

Abstract
Introduction
Mutations in the gene encoding the type II collagen gene (COL2A1) have been found to affect the entire skeletal system. Recently, inheritable skeletal dysplasia caused by novel COL2A1 mutations has been linked to an inherited disease of the hip joint that neither involves the entire skeletal system nor is characterized by the presence of concomitant disorders, such as spinal or ocular abnormalities.
Case presentation
A 27-year-old Japanese woman previously diagnosed with avasucular necrosis (AVN) of the femoral head on the basis of radiological findings was referred to the study site for surgical management of a painful hip joint. She had no history of disease but suffered from bilateral hip joint lesions. Analysis of her pedigree revealed that bilateral hip joint lesions affected more than three generations of her family. Based on these findings, haplotype analysis of her and her family members was performed by examining select candidate genes from the critical interval for epiphyseal dysplasia of the femoral head on 12q13 and sequencing the promoter and exonic regions of COL2A1.
Conclusion
A novel COL2A1 mutation (c.1744G>A) was identified within one Japanese family.
Keywords: COL2A1 mutation, Avascular necrosis of the femoral head, Skeletal dysplasia
Introduction
Mutations in the gene encoding the type II collagen gene (COL2A1) cause a series of type II collagenopathies that manifest as inheritable skeletal disorders, including achondrogenesis type 2 (ACG2; Langer-Saldino type), hypochondrogenesis, platyspondylic (Torrance type), congenital spondyloepiphyseal (SEDC), spondyloepimetaphyseal (SEMD) and Kniest dysplasia, Stickler syndrome type 1 and Stickler-like syndrome. These disorders often affect the entire skeletal system. Recently, such skeletal disorders have been reported to cause premature hip osteoarthritis (OA) [1], avascular necrosis (AVN) of the femoral head [2], and Legg-Calvé-Perthes disease (LCP) [3], and have been linked to an inherited disease of the hip joint that neither involves the entire skeletal system nor is characterized by the presence of concomitant disorders, such as spinal or ocular abnormalities.
Case presentation
A 27-year-old Japanese woman was admitted to a local hospital for treatment of a painful bilateral hip joint. Based on assessment of the radiological findings, she was diagnosed with AVN and referred to the study site for surgical management of her condition. The patient had no history of disease (for example, systemic lupus erythematosus (SLE) or human immunodeficiency virus (HIV)), alcohol abuse, steroid use, trauma or diving (causing dysbarism), and her laboratory findings were normal. However, as shown in her family pedigree, and her family clinical findings, more than three generations of her family have hip joint lesions: the proband (IV-1), her father (III-20) and her grandmother (II-9), II-1, 3, 5, III-3, 6, 11, 15 (Figure 1, Table 1). These findings suggest the presence of autosomal dominant inheritance with a high level of penetrance within this family (not shown).Although our patient’s radiological findings indicated osteonecrosis-like lesions of both femoral heads, the plain radiographic (Figure 2), magnetic resonance imaging (MRI) (Figure 3), and computed tomography (CT) (Figure 4) findings were negative for AVN induced by steroids and alcohol. Our patient’s anteroposterior (AP) radiographs of the other sites (knees, ankles, spine, hands, elbows) revealed almost normal findings (Figures 5 and 6).
Figure 1.
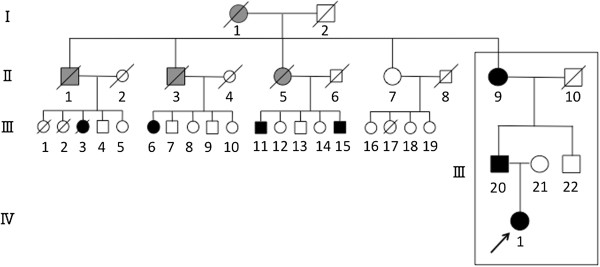
Pedigree of our patient’s family. Members I-1, II-1, 3, 5 (gray) had complained of coxalgia during their lifetimes and are suspected of having had hip joint disease. Members II-9, III-3, 20 had been diagnosed with end-stage OA and member III-3, 20 had required total hip arthroplasty. Members III-6, 11, 15 had been diagnosed with AVN and required trochanteric osteotomy. The proband (IV-1) is indicated by the black arrow.
Table 1.
Clinical findings of patient and family with lesions of the femoral head
| Sex | Height (cm) | Body weight (Kg) | BMI | Diagnosis | Treatment | |
|---|---|---|---|---|---|---|
|
Grandmother |
Female |
135 |
35 |
19.2 |
Osteoarthritis |
conservative treatment |
|
(II-9) | ||||||
|
Father |
Male |
156 |
62 |
25.5 |
Osteoarthritis |
THA |
|
(III-20) | ||||||
|
Pedigree |
Female |
149 |
39 |
17.6 |
Skeletal dysplasia |
Trochanteric curved varus osteotomy |
|
(IV-1) | ||||||
|
II-3 |
Female |
150 |
50 |
22.2 |
Osteoarthritis |
THA |
|
III-6 |
Female |
143 |
42 |
20.6 |
ION |
Trochanteric osteotomy |
|
III-11 |
Male |
150 |
50 |
22.2 |
ION |
Trochanteric osteotomy |
| III-15 | Male | 158 | 67 | 26.8 | ION | Trochanteric osteotomy |
Table 1 shows the clinical findings of more than three generations of the patient’s family who have hip joint lesions: the proband (IV-1), her father (III-20) and her grandmother (II-9), II-1, 3, 5, III-3, 6, 11, 15. *1 BMI, Body mass index; *2 THA, Total hip arthroplasty; *3 AVN, Avascular necrosis.
Figure 2.
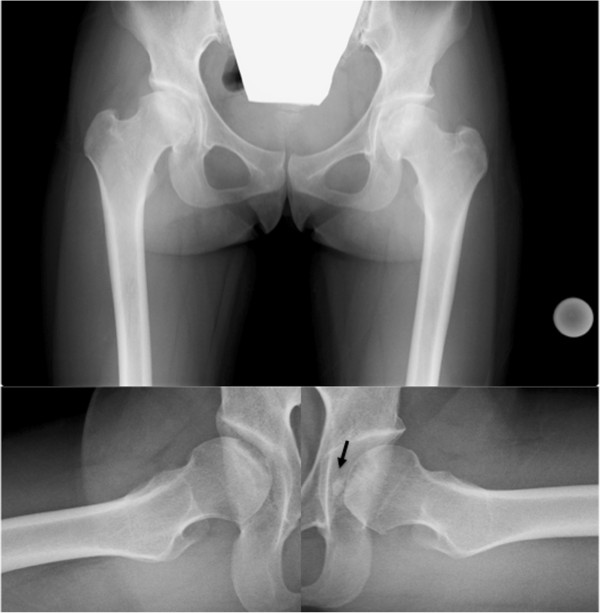
Plain radiographic findings of the femoral head. Anteroposterior (AP) and lateral radiographs (Sugioka) obtained at the time of the onset of pain show the crescent sign, collapse of femoral head, and no joint space narrowing in either femoral head. The lateral view (Sugioka) of left hip shows the presence of a free body (black arrow).
Figure 3.
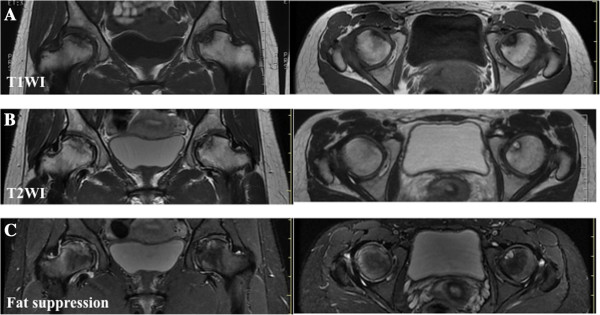
Magnetic resonance imaging findings of the femoral head. A) Coronal and axial slices of T1-weighted image show diffuse low-signal intensities in the femoral head. B) Coronal and axial slices of T2-weighted image show a combination of high- and low-signal intensities in the femoral head. C) This is a fat-suppression MRI in which part of the proximal portion beyond the low-intensity band shows contrast enhancement, an indication of perfusion.
Figure 4.
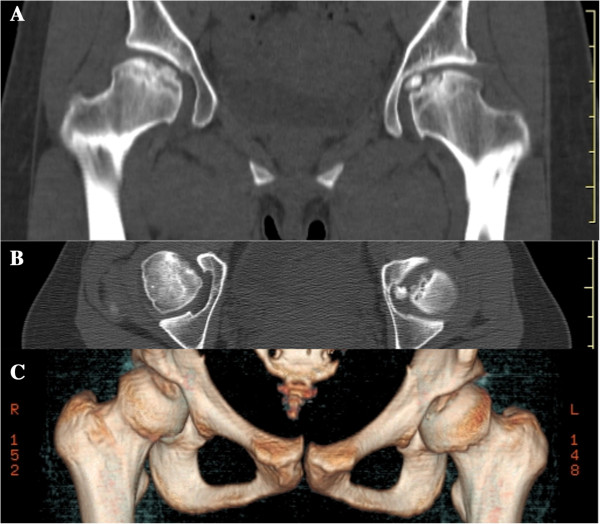
Computed tomography findings of the femoral head. Coronal (A) and axial (B and C) CT images of the right femoral head show a concave articular surface and a free body in the hip joint of the left femoral head. The free body is spherical and has a smooth surface.
Figure 5.
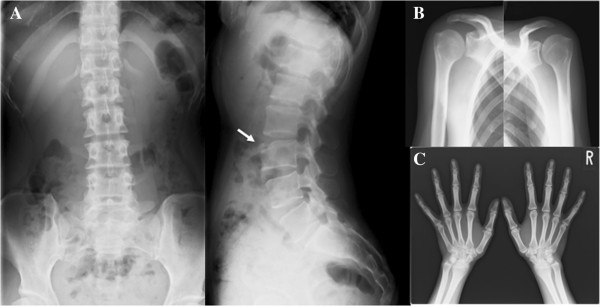
Plain radiographic findings of the lumbar spine, shoulder and hand. AP and lateral radiographs of the spine (A) show the third lumbar anterior goniodialysis (white arrow and circle) while AP radiographs of the shoulder (B) and hand (C) reveal almost normal findings.
Figure 6.
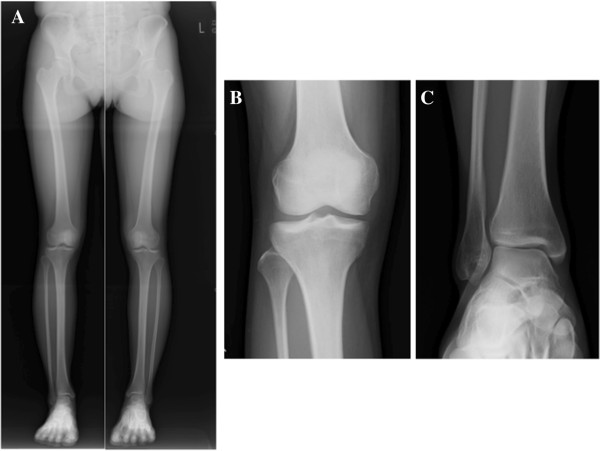
Plain radiographic findings of the full length of the lower limbs, knee and ankle. AP radiographs of the full length of the lower limbs (A), knee (B), and ankle (C) reveal almost normal findings with no limb shortening.
Haplotype analysis of the family members was performed by examining select candidate genes from the critical interval for epiphyseal dysplasia of the femoral head on 12q13. The entire coding regions of COL2A1, along with the flanking intronic regions, were amplified by polymerase chain reaction (PCR) using the Ex Taq™ system with an ABI PRISM® 3100 Genetic Analyzer (Applied Biosystems, Foster, CA, USA) and a BigDye® Terminator Cycle Sequencing Kit (Applied Biosystems). The complementary deoxyribonucleic acid (cDNA) sequence of COL2A1 was obtained from GenBank (accession no. NM_001844.4). For cDNA numbering, +1 corresponds to the A of the ATG translation initiation codon 1 in the reference sequence (Figure 7). A G→A transition of COL2A1 (c.1744G>A (p.Gly582Ser)) was detected in the affected member in each of the three generations of the family (Figures 8 and 9). This transition predicts the replacement of glycine with serine in COL2A1. Based on this finding, members of the family were diagnosed with inherited epiphyseal dysplasia located on the femoral head. For those three affected members in whom autosomal dominant inheritance of the disease was strongly suspected (IV-1, III-20, II-9), the chromosomal position of the gene to COL2A1 (c.1744G>A (p.Gly582Ser)) was mapped. No mutation was detected in the COL2A1 coding region of the mother or in a normal control (Figure 9).
Figure 7.
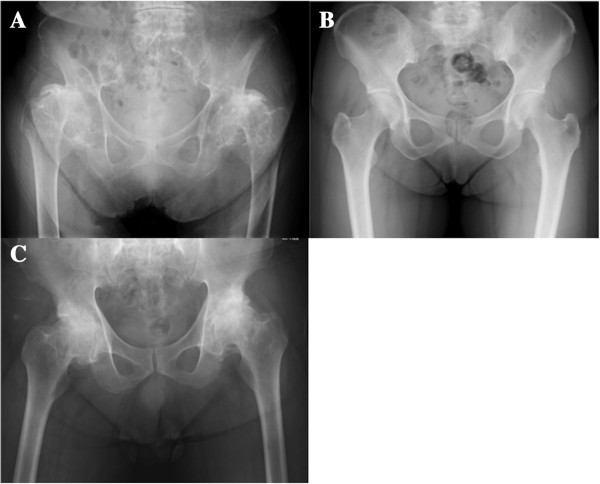
Plain radiologic findings of our patient’s family. Plain radiographs of our patient’s grandmother (A) and father (B) show end-stage OA, while a radiograph of our patient’s mother (C) appears almost normal.
Figure 8.
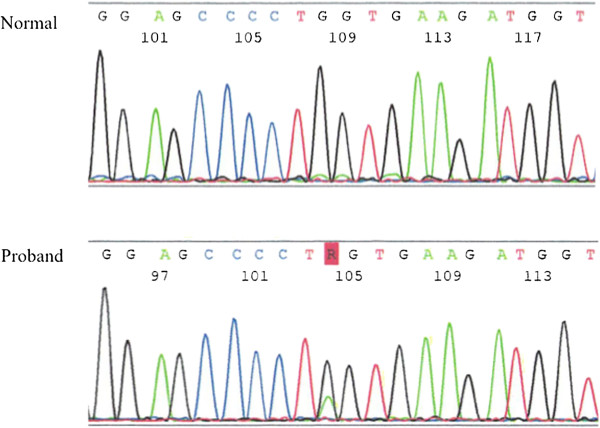
COL2A1mutations in our patient. Sequencing analysis shows a G→A transition of COL2A1 (c.1744G>A (p.Gly582Ser)). This transition predicts the replacement of glycine with serine at codon 382 in COL2A1.
Figure 9.
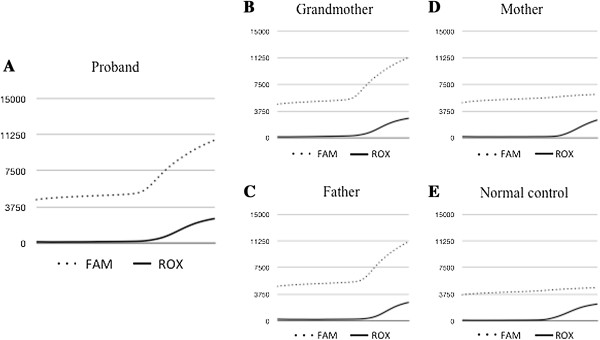
Real-time polymerase chain reaction analysis of our patient’s family and a normal control. Probe-adapted real-time PCR analysis using cycling probe technology led to the detection of a single nucleic acid polymorphism (SNP) in a target DNA sequence in our patient’s father and grandmother. For SNP typing, two cycling probes labeled with two different fluorescent dyes (6-carboxyfluorescein (FAM) or 6-carboxy-X-rhodamine (ROX)) were used, with each probe harboring ribonucleic acid (RNA) corresponding to the wild-type nucleotide or the nucleotide with a mutation at the SNP position. Expression analysis of our patient (A), her father (B) and her grandmother (C) shows a G→A transition of COL2A1 (c.1744G>A), while expression analysis of her mother (D) and a normal control (E) shows no mutation in the COL2A1 coding region.
Discussion
In this study, we identified a novel COL2A1 mutation (c.1744G>A (p.Gly582Ser)) from the chromosome 12q13 region within one Japanese family. A history of hip disorder and typical radiologic findings lend support to the idea that the identified mutation in the COL2A1 gene transition might be the cause of the disease in this family.
COL2A1 mutations typically manifest as inheritable skeletal disorders often affecting the entire skeletal system [4]. Except for the hip joint, the affected members of this family were otherwise normal, with normal spine development. Moreover, they had no anomalies of the ocular or auditory system.
Recently, inheritable epiphyseal dysplasia caused by novel COL2A1 mutations has been linked to three inherited diseases of the hip joint. Kannu et al. reported premature osteoarthritis as well as LCP as presenting with COL2A1 mutation [1,5]. On the other hand, Liu et al. reported three families with AVN which were carriers of the COL2A1 mutation [2]. Recently, Su et al. reported that age at onset in relation to closure of the femoral head epiphysis is a critical factor in determining the disease pattern [6]. However, there is no clear consensus as to inherited hip disease induced by COL2A1 mutations. In this study, the father of our patient who, along with her grandmother, had been diagnosed with end-stage OA, had required total hip arthroplasty, while several of her more distant relatives diagnosed with AVN had required trochanteric osteotomy and total hip arthroplasty.
Osteoarthritis, AVN and LCP are difficult to distinguish, as both symptoms and clinical findings are similar. We believe that we should recognize familial hip dysplasia induced by COL2A1 mutation. However, the diagnosis will differ depending on the age at presentation to hospital. It is predicted that cartilaginous structure abnormality is already in existence at the time of the growth spurt. In addition, in joint cartilage, type 2 collagen forms a complex with proteoglycan. A decrease in this type II proteoglycan complex results in a decrease in cartilaginous elasticity. When there is disease progression without symptoms, cartilaginous abrasion and damage and subchondral bone exposure occurs until, ultimately, severe OA results. Nishimura et al. reported that glycine to serine substitutions resulted in alternating zones that produce severe and milder skeletal phenotype [4]. Our result was similar, as this transition predicted the replacement of glycine with serine in COL2A1 (p.Gly582Ser). The patient, whose lesions were localized to the hip joint, had neither a remarkable clinical history nor remarkable radiological (Computed radiography (CR), MRI, and CT) characteristics. As her family findings indicated, the phenotypic spectrum of COL2A1 mutations may be difficult to diagnose, leading to concern regarding the possibility of misdiagnosis as type II collagenopathy presenting as LCP [3] (in patients 0 to 18 years of age), AVN [2] (in patients 18 to 45 years of age) or OA (in patients 45 years of age and older). The findings of this study also indicate that the severity of the disease might be affected by the site of the COL2A1 mutation.
We believe that new discoveries in molecular genetics have allowed for identification of the genes causing a large number of skeletal disorders, improving the means of their classification and differentiation.
Conclusion
A novel COL2A1 mutation (c.1744G>A (p.Gly582Ser)) was identified within one Japanese family with epiphyseal dysplasia. Genetic analysis revealed that more than three generations carried the COL2A1 mutation causing this disorder.
Consent
This study was approved by the Ethics Committee of Hirosaki University Graduate School of Medicine and conducted according to the principles of the Declaration of Helsinki. Written informed consent was obtained from our patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Abbreviations
ACG2: Achondrogenesis type 2; AVN: Avascular necrosis; COL2A1: Collagen, type II, alpha 1; HIV: Human lummunodeficiency virus; LCP: Legg-Calvé-Perthes disease; OA: Osteoarthritis; SEDC: Spondyloepiphyseal dysplasia congenita; SEMD: Spondyloepimetaphyseal dysplasia; SLE: Systemic lupus erythematosus; SNP: Single nucleic acid polymorphism.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MK analyzed and interpreted the patient data regarding the skeletal dysplasia. YN, HO and KF were involved in reviewing the literature and proofreading the manuscript, and were major contributors in writing the manuscript. YN performed the final revisions of the manuscript. YI is the senior author and was responsible for final proofreading of the article. All authors read and approved the final manuscript.
Contributor Information
Masaki Kishiya, Email: kishiya@isis.ocn.ne.jp.
Yoshihide Nakamura, Email: yoshi-n@cc.hirosaki-u.ac.jp.
Hirotaka Ohishi, Email: ohishi@cc.hirosaki-u.ac.jp.
Ken-Ichi Furukawa, Email: furukawa@cc.hirosaki-u.ac.jp.
Yasuyuki Ishibashi, Email: yasuyuki@cc.hirosaki-u.ac.jp.
Acknowledgements
We thank Shiro Ikegawa for his helpful discussion regarding the subject of epiphyseal dysplasia and assistance in sequencing all the coding regions of COL2A1.
References
- Kannu P, Bateman JF, Randle S, Cowie S, du Sart D, McGrath S, Edwards M, Savarirayan R. Premature arthritis is a distinct type II collagen phenotype. Arthritis Rheum. 2010;62:1421–1430. doi: 10.1002/art.27354. [DOI] [PubMed] [Google Scholar]
- Liu YF, Chen WM, Lin YF, Yang RC, Lin MW, Li LH, Chang YH, Jou YS, Lin PY, Su JS, Huang SF, Hsiao KJ, Fann CS, Hwang HW, Chen YT, Tsai SF. Type II collagen gene variants and inherited osteonecrosis of the femoral head. N Engl J Med. 2005;352:2294–2301. doi: 10.1056/NEJMoa042480. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Matsuda T, Kitoh H, Haga N, Ohashi H, Nishimura G, Ikegawa S. A recurrent mutation in type II collagen gene causes Legg-Calve-Perthes disease in a Japanese family. Hum Genet. 2007;121:625–629. doi: 10.1007/s00439-007-0354-y. [DOI] [PubMed] [Google Scholar]
- Nishimura G, Haga N, Kitoh H, Tanaka Y, Sonoda T, Kitamura M, Shirahama S, Itoh T, Nakashima E, Ohashi H, Ikegawa S. The phenotypic spectrum of COL2A1 mutations. Hum Mutat. 2005;26:36–43. doi: 10.1002/humu.20179. [DOI] [PubMed] [Google Scholar]
- Kannu P, Irving M, Aftimos S, Savarirayan R. Two novel COL2A1 mutations associated with a Legg-Calve-Perthes disease-like presentation. Clin Orthop Relat Res. 2011;469:1785–1790. doi: 10.1007/s11999-011-1850-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su P, Zhang L, Peng Y, Liang A, Du K, Huang D. A histological and ultrastructural study of femoral head cartilage in a new type II collagenopathy. Int Orthop. 2010;34:1333–1339. doi: 10.1007/s00264-010-0985-9. [DOI] [PMC free article] [PubMed] [Google Scholar]