DOI] [PubMed] [Google Scholar]
- 47.Whitlock MC. 1995. Variance-induced peak shifts. Evolution 49, 252–259. ( 10.2307/2410335) [DOI] [PubMed] [Google Scholar]
- 48.Whitlock MC. 1997. Founder effects and peak shifts without genetic drift: adaptive peak shifts occur easily when environments fluctuate slightly. Evolution 51, 1044–1048. ( 10.2307/2411033) [DOI] [PubMed] [Google Scholar]
- 49.Hadany L. 2003. Adaptive peak shifts in a heterogenous environment. Theor. Popul. Biol. 63, 41–51. ( 10.1016/S0040-5809(02)00011-4) [DOI] [PubMed] [Google Scholar]
- 50.Hadany L, Beker T. 2003. Fitness-associated recombination on rugged adaptive landscapes. J. Evol. Biol. 16, 862–870. ( 10.1046/j.1420-9101.2003.00586.x) [DOI] [PubMed] [Google Scholar]
- 51.Weissman DB, Desai MM, Fisher DS, Feldman MW. 2009. The rate at which asexual populations cross fitness valleys. Theor. Popul. Biol. 75, 286–300. ( 10.1016/j.tpb.2009.02.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weissman DB, Feldman MW, Fisher DS. 2010. The rate of fitness-valley crossing in sexual populations. Genetics 186, 1389–1410. ( 10.1534/genetics.110.123240) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gibson JL, et al. 2010. The σE stress response is required for stress-induced mutation and amplification in Escherichia coli. Mol. Microbiol. 77, 415–430. ( 10.1111/j.1365-2958.2010.07213.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haigh J. 1978. The accumulation of deleterious genes in a population: Muller's Ratchet. Theor. Popul. Biol. 14, 251–267. ( 10.1016/0040-5809(78)90027-8) [DOI] [PubMed] [Google Scholar]
- 55.Gessler DDG. 1995. The constraints of finite size in asexual populations and the rate of the ratchet. Genet. Res. 66, 241 ( 10.1017/S0016672300034686) [DOI] [PubMed] [Google Scholar]
- 56.Gordo I, Dionisio F. 2005. Nonequilibrium model for estimating parameters of deleterious mutations. Phys. Rev. E 71, 18–21. ( 10.1103/PhysRevE.71.031907) [DOI] [PubMed] [Google Scholar]
- 57.Kibota TT, Lynch M. 1996. Estimate of the genomic mutation rate deleterious to overall fitness in E. coli. Nature 381, 694–696. ( 10.1038/381694a0) [DOI] [PubMed] [Google Scholar]
- 58.Gordo I, Perfeito L, Sousa A. 2011. Fitness effects of mutations in bacteria. J. Mol. Microbiol. Biotechnol. 21, 20–35. ( 10.1159/000332747) [DOI] [PubMed] [Google Scholar]
- 59.Drake JW, Charlesworth B, Charlesworth D, Crow JF. 1998. Rates of spontaneous mutation. Genetics 148, 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wielgoss S, Barrick JE, Tenaillon O, Cruveiller S, Chane-Woon-Ming B, Médigue C, Lenski RE, Schneider D. 2011. Mutation rate inferred from synonymous substitutions in a long-term evolution experiment with Escherichia coli. Genes Genome Genet. 1, 183–186. ( 10.1534/g3.111.000406) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hall LMC, Henderson-Begg SK. 2006. Hypermutable bacteria isolated from humans: a critical analysis. Microbiology 152, 2505–2514. ( 10.1099/mic.0.29079-0) [DOI] [PubMed] [Google Scholar]
- 62.Pupo GM, Richardson BJ. 1995. Biochemical genetics of a natural population of Escherichia coli: seasonal changes in alleles and haplotypes. Microbiology 141, 1037–1044. ( 10.1099/13500872-141-4-1037) [DOI] [PubMed] [Google Scholar]
- 63.Berg OG. 1996. Selection intensity for codon bias and the effective population size of Escherichia coli. Genetics 142, 1379–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eshel I. 1981. On the survival probability of a slightly advantageous mutant gene with a general distribution of progeny size: a branching process model. J. Math. Biol. 12, 355–362. ( 10.1007/BF00276922) [DOI] [PubMed] [Google Scholar]
- 65.Fisher RA. 1930. The genetical theory of natural selection. Oxford, UK: Clarendon Press. [Google Scholar]
- 66.Patwa Z, Wahl LM. 2008. The fixation probability of beneficial mutations. J. R. Soc. Interface 5, 1279–1289. ( 10.1098/rsif.2008.0248) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weinreich DM, Chao L. 2005. Rapid evolutionary escape by large populations from local fitness peaks is likely in nature. Evolution 59, 1175–1182. ( 10.1111/j.0014-3820.2005.tb01769.x) [DOI] [PubMed] [Google Scholar]
- 68.Maynard Smith J, Haigh J. 1974. The hitch-hiking effect of a favourable gene. Genet. Res. 23, 23–35. ( 10.1017/S0016672300014634) [DOI] [PubMed] [Google Scholar]
- 69.Lynch M, Bürger R, Butcher D, Gabriel W. 1993. The mutational meltdown in asexual populations. J. Hered. 84, 339–344. [DOI] [PubMed] [Google Scholar]
- 70.Obolski U, Hadany L. 2012. Implications of stress-induced genetic variation for minimizing multidrug resistance in bacteria. BMC Med. 10, 1–30. ( 10.1186/1741-7015-10-89) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shor E, Fox CA, Broach JR. 2013. The yeast environmental stress response regulates mutagenesis induced by proteotoxic stress. PLoS Genet. 9, e1003680 ( 10.1371/journal.pgen.1003680) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karpinets T, Greenwood D, Pogribny I, Samatova N. 2006. Bacterial stationary-state mutagenesis and mammalian tumorigenesis as stress-induced cellular adaptations and the role of epigenetics. Curr. Genomics 7, 481–496. ( 10.2174/138920206779315764) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Torres-Barceló C, Cabot G, Oliver A, Buckling A, MacLean RC. 2013. A trade-off between oxidative stress resistance and DNA repair plays a role in the evolution of elevated mutation rates in bacteria. Proc. R. Soc. B 280, 20130007 ( 10.1098/rspb.2013.0007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dawson KJ. 1998. Evolutionarily stable mutation rates. J. Theor. Biol. 194, 143–157. ( 10.1006/jtbi.1998.0752) [DOI] [PubMed] [Google Scholar]
- 75.Giraud A, Matic I, Tenaillon O, Clara A, Radman M, Fons M, Taddei F. 2001. Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science 291, 2606–2608. ( 10.1126/science.1056421) [DOI] [PubMed] [Google Scholar]
- 76.Loh E, Salk JJ, Loeb LA. 2010. Optimization of DNA polymerase mutation rates during bacterial evolution. Proc. Natl Acad. Sci. USA 107, 1154–1159. ( 10.1073/pnas.0912451107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gentile CF, Yu S-C, Serrano SA, Gerrish PJ, Sniegowski PD. 2011. Competition between high- and higher-mutating strains of Escherichia coli. Biol. Lett. 7, 422–424. ( 10.1098/rsbl.2010.1036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shee C, Gibson JL, Darrow MC, Gonzalez C, Rosenberg SM. 2011. Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc. Natl Acad. Sci. USA 108, 13 659–13 664. ( 10.1073/pnas.1104681108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tenaillon O, Le Nagard H, Godelle B, Taddei F. 2000. Mutators and sex in bacteria: conflict between adaptive strategies. Proc. Natl Acad. Sci. USA 97, 10 465–10 470. ( 10.1073/pnas.180063397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Johnson T, Barton NH. 2002. The effect of deleterious alleles on adaptation in asexual populations. Genetics 162, 395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leigh EGJ. 1973. The evolution of mutation rates. Genetics 73(Suppl. 73), 1–18. [PubMed] [Google Scholar]
- 82.Komarova NL, Wodarz D. 2004. The optimal rate of chromosome loss for the inactivation of tumor suppressor genes in cancer. Proc. Natl Acad. Sci. USA 101, 7017–7021. ( 10.1073/pnas.0401943101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Komarova NL, Sadovsky AV, Wan FYM. 2008. Selective pressures for and against genetic instability in cancer: a time-dependent problem. J. R. Soc. Interface 5, 105–121. ( 10.1098/rsif.2007.1054) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Agur Z, Mazor G, Meilijson I. 1991. Maturation of the humoral immune response as an optimization problem. Proc. R. Soc. Lond. B 245, 147–150. ( 10.1098/rspb.1991.0101) [DOI] [PubMed] [Google Scholar]
- 85.Jackson AL, Loeb LA. 1998. The mutation rate and cancer. Genetics 148, 1483–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bielas JH, Loeb KR, Rubin BP, True LD, Loeb LA. 2006. Human cancers express a mutator phenotype. Proc. Natl Acad. Sci. USA 103, 18 238–18 242. ( 10.1073/pnas.0607057103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lambert G, Estévez-Salmeron L, Oh S, Liao D, Emerson BM, Tlsty TD, Austin RH. 2011. An analogy between the evolution of drug resistance in bacterial communities and malignant tissues. Nat. Rev. Cancer 11, 375–382. ( 10.1038/nrc3039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ruan K, Song G, Ouyang G. 2009. Role of hypoxia in the hallmarks of human cancer. J. Cell. Biochem. 107, 1053–1062. ( 10.1002/jcb.22214) [DOI] [PubMed] [Google Scholar]
- 89.Bindra RS, Crosby ME, Glazer PM. 2007. Regulation of DNA repair in hypoxic cancer cells. Cancer Metastasis Rev. 26, 249–260. ( 10.1007/s10555-007-9061-3) [DOI] [PubMed] [Google Scholar]
- 90.Podlaha O, Riester M, De S, Michor F. 2012. Evolution of the cancer genome. Trends Genet. 28, 155–163. ( 10.1016/j.tig.2012.01.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schrag SJ, Perrot V, Levin BR. 1997. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc. R. Soc. Lond. B 264, 1287–1291. ( 10.1098/rspb.1997.0178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perez F, Granger BE. 2007. IPython: a system for interactive scientific computing. Comput. Sci. Eng. 9, 21–29. ( 10.1109/MCSE.2007.53) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The simulation data are deposited on Dryad (doi:10.5061/dryad.3066j). The IPython [92] code used to produce the figures is deposited as electronic supplementary material.