

Abstract
Treatment of chronic inflammatory diseases with tumor necrosis factor alpha (TNF-α) antagonists has been associated with increased risk of tuberculosis (TB). We examined the usefulness of the rabbit model of active pulmonary TB for studying the impact of the human immune modulatory reagent etanercept on the host immune response. Control of Mycobacterium tuberculosis (Mtb) infection, disease pathology, and the global transcriptional response in Mtb-infected lungs of rabbits were studied. Etanercept treatment exacerbated disease pathology and reduced bacillary control in the lungs, compared with infected untreated animals. Reduced collagen and fibrin deposition in the granulomas was associated with significant downregulation of the collagen metabolism and fibrosis network genes and upregulation of genes in the inflammatory response and cell recruitment networks in the lungs of etanercept treated, compared with untreated rabbits. Our results suggest that targeting the TNF-α signaling pathway disrupts the tissue remodeling process, which is required for the formation and maintenance of well-differentiated granulomas and for control of Mtb growth in the lungs. These results validate the use of the rabbit model for investigating the impact of selected human immune modulatory drugs, such as a TNF-α antagonist, on the host immune response and pathogenesis in TB.
Introduction
Tumor necrosis factor alpha (TNF-α) antagonists have greatly improved the prognosis of human autoimmune inflammatory diseases, such as rheumatoid arthritis, psoriasis, and Crohn's disease. However, the use of TNF-α-targeted therapeutics has been associated with elevated risk of tuberculosis (TB), consistent with the crucial role played by TNF-α in regulating the immune response to Mycobacterium tuberculosis (Mtb) infection (Klausner and others 1996; Roach and others 2002; Clay and others 2008). While the introduction of routine prescreening for latent Mtb infection (LTBI) and prophylactic TB treatment have reduced the incidence of TB reactivation during treatment with TNF-α antagonists, these adverse events have not been completely eliminated (Mohan and others 2004; Brassard and others 2006; Wallis 2008; Prieto-Pérez and others 2013). This observation, together with results of a modeling study suggest that TNF-α antagonists may also increase the risk of progression of new TB infections to disease and worsening of clinical manifestations in patients with preexisting TB (Wallis 2008). Thus, a better understanding of the impact of TNF-α antagonists on the granulomatous response during Mtb infection and the mechanisms underlying their ability to exacerbate active TB disease, in addition to reactivation of LTBI, is needed (Wallis and Ehlers 2005).
The most common classes of TNF-α inhibitors approved by the FDA for clinical use include neutralizing monoclonal antibodies (infliximab, adalimumab, and certolizumab pegol), and soluble TNF-α receptors (TNFR), such as etanercept (Enbrel). Several groups, including our own, have shown that treatment of Mtb-infected mice with neutralizing anti-TNF-α antibody leads to exacerbated disease pathology, increased lung bacillary load, and reduced survival of the infected animals (Flynn and others 1995; Plessner and others 2007; Koo and others 2011). Similar results have been shown in nonhuman primates (NHPs), where administration of adalimumab, prior to Mtb infection, led to more aggressive and invasive disease in the lungs and other organs (Plessner and others 2007). Compared to monoclonal anti-TNF-α antibodies, receptor targeted antagonists have shown a lower risk of TB disease in patients (Brassard and others 2006; Plessner and others 2007; Wallis 2008; Tubach and others 2009; Wallis and others 2009; Li 2011; Winthrop and others 2013). In mice, administration of murine TNF-α receptor Fc fusion molecule prior to Mtb infection did not affect bacillary burden or survival, while initiation of treatment after 4 months of Mtb infection led to uncontrolled disease and reduced survival (Plessner and others 2007). In an NHP model of LTBI, administration of soluble TNF-α (p55-TNF-αR1) caused reactivation of the infection, primarily manifested as extra pulmonary TB with limited lung involvement (Lin and others 2010). However, the impact of TNF-α receptor antagonists on models of active pulmonary TB that recapitulate the spectrum of granulomatous pathology seen in human disease has not been well explored.
We have characterized a rabbit model of progressive pulmonary TB generated by aerosol infection with Mtb HN878 (Flynn and others 2008; Kaplan and Tsenova 2010; Subbian and others 2011c). Extensive work by our group and others has shown that the rabbit model of pulmonary TB recapitulates the disease pathology and granuloma evolution, including hypoxic necrotic center and cavity formation as seen in human pulmonary TB (Flynn and others 2008; Manabe and others 2008; Via and others 2008; Kaplan and Tsenova 2010; Subbian and others 2011c). Using the rabbit model, we previously showed that treatment with a phosphodiesterase-4 (PDE4) inhibitor partially inhibited TNF-α production without causing general immune suppression. PDE4-inhibitor-treated rabbits showed similar granuloma structure and unchanged bacillary loads in the lungs, compared to untreated infected rabbits (Subbian and others 2011b). Global transcriptome analysis of the rabbit lungs showed significant changes in host gene expression profiles during treatment that demonstrated a link between PDE4 inhibition and specific downregulation of innate immunity networks (Subbian and others 2011a).
In the present study, we examined the impact of treatment with etanercept, a soluble TNF-α receptor (R2) Fc fusion protein (TNFR2-Fc), on active pulmonary TB in the rabbit model. We analyzed the genome-wide lung transcriptional response of infected rabbits treated with etanercept, compared to untreated animals, and correlated it with the extent and nature of the pathology in the lungs.
Materials and Methods
Bacteria and chemicals
Mtb HN878 was grown as described (Koo and others 2012). Etanercept was obtained from Amgen, Inc. and Wyeth Pharmaceuticals. All other chemicals were from Sigma unless otherwise stated.
Infection and treatment of rabbits
Specific pathogen-free, New Zealand White rabbits, ∼2.5 kg (Millbrook Farms) were infected with aerosolized Mtb HN878 (CH Technologies, Inc.) as described (Tsenova and others 2006). At 3 h postexposure, 2 animals were euthanized to enumerate colony forming units (CFU) in the lungs (expressed as CFU per whole lung). Treatment with etanercept at 8 mg/kg (human adult dose), administered subcutaneously once weekly, was started at 4 weeks postinfection and continued for 8 weeks (Fig. 1A). Mtb-infected, untreated rabbits served as controls. Changes in animal body and organ (lung, liver, and spleen) weights were monitored to evaluate extent of disease. At defined time points (0, 4, and 8 weeks post-treatment initiation), rabbits (n=4–6 per time point/group) were euthanized; lungs were used for CFU enumeration, histopathology examination, and RNA isolation. Numbers of dorsal sub-pleural lesions were counted as a measure of gross pathology. All procedures were performed in Biosafety Level-3 facilities, according to protocols approved by the Rutgers University IACUC.
FIG. 1.

Experimental design and the effect of etanercept on the response of rabbits to Mtb infection. (A) Schema showing the design of rabbit infection, treatment, and sample collection time points. (B) Bacillary load (CFU) in the lungs. *Statistically significant difference between etanercept-treated and untreated rabbits after 8 weeks of treatment (ie, 12 weeks postinfection). (C) Number of dorsal subpleural lesions (left axis, hatched bars) and percent of the lung sections occupied by granulomatous lesions (right axis, gray bars) at 4 and 12 weeks postinfection. Statistically significant differences in the numbers of lesions (*) and lung involvement (**) were observed between etanercept-treated and control rabbits after 8 weeks of treatment (ie, 12 weeks postinfection). CFU, colony forming units; Mtb, Mycobacterium tuberculosis.
Histology and morphometric analysis
Formalin-fixed paraffin-embedded sections of rabbit lung tissue were stained with hematoxylin-eosin, Ziehl-Nielsen [for acid-fast bacilli (AFB)], and Masson's trichrome and Van Gieson stains (for collagen/elastin) (IDEXX-RADIL Laboratories, Inc.). Semi-quantitative evaluation of pathology was based on the following scoring: 0-intact lung; 1-increased cellularity; 2-granuloma formation; 3-granulomas with central necrosis; 4-large coalescent lesions with necrosis; 5-partial liquefaction; 6-extensive liquefaction; 7-cavity. Morphometric analysis of the percentage of lung area involved in pathology was made using Sigmascan Pro Software (Systat Softwares, Inc.).
Lung total RNA isolation
Total RNA from rabbit lungs was isolated, as described (Subbian and others 2013b). Briefly, tissue samples were homogenized in TRIzol reagent (Life Technologies), extracted with bromo-chloro-propane and precipitated with isopropanol; RNA was purified using Nucleospin RNA-II (Macherey-Nagel).
Microarray and gene network/pathway analysis
Total RNA was subjected to transcriptome profiling using 4X44k rabbit whole genome microarrays (Agilent Technologies), as described (Subbian and others 2013a). Briefly, arrays were hybridized with a mixture of Cy3- or Cy5-labeled cDNA, washed, scanned, and data extracted. Background-corrected, normalized data were analyzed by one-way Anova using Partek Genomics Suite (Partek, Inc.); an unadjusted P value <0.05 was used to select statistically significantly differentially expressed genes (SDEG). The SDEG were uploaded to Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Inc.) to determine gene ontology and derive networks and pathways, as described (Subbian and others 2013b). The SDEG were further analyzed using a z-score algorithm from IPA, which predicts the impact of activation/inhibition status of transcriptional regulators and associated genes on cellular functions (z-score of ≥+2 predicts activation; ≥−2 predicts inhibition).
Quantitative polymerase chain reaction analysis
To validate microarray transcript, quantitative polymerase chain reaction (qPCR) was performed on selected gene targets, as described (Subbian and others 2011a). Briefly, cDNA was synthesized from total RNA samples, using Sprint RT Complete kit (Clontech Laboratories, Inc.); second strand synthesis was performed with rabbit gene-specific primer pairs (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/jir) and SYBR green qPCR SuperMix Universal (Life technologies).
Statistical analysis
The independent Student t-test from GraphPad Prism software (GraphPad Software, Inc.) was used for parametric statistical analysis of qPCR and other experiments. For all parameters, P≤0.05 was considered significant.
Results
Effect of etanercept on the lung bacillary load
Aerosol infection of rabbits with Mtb HN878 resulted in implantation of 3.2 log10 bacilli (on day 0) into the lungs, followed by exponential bacterial growth, reaching 6.8±0.2 log10 CFU by 4 weeks of infection (Fig. 1B). From 4 to 8 weeks postinfection, the bacillary load remained stable (P=0.11), then moderately decreased to 5.7±1.0 log10 CFU by 12 weeks of infection in untreated animals. In the untreated animals, there were no significant differences in lung CFU between 4 and 8 or 12 weeks postinfection (P=0.63 and 0.11, respectively). Four weeks of etanercept treatment, starting at 4 weeks postinfection, did not significantly affect CFU numbers (P=0.78). However, after 8 weeks of treatment, the bacillary load had significantly increased to 7±0.9 log10 CFU (P=0.04), indicating that long-term exposure to etanercept had reduced the control of Mtb growth in the lungs of chronically-infected rabbits. No significant difference in liver bacterial load was noted between untreated and etanercept-treated rabbits at any time (not shown). Similarly, no significant differences in body and organ weights were noted (not shown).
Effect of etanercept treatment on lung pathology
Whole lungs were examined for gross pathology and enumeration of visible dorsal sub-pleural lesions at 4 and 12 weeks postinfection (Fig. 1C). In untreated rabbit lungs, an average of 78 sub-pleural lesions was observed at 4 weeks postinfection; by 12 weeks the mean lesion count had dropped somewhat reaching 64 in infected lungs. After 4 weeks of etanercept treatment (8 weeks postinfection), there was no significant difference in the number of sub-pleural lesions (not shown). However, significantly higher numbers of lung lesions (86; P=0.014) were noted after 8 weeks of etanercept treatment, compared with the untreated animals.
Histologic examination of untreated rabbit lungs at 4 weeks of infection showed multiple, well-organized granulomas (0.5–1.5 mm diameter), some of which had limited central necrosis (Fig. 2A–C). By 8 weeks, the granulomas were larger and more necrotic, with scattered AFB. At 12 weeks postinfection, coalescent lesions (about 2 mm diameter) with central necrosis, surrounded by macrophages and lymphocytes, were apparent (Fig. 2D–F), with some granulomas showing mineralization (calcification). After 4 weeks of etanercept treatment, no significant differences in histopathology were noted between treated and untreated animals (not shown). However, rabbits treated with etanercept for 8 weeks had larger (3–4 mm diameter) confluent granulomas with extensive necrosis, polymorphonuclear (PMN) leukocyte infiltration, and liquefaction (Fig. 2G–I). Some lesions had progressed to form cavities, surrounded by concentric layers of necrosis and a highly cellular layer, composed of macrophages, lymphocytes, and PMNs (Fig. 2J–L). Acid-fast staining revealed numerous bacilli at the cavity surface and in the necrotic zones (not shown). These findings were confirmed by a semi-quantitative evaluation of lung pathology (See Materials and Methods). The average disease score for animals treated with etanercept for 8 weeks was more than twice that of untreated animals (5.6 and 2.3, respectively). No significant difference was noted in the morphometric analysis between the untreated and etanercept-treated rabbit lungs after 4 weeks of treatment. However, significantly greater lung area involvement was observed after 8 weeks of etanercept treatment, compared with untreated infected rabbits (P=0.03) (Fig. 1C).
FIG. 2.

Histopathologic evaluation of Mtb-infected and etanercept treated or untreated rabbit lungs. (A–C) Lungs of infected rabbit at 4 weeks postinfection. Well-organized granulomas (0.5–1.5 mm diameter) with central necrosis, surrounded by macrophages (M) and lymphocyte (L) cuff. (D–F) Lungs of infected untreated rabbit at 12 weeks postinfection. Large coalescent granulomas (about 2 mm diameter), with central necrosis (N) and mineralization, and high numbers of macrophages (M) and lymphocytes (L). (G–I) Lungs of 8 weeks-etanercept-treated (ie, 12 weeks postinfection) rabbit. Much larger lesions (3–4 mm diameter) with extensive necrosis (N) and liquefaction surrounded by PMNs, foamy macrophages (M), and lymphocytic (L) aggregates. (J–L) Etanercept-treated rabbit lung with a cavity (cav) at 8 weeks treatment (ie, 12 weeks postinfection). Note the layers around the cavity lumen: an extensive necrotic zone (N), with numerous PMNs (P) and macrophages (M). H&E stain. Original magnification:×4 (A, D, G, J),×10 (B, E, H),×40 (C, F, I, K, L). H&E, hematoxylin-eosin; PMN, polymorphonuclear.
Effect of etanercept treatment on lung transcriptome of Mtb-infected rabbits
Global transcriptional changes were evaluated by microarray, and genes that were SDEG in the lungs of etanercept-treated, compared to untreated, Mtb-infected rabbits (Fig. 3A) were selected using a P value of significance (P<0.05). A total of 3,407 SDEG (1,306 up and 2,101 down) were identified following 4 weeks of etanercept treatment, which increased to 7,724 (2,221 up and 5,503 down) after 8 weeks of treatment, with 912 SDEG common to both time points (Fig. 3A, B). A majority of genes upregulated in the Mtb-infected rabbit lungs showed dampened expression in response to etanercept treatment.
FIG. 3.
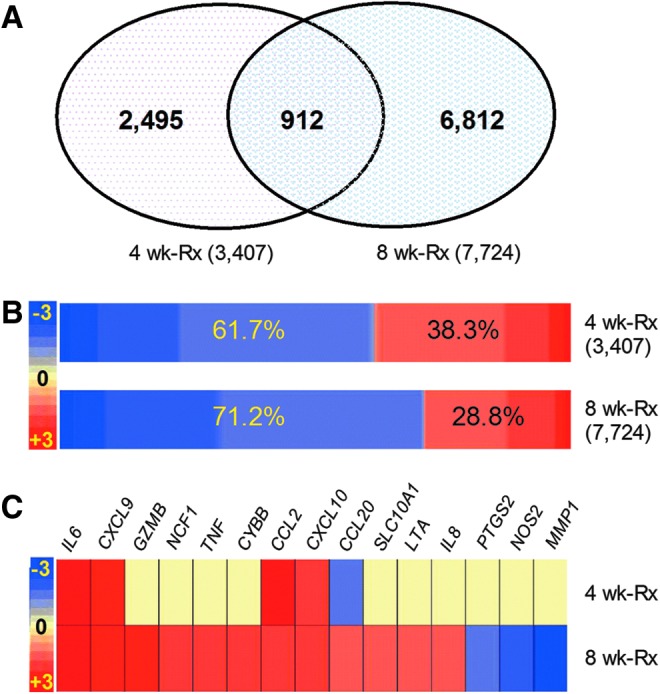
Genomewide transcriptional analysis of Mtb-infected etanercept-treated rabbit lungs. (A) Venn diagram showing significantly differentially expressed (P<0.05) genes (SDEG) in the etanercept treated, compared to untreated rabbit lungs at 4 and 8 weeks of treatment. (B) Intensity map showing the expression pattern of SDEG in the etanercept-treated rabbits at 4 and 8 weeks of treatment. The percentages in the boxes are derived from the number of up (red) or down (blue) regulated genes versus total number of genes at each time point. The color scale bar ranges from −3 (blue; downregulation) to +3 (red; upregulation). (C) Intensity plot showing the expression level of 15 SDEG involved in etanercept interaction network in the lungs of etanercept treated, compared to untreated rabbits at 4 and 8 weeks of treatment. The color scale bar ranges from −3 (blue; downregulation) to +3 (red; upregulation) and the gradation in color corresponds to respective level of expression. Yellow color denotes no significant change between the untreated and etanercept-treated samples.
Validation of microarray gene expression by qPCR
To validate the gene expression data obtained by microarray analysis, we measured transcript levels by qPCR of 16 genes, selected for their role in host immunity against Mtb infection, using the same RNA samples (Table 1). The patterns and direction of expression, as measured by qPCR, were consistent with the microarray data; no significant differences in expression levels, determined by both methods, were observed (Table 1).
Table 1.
Validation of Microarray Gene Expression by Quantitative Polymerase Chain Reaction
| 4 weeks treatment | 8 weeks treatment | |||
|---|---|---|---|---|
| Genes | Microarray | qPCR (se) | Microarray | qPCR (se) |
| ARG1 | 2.5 | 1.2 (0.04) | 1.9 | 1.3 (0.11) |
| VCAM1 | 1 | 1.6 (0.08) | −1.4 | −1.8 (0.1) |
| TIMP1 | 1.4 | 1.4 (0.02) | 1.2 | 1.8 (0.06) |
| CD28 | 1.1 | 1.8 (0.07) | 1.1 | 1.5 (0.02) |
| IL13 | 1.2 | 2.2 (0.93) | 1.3 | 1.7 (0.16) |
| SELL | −2.04 | −2.8 (0.94) | 1.2 | 2.6 (0.48) |
| LGALS3 | 1.3 | 1.3 (0.10) | 1.7 | 2.0 (0.25) |
| TLR2 | −1 | −1.9 (0.31) | 1.8 | 1.9 (0.16) |
| TNFA | 1 | −2.4 (0.34) | 1.8 | 1.4 (0.28) |
| MMP1 | −1.2 | −1.4 (1.08) | −2.2 | −2.9 (0.1) |
| IL6 | 2.4 | 1.3 (0.21) | 2.5 | 2.4 (0.52) |
| NOS2 | 1 | 2.2 (0.94) | −1.7 | −2.3 (0.57) |
| SPP1 | 1.6 | 3.0 (1.81) | 2.3 | 3.6 (0.79) |
| CCL2 | 1.9 | 1.3 (0.13) | 1.8 | 1.2 (0.08) |
| CXCL10 | 1.7 | 2.3 (0.84) | 1.7 | 1.7 (0.24) |
| MSR1 | 2.1 | 1.6 (0.25) | 1.8 | 1.3 (0.15) |
se, standard error; qPCR, quantitative polymerase chain reaction.
Biological functions affected by etanercept treatment
We interrogated the SDEG using IPA to evaluate cellular functions perturbed by etanercept treatment. Following 4 weeks of etanercept treatment, our z-score-based gene ontology analysis revealed a significant enrichment of genes associated with increased activation of organismal (host) death, altered cell morphology, inhibition of cell viability, survival and proliferation, and cytoskeleton formation-associated functions (Table 2). After 8 weeks of etanercept treatment, SDEG associated with biological functions such as organismal death, cell death, apoptosis and necrosis networks/pathways, were significantly activated, while those involved in cell survival, viability, proliferation, migration, and movement, and cell cycle progression and endocytosis, were inhibited (Table 2). Thus, ontology analysis of the SDEG suggests that etanercept treatment resulted in the dysregulation of normal cellular functions and viability by 4 weeks, with expanded and more prominent impairments of cell functions and tissue disintegration after 8 weeks of treatment, consistent with the increased tissue necrosis seen histologically in rabbit lungs.
Table 2.
z-Score-Based Cellular/Biological Function Analysis of Significantly Differentially Expressed Genes in the Etanercept-Treated Rabbit Lungs
| Cell functions | P value | Activation state | z-Score | Molecules |
|---|---|---|---|---|
| 4 weeks treatment | ||||
| Organismal (host) death | 7.26E-09 | Increased | 5.93 | 384 |
| Morphology of cells | 2.96E-03 | Increased | 2.152 | 206 |
| Formation of cytoskeleton | 1.10E-03 | Decreased | −2.057 | 58 |
| Proliferation of cells | 4.73E-05 | Decreased | −2.883 | 509 |
| Cell viability | 6.97E-07 | Decreased | −3.297 | 211 |
| Cell survival | 4.14E-06 | Decreased | −3.496 | 222 |
| 8 weeks treatment | ||||
| Organismal (host) death | 6.62E-20 | Increased | 9.337 | 747 |
| Cell death | 6.27E-21 | Increased | 3.706 | 1012 |
| Apoptosis | 2.84E-21 | Increased | 3.042 | 839 |
| Necrosis | 7.81E-15 | Increased | 2.437 | 764 |
| Endocytosis | 3.14E-08 | Decreased | −2.004 | 94 |
| Cell cycle progression | 5.57E-17 | Decreased | −2.125 | 364 |
| Cell movement | 1.86E-16 | Decreased | −2.213 | 620 |
| Migration of cells | 4.53E-16 | Decreased | −2.23 | 562 |
| Proliferation of cells | 1.46E-20 | Decreased | −2.292 | 1051 |
| Cell survival | 1.04E-13 | Decreased | −2.556 | 436 |
| Cell viability | 1.04E-12 | Decreased | −2.616 | 401 |
Analysis of selected networks in etanercept-treated Mtb-infected rabbit lungs
To investigate the impact of etanercept treatment on the rabbit immune response, we examined the SDEG associated specifically with these networks/pathways.
Etanercept interaction network
The SDEG included 15 genes, encoding proinflammatory cytokines, chemokines, and effector molecules, reported to interact with etanercept in humans, according to the IPA knowledgebase (Fig. 3C). Of these, 4 were upregulated, one was downregulated and expression of the other 10 was unaffected by 4 weeks of etanercept treatment, while 12 and 3 were up- and downregulated, respectively, at 8 weeks (Fig. 3C). In particular, MMP1 that encodes matrix metalloprotease-1, a key enzyme involved in tissue destruction/remodeling, and NOS2 that encodes nitric oxide synthase, an important mediator of antibacterial defense by phagocytes, were both significantly downregulated after 8 weeks of etanercept treatment. These observations confirmed the activity of the human reagent in the rabbit infection model and may in part explain the increased bacillary load seen in the lungs of etanercept-treated, relative to untreated, rabbits.
Inflammatory response network
A subset of 196 SDEG identified at both 4 and 8 weeks of etanercept treatment was associated with the inflammatory response network (Supplementary Table S2). Of these, 43 and 41 SDEG were up- and downregulated, respectively, at 4 weeks of treatment; while 101 and 58 were up- and downregulated, respectively, at 8 weeks. Notably, key proinflammatory genes (CCL2, IL6, SPP1, CXCR10, LPAR2, ARG1, LIPA, LCN2, FAS, CD69, CTSC, CLEC7A, ANXA3, LY96, and TNFAIP6) were significantly upregulated at both time points, while other inflammatory response genes (TNFA, LTA, IL8, NCF1, CCL20, TP53, CCL24, and GPX1) were upregulated only at 8 weeks of treatment. The expression pattern of the inflammatory response network is consistent with the histological observations in the lungs showing enlarged and dysregulated granulomas, with increased inflammation, following 8 weeks of etanercept treatment.
Leukocyte necrosis and cytotoxicity network
The z-score predicted that cell death and necrosis were dysregulated in rabbit lungs after 8 weeks of etanercept. As these networks/pathways are known to be involved in inflammation, we examined the SDEG involved in the cell necrosis and cytotoxicity network (Supplementary Fig S1). Among 111 SDEG in this network, 15 and 26 were up- and downregulated, respectively, after 4 weeks of treatment, while 53 and 40 SDEG were up- and downregulated, respectively, at 8 weeks. A subset of 18 SDEG in this network are also associated with the inflammatory response network (Supplementary Table S2). Of these, 8 and 12 SDEG were upregulated after 4 and 8 weeks of treatment, respectively. Taken together, the expression pattern of SDEG in the leukocyte necrosis and cytotoxicity network further supports a role for cell necrosis and inflammation in the exacerbation of lung pathology in Mtb-infected rabbits following etanercept treatment.
Collagenase interaction and tissue fibrosis network
Histological analysis showed larger confluent lesions in rabbit lungs after 8 weeks of etanercept treatment, compared with well-defined compact granulomas in untreated animals. To explore underlying molecular factors contributing to this differential granuloma architecture, we examined SDEG involved in the collagenase interaction and tissue fibrosis networks (Fig. 4). Of 69 SDEG in the collagenase interaction network, 10 and 12 SDEG were up- and downregulated, respectively, after 4 weeks of etanercept, while only 13 of the 69 SDEG were upregulated, and 54 (>78%) were downregulated after 8 weeks of treatment (Fig. 4A). The majority of these downregulated SDEG belongs to the collagenase, matrix metalloprotease, integrin, and fibrin gene families; although, a few members of these gene families (MMP23B, MMP27, FBLN2, ITGAB5, and LGALS3) were upregulated by etanercept. Similarly, of the 90 SDEG in the tissue fibrosis network, 11 were downregulated at 4 weeks, and 72 were downregulated at 8 weeks of treatment, while only 7 and 28 SDEG were upregulated at 4 and 8 weeks, respectively (Fig. 4B). Taken together, down regulation of the majority of genes in the collagenase interaction and tissue fibrosis networks suggest significant dysregulation of tissue remodeling in the lungs of Mtb-infected rabbits by etanercept treatment.
FIG. 4.

Intensity plots of gene expression in selected IPA networks in the lungs of Mtb-infected etanercept-treated rabbits. (A) Intensity plot of 69 genes involved in the collagenase interaction network. (B) Intensity plot of 90 genes involved in the tissue fibrosis network. The values plotted are etanercept treated compared to untreated rabbits at 4 and 8 weeks of treatment. The color scale bar ranges from −3 (blue; downregulation) to +3 (red; upregulation) and the gradation in color corresponds to respective level of expression. Yellow color denotes no significant change between the untreated and etanercept-treated samples. IPA, Ingenuity Pathway Analysis.
Effect of etanercept treatment on lung fibrosis
Lung sections from treated and untreated infected rabbits were stained using Masson's trichrome and Van Gieson methods, which are specific for fibrosis, collagen deposition (Fig. 5). Microscopic examination revealed little to no fibrosis in the lungs after 4 weeks of etanercept treatment (not shown). Eight weeks of etanercept treatment resulted in significantly diminished and more diffuse collagen deposition and fibrosis around and within lung granulomas (Fig. 5D–F), compared with the untreated Mtb-infected rabbits (Fig. 5A–C). Taken together, the impact of etanercept on collagen deposition and tissue fibrosis in the lungs seen by histopathology analysis was consistent with our network/pathway analysis, thus strengthening our finding that etanercept treatment was associated with significant dysregulation of tissue healing/remodeling.
FIG. 5.

Extent of fibrosis in Mtb-infected untreated (A–C) or etanercept-treated (D–F) rabbit lungs at 8 weeks of treatment. (A–C) fine network of extracellular matrix components such as collagen can be seen by the blue staining in the Masson's trichrome and the pink staining in the Van Gieson stained sections (C) (shown with arrowheads) of lungs from untreated animals, infected with Mtb. Note the elastin, stained in black (long arrows in C). (D–F) significantly less staining is seen in the granulomas of the etanercept-treated rabbits. Masson's trichrome stain (A, B, D, E); Van Gieson stain (C, F). Original magnification:×10 (A, D),×40 (B, C, E, F).
Discussion
Using a rabbit model of progressive pulmonary TB, we show that etanercept treatment exacerbates infection and disease pathology in the lungs, resulting in increased bacillary load and larger granulomas, with more extensive necrosis and cavity formation. While the extent of lung pathology was increased by etanercept treatment, the treated rabbits did not show complete immune suppression, as indicated by the presence of intact granulomatous structures, consistent with other reports on the effects of this TNF-α antagonists. In humans with chronic inflammatory and autoimmune diseases, treatment with TNF-α antagonist has been associated with a range of 1.6 to 25.1 fold increased risk for reactivation TB (Solovic and others 2010). The reported risk was higher among patients treated with anti-TNF-α monoclonal antibodies than those who received soluble TNF-α receptors (Solovic and others 2010). The differential abilities of these 2 classes of TNF-α antagonists to reactivate TB in humans, has been attributed to their differential pharmacokinetic properties, their relative capacities to inhibit soluble and membrane bound TNF-α, and their variation in inducing immune cell apoptosis (Solovic and others 2010). However, a systematic analysis of the granulomatous response during etanercept or other immune modulators treatment of active pulmonary TB, in a model animal that is relevant to human disease, has not been reported. Our present study is the first to examine the impact of etanercept during Mtb infection and disease progression in the rabbit model, previously shown by us and others to closely mimic human pulmonary TB (Flynn and others 2008; Manabe and others 2008; Via and others 2008; Kaplan and Tsenova 2010; Subbian and others 2011c). Our results demonstrated that, although etanercept did not reduce TNF-α mRNA expression, the network genes that regulate effector functions downstream of TNF-α were significantly perturbed by treatment. This finding is consistent with the mode of action of etanercept, which binds to TNF-α, as shown both in experimental animal models and in treated humans. Studies conducted by Eli Lilly and Company using a Biacore analysis demonstrated high affinity binding of etanercept to rabbit TNF-α (KD <2.5 pM), comparable to that with human TNF-α (KD <1.6 pM), (Liana Tsenova, personal communication). This observation demonstrates the functionality of etanercept, a soluble TNF-α receptor (R2) Fc fusion protein (TNFR2-Fc), in blocking the effects of TNF-α in the rabbit model. Our observations complement studies with TNF-α-neutralization in NHPs and humanized mice (Lin and others 2010; Heuts and others 2013). In Mtb-infected cynomolgus monkeys, TNF-α neutralization resulted in apparently normal granuloma formation; however, the host-protective function of the granulomas was impaired, since the disease in treated NHPs was more aggressive and the bacilli disseminated (Lin and others 2010). Similarly, etanercept treatment of Mycobacterium bovis BCG- infected humanized mice resulted in structurally normal granulomas in the lungs and liver, although the bacterial load was higher than in untreated animals (Heuts and others 2013). In contrast, treatment of mice with TNF-α antagonists during Mtb infection leads to highly disorganized granulomas and exacerbated disease pathology (Plessner and others 2007).
Our gene expression analysis of the immune regulatory networks suggested that the impaired granulomatous response induced by etanercept treatment was associated with reduced fibrosis and tissue remodeling. In addition, we found a striking reduction in the expression of many host genes involved in collagen metabolism and deposition, including matrix metalloproteinase (MMPs), with etanercept treatment (Fig. 4B). Moreover, histologic analysis revealed corresponding differences in the lung lesions of etanercept-treated rabbits, characterized by reduced collagen deposition and diminished fibrotic encapsulation of the granulomas, compared with untreated infected animals. Thus, the exacerbation of disease pathology in the lungs of Mtb-infected etanercept-treated rabbits appears to be associated with subversion of the granulomatous maturation processes, such as fibrosis and tissue remodeling. This suggests that the fibrotic process, in itself, is likely to be a contributor to the effective containment of Mtb in the lungs, and that the extent of disease depends on the regulation of fibrosis and granuloma differentiation at the site of infection. Previous studies have shown that TNF-α and other cytokines induce production of MMP-1, MMP-3, and MMP-13, in addition to type II collagen, which are involved in tissue remodeling and wound healing (Koshy and others 2002; Elkington and others 2011). In addition, TNF-α antagonists have been shown to reduce the concentrations of MMP-1, MMP-3, or their precursors in the serum of rheumatoid arthritis patients (Brennan and others 1997; Weinblatt and others 2003; Tracey and others 2008). In an NHP model of SIV-infection, treatment with anti-TNF-α antibody (adalimumab) significantly reduced the deposition of type-I collagen and fibronectin, and limited fibrosis in lymphoid tissue (lymph nodes), compared with untreated animals (Tabb and others 2013). Further, well-organized mature granulomas have been shown to efficiently control bacillary growth in the lungs of CBA/J IL-10−/− mice, compared with poorly-organized larger granulomas with elevated cellular accumulation at the infection foci observed in TNF-α antagonist-treated animals (Cyktor and others 2013). Taken together, these observations support a critical role for TNF-α-mediated regulation of tissue remodeling in the control of Mtb infection and disease.
Several reports using different mouse models have shown that anti-TNF-α therapy during Mtb infection interferes with granuloma formation and causes disease exacerbation (Egen and others 2008; Bourigault and others 2013). However, the disease pathology, type of granulomatous response, and the kinetics of disease progression following Mtb infection are significantly different between mice and human. For example, Mtb infection in the lungs of most of the mouse strains does not result in the formation of typical granuloma and thus the lung lesions do not undergo necrosis, caseum formation, or cavitation, all salient features seen in human lung lesions (Cooper and Flynn 1995; Sakamoto 2012). In contrast to the guinea pig model, outbred rabbits and humans are both relatively more resistant to Mtb infection. The pulmonary pathology in the Mtb-infected rabbits mimics the lung lesions found in human disease, including hypoxia in the necrotic center, caseum formation, liquefaction, fibrotic and calcified nodules, and cavitation of the granulomas that allows dissemination of the infecting bacilli (Flynn and others 2008). In addition, similar to humans, individual lung granulomas in Mtb-infected rabbits evolve and mature independently of each other, within the same animal. These heterogeneous lung lesions significantly and differentially impact the bacillary physiology and metabolism, which are crucial to the host–pathogen interactions and the outcome of Mtb infection with or without various treatment modalities. Further, mice are more resistant than rabbits to the effects of human TNF-α modulators, due to limited cross-reactivity between mouse and human TNF-α (Dinges and Schlievert 2001). Rabbits, similar to humans, are relatively more sensitive to the toxicity of TNF-α induced by LPS, than mice (Redl and others 1993). Since the rabbit model of pulmonary TB shares most of the clinical, pathological, and cellular features observed in humans, it has an advantage over the mouse models, and it is a more relevant model to evaluate human reagents. The present study provides novel findings on the impact of etanercept treatment on inflammation and tissue remodeling during Mtb infection, thereby demonstrating the value of the rabbit model for studies of TB pathogenesis. This report is the first to investigate the mechanism underlying the effect of soluble TNF-α receptor treatment during active pulmonary tuberculosis in the rabbit.
Studies with TNF-α antagonists have demonstrated an essential role for this cytokine in the regulation of several proinflammatory cytokine and chemokine networks (Cooper and Flynn 1995; Flynn and others 1995; Roach and others 2002; Ehlers 2003; Algood and others 2005; Ly and others 2009). Mtb infection of TNF-α-deficient mice, and treatment of chronically infected mice with anti-TNF-α antibody, showed exacerbated bacillary growth and delayed, dysregulated granuloma formation (Flynn and others 1995; Bean and others 1999; Koo and others 2011). In Mtb-infected macaques, TNF-α neutralization altered chemokine receptor expression, impaired cellular recruitment to disease sites, and resulted in a disproportionate degree of extrapulmonary disease (Lin and others 2010). Control of Mtb infection requires the recruitment and activation of phagocytes and T cells to the site of infection and inhibition of bacillary dissemination from the lungs (Tracey and others 2008). Killing of intracellular Mtb within activated macrophages is primarily mediated by reactive oxygen species (ROS) and reactive nitrogen species (RNS), produced by PHOX/NOX and NOS2/iNOS, respectively; defects in the production of ROS and/or RNS in mice and in humans are associated with susceptibility to infection (Chan and others 1992; MacMicking and others 1997; Lambeth 2004; Lee and others 2008). Importantly, these antibacterial mechanisms are regulated by TNF-α (Flynn and others 1995; Roca and Ramakrishnan 2013). In our study, the transcript level of network genes involved in the ROS and RNS-mediated host defense, including NOS2 and NOX4, in addition to cell survival and proliferation, such as integrins ITGA2, ITGA3, ITGB1, ITGB3, ITGAV, and VCAM were significantly down modulated in the etanercept-treated rabbits (Zhang and Wang 2012).
A common observation in the treatment of chronic inflammatory diseases with TNF-α antagonists is the rapid reduction in cellularity at the site of inflammation (Feldmann and others 1996; Baeten and others 2002). However, the relative contributions of host cell functions, such as apoptosis and cytotoxicity, in regulating immune cell influx remain to be elucidated. In contrast, in Mtb-infected rabbits, etanercept treatment was associated with larger and more cellular granulomas. This discrepancy may be due to the concomitant loss of control of bacillary growth in the rabbits, which drives the inflammatory process. The elevated level of TNF-α expression after 8 weeks of etanercept treatment in Mtb-infected rabbits supports this explanation and suggests that a high mycobacterial load can reverse the anti-inflammatory activity of etanercept. In addition, although TNF-α is known to regulate the expression of chemokines, including CXCL10, CXCL9, and CXCR3, some of which are important for cell migration during Mtb infection, neutralization of TNF-α in Mtb-infected murine macrophages did not completely abolish expression of these molecules (Algood and others 2005; Tracey and others 2008). This suggests that the production of these chemokines is, at least in part, regulated by a TNF-α-independent mechanism, which is capable of mediating immune cell recruitment to the site of infection.
Conclusion
We show that an optimally regulated fibrotic process is required for development and maintenance of well-differentiated granulomas that control Mtb growth and limit the extent of disease pathology in the lungs. When the TNF-α signaling pathway is inhibited by etanercept treatment in the context of active Mtb infection, this protective granulomatous response is dysregulated, resulting in a worsening of inflammation, rather than an improvement. To the extent that TNF-α inhibitors and other host immune targeted interventions will be useful in treatment of TB, their effects on TNF-α production must be limited and tightly regulated. In addition, such interventions must be administered in combination with antibiotics to ensure that any loss of antimicrobial activity does not result in increased bacillary loads in the treated patients. Finally, our study paves the way for future research using the rabbit model of pulmonary TB to evaluate the impact of other immune modulators used to treat human disease. Their safety and efficacy and mechanisms of action can be tested in this animal model.
Supplementary Material
Acknowledgments
We thank Sabrina Dalton for secretarial assistance and Kristy Kikly from Eli Lilly for her valuable suggestions and assistance with funding the project. This study was supported by grants from the Bill & Melinda Gates Foundation (TB Drug Accelerator), the National Institutes of Health (RO1 AI054338 to G.K.), and funding from Eli Lilly and Company.
Author Disclosure Statement
This study has not been previously presented in any meetings/conferences. The authors declare that no competing financial interests exist.
References
- Algood HM, Lin PL, Flynn JL. 2005. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis 41Suppl 3:S189–S193 [DOI] [PubMed] [Google Scholar]
- Baeten D, Demetter P, Cuvelier CA, Kruithof E, Van Damme N, De Vos M, Veys EM, De Keyser F. 2002. Macrophages expressing the scavenger receptor CD163: a link between immune alterations of the gut and synovial inflammation in spondyloarthropathy. J Pathol 196(3):343–350 [DOI] [PubMed] [Google Scholar]
- Bean AG, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, Britton WJ. 1999. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J Immunol 162(6):3504–3511 [PubMed] [Google Scholar]
- Bourigault ML, Vacher R, Rose S, Olleros ML, Janssens JP, Quesniaux VF, Garcia I. 2013. Tumor necrosis factor neutralization combined with chemotherapy enhances Mycobacterium tuberculosis clearance and reduces lung pathology. Am J Clin Exp Immunol 2(1):124–134 [PMC free article] [PubMed] [Google Scholar]
- Brassard P, Kezouh A, Suissa S. 2006. Antirheumatic drugs and the risk of tuberculosis. Clin Infect Dis 43(6):717–722 [DOI] [PubMed] [Google Scholar]
- Brennan FM, Browne KA, Green PA, Jaspar JM, Maini RN, Feldmann M. 1997. Reduction of serum matrix metalloproteinase 1 and matrix metalloproteinase 3 in rheumatoid arthritis patients following anti-tumour necrosis factor-alpha (cA2) therapy. Br J Rheumatol 36(6):643–650 [DOI] [PubMed] [Google Scholar]
- Chan J, Xing Y, Magliozzo RS, Bloom BR. 1992. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J Exp Med 175(4):1111–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay H, Volkman HE, Ramakrishnan L. 2008. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity 29(2):283–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AM, Flynn JL. 1995. The protective immune response to Mycobacterium tuberculosis. Curr Opin Immunol 7(4):512–516 [DOI] [PubMed] [Google Scholar]
- Cyktor JC, Carruthers B, Kominsky RA, Beamer GL, Stromberg P, Turner J. 2013. IL-10 inhibits mature fibrotic granuloma formation during Mycobacterium tuberculosis infection. J Immunol 190(6):2778–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinges MM, Schlievert PM. 2001. Comparative analysis of lipopolysaccharide-induced tumor necrosis factor alpha activity in serum and lethality in mice and rabbits pretreated with the staphylococcal superantigen toxic shock syndrome toxin 1. Infect Immun 69(11):7169–7172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egen JG, Rothfuchs AG, Feng CG, Winter N, Sher A, Germain RN. 2008. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity 28(2):271–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers S. 2003. Role of tumour necrosis factor (TNF) in host defence against tuberculosis: implications for immunotherapies targeting TNF. Ann Rheum Dis 62Suppl 2:ii37–ii42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkington PT, Ugarte-Gil CA, Friedland JS. 2011. Matrix metalloproteinases in tuberculosis. Eur Respir J 38(2):456–464 [DOI] [PubMed] [Google Scholar]
- Feldmann M, Brennan FM, Maini RN. 1996. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol 14:397–440 [DOI] [PubMed] [Google Scholar]
- Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR. 1995. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2(6):561–572 [DOI] [PubMed] [Google Scholar]
- Flynn JL, Tsenova L, Izzo A, Kaplan G. 2008. Experimental animal models of tuberculosis. In: Kaufmann SHE, Britton WJ, eds. Handbook of tuberculosis: immunology and cell biology. Weinheim: WILEY-VCH Verlag GmbH & Co. pp 389–426 [Google Scholar]
- Heuts F, Gavier-Widen D, Carow B, Juarez J, Wigzell H, Rottenberg ME. 2013. CD4+ cell-dependent granuloma formation in humanized mice infected with mycobacteria. Proc Natl Acad Sci U S A 110(16):6482–6487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan G, Tsenova L. 2010. Pulmonary tuberculosis in the rabbit. In: Leong FJ, Dartois V, Dick T, editors. A colour atlas of comparative pulmonary tuberculosis histopathology. Boca Raton, London, New York: CRC Press, Taylor &Francis Group; pp 107–130 [Google Scholar]
- Klausner JD, Freedman VH, Kaplan G. 1996. Thalidomide as an anti-TNF-alpha inhibitor: implications for clinical use. Clin Immunol Immunopathol 81(3):219–223 [DOI] [PubMed] [Google Scholar]
- Koo MS, Manca C, Yang G, O'Brien P, Sung N, Tsenova L, Subbian S, Fallows D, Muller G, Ehrt S, Kaplan G. 2011. Phosphodiesterase 4 inhibition reduces innate immunity and improves isoniazid clearance of Mycobacterium tuberculosis in the lungs of infected mice. PLoS One 6(2):e17091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo MS, Subbian S, Kaplan G. 2012. Strain specific transcriptional response in Mycobacterium tuberculosis infected macrophages. Cell Commun Signal 10(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshy PJ, Lundy CJ, Rowan AD, Porter S, Edwards DR, Hogan A, Clark IM, Cawston TE. 2002. The modulation of matrix metalloproteinase and ADAM gene expression in human chondrocytes by interleukin-1 and oncostatin M: a time-course study using real-time quantitative reverse transcription-polymerase chain reaction. Arthritis Rheum 46(4):961–967 [DOI] [PubMed] [Google Scholar]
- Lambeth JD. 2004. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4(3):181–189 [DOI] [PubMed] [Google Scholar]
- Lee PP, Chan KW, Jiang L, Chen T, Li C, Lee TL, Mak PH, Fok SF, Yang X, Lau YL. 2008. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: a review of 17 patients living in a region endemic for tuberculosis. Pediatr Infect Dis J 27(3):224–230 [DOI] [PubMed] [Google Scholar]
- Li SG. 2011. Clinical image: development of miliary tuberculosis following one intraarticular injection of etanercept. Arthritis Rheum 63(5):1364. [DOI] [PubMed] [Google Scholar]
- Lin PL, Myers A, Smith L, Bigbee C, Bigbee M, Fuhrman C, Grieser H, Chiosea I, Voitenek NN, Capuano SV, Klein E, Flynn JL. 2010. Tumor necrosis factor neutralization results in disseminated disease in acute and latent Mycobacterium tuberculosis infection with normal granuloma structure in a cynomolgus macaque model. Arthritis Rheum 62(2):340–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly LH, Jeevan A, McMurray DN. 2009. Neutralization of TNFalpha alters inflammation in guinea pig tuberculous pleuritis. Microbes Infect 11(6–7):680–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. 1997. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci U S A 94(10):5243–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe YC, Kesavan AK, Lopez-Molina J, Hatem CL, Brooks M, Fujiwara R, Hochstein K, Pitt ML, Tufariello J, Chan J, McMurray DN, Bishai WR, Dannenberg AM, Jr, Mendez S. 2008. The aerosol rabbit model of TB latency, reactivation and immune reconstitution inflammatory syndrome. Tuberculosis (Edinb) 88(3):187–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan AK, Cote TR, Block JA, Manadan AM, Siegel JN, Braun MM. 2004. Tuberculosis following the use of etanercept, a tumor necrosis factor inhibitor. Clin Infect Dis 39(3):295–299 [DOI] [PubMed] [Google Scholar]
- Plessner HL, Lin PL, Kohno T, Louie JS, Kirschner D, Chan J, Flynn JL. 2007. Neutralization of tumor necrosis factor (TNF) by antibody but not TNF receptor fusion molecule exacerbates chronic murine tuberculosis. J Infect Dis 195(11):1643–1650 [DOI] [PubMed] [Google Scholar]
- Prieto-Pérez R, Cabaleiro T, Daudén E, Abad-Santos F. 2013. Gene polymorphisms that can predict response to anti-TNF therapy in patients with psoriasis and related autoimmune diseases. Pharmacogenomics J 13:297–305 [DOI] [PubMed] [Google Scholar]
- Redl H, Bahrami S, Schlag G, Traber DL. 1993. Clinical detection of LPS and animal models of endotoxemia. Immunobiology 187(3–5):330–345 [DOI] [PubMed] [Google Scholar]
- Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. 2002. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 168(9):4620–4627 [DOI] [PubMed] [Google Scholar]
- Roca FJ, Ramakrishnan L. 2013. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 153(3):521–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K. 2012. The pathology of Mycobacterium tuberculosis infection. Vet Pathol 49(3):423–439 [DOI] [PubMed] [Google Scholar]
- Solovic I, Sester M, Gomez-Reino JJ, Rieder HL, Ehlers S, Milburn HJ, Kampmann B, Hellmich B, Groves R, Schreiber S, Wallis RS, Sotgiu G, Scholvinck EH, Goletti D, Zellweger JP, Diel R, Carmona L, Bartalesi F, Ravn P, Bossink A, Duarte R, Erkens C, Clark J, Migliori GB, Lange C. 2010. The risk of tuberculosis related to tumour necrosis factor antagonist therapies: a TBNET consensus statement. Eur Respir J 36(5):1185–1206 [DOI] [PubMed] [Google Scholar]
- Subbian S, Bandyopadhyay N, Tsenova L.P OB, Khetani V, Kushner NL, Peixoto B, Soteropoulos P, Bader JS, Karakousis PC, Fallows D, Kaplan G. 2013a. Early innate immunity determines outcome of Mycobacterium tuberculosis pulmonary infection in rabbits. Cell Commun Signal 11(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbian S, O'Brien P, Kushner NL, Yang G, Tsenova L, Peixoto B, Bandyopadhyay N, Bader JS, Karakousis PC, Fallows D, Kaplan G. 2013b. Molecular immunologic correlates of spontaneous latency in a rabbit model of pulmonary tuberculosis. Cell Commun Signal 11(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbian S, Tsenova L, O'Brien P, Yang G, Koo MS, Peixoto B, Fallows D, Dartois V, Muller G, Kaplan G. 2011a. Phosphodiesterase-4 inhibition alters gene expression and improves isoniazid—mediated clearance of Mycobacterium tuberculosis in rabbit lungs. PLoS Pathog 7(9):e1002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbian S, Tsenova L, O'Brien P, Yang G, Koo MS, Peixoto B, Fallows D, Zeldis JB, Muller G, Kaplan G. 2011b. Phosphodiesterase-4 inhibition combined with isoniazid treatment of rabbits with pulmonary tuberculosis reduces macrophage activation and lung pathology. Am J Pathol 179(1):289–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbian S, Tsenova L, Yang G, O'Brien P, Parsons S, Peixoto B, Taylor L, Fallows D, Kaplan G. 2011c. Chronic pulmonary cavitary tuberculosis in rabbits: a failed host immune response. Open Biol 1(4):110016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb B, Morcock DR, Trubey CM, Quinones OA, Hao XP, Smedley J, Macallister R, Piatak M, Jr., Harris LD, Paiardini M, Silvestri G, Brenchley JM, Alvord WG, Lifson JD, Estes JD. 2013. Reduced inflammation and lymphoid tissue immunopathology in rhesus macaques receiving anti-tumor necrosis factor treatment during primary simian immunodeficiency virus infection. J Infect Dis 207(6):880–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. 2008. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 117(2):244–279 [DOI] [PubMed] [Google Scholar]
- Tsenova L, Harbacheusky R, Ellison E, Manca C, Kaplan G. 2006. Aerosol Exposure system for rabbits: application to M. tuberculosis infection. Appl Biosaf 11(1):7–14 [Google Scholar]
- Tubach F, Salmon D, Ravaud P, Allanore Y, Goupille P, Breban M, Pallot-Prades B, Pouplin S, Sacchi A, Chichemanian RM, Bretagne S, Emilie D, Lemann M, Lortholary O, Mariette X. 2009. Risk of tuberculosis is higher with anti-tumor necrosis factor monoclonal antibody therapy than with soluble tumor necrosis factor receptor therapy: the three-year prospective French Research Axed on Tolerance of Biotherapies registry. Arthritis Rheum 60(7):1884–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via LE, Lin PL, Ray SM, Carrillo J, Allen SS, Eum SY, Taylor K, Klein E, Manjunatha U, Gonzales J, Lee EG, Park SK, Raleigh JA, Cho SN, McMurray DN, Flynn JL, Barry CE, 3rd. 2008. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun 76(6):2333–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis RS. 2008. Tumour necrosis factor antagonists: structure, function, and tuberculosis risks. Lancet Infect Dis 8(10):601–611 [DOI] [PubMed] [Google Scholar]
- Wallis RS, Ehlers S. 2005. Tumor necrosis factor and granuloma biology: explaining the differential infection risk of etanercept and infliximab. Semin Arthritis Rheum 34(5 Suppl1):34–38 [DOI] [PubMed] [Google Scholar]
- Wallis RS, van Vuuren C, Potgieter S. 2009. Adalimumab treatment of life-threatening tuberculosis. Clin Infect Dis 48(10):1429–1432 [DOI] [PubMed] [Google Scholar]
- Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, Teoh LA, Fischkoff SA, Chartash EK. 2003. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum 48(1):35–45 [DOI] [PubMed] [Google Scholar]
- Winthrop KL, Baxter R, Liu L, Varley CD, Curtis JR, Baddley JW, McFarland B, Austin D, Radcliffe L, Suhler E, Choi D, Rosenbaum JT, Herrinton LJ. 2013. Mycobacterial diseases and antitumour necrosis factor therapy in USA. Ann Rheum Dis 72(1):37–42 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang H. 2012. Integrin signalling and function in immune cells. Immunology 135(4):268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.