

Abstract
Genome-wide investigations have dramatically increased our understanding of nucleosome positioning and the role of chromatin in gene regulation, yet some genomic regions have been poorly represented in human nucleosome maps. One such region is represented by human chromosome 9p21–22, which contains the type I interferon gene cluster that includes 16 interferon alpha genes and the single interferon beta, interferon epsilon, and interferon omega genes. A high-density nucleosome mapping strategy was used to generate locus-wide maps of the nucleosome organization of this biomedically important locus at a steady state and during a time course of infection with Sendai virus, an inducer of interferon gene expression. Detailed statistical and computational analysis illustrates that nucleosomes in this locus exhibit preferences for particular dinucleotide and oligomer DNA sequence motifs in vivo, which are similar to those reported for lower eukaryotic nucleosome–DNA interactions. These data were used to visualize the region's chromatin architecture and reveal features that are common to the organization of all the type I interferon genes, indicating a common nucleosome-mediated gene regulatory paradigm. Additionally, this study clarifies aspects of the dynamic changes that occur with the nucleosome occupying the transcriptional start site of the interferon beta gene after virus infection.
Introduction
Eukaryotic genomic DNA is packaged by nucleosomes to compact the DNA, control DNA replication, and regulate gene expression (Kornberg and Stryer 1988). Genome-wide investigations in human cells and model organisms have recognized fundamental features of nucleosome organization important for gene regulation (Bernstein and others 2004; Yuan and others 2005; Johnson and others 2006; Lee and others 2007; Schones and others 2008; Kaplan and others 2009; Valouev and others 2011; Brogaard and others 2012).
Genome-wide studies of nucleosome positions have been greatly facilitated by advancements in next-generation sequencing coupled with powerful statistical and computational analysis tools for mapping nucleosomes to reference genomes (Zhang and Pugh 2011). These approaches have been able to provide nucleosome positioning information of at least moderate coverage for entire model organism genomes and much of the human genome, yet some regions, especially those with low complexity and genetic redundancy, have been poorly represented in human nucleosome occupancy maps. Moreover, as the expression of many biomedically relevant genes is rapidly and transiently induced in response to specific extracellular or intracellular stimuli, little information is available on specific nucleosome-level dynamic changes that may accompany the expression of acutely inducible genes, gene families, or gene regulatory networks.
Most transcribed genes contain a nucleosome-depleted region (NDR) at their 5′ promoter and 3′ transcriptional end site, while some quiescent genes have regulatory nucleosomes positioned directly over their promoter preventing polymerase access to the transcription start site (TSS). Genomic-scale examination of chromatin architecture revealed that both underlying DNA sequence preferences and ATP-dependent chromatin remodeling processes guide nucleosomes to their proper positions where they can alter DNA accessibility to transcription factors and positively or negatively influence gene expression (Kornberg and Lorch 1999; Lomvardas and Thanos 2002; Richmond and Davey 2003; Whitehouse and others 2007; Badis and others 2008; Hartley and Madhani 2009; Kaplan and others 2009; Zhang and others 2011; Brogaard and others 2012; Yen and others 2012).
Interferon (IFN) refers to a group of cytokines that were initially characterized as agents that interfere with virus replication, but are now recognized as a diverse class of related and unrelated anti-microbial and immune regulatory signaling molecules (Isaacs and Lindenmann 1957; Borden and others 2007). The human type I IFN genes are clustered within ∼450 kb on the short arm of human chromosome 9 (Fig. 1A) (Diaz and others 1994; Genin and others 2009). The 16 members of this cluster are the IFNA genes (numbered A1, A2, A4, A5, A6, A7, A8, A10, A13, A14, A16, A17, and A21) and the single IFNB1, IFNE, and IFNW1 genes, which are 53%–97% identical at the nucleotide level. All the type I IFN genes are transcriptionally inactive unless induced by innate immune signaling systems downstream of acute virus infections, and they encode intronless mRNAs that produce related IFN proteins that are >20% identical, yet differ in their tissue-specific expression and signal transduction potency (Thomas and others 2011). The difficulty in accurately mapping short single-ended sequence reads to regions of significant genetic similarity, and the absence of an appropriate activating stimulus, such as virus infection, leaves the IFN cluster poorly represented in genome-wide nucleosome maps.
FIG. 1.

Virus-induced expression of the type I IFN gene cluster. (A) Illustration of the human chromosome on 9p21–22 depicting ∼450-kb type I IFN gene cluster. The type I IFN genes and KLHL9, the sole non-IFN gene in the region, are indicated. (B) Type I IFN gene induction by Sendai virus infection in Namalwa cells. Cells were mock-infected or infected (5 pfu/cell), and 5×106 cells were harvested at each time point for RNA isolation, and RT-PCR was performed with IFN gene-specific primer pairs as indicated or GAPDH for normalization. Error bars denote standard deviation for triplicate PCRs. See also Supplementary Fig. S2. (Supplementary Data are available online at www.liebertpub.com/jir).
One of the type I IFN genes, encoding IFNB1, is among the best-characterized inducible genes in higher eukaryotes and provides an excellent model of virus-inducible transcription regulation. Accumulated evidence has shown that at steady state the IFNB1 promoter contains a (+1) nucleosome positioned over the TSS, obscuring access to the TATA box, adjacent to an NDR that harbors positive regulatory domains (Zinn and others 1983; Goodbourn and others 1985). Coordinated and cooperative actions of virus-induced transcription factors, including ATF2/c-jun, NFκB, IRF3, and IRF7, recruit transcriptional co-activators, including histone-modifying enzymes CBP, p300, and GCN5, and chromatin remodeling machines Swi/Snf and BRG1, to the NDR. These co-activators mediate reorganization of the (+1) nucleosome, which has been inferred to “slide” to a position 36 nucleotides downstream in response to virus infection (Lomvardas and Thanos 2001). This nucleosome remodeling allows the de novo assembly of the RNA polymerase II (Pol II) holoenzyme at the TSS (Agalioti and others 2000; Lomvardas and Thanos 2002; Ford and Thanos 2010). The transcriptional regulation of IFNB1 biosynthesis underscores the importance of local chromatin architecture and dynamics in regulating gene expression in the innate antiviral response. The nucleosome positions of the remaining type I IFN family members have not been described.
To investigate the organization of this biomedically important cytokine gene cluster, we generated a high-density nucleosome profile at steady state and during a time course of Sendai virus infection. The resulting nucleosome maps provide a visualization of chromatin architecture in this locus and reveal that nucleosome obstruction of the TSS to be a conserved feature of type I IFN promoters. Moreover, these maps were of sufficient depth to reveal that well-positioned nucleosomes in this locus reflect particular dinucleotide and oligomer DNA sequence motif-binding preferences observed in lower eukaryotes and model organisms.
Materials and Methods
Cell culture and virus infection
Human Namalwa cells (ATCC CRL-1432) were cultured in RPMI 1640 medium (Life Technologies) containing 10% cosmic calf serum (Hyclone), glutamine, 1 mM sodium pyruvate (Cellgro), and 1% penicillin/streptomycin (Life Technologies). Infection of cells with Sendai virus (Cantell strain, Sendai virus, 3×108 pfu/mL) was performed at 5 plaque-forming units per cell (pfu/cell) in serum-free media. After 1 h, cells were lightly spun down, washed, placed in growth media supplemented with 2% CCS, and, finally, harvested at different time points—up to 20 h—following infection.
Expression analysis
Namalwa cells were infected with Sendai virus (5 pfu/cell) and 5×106 cells were harvested at each time point. Total RNA was extracted using TRIzol Reagent (Invitrogen). Samples were treated with DNase I (Life Technologies), and 1 μg of RNA was subjected to reverse transcriptase Superscript III (Life Technologies) for cDNA synthesis. Quantitative real-time PCR was carried out using the MX3000P SYBR Green real-time PCR system (Agilent). Expression was normalized against GAPDH and displayed as fold change over mock conditions. The oligos used for this analysis are listed in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/jir).
Isolation of mononucleosome core DNA fragments
To isolate mononucleosome core DNA, a crude nuclear fraction was separated by washing 6×107 cells 3 times with MC lysis buffer [10 mM Tris-HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2, and 0.5% (v/v) Igepal] at 4°C. Genomic DNA in MNase reaction buffer [10 mM Tris-HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2, 1 mM CaCl2, 4% (v/v) Igepal, and 1 mM PMSF] was digested with 25 units of MNase (USB; 0.025 U/μL final concentration) for 10 min at 37°C. The digestion was stopped by the addition of EDTA, followed by phenol/chloroform extraction and ethanol precipitation. The digested DNA was run on a 3.3% agarose gel (Lonza) and the smear of DNA fragments from ∼135 to 170 bp was excised from the gel. The gel slabs were sliced into small pieces and diluted in crush-and-soak buffer (300 mM sodium acetate, 1 mM EDTA pH 8.0, 0.1% SDS). Once in solution, the agarose pieces were rotated gently at room temperature for 48 h. Subsequently, the crush-and-soak buffer was collected and filtered with an Amicon Ultra-15 filtration device (Millipore). The DNA was concentrated and filtrated with Amicon Ultra Centrifugal Filters (Millipore) and cleaned up using a QIAquick PCR Kit (Qiagen) following the manufacturer's protocol with minor modifications.
Preparation of mapped DNA fragments for ABI SOLiD sequencing
The ends of isolated mononucleosomal-length DNAs were processed using the DNATerminator Repair Kit (Lucigen) according to the manufacturer's protocol. Following blunt-ending, DNA was purified using a QIAquick PCR Kit (Qiagen) as noted above. ABI's double-stranded P1 and P2 adaptors (for SOLiD 4 System) were ligated to the end-repaired DNA with Quick Ligase (NEB) during a 5-min reaction at room temperature. After adaptor ligation, DNA was purified using a QIAquick PCR Kit (Qiagen) as noted previously. The ligation reactions were separated on a 6% native polyacrylamide gel, and the relevant bands were isolated and purified as previously described.
Library amplification
The eluted adapter-ligated libraries were amplified with 17–18 cycles of PCR using the SOLiD P1 and SOLiD P2 primer. The number of cycles used in the PCR amplification was monitored and selected as described (Gu and Fire 2010). In summary, a series of PCRs with increasing cycle numbers were carried out, and cycle numbers were carefully chosen for which product levels have not saturated (i.e., product levels are still able to increase substantially with additional cycles). This ensured that the majority of amplified segments were still annealed to a true complement and avoided reannealing distortion in the resulting sequence libraries (Parameswaran and others 2007). PCR products were separated and purified on a 2% agarose gel (Lonza), eluted in the crush-and-soak buffer, and purified with the QIAquick PCR kit as described above.
DNA sequencing and mapping
Nucleosome-protected DNA libraries were paired-end sequenced using ABI's SOLiD next-generation sequencing system to produce 35-bp F3 reads and 25-bp F5-P2 reads. All sequence data were mapped using the SOLiD software pipeline against the human hg19 assembly using the first 30 bp from each F3 read and all 25 bp of the F5-P2 reads. This was done to maximize the number of reference-mapped reads as the quality of base calling dropped off farther into the read. For all downstream analysis, only uniquely mapped reads were used to avoid ambiguity.
Bacterial artificial chromosome labeling and hybridization
Essentially, bacterial artificial chromosome (BAC) DNA labeling was performed as described previously (Bashiardes and others 2005). In summary, the BAC DNA used (∼150 ng) for the enrichment procedure was isolated using a NucleoBond BAC 100 kit (Clontech) following the manufacturer's protocol. To incorporate biotinylated residues into the BAC DNA, a nick translation kit (Roche; according to manufacturer's protocol), biotin-dUTP (Enzo), and (A-32P) dCTP were used. The isotope was included in a parallel reaction as a tracer to confirm that the biotinylation reaction proceeded efficiently and to confirm binding of the BAC DNA to streptavidin-coated magnetic beads.
The biotinylated products were purified through a Sephadex G-50 spin column (GE) and the resulting solution was lyophilized to dryness. The biotin-labeled DNA pellet was then dissolved in Cot-1 DNA (2 μg/μL; Invitrogen) and transferred to a 200-μL PCR tube. The human genome is composed of many repetitive elements, and the use of Cot-1 DNA (placental DNA that is predominantly 50–300 bp in size and enriched for repetitive DNA sequences such as the Alu and Kpn family members) drastically reduces the number of these nonspecific DNA fragments from being hybridized and subsequently sequenced.
The solution was overlayed with mineral oil (Sigma), denatured by incubation at 95°C for 5 min, and incubated at 65°C for 15 min. About 5 μL of 2× hybridization buffer (1.5 M NaCl, 40 mM sodium phosphate buffer pH 7.2, 10 mM EDTA pH 8, 10× Denhardt's, 0.2% SDS) was added and incubated further at 65°C for 6 h.
To select and capture the hybrids, 2 μg of adapter-ligated genomic DNA was delivered in 5 μL of water into a 200-μL PCR tube and overlayed with mineral oil (Sigma). The DNA was denatured at 95°C for 5 min and incubated at 65°C for 15 min. About 5 μL of 2× hybridization buffer was added, and then the entire sample was transferred to the tube containing the Cot-1-blocked BAC DNA. Following transfer, the hybridization reaction was incubated at 65°C for a further 72 h. In a microcentrifuge tube, 100 μL of streptavidin-coated beads (Invitrogen) was washed twice in 200 μL Streptavidin bead binding buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA pH 8, and 1 M NaCl). The beads were resuspended in 150 μL Streptavidin bead-binding buffer, and then the hybridization buffer was added to the bead suspension. Binding was carried out at 25°C for 30 min with periodic gentle mixing to ensure that the beads remained evenly suspended. Beads were removed from the binding buffer and washed once, at 25°C for 15 min, in 1 mL of 1× SSC with 0.1% SDS, and then 3 times, each at 65°C for 15 min, in 1 mL 0.1× SSC with 0.1% SDS. The genomic DNA hybridized to the BAC was eluted with the addition of 100 μL of 0.1 M NaOH to the beads and incubation at 25°C for 10 min. The beads were removed from the elution mixture, and then the supernatant was transferred to a fresh microcentrifuge tube containing 100 μL of 1 M Tris-HCl (pH 7.5) and desalted through a Sephadex G-50 resin.
Nucleosome data analysis
The nucleosome DNA sequences from MNase mapping tend to have large variability in length (typically range from 100 to 200 bp) because of MNase specificity and partial unwrapping of nucleosomes. The variability in the length can be problematic when trying to identify the precise nucleosome centers, causing nucleosome boundaries to be poorly defined. Reads lengths between 127 and 167 bp were used to construct the nucleosome occupancy maps to achieve better accuracy in defining nucleosome positions. In addition, a center-weighted nucleosome occupancy curve was constructed as follows: If a nucleosome DNA sequence is of odd length, a Gaussian weight of 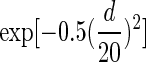 is assigned to a position d bp away from the center of the sequence for d≤73. If a sequence length is even, then the central 2 positions will be treated as the center in turn to assign a weight
is assigned to a position d bp away from the center of the sequence for d≤73. If a sequence length is even, then the central 2 positions will be treated as the center in turn to assign a weight 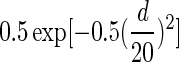 for a position d bp away from the center (Brogaard and others 2012).
for a position d bp away from the center (Brogaard and others 2012).
Density dot plot
For each data set, the nucleosome's occupancy is based on 127–167-bp fragments. For each fragment, nucleotides ±63 bp from the fragment center were given an even score (i.e., 1). The occupancy scores, therefore, show how many fragments covered each position. Only the core 127 bp of each fragment was considered to avoid biases in nucleotide distributions that may arise from the sequence specificity of the MNase used to isolate the nucleosome. This way we do not include statistics that are taken from the cut site of the nuclease.
Nucleosome dyad coordinates for rotational positioning analysis
The peaks selected represent the approximate nucleosome centers. However, with the well-understood sequence preferences of MNase, there may be systematic errors. If we aligned DNAs solely from these peak positions, the preferential dinucleotide and oligomer signals would either become negligible or even completely dissolve. To achieve better signal, we effectively used a previously developed strategy (Brogaard and others 2012). In short, we examined for a 147-bp sequence nearest to the peak position within a ±10-bp region. If no such sequence existed, we sequentially examined for sequences of lengths 148, 146, 149, 145, and 150 bp within a ±10-bp region until the first sequence was identified. The center of the identified sequence was treated as the nucleosome center to generate the AA/TT/TA/AT and GG/CC/GC/CG frequency plots and the oligomeric and dinucleotide preference k-mer bias plots. If no sequence was identified in ±10-bp region, the peak position was treated as the true nucleosome center to generate the alignment.
Results
Sendai virus infection induces type I IFN gene expression
Nearly every nucleated cell in the human body is able to produce type I IFNs in response to virus infections, but the expression of some type I IFN genes is restricted to specific lymphocytes (Pestka and others 2004). In order to maximize the expression of diverse type I IFN genes, the Namalwa B lymphoid cell line was infected with the Cantell strain of Sendai virus, a well-documented system to study the type I IFN response (Sehgal and others 1982; Strahle and others 2006; Genin and others 2009).
Quantitative RT-PCR (RT-qPCR) analysis revealed the immediate and robust expression of IFNB1, IFNW1, and a set of tested IFNA genes in response to Sendai virus infection (Fig. 1B). All genes examined were induced within 2 h of infection with maximum mRNA abundance observed by 4 h post-infection (h.p.i.) after which the abundance of the mRNAs gradually decreased. These observations confirmed the rapid induction of diverse type I IFN genes in Namalwa cells by Sendai virus infection and established that the type I IFN genes are maximally transcribed before 4 h.p.i. It is noteworthy that while all the type I IFN genes were induced with a similar time course, the IFNβ mRNA was detected at much higher levels, peaking at ≥2 orders of magnitude higher than the other mRNAs.
Nucleosome mapping by direct selection of protected genomic fragments
To determine the nucleosome landscape of the human type I IFN locus before and after Sendai virus infection, nucleosome-protected DNA fragments from this region were specifically and significantly enriched by hybridization, and then subjected to next-generation sequencing (Lovett and others 1991; Bashiardes and others 2005; Yigit and others 2013a, 2013b). Cells (6×107 per condition) were mock-infected or infected with Sendai virus for 1, 2, and 4 h, and then subjected to nuclear fractionation and limited digestion with micrococcal nuclease (MNase) to produce 80%–90% mononucleosome-length DNA. The mono-nucleosome-protected DNA was hybridized to an affinity matrix consisting of type I IFN locus-specific DNA derived from BACs. Three BACs, RP11-1P8 (Hu Chr 9: 21,073,242–21,226,920), RP11-956G20 (Hu Chr 9: 21,207,896–21,411,355), and RP11-158I17 (Hu Chr 9: 21,342,234–21,524,030), were used to span the entire type I IFN locus. The enriched mono-nucleosome DNA was used to prepare libraries for paired-end sequencing using the ABI SOLiD platform (Supplementary Fig. S1).
Analysis of the uniquely mapped sequenced reads verified the quality of each of the sequencing data sets (Fig. 2A) and ensured a significant enrichment of nucleosome-protected DNA fragments for the type I IFN locus. A great percentage of sequence tags (∼61%–85%) mapped to our region of interest (Chr 9: 21,073,242–21,524,030), resulting in an ∼4,200–5,800-fold enrichment, with an average depth of coverage in the target region of 1,850–2,200 tags per nucleotide. Additionally, the paired-end libraries allowed us to precisely determine that >99.9% of the sequence tags have an insert length between 120 and 180 bp (Fig. 2B). Each sample has a similar range and frequency of insert length with a summit observed at ∼150 bp, demonstrating that comparably sized nucleosome cores were purified from each sample.
FIG. 2.

Enrichment and sequencing of nucleosome protected DNA fragments in the type I IFN gene cluster. (A) Statistical analysis of sequence data. See text for details. (B) Plot of the insert length distributions for the nucleosome protected sequences with length between 120 and 180 bp in each sample.
Global nucleosome landscape at the type I IFN gene cluster
To determine if nucleosomes in the IFN locus are positioned with primary sequence preferences, observed nucleosome occupancies were compared with a predictive model (Xi and others 2010) (Supplementary Fig. S2B). A good correlation between the predicted and observed nucleosome occupancy per base pair was observed in all samples (mock infected, r=0.51; 1 h.p.i., r=0.47; 2 h.p.i., r=0.54; and 4 h.p.i., r=0.50).
To systematically measure covariance across the type I IFN locus, the Pearson correlation coefficient (r) was calculated between the different nucleosome profiles. The nucleosome organization of the mock-infected and Sendai virus-infected cells (1, 2, and 4 h) is comparable, although not absolutely indistinguishable (Fig. 3A), with correlations of 0.83, 0.92, and 0.96, respectively, between the nucleosome occupancy per base pair. Additionally, the correlation coefficients are similar when comparing the 1-h-infected to the 2-h- and 4-h-infected maps (r=0.72 and 0.93, respectively) and the 2-h-infected to the 4-h-infected maps (r=0.85). The high degree of similarity between all of the maps indicates that throughout the course of infection, when all type I IFN genes are transcriptionally active, most nucleosomes throughout the ∼450-kb locus remain unaltered. However, in a number of the comparisons with the sample collected at 1 h.p.i., 2 distinguishable nucleosome occupancy profiles appear. This suggests that, at this point after infection, a subset of nucleosomes alters their occupancy profiles, consistent with heterogeneous transcriptional responses among the population of infected cells.
FIG. 3.

Comparison of nucleosome maps reveals distinct chromatin organization in constitutive versus virus-activated genes. (A) Density plots comparing the normalized nucleosome occupancy per base pair in the mock-infected cells versus the infected cells. Color is used to represent the number of base pairs with occupancy scores mapping to that point in the graph. Values above zero indicate nucleosome enrichment relative to the locus-wide average. The Pearson correlation (R) between the maps is indicated. (B) Nucleosome occupancy in the human type I IFN gene cluster. Individual plots of nucleosome occupancy per base pair across the ∼450-kb locus are shown for the mock-infected cells and for the Sendai virus-infected cells at 1, 2, and 4 h.p.i. A scale bar representing the positions of relevant genes is illustrated above. (C) Magnified view of the KLHL9 region. The KLHL9 gene is illustrated as a solid line, the TSS and directionality indicated by an arrow, and the gene body indicated with a box. Each row below depicts nucleosome occupancy per base pair for mock infected and 1, 2, and 4 h SeV infected. Positions of 5′ and 3′ NDRs and +1 and −1 nucleosomes flanking the TSS are indicated. (D) Magnified view of the IFNB1 region. The IFNB1 gene is illustrated as a solid line, the TSS and directionality indicated by an arrow, and the gene body indicated with a box. Each row below depicts nucleosome occupancy per base pair. Positions of 5′ and 3′ NDRs and +1, −1, and −2 nucleosomes flanking the TSS are indicated. (E) Common nucleosome organization in the type I IFN gene promoters. Normalized nucleosome occupancy scores are plotted with the TSS designated “0” for the IFNB1, IFNW1, and 13 IFNA genes. See also Supplementary Fig. S2.
Local nucleosome landscape at the type I IFN gene cluster
Nucleosome arrangements around specific genes in the mapped locus were examined in greater detail. In addition to the virus-induced type I IFN genes, a single constitutively expressed gene, KLHL9, is encoded in this region. KLHL9 mRNA levels remain constant during Sendai virus infection (Supplementary Fig. S2A). The nucleosome occupancy in and around the KLHL9 gene is similar to that observed for most transcribed human genes (Schones and others 2008; Valouev and others 2011); an NDR is observed at the 5′ and 3′ ends of the gene, and well-positioned (+1) and (−1) nucleosomes flank the NDR surrounding the TSS. This nucleosome architecture does not change in response to Sendai virus infection (Fig. 3C).
Comparison of the nucleosome occupancy patterns at the KLHL9 gene (Fig. 3C) with that of the IFNB1 gene (Fig. 3D) highlights 2 distinctly different nucleosome organization strategies. In the absence of virus activation, the nucleosome organization around the IFNB1 gene looks similar to that of KLHL9, but there are some key differences in the positioning of nucleosomes relevant to its expression. When the IFNB1 gene is inactive, we observe 5′ and 3′ flanked by well-positioned (+1) and (−1) nucleosomes. Distinct from KLHL9, the well-positioned (+1) nucleosome tightly clusters over ∼160-bp encompassing nucleotide positions −84 to +76, completely obscuring access to the TSS. The (−1) nucleosome is more heterogeneously positioned, and appears to span ∼190-bp encompassing nucleotide positions −386 to −196. In between the 2 nucleosomes is an NDR that spans ∼100 bp from nucleotide position −192 to −92. This pattern is maintained up to 1 h following virus infection. By 2 h.p.i., as the virus infection is detected by host signaling apparatus, the IFNB1 gene becomes transcriptionally active. At this time, the (+1) nucleosome becomes more heterogeneous and begins to occupy a larger DNA footprint of ∼190 bp, expanding its footprint to nucleotide positions −84 to +106. This heterogeneity is suggestive of either population diversity or dynamic position averaging, which means that a given nucleosome in a cell can potentially query several different local genomic positions in the cell population. Dramatically, this TSS-obscuring nucleosome largely disappears by 4 h.p.i., demonstrating that the (+1) nucleosome is evicted to allow for—or as a consequence of—robust IFNB1 gene expression in the virus-infected cell (Fig. 3D, bottom row).
The features of nucleosome organization surrounding the IFNB1 gene promoter led us to explore the patterning around the remainder of the type I IFN genes (Fig. 3E). In uninfected cells all of the type I IFN genes, except IFNE, have similar nucleosome arrangements, with 5′ and 3′ NDRs flanked by positioned nucleosomes. Like IFNB1, they all feature a well-positioned (+1) nucleosome that is positioned over their TSS. However, no specific alterations in nucleosome protection patterns, evidence of nucleosome eviction, or other disruption in the apparent position of this nucleosome was observed as a result of virus infection (see individual examples in Supplementary Fig. S2C and D). This observation is consistent with the observed low mRNA detection levels among the IFNα genes, and is in agreement with the concept of incomplete penetrance or heterogeneity within the population of infected cells.
Sequence features related to nucleosome organization in the type I IFN locus
In vivo, nucleosomes are enriched for specific DNA sequence motifs, which span the length of nucleosomal DNA. At approximately every 10th bp, when the DNA backbone faces the histone core, AA/TT/AT/TA dinucleotides are favored, and when the DNA backbone faces outwards, away from the histone core, CC/CG/GC/GG dinucleotides are preferred (Bernstein and others 2004; Hughes and Rando 2009; Tillo and others 2010). To characterize DNA sequences contributing to the consistent positioning of nucleosomes in the type I IFN locus, we used the center-weighted score data from each sample to identify and pool together 3,996 well-positioned nucleosomes for analysis. The weakest nucleosome affinity was found in DNA regions enriched in A-polymers (AAA), while the strongest nucleosome preferences were exhibited by C-polymers (CCC), in agreement with the general understanding that these sequences play key roles in nucleosome rotational positioning (Fig. 4A, and Supplementary Fig. S3A and B). Likewise, AT and TA dinucleotides displayed a negative preference for nucleosomes, whereas GG, GC, CG, and CC dinucleotides promoted the greatest nucleosome affinity. These sequence motifs were observed with 10-bp periodicity (Fig. 4B and C, and Supplementary Fig. S3B) that is reflective of the helical preferences in nucleosome–DNA interactions observed in model organism genomes (Segal and others 2006; Kaplan and others 2009; Brogaard and others 2012), supporting evolutionary preservation of intrinsic nucleosome positioning signals (Gaffney and others 2012).
FIG. 4.

Rotational positioning and nucleotide preferences of nucleosomes in the type I IFN gene cluster. (A) Frequency of dinucleotide and trinucleotide occurrences as a function of distance relative to the calculated nucleosome center, revealing signatures of rotational positioning in vivo. The y-axis represents the frequency of nucleosome dyads at a given distance normalized to expected nucleotide frequency. The 10-bp-spaced peaks represent helical rotational preferences of oligomers relative to the nucleosome surface. (B) Position-dependent dinucleotide sequence preferences in the type I IFN gene cluster. Well-positioned nucleosomes (3,996) determined from center-weighted scores were analyzed for the occurrence of AA/AT/TT/TA (black) and CC/CG/GC/GG (red) dinucleotides at each position of the alignment with a 3-bp moving average. (C) Representation of the most influential individual dinucleotide preferences in the type I IFN gene cluster. The frequency of nucleotide dimers is plotted relative to a nucleosome dyad inferred from mapped sequence reads. See also Supplementary Fig. S3.
Discussion
This report describes high-density nucleosome position analysis of human chromosome 9p21–22, providing a detailed view of the chromatin packaging throughout the human type I IFN gene cluster. Comprehensive quantitative and statistical analysis has demonstrated features of nucleosome organization in this locus that are shared with lower eukaryotes, and both confirm and expand our understanding of the packaging of the human IFNB1 gene in vivo, before and after virus infection. The IFNB1 proximal promoter contains an NDR flanked by nucleosomes including a well-positioned (+1) nucleosome that occupies nucleotides −84 to +76 of the IFNB1 promoter at steady state, preventing transcriptional activation in general agreement with prior molecular biological studies. An identical arrangement of promoter-obscuring (+1) nucleosomes was found for every type I IFN gene, indicating that this is a universal regulatory feature that has been conserved during IFN gene duplication. Results demonstrate a common mode of nucleosome positioning for all the type I IFN genes throughout the locus, suggesting a requirement for nucleosome displacement for expression of these related cytokine genes.
These high-resolution nucleosome occupancy maps allowed the observation of some previously unrecognized features of the IFNB1 gene promoter. Notably, while the (+1) nucleosome obscuring the TSS adjacent to the TATA box has been portrayed to slide 36 bp to allow for appropriate IFNB1 gene expression (Lomvardas and Thanos 2001, 2002), no evidence for nucleosome sliding to a new position was found. Instead, the data are consistent with a model in which the (+1) nucleosome is evicted to allow for Pol II loading and productive transcription. Additionally, we found that the 5′ NDR and the (+1) and (−1) nucleosome are not as rigidly positioned as previously defined (Lomvardas and Thanos 2001). Rather, in a population of cells these nucleosomes query a subset of different positions, with the (−1) nucleosome appearing to be more promiscuous than the (+1) nucleosome, which is more tightly positioned.
Dramatic (+1) nucleosome rearrangement was observed only in IFNB1 4 h.p.i. despite the robust mRNA increases detected by RT-qPCR for each type I IFN gene during a majority of the initial infection (Fig. 1). The type I IFN genes, like other cytokines, are well known to display a heterogeneous cellular response, even in clonal cell lines, resulting in only 10%–30% of the cells with active IFN gene transcription despite uniform and high-multiplicity virus infection of every cell (Kelly and Locksley 2000; Senger and others 2000; Calado and others 2006; Hu and others 2007; Apostolou and Thanos 2008; Zhao and others 2012). The lack of nucleosome movement in the bulk of the cell population represents a large majority of IFN loci in a quiescent state, masking the minority fraction of active IFN genes. Additionally, each IFN promoter carries its own intrinsic binding specificity for virus-inducible transcription factors, ultimately affecting the robustness of their transcriptional response (Genin and others 2009). RT-qPCR (Fig. 1B) demonstrates that IFNB1 mRNA level increases 1–3 orders of magnitude higher than the other type I IFN mRNAs investigated. This observation indicates that a much greater proportion of cells expressed IFNB1 relative to the other type I IFN gene family members (Apostolou and Thanos 2008)—most significantly at 4 h.p.i.—and, in turn, nucleosome rearrangements are more easily observed in the IFNB1 gene at this time.
The region's sole constitutively expressed gene, KLHL9, also has well-positioned (+1) and (−1) nucleosomes. Unlike the IFN genes, these nucleosomes do not obscure the transcriptional start site, and an appropriate NDR is maintained that allows unhindered Pol II engagement. The mechanisms governing nucleosome positioning have been intensely studied and debated (Segal and Widom 2009; Iyer 2012), but current models acknowledge that both intrinsic nucleosome DNA sequence preferences (Brogaard and others 2012; Gaffney and others 2012) and sequence-independent processes (Zhang and Pugh 2011; Yen and others 2012) participate in guiding nucleosomes to and from DNA destinations. The IFN locus reflects both of these phenomena. Periodic dinucleotide signatures known to influence nucleosome occupancy of lower eukaryotic genomes are readily observed in our in vivo analysis in human lymphocytes. These sequence preferences may have been overlooked in previous analyses, but the combination of target enrichment, paired-end sequencing, and rigorous computational analysis used here resulted in sufficient depth of coverage to reveal characteristic nucleosome rotational preference signatures. Even with identifiable nucleosome sequence preferences, the lower analytical performance of the human maps compared with less complex eukaryotes is most likely explained by the actions of more diverse chromatin remodelers, transcription factors, or other ATP-facilitated mechanisms that have been shown to make significant contributions to nucleosome organization that often supersede intrinsic sequence preferences (Zhang and others 2011; Yen and others 2012).
The steady-state nucleosome maps of the IFNA and IFNW1 gene promoters clearly resemble those of the IFNB1 gene, suggesting that they all use chromatin to inhibit transcription in the absence of activating signal. As such, we predict that, like IFNB1, transcriptional activation of these genes requires promoter-specific recruitment of general and gene-specific remodeling factors that will facilitate Pol II intiation and elongation. The benefit of such a multilayered control system is to provide substantial regulatory hurdles in the form of nucleosome remodeling and Pol II recruitment, which will prevent inappropriate transcriptional activation of antiviral responses that lead to poor health outcomes such as autoimmune disorders (Hall and Rosen 2010), and enable efficient re-establishment of the steady-state quiescence following signal resolution.
Data Access
Sequencing data have been submitted to the NCBI short reads archive under project accession No. SRP015871 and alias PRJNA175783.
Supplementary Material
Acknowledgment
The authors are grateful to the Northwestern University Genomics Core for all ABI SOLiD sequencing and sequence alignment completed for this project.
Author Disclosure Statement
No competing financial interests exist.
References
- Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D. 2000. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell 103(4):667–678 [DOI] [PubMed] [Google Scholar]
- Apostolou E, Thanos D. 2008. Virus infection induces NF-kappaB-dependent interchromosomal associations mediating monoallelic IFN-beta gene expression. Cell 134(1):85–96 [DOI] [PubMed] [Google Scholar]
- Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, Carlson CD, Gossett AJ, Hasinoff MJ, Warren CL, Gebbia M, Talukder S, Yang A, Mnaimneh S, Terterov D, Coburn D, Li Yeo A, Yeo ZX, Clarke ND, Lieb JD, Ansari AZ, Nislow C, Hughes TR. 2008. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell 32(6):878–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashiardes S, Veile R, Helms C, Mardis ER, Bowcock AM, Lovett M. 2005. Direct genomic selection. Nat Methods 2(1):63–69 [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL. 2004. Global nucleosome occupancy in yeast. Genome Biol 5(9):R62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. 2007. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 6(12):975–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogaard K, Xi L, Wang J-P, Widom J. 2012. A map of nucleosome positions in yeast at base-pair resolution. Nature 486(7404):496–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado DP, Paixao T, Holmberg D, Haury M. 2006. Stochastic monoallelic expression of IL-10 in T cells. J Immunol 177(8):5358–5364 [DOI] [PubMed] [Google Scholar]
- Diaz MO, Pomykala HM, Bohlander SK, Maltepe E, Malik K, Brownstein B, Olopade OI. 1994. Structure of the human type-I interferon gene cluster determined from a YAC clone contig. Genomics 22(3):540–552 [DOI] [PubMed] [Google Scholar]
- Ford E, Thanos D. 2010. The transcriptional code of human IFN-beta gene expression. Biochim Biophys Acta 1799(3–4):328–336 [DOI] [PubMed] [Google Scholar]
- Gaffney DJ, McVicker G, Pai AA, Fondufe-Mittendorf YN, Lewellen N, Michelini K, Widom J, Gilad Y, Pritchard JK. 2012. Controls of nucleosome positioning in the human genome. PLoS Genet 8(11):e1003036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genin P, Vaccaro A, Civas A. 2009. The role of differential expression of human interferon—a genes in antiviral immunity. Cytokine Growth Factor Rev 20(4):283–295 [DOI] [PubMed] [Google Scholar]
- Goodbourn S, Zinn K, Maniatis T. 1985. Human beta-interferon gene expression is regulated by an inducible enhancer element. Cell 41(2):509–520 [DOI] [PubMed] [Google Scholar]
- Gu SG, Fire A. 2010. Partitioning the C. elegans genome by nucleosome modification, occupancy, and positioning. Chromosoma 119(1):73–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JC, Rosen A. 2010. Type I interferons: crucial participants in disease amplification in autoimmunity. Nat Rev Rheumatol 6(1):40–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley PD, Madhani HD. 2009. Mechanisms that specify promoter nucleosome location and identity. Cell 137(3):445–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Sealfon SC, Hayot F, Jayaprakash C, Kumar M, Pendleton AC, Ganee A, Fernandez-Sesma A, Moran TM, Wetmur JG. 2007. Chromosome-specific and noisy IFNB1 transcription in individual virus-infected human primary dendritic cells. Nucleic Acids Res 35(15):5232–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A, Rando OJ. 2009. Chromatin “programming” by sequence—is there more to the nucleosome code than %GC? J Biol 8(11):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs A, Lindenmann J. 1957. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 147(927):258–267 [DOI] [PubMed] [Google Scholar]
- Iyer VR. 2012. Nucleosome positioning: bringing order to the eukaryotic genome. Trends Cell Biol 22(5):250–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SM, Tan FJ, McCullough HL, Riordan DP, Fire AZ. 2006. Flexibility and constraint in the nucleosome core landscape of Caenorhabditis elegans chromatin. Genome Res 16(12):1505–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, Segal E. 2009. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458(7236):362–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BL, Locksley RM. 2000. Coordinate regulation of the IL-4, IL-13, and IL-5 cytokine cluster in Th2 clones revealed by allelic expression patterns. J Immunol 165(6):2982–2986 [DOI] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y. 1999. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98(3):285–294 [DOI] [PubMed] [Google Scholar]
- Kornberg RD, Stryer L. 1988. Statistical distributions of nucleosomes: nonrandom locations by a stochastic mechanism. Nucleic Acids Res 16(14A):6677–6690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C. 2007. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet 39(10):1235–1244 [DOI] [PubMed] [Google Scholar]
- Lomvardas S, Thanos D. 2001. Nucleosome sliding via TBP DNA binding in vivo. Cell 106(6):685–696 [DOI] [PubMed] [Google Scholar]
- Lomvardas S, Thanos D. 2002. Modifying gene expression programs by altering core promoter chromatin architecture. Cell 110(2):261–271 [DOI] [PubMed] [Google Scholar]
- Lovett M, Kere J, Hinton LM. 1991. Direct selection: a method for the isolation of cDNAs encoded by large genomic regions. Proc Natl Acad Sci USA 88(21):9628–9632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameswaran P, Jalili R, Tao L, Shokralla S, Gharizadeh B, Ronaghi M, Fire AZ. 2007. A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic Acids Res 35(19):e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S, Krause CD, Walter MR. 2004. Interferons, interferon-like cytokines, and their receptors. Immunol Rev 202:8–32 [DOI] [PubMed] [Google Scholar]
- Richmond TJ, Davey CA. 2003. The structure of DNA in the nucleosome core. Nature 423(6936):145–150 [DOI] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. 2008. Dynamic regulation of nucleosome positioning in the human genome. Cell 132(5):887–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang JP, Widom J. 2006. A genomic code for nucleosome positioning. Nature 442(7104):772–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Widom J. 2009. What controls nucleosome positions? Trends in genetics: TIG 25(8):335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal PB, May LT, LaForge KS, Inouye M. 1982. Unusually long mRNA species coding for human alpha and beta interferons. Proc Natl Acad Sci USA 79(22):6932–6936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger K, Merika M, Agalioti T, Yie J, Escalante CR, Chen G, Aggarwal AK, Thanos D. 2000. Gene repression by coactivator repulsion. Mol Cell 6(4):931–937 [DOI] [PubMed] [Google Scholar]
- Strahle L, Garcin D, Kolakofsky D. 2006. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351(1):101–111 [DOI] [PubMed] [Google Scholar]
- Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, Lee C, Yarden G, Vleck SE, Glenn JS, Nolan GP, Piehler J, Schreiber G, Garcia KC. 2011. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 146(4):621–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillo D, Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Field Y, Lieb JD, Widom J, Segal E, Hughes TR. 2010. High nucleosome occupancy is encoded at human regulatory sequences. PloS ONE 5(2):e9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. 2011. Determinants of nucleosome organization in primary human cells. Nature 474(7352):516–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. 2007. Chromatin remodelling at promoters suppresses antisense transcription. Nature 450(7172):1031–1035 [DOI] [PubMed] [Google Scholar]
- Xi L, Fondufe-Mittendorf Y, Xia L, Flatow J, Widom J, Wang JP. 2010. Predicting nucleosome positioning using a duration Hidden Markov Model. BMC Bioinform 11:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. 2012. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell 149(7):1461–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yigit E, Bischof JM, Zhang Z, Ott CJ, Kerschner JL, Leir SH, Buitrago-Delgado E, Zhang Q, Wang JP, Widom J, Harris A. 2013a. Nucleosome mapping across the CFTR locus identifies novel regulatory factors. Nucleic Acids Res 41(5):2857–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yigit E, Zhang Q, Xi L, Grilley D, Widom J, Wang JP, Rao A, Pipkin ME. 2013b. High-resolution nucleosome mapping of targeted regions using BAC-based enrichment. Nucleic Acids Res 41(7):e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. 2005. Genome-scale identification of nucleosome positions in S. cerevisiae. Science 309(5734):626–630 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Pugh BF. 2011. High-resolution genome-wide mapping of the primary structure of chromatin. Cell 144(2):175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wippo CJ, Wal M, Ward E, Korber P, Pugh BF. 2011. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science 332(6032):977–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Zhang J, Phatnani H, Scheu S, Maniatis T. 2012. Stochastic expression of the interferon-beta gene. PLoS Biol 10(1):e1001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinn K, DiMaio D, Maniatis T. 1983. Identification of two distinct regulatory regions adjacent to the human beta-interferon gene. Cell 34(3):865–879 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.