

Abstract
Regarding oral squamous cell carcinoma (OSCC) development, chewing areca is known to be a strong risk factor in many Asian cultures. Therefore, we established an OSCC induced mouse model by 4-nitroquinoline-1-oxide (4-NQO), or arecoline, or both treatments, respectively. These are the main two components of the areca nut that could increase the occurrence of OSCC. We examined the effects with the noncommercial MCGI (mouse CpG islands) microarray for genome-wide screening the DNA methylation aberrant in induced OSCC mice. The microarray results showed 34 hypermethylated genes in 4-NQO plus arecoline induced OSCC mice tongue tissues. The examinations also used methylation-specific polymerase chain reaction (MS-PCR) and bisulfite sequencing to realize the methylation pattern in collected mouse tongue tissues and human OSCC cell lines of different grades, respectively. These results showed that retinoic acid receptor β (RARB) was indicated in hypermethylation at the promoter region and the loss of expression during cancer development. According to the results of real-time PCR, it was shown that de novo DNA methyltransferases were involved in gene epigenetic alternations of OSCC. Collectively, our results showed that RARB hypermethylation was involved in the areca-associated oral carcinogenesis.
1. Introduction
Throughout the world, oral squamous cell carcinoma (OSCC) is one of the most common types of cancers. It has a high cure rate for small primary tumors and involves the development of second primary tumors and the long-term survival rate is <60% [1]. Furthermore, in Taiwan, according to statistics from the Department of Health, Executive Yuan, Taiwan, OSCC ranks as fourth among the ten leading causes of cancer among males and is the fourth leading cancer in the male population and the number of deaths increases every year [2]. The main risk factor for developing OSCC is chewing areca, especially in many Asian cultures.
In a clinical study, the incidence of oral cancer was elevated 28 times for betel quid users as compared to nonbetel quid users [3]. Cigarette smoking has synergistic effect with areca chewing, and such users have an 89 times higher incidence rate than nonusers. If one has the habit of drinking, smoking, and betel quid chewing combined, there will be a 123 times higher incidence rate of having oral cancer than those average individuals in the general population that are nonusers. There is the longitudinal cohort study on the alcohol, betel quid, and smoking, to the oral cancer risk. The betel quid partook the significant higher hazard risk to the oral carcinogenesis [4]. The most tumorigenic part of betel quid is the Piper longum L. and the calcium hydroxide (slaked lime) which will cause the oral cavity to develop into an alkaline condition which will promote the tumorigenic effect of the Safrole in the Piper longum L. The fibers of areca also cause oral mucosa damage and increased mucosa to be exposed to the tumorigenic material in the betel quid. In a clinical survey, oral cancer with areca chewing had far more incidence of oral submucosa fibrosis and erythroplakia; furthermore, the pathologic findings also show severe hyperkeratosis, caries, and gingivitis as compared to nonareca users. The oral cancer patients who had the habit of betel quid chewing were also found to have a higher percentage of dysplastic change surrounding the tumor margin, and skip cancer lesion was frequently noted in the upper aerodigestive tract (tongue, hypopharynx, and esophagus). The condemned mucosa even reached the esophagus. In an average clinical survey, 18% of cases were found to have esophagus cancer diagnosed at the same time when oral cancer presented [5]. Synchronous double cancer (second primary cancer) in the upper aerodigestive tract is frequently noted [6].
There was a stimulating effect when areca nut is chewed along with betel leaf [7]. Furthermore, in Chiang et al. [8], they used areca nut extract (ANE) and saliva-reacted ANE (sANE) to treat three oral carcinoma cell lines, KB (epidermoid carcinoma), SAS (tongue carcinoma), and Ca9-22 (gingival carcinoma). The higher cytotoxic effects involving cell morphologic changes and upregulation of inflammatory signaling in mRNA expression levels were observed in these treatments. In addition, arecoline is the major alkaloid in areca nut extracts and betel quid. It is the primary active ingredient responsible for the central nervous system simulation that is roughly comparable to that of nicotine, which has a similar chemical structure [9–12]. There is also another carcinogen, 4-nitroquinoline 1-oxide (4-NQO), which effectively induces oral and esophageal cancers that closely resemble early human lesions in mice and rats [13, 14]. In this study, we followed Chang et al. [15] who established an effective mouse model of oral cancer and used this model to identify potential markers of oral tumor progression by utilizing a noncommercial methylation microarray.
The promoter hypermethylation now has a key role for research in the area of human multistage carcinogenesis. Silencing of certain tumor suppressor genes may occur in the absence of genetic change, via aberrant methylation of CpG islands [16–18]. OSCC is believed to arise through the accumulation of numerous genetic and epigenetic alterations [19–21]. There are several methods to determine whether promoter methylation has been developed including combination of bisulfite restriction assay (COBRA) [22], genomic bisulfate sequencing [23], methylation-specific PCR (MS-PCR) [24], and microarray-based methylation analysis [25]. Methylation microarray is a high throughput tool for genome-wide methylation analysis [26–31]. To identify and characterize potential targets for treating oral cancer, a genome-wide approach was taken to quantitatively measure genomic alterations in OSCC [32, 33]. Consequently, we used home-made mouse CpG island microarray to understand aberrant methylation profile during OSCC tumorigenesis in this study and validated the methylation status by MS-PCR, bisulfite sequencing, and real-time PCR.
In many previous studies, retinoid acid suppresses carcinogenesis and inhibits the growth of human head and neck squamous cell carcinoma (HNSCC) [34, 35]. Loss of retinoids and their receptors has been associated with malignant progression in HNSCC [36]. Their receptors (RAR) are central regulators to the normal growth and differentiation of a variety of epithelial cells. RAR changes have been associated with cell immortalization, and re-expression of RAR-beta (RARB) leads to growth inhibition in some circumstances [19]. Loss of RARB expression is associated with a change in proliferative life span potential from mortality to immortality in HNSCC [37–39]. The promoter hypermethylation of RARB could inhibit the gene expression when added to the methylation inhibitor and deacetylation inhibitor like 5′-aza-2′-deoxycytidine (5-aza-dC) and trichostatin A (TSA) which could recover the gene expression and inhibit tumor cells growth [36, 39].
In the present study, we investigated the role of RARB hypermethylation of CpG islands in OSCC mouse model and its association with RARB expression in human oral cancer cell lines. In addition, we examined whether the repression of RARB transcription could be reversed by 5-aza-dC in human oral cancer cell lines, and finally, we evaluated the three main DNA methyltransferases that were involved in RARB hypermethylation.
2. Materials and Methods
2.1. Mouse Model for Oral Cancer
The mouse model development was modified as highlighted by Chang et al. [15, 40]. Briefly, the OSCC model was established by treating arecoline (Sigma, St. Louis, MO), as well as in combination with 4-NQO (Fluka, St. Louis, MO) in 4-5 week age old of C57BL/6JNarl male mice. The conditions for OSCC formation are 500 μg/mL arecoline (A), 200 μg/mL 4-NQO (N), and 4-NQO (200 μg/mL) combined with arecoline (500 μg/mL) (NA) in the drinking water for 8 weeks. The drinking water was changed every day, and mice were allowed access to the drinking water at all times while receiving treatment. After the treatment, the drinking water was changed to ddH2O and mice were sacrificed at week 8, 12, 14, 18, 20, 26, and 28, respectively. The tongues were collected and classified into tumor parts (T) and nontumor parts (NT) for mouse CpG island microarray analysis.
2.2. Cell Culture and 5-aza-dC Treatment
Normal human oral keratinocytes (NHOK) were cultured in Keratinocyte Growth Medium (KGM, GIBCO, CA, USA). Oral cancer cell lines, DOK, OC2, and Ca9-22, were cultured in DMEM (GIBCO, CA, USA). HSC3 and TW2.6 were cultured in DMEM-F12 (GIBCO, CA, USA). All cells supplemented with 10% fetal calf serum and 1% penicillin-streptomycin and cultured at 37°C with 5% CO2. Oral cancer cells were treated by 5-aza-dC at 2 μM to reverse the methylation status as described in [36, 41].
2.3. DNA and RNA Extraction
The genomic DNA extraction was conducted as noted in our previous report [42]. We used collected nontumor parts (NT) and tumor parts (T) of tongues for DNA and RNA extractions.
2.4. RT-PCR and Real-Time PCR
Total RNA was prepared using the TRI REAGENT (Invitrogen, CA, USA). One microgram of total RNA was treated with 10 units of RQ1 RNase-Free DNase (Prome ga, WI, USA) and extracted with phenol-chloroform. DNase that treated total RNA (1 μg) was reverse transcribed with the ImProm II Reverse Transcription System (Promega, WI, USA). For RT-PCR amplification was performed with 2720 thermal cycler (Applied Biosystems Inc., CA, USA) and Real-Time PCR amplification was performed with Rotor-Gene 6000 (Corbett, CA, USA). The amplification of RT-PCR was repeated for 28 cycles as follows: 95°C, 30 sec for denature of the annealing temperature depending on the pair of gene specific primer sets (Table 1) for 30 sec and 72°C, 30 sec for extension. PCR reactions were performed in triplicate and the transcription level was normalized with the GAPDH. For Real-Time PCR, the calculated gene expression fold from CT value was performed according to the previously mentioned study, with a P value of less than 0.05 exhibiting an obviously significant difference.
Table 1.
Primer sets used for RT-PCR, MS-PCR, and bisulfite sequencing.
| Primer sets | Sense primer (5′→3′) | Antisense primer (5′→3′) | Tm (°C) | PCR size (bp) |
|---|---|---|---|---|
| RARB-Hu-RT | AGGAGACTTCGAAGCAAG | GTCAAGGGTTCATGTCCTTC | 60 | 771 |
| Dnmt1-Hu-RT | TACCTGGACGACCCTGACCTC | CGTTGGCATCAAAGATGGACA | 60 | 102 |
| Dnmt3a-Hu-RT | TATTGATGAGCGCACAAGAGAGC | GGGTGTTCCACCCTAACATTGAG | 64 | 110 |
| Dnmt3b-Hu-RT | GGCAAGTTCTCCGAGGTCTCTG | TGGTACATGGCTTTTCGATAGGA | 62 | 112 |
| RARB-Mo-M | GGATTAGAGTTTTCGTGCGTCG | TACCCCGCCGATACCCAAACG | 65 | 90 |
| RARB-Mo-U | GGATTAGAGTTTTTGTGTGTTG | TACCCCACCAATACCCAAACA | 62 | 90 |
| RARB-Mo-BS | CCACCCAACTCCATCAAACTC | CCATACAATCAAACATAATCTC | 58 | 476 |
| RARB-Hu-M | ATGTCGAGAACGCGAGCGATTC | CTCGACCAATCCAACCGAAACG | 64 | 151 |
| RARB-Hu-U | GGATGTTGAGAATGTGAGTGATTT | TACTCAACCAATCCAACCAAAACA | 62 | 155 |
| RARB-Hu-BS | GTGTGATAGAAGTAGTAGGAAG | GTGATAGAAGTGGTAGGAAG | 55 | 401 |
∗Hu: human, Mo: mouse, RT: real-time RT-PCR, M: methylated set, U: unmethylated set, BS: bisulfite sequencing.
2.5. Preparation of Mouse CpG Island Microarray and Amplicon Generation
The mouse CpG island microarray was based on previously described human CpG island microarrays [43–46]. A total 2,304 mouse CpG islands library (mCGI) clones were spotted on UltraGAPS Coated Slides (Corning, MA, USA) by the BioDot AD1500 (BIODOT, CA, USA). The amplicons for methylation analysis were prepared as previously described [47, 48].
2.6. Microarray Hybridization and Data Analysis
The purified amplicons (5 μg) were conducted using the BioPrime DNA labeling system (Invitrogen, CA, USA). Cyanine 5-ddUTP (Cy5-ddUTP) and Cyanine 3-ddUTP (Cy3-ddUTP) (Perkin-Elmer Life Sciences, NJ, USA) fluorescent dyes were coupled to tumor (T) and normal (NT) amplicons, respectively, and cohybridized to the microarray panel. The combined tumor/normal control pair, with 8 μg DNA, more than 180 pmol Cy5, and 150 pmol Cy3, would give strong hybridization signals. The hybridization of 4,608 spots is carried out under a 24 × 50 mm cover glass sealed tightly within a moistened hybridization chamber, GeneMachines HybChambers (Genomic Solutions, MI, USA), in a 65°C water bath from 12 to 16 h. The posthybridization washing steps are essentially those described by UltraGAPS Coated Slides instruction Manual. The hybridized slides were scanned with the GenePix 4000B scanner (Axon, CA, USA) and the acquired images were analyzed with the software GenePix Pro 4.0 (Axon, CA, USA). The microarray data was analyzed as described previously [43–46, 48, 49]. Briefly, the Cy5/Cy3 ratio and the hybridization intensity from the tumor amplicons to the hybridization intensity from the normal amplicons, from each image, are normally guided by both the average global Cy5/Cy3 ratio from each image and the Cy5/Cy3 ratios from 9 internal controls (clones without restriction cutting sites whose copy numbers remain the same in tumors and normal samples). Yellow spots (normalized Cy5/Cy3 = 1) represent equal amounts of bound DNA from each amplicon, indicating no methylation differences between tumor (T) and nontumor (NT) genomes. The analyzed data were using hierarchical clustering to classify the relationships of all genes between collected T and NT samples. A hierarchical clustering algorithm was used to investigate relationships among tumor samples. The complete linkage and the dissimilarity measure (1 minus the Pearson correlation coefficient of the log-adjusted Cy5 : Cy3 ratios) were used for the analysis. The resultant dendrogram showed linked closely related colorectal tumors into a phylogenetic tree whose branch lengths represented the degree of similarity between these tumors.
2.7. Bisulfite Sequencing and Methylation-Specific PCR (MS-PCR) for Methylation Status Analysis
Genomic DNA (~0.5 μg) was treated with sodium bisulfite according to the manufacture's recommendations (EZ DNA Methylation Kit; Zymo Research, CA, USA). All selected genes methylation statuses were examined by methylation-specific PCR (MS-PCR) and sodium bisulfite genomic sequencing. The PCR reaction was as follows: 95°C for 5 min, followed by 45 cycles of 95°C, 30 sec, Tm for 30 sec (Table 1), 72°C for 45 sec and ended with an extension of 72°C for 5 min and quick chill to 4°C on a Geneamp2400 PCR system (Applied Biosystems, CA, USA). For bisulfate sequencing analysis, each PCR product was subcloned into the pGEM-T Easy Vector (Promega, WI, USA) and performed 5–10 clones in each selection, respectively. Each colony was sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit and the automated ABI PRISM 3100 Genetic Analyzed (Applied Biosystems, CA, USA).
2.8. Statistical Analysis
Statistical analysis was performed using t-test to examine the association between NHOK and other cell lines. One-sided testing was used to calculate the P, and P < 0.05 was considered statistically significant.
3. Results
3.1. OSCC Mouse Model Induced by Arecoline and 4-NQO
To evaluate the efficiency of mouse model involving cotreating with arecoline and 4-NQO that mimic the etiology for OSCC tumor growth, the percentage of mice exhibiting carcinogenesis was calculated in Figure 1(a). Tumor development was assessed when treated with 4-NQO (N) and 4-NQO plus arecoline (NA) but not in arecoline. Mice were also sacrificed at 18, 26, and 28 weeks, and tongues with tumors were excised, fixed, embedded, and sectioned for H&E staining (Figure 1(b)). The H&E staining also showed that the tumor progresses were dealing with time and according to the treatments. According to the results, the incidence of tongue carcinogenesis in NA group was significantly higher than N group and arecoline group. Taken together, the treatment of NA at week 28 was much more serious than other weeks. These results suggest that arecoline promotes 4-NQO carcinogenesis in damaged oral epithelia cells.
Figure 1.
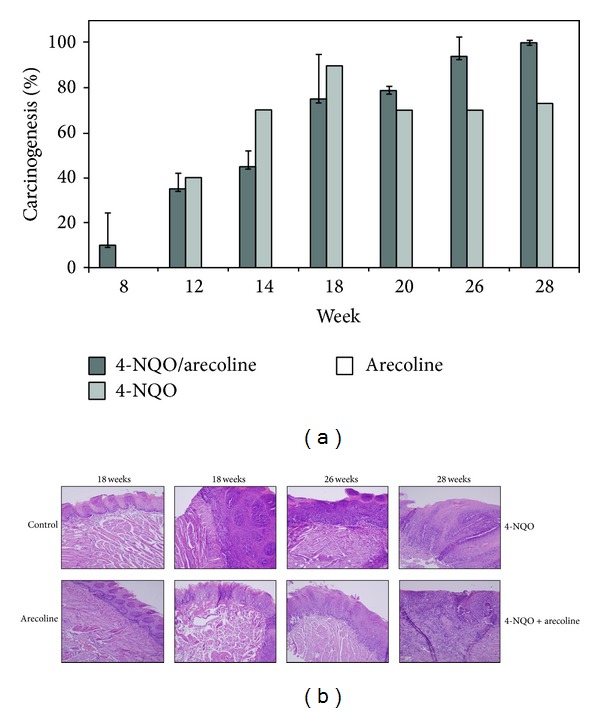
The progression of mouse model development for OSCC. (a) The ratio of carcinogenesis in mouse OSCC model. There were three treatments, 4-NQO/arecoline, 4-NQO, and arecoline. Mice were sacrificed at weeks 8, 12, 14, 18, 20, 26, and 28, respectively. The scoring criteria for mouse OSCC model are described in Section 2. (b) OSCC tongue tissues with tumors were excised, fixed, embedded, and sectioned for H&E staining. The mice that were treated with 4-NQO + arecoline would induce more serious OSCC formation than 4-NQO only and arecoline only. The order of severity was followed the time of treatment.
3.2. Identification of Hypermethylation Genes from OSCC Tissues by CpG Island Microarray
According to the tumor progression percentages and H&E staining results, we selected the tongues tissues to be targets from OSCC mouse model with N and NA at 26 and 28 weeks for MCGI microarray screening. Figure 2(a) depicts representative data from the OSCC study. The expanded hybridization views showed the usefulness of the MCGI microarrays cohybridized with fluorescently labeled T (tumor part) and NT (nontumor part) with N at week 28 and NA at weeks 26 and 28, respectively. Spots hybridized predominantly with tumor amplicon, but not with nontumor amplicon, would appear red and are indicative of hypermethylated CpG island loci, present in the tumor genome. The hybridization results showed that treatment with NA at week 28 represented much more spots that were obviously more hypermethylated than other treatments. Yellow spots (Cy5 : Cy3 = 1) represent equal amounts of bound DNA from each amplicon, an indication of no methylation differences between tumor and nontumor genomes. Selection of genes was based on the criteria described in the Materials and Methods. Figures 6(b) and 6(c) showed specific genes of selection results in hypermethylation and hypomethylation, respectively. We conducted a confirmation study to determine whether the cutoff ratio (≧2) could accurately identify hypermethylation. The hierarchical clustering presented the 109 gene loci of hypermethylation in the classifier in N and NA (Figure 2(b)). This methylation profile analysis has led to the identification of CpG island clusters that could evaluate many new genes correlating with OSCC progression in mouse model. These newly collected genes are shown in detail in Table 2. Upon further examination, we selected the RARB gene to examine in greater detail by MS-PCR bisulfite sequencing and semiquantitative RT-PCR. The locations of CpG islands in mouse and human RARB genes were predicted using MethPrimer (http://www.urogene.org/methprimer/index1.html), respectively (Figures 3(a) and 4(a)).
Figure 2.
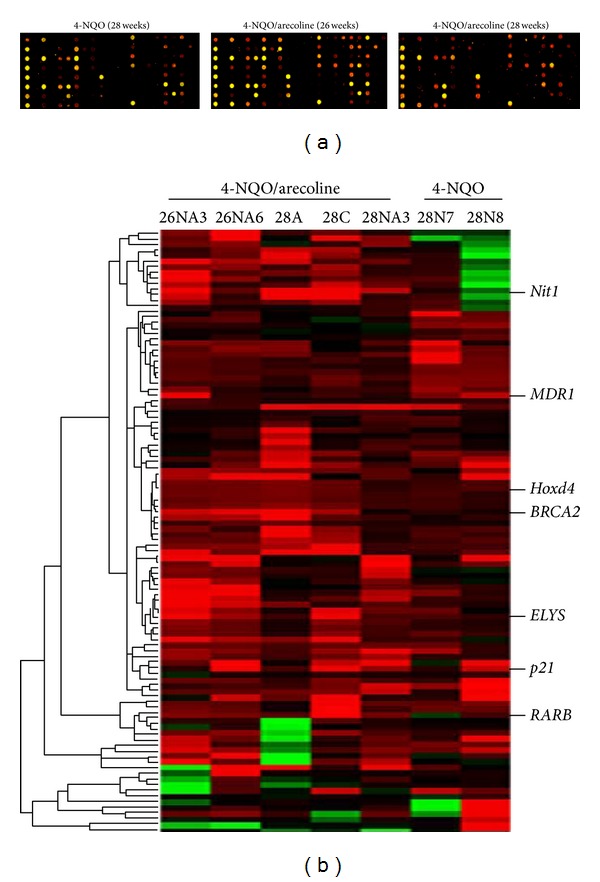
Presenting of MCGI microarray hybridization results and hierarchical clustering of methylation data of OSCC model mice. (a) MCGI microarray hybridization panel contained 4,608 duplicated CpG island tags. The expanded hybridization views showed the usefulness of the MCGI microarrays cohybridized with fluorescently labeled T (tumor part) and NT (nontumor part) with N at 28 weeks and NA at 26 and 28 weeks. Spots hybridized predominantly with tumor amplicon but not with normal amplicon would appear red and are indicative of hypermethylated CpG island loci present in the tumor genome. (b) Hierarchical clustering of N (4-NQO) and NA (4-NQO/arecoline) samples. At the top lists the 5 NA and 2 N studied. The row corresponds to each of 109 CpG island loci selected for methylation analysis. CpG islands (the normalized Cy5 : Cy3 ratios are ≧2) are those with hypermethylation in tumor DNA. The selected hypermethylated candidates were showed on the right sides. They are described in greater detail in Table 2.
Figure 6.
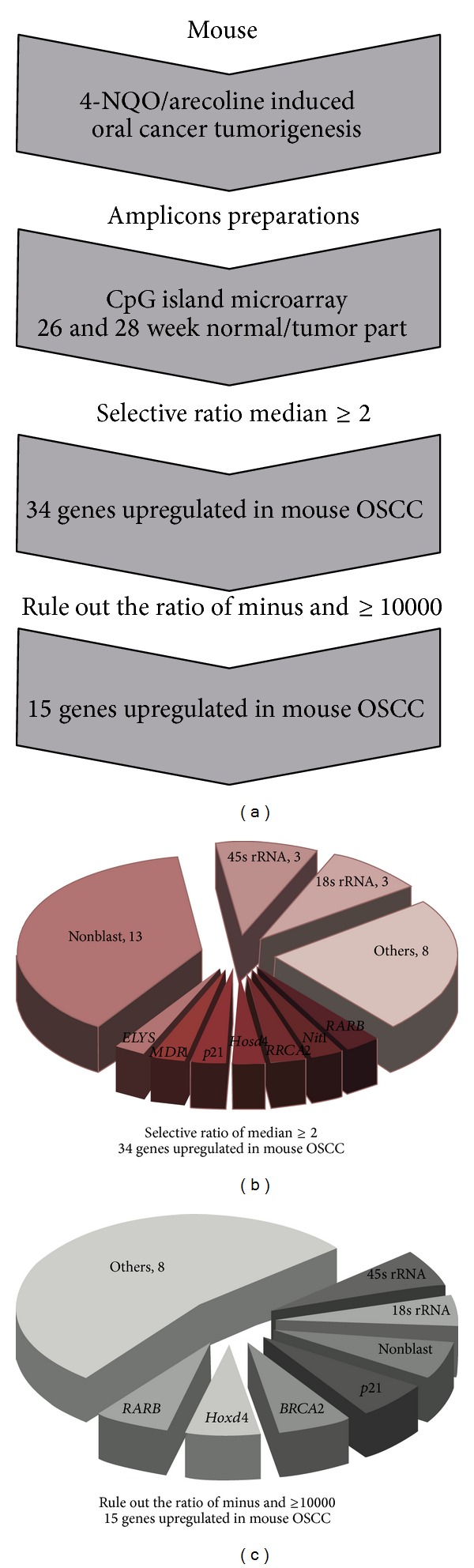
Outline of this study. (a) The flowchart of research project. (b) The selection from MCGI microarray hybridization results by the ratio higher or equal two. There were 34 genes that were hypermethylated in OSCC mouse model. (c) The selection from MCGI microarray hybridization results by ruling out the ratio was minus and higher or equal to 10,000. There were 15 genes that were hypermethylated in OSCC mouse model.
Table 2.
The selected gene list of hypermethylation.
| Accession number | Gene symbol | Description | Functions | Related carcinoma types |
|---|---|---|---|---|
| S80555 | RARB | Retinoic acid receptor-beta | DNA binding, ligand-dependent nuclear receptor activity | Nonsmall cell lung cancer, hepatoma, bladder cancer, rectal cancer, breast cancer, head and neck squamous cancer |
| AF069985 | Nit1 | Nitrilase homolog I | Hydrolase activity | Oral squamous cell carcinoma (in this study) |
| AL355176 | BRCA2 | Breast cancer II gene | Protein binding | Bladder cancer, breast cancer, ovarian cancer |
| AL928664 | Hoxd4 | Homeo box D4 gene | DNA binding | Breast cancer, leukemia, neuroblastoma |
| AF457187 | p21 | Cyclin-dependent kinase inhibitor IA | Cyclin-dependent protein kinase inhibitor activity | Nonsmall cell lung cancer, bladder cancer, breast cancer, ovarian cancer, medulloblastoma, hepatoma |
| M60348 | MDR1 | Multidrug-resistance protein gene | ATP binding, ATPase activity | Pancreatic cancer, breast cancer, colorectal cancer, glioblastoma, leukemia, laryngeal cancer cell |
| AB081498 | ELYS | Embryonic large molecule derived from yolk sac | DNA binding | Oral squamous cell carcinoma (in this study) |
Figure 3.
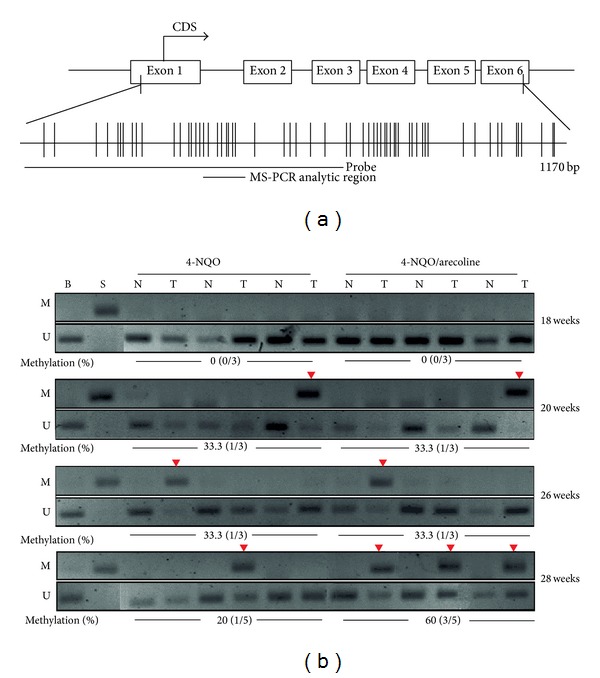
Methylation status of RARB gene in mouse OSCC model. (a) The CpG island diagram of RARB gene. The hybridized probe used to microarray located from exon one to exon three. We designed the MS-PCR primer sets located within this region. (b) The designed MS-PCR analysis results in 4-NQO and 4-NQO/arecoline at 18, 20, 26, and 28 weeks, respectively. The RARB gene showed hypermethylation (60%) in 4-NQO/arecoline at 28 week. M: methylated set, U: unmethylated set, B: blood DNA for methylation negative control, S: SssI treated DNA for methylation positive control, and %methylation: the percentages of methylation. The red inverted triangle showed that the methylated RARB gene could be amplified.
Figure 4.
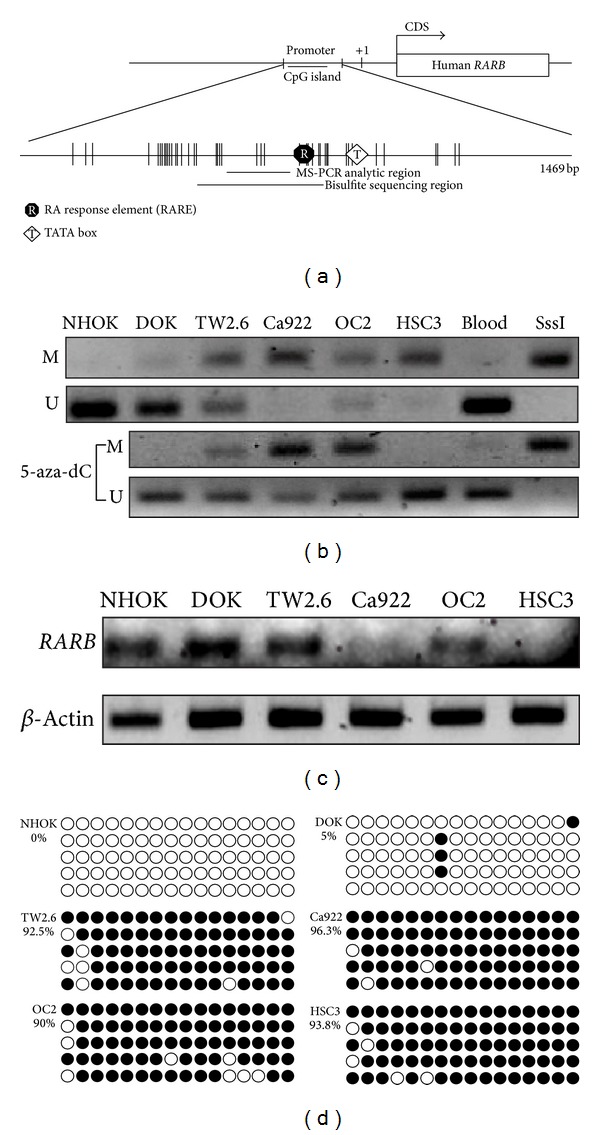
Methylation status in human oral cancer cell lines of RARB. (a) The schematic for CpG island presentation of RARB. There was a CpG island located in promoter region. We designed the MS-PCR and bisulfite sequencing primer sets in this region. (b) The RARB methylation status in different human oral cancer cell lines. The cell lines were also treated with 5′-aza-dC. The methylation status was recovered because of 5′-aza-dC treatment. (c) The RARB mRNA expression in different human oral cancer cell lines. The RARB was not expressed in Ca922, OC2, and HSC3 obviously. (d) The RARB bisulfite sequencing in different human oral cancer cell lines. There were more methylated RARB in TW2.6, Ca922, OC2, and HSC3 than in NHOK and DOK. The hollow circle is the unmethylated CpG site and the full circle is the methylated CpG site.
3.3. Verification of Methylated Genes by MS-PCR and Bisulfite Sequencing
The RARB gene was a candidate target to verify the methylation status in these mouse OSCC tongues tissues and in human oral cancer cell lines that were also treated by 5′-aza-2′-deoxycytidine (5-aza-dC). Interestingly, RARB was hypermethylated in mouse OSCC and reexpression by 5-aza-dC treatment in human oral cancer cell lines. In Figure 3(b), RARB was hypermethylated in mouse OSCC tumor parts of N (4-NQO) and NA (4-NQO + Arecoline) compared to normal parts. Furthermore, the methylation ratio of RARB in NA treatment at 28 week was 60%. It was much higher than the 4-NQO treatment (20%) at 28 week. These results correlated with the microarray analysis data. RARB also investigated the methylation status in human oral cancer lines (Figure 4(b)). The results showed that Ca922, TW2.6, and HSC3 were hypermethylated than in NHOK. It also lost expressions in Ca922, TW2.6, and HSC3 but not in NHOK (Figure 4(c)). In Figure 4(d), the bisulfite sequencing showed the hypermethylation in TW2.6 (92.5%), Ca922 (96.3%), OC2 (90%), and HSC3 (93.8%), but in the NHOK and DOK, the normal and precancer cell lines were not methylated in bisulfite sequencing results. Taken together, these results showed that the promoter methylation of RARB plays the main role in OSCC progression.
3.4. Methyltransferase Expressions in Human OSCC Cell Lines
DNA methylation is catalyzed by the family of DNA methyltransferases (DNMT) including Dnmt1, Dnmt3a, and Dnmt3b. Figure 5 shows the measurement of gene expressions via real-time PCR on Dnmt1, Dnmt3a, and Dnmt3b in human oral cancer lines. For Dnmt1, there were no expression differences between NHOK and other cell lines (Figure 5(a)). However, Dnmt3a and Dnmt3b showed significantly higher expression levels in TW2.6 and Ca922 than others (Figures 5(b) and 5(c)).
Figure 5.
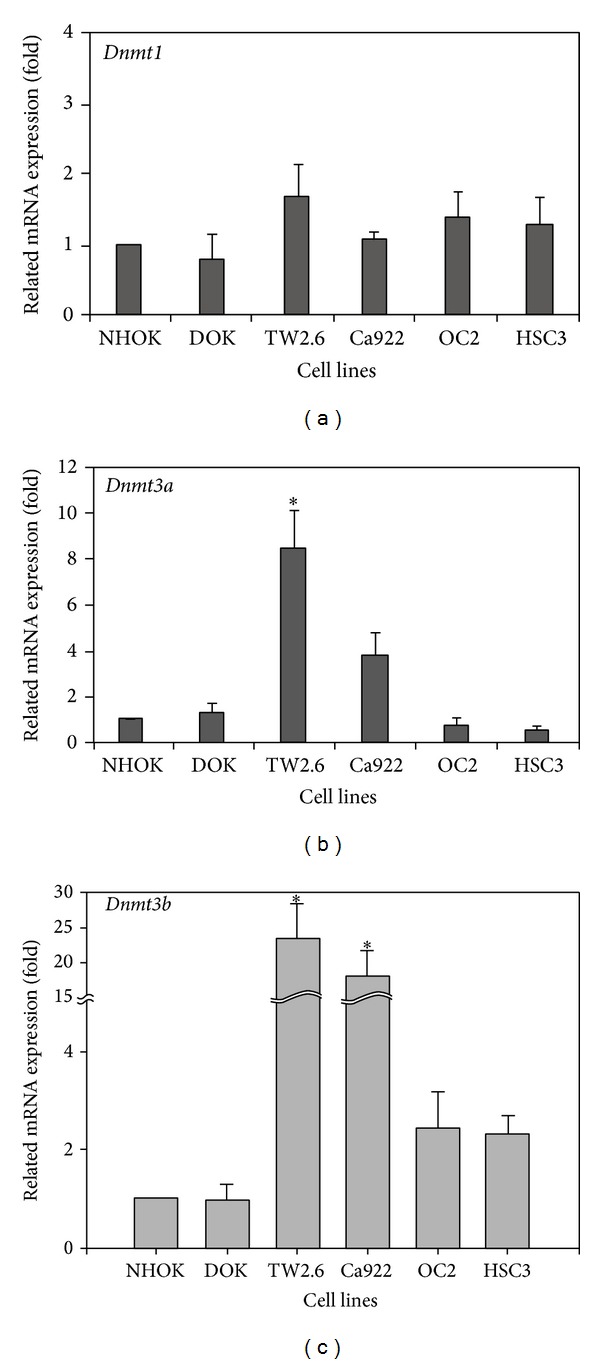
DNMTs RNA expression in human oral cancer cell lines. The measurement of DNMTs expression levels by real-time PCR was showed in (a), (b), and (c), respectively. (a) The Dnmt1 expression was not changed in all human oral cancer cell lines. (b) and (c) TW2.6 and Ca922 were higher expression in Dnmt3a and Dnmt3b than others. The figures shown are the mean of three experiments where all of the samples were analyzed in triplicate. The start sign showed the statistically significant (P < 0.05).
4. Discussion
OSCC is the most common head and neck neoplasm, affecting 270,000 people worldwide each year [20, 32]. According to related research, patients who smoke, drink, and chew betel quid experience a 5.32-fold increased likelihood of death as compared to those without any oral habits [50]. In Taiwan and other Southeast Asian countries, betel quid chewing is one of the most important risk factors for oral cancer patients and associates as the main cause between betel quid chewing and oral cancer development [51]. 4-NQO is quinoline derivative and a tumorigenic compound which can induce DNA lesions. Quinone oxidoreducatase is one of the major enzymes that convert 4-NQO to the more active metabolite, 3-hydroxyaminoquinoline 1-oxide [52]. This oxidoreducatase can be produced from the mucosal of the sublingual in humans and mice. Arecoline is a natural alkaloid product found in the areca nut. In this study, these two compounds were used to induce oral carcinogenesis in a mouse model. When we added 4-NQO plus Arecoline to the drinking water of mice, the results showed that mice were shown to have induced carcinogenesis at 100% at week 26 and 28 (Figure 1(a)). In H&E staining, resected oral tissue at the end of week 28 (Figure 1(b)) also showed that NA and N treatment could induce squamous cell carcinoma, respectively. However, treatment with arecoline alone revealed that it did not induce tumorigenesis after 28 weeks.
Methylation is important in the development of OSCC and many tumor suppressor genes targeted by promoter methylation will by no doubt be described in the future. The techniques used at present to detect methylation provide good sensitivity, specificity, and speed. There are many types of methylation arrays for a genome-wide approach to realize methylation profile. In this study, we applied a home-made high throughput MCGI array to analyze DNA methylation across the entire genome in the OSCC mouse tongue tissues. This home-made MCGI array not only could identify methylation profile but also could detect mRNA expression (in our previous studies). The chip was cohybridized with mouse Cot-I DNA and total RNA mixture to evaluate the quality and the exon-containing portions can be used to measure levels of gene expression. According to the previous research, the methylation status and mRNA expression levels could be verified by MCGI array.
According to the depicted representative data from previous studies, RARB expression is thought to be associated with cellular sensitivity to retinoid in numerous cancer cells, including HNSCC cells, breast cancer cells, lymphocytic leukemia, and lung cancer cells [53–57]. Methylation of RARB was identified which had a correlation with primary oral malignant diseases [58, 59]. The methylation array of 4,608 genes (duplicate on chip) that we used in this study included the majority of genes which have previously been associated with head and neck cancer (e.g., DAPK, MGMT, and CDH1, etc.). However, only RARB in previously studied genes were shown to be positive for methylation. To explain the possibility of this discrepancy, the hybridized probes maybe located on different regions between our array and previous methylation studies. However, we found some interesting genes and described this in greater detail in Table 2 as they were shown to have hypermethylation on the chip, but this was not reported before in OSCC.
In our study, DNMT1 does not affect the expression in human OSCC cell lines. Nevertheless, DNMT3a and DNMT3b did affect the expressions in human OSCC cell lines (Figure 5). These results showed that DNMT3a and DNMT3b were higher expressions in TW2.6 and Ca922, the primary oral cancer cell lines, but not in OC2 and HSC3. As compared to other tumors, no correlation was seen between DNMT upregulation and promoter hypermethylation-induced inactivation of tumor-related genes. The exact mechanisms of DNMT upregulation remain unclear, but it is suggested that aberrant DNMT activity, especially with regard to DNMT1, is due to a rapid proliferation of cancer cells because DNMT1 binds to proliferating cell nuclear antigen (PCNA) [60]. Overexpression of all the DNMTs at the mRNA level has been shown for several cancers [45, 61, 62]. However, DNMT3a and DNMT3b were the de novo methyltransferases. In this result (Figures 5(b) and 5(c)), we thought DNMT3a and DNMT3b were added to new methyl groups to DNA to cause DNA methylation aberrant at the early stage of OSCC progression.
We used some different approaches to deal with the sparseness of data. To begin with we used a methylation array of 4,086 genes (duplicated on chip), which provided more comprehensive data. Here we also suggest that this study could offer a basic evidence that promoter hypermethylation of RARB is correlated with the occurrence of betel-related OSCC.
Isotretinoin (13-cis-retinoic acid) is considered to have the effect of preventing second primary cancers and local or regional recurrence after head and neck cancer is treated. However, in a previous clinical trial, chemoprevention therapy involving retinoid acid does not cause significant differences in early head and neck cancers. The nonsignificant benefit result is due to the small number of patients. The prospective study rendered a small percentage of patients that have second primary cancers and local regional recurrence, thus causing the results to be less significant even though there was trend to have reduced second primary cancers developed and less local regional recurrence in prior studies [63]. However, there was another clinical research using the isotretinoin (13-cis-retinoic acid) (50 to 100 mg per square meter of body-surface area per day) as compared with placebo, to be taken daily for 12 months. They offer significantly reduced second primary cancer development after 32 months of follow-up, and multiple second primary tumors developed in the placebo group [64]. Therefore, they suggest that isotretinoin still has the ability to prevent second primary cancers, but there was less use in preventing the primary site recurrence [64]. There was another human cohort study using an in situ hybridization (ISH) analysis checker with 38 pairs of surgical specimens of primary OSCC and noncancerous matched normal control to compare the cellular expression level of RARB. They found the loss of RARB in the advanced OSCCs especially when they are betel quid users [65]. This prominent result also give us an understanding that RARB is really an important factor for chemoprevention for tumor progression. Therefore, betel quid related hypermethylation of RARB will really increase the tumorigenesis and poor treatment outcome of oral cancer. Concerning the mechanisms of the chemoprevention function, there was another study that revealed that the retinoids could suppress basal expression of Cox-2 or EGF-mediated induction of Cox-2 in human oral squamous carcinoma cells [66]. Thus, RARB not only has cell cycle inhibition and tumor suppressant effects, but also anti-inflammatory effects that cease COX-2 related cancerization effects on the oral mucosa.
This is pilot study that talked about the detailed mechanisms of retinoid acid function affected by areca. In the future, we should consider retinoid acid use or RARB related drugs for areca users who are found to have oral tumors or oral cancer to reduce the incidence of oral cancer and to provide a better treatment outcome.
Acknowledgments
This research was supported in part by Grants NSC-98-2313-B-005-012 and NSC-101-2313-B-005-014-MY3 from the National Science Council, Grant COA-97-6.2.1-U1(9) from the Council of Agriculture, and grant from the Ministry of Education, Taiwan, under the Aiming for the Top University plan (ATU-101-s0508). This work was supported by Grants CMU95-302, CMU97-098, and CMU97-099 from China Medical University, Taiwan (J.-C. Cheng) and DMR-103-025 from China Medical University Hospital (Y.-A. Tsou). The authors thank Dr. Dar-Bin, Shieh (National Cheng Kung University, Taiwan) to support DOK cells. The authors also thank technique support from Ms. Chia-Fan Lin.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Zi-Lun Lai, Yung-An Tsou, and Shin-Ru Fan contributed equally to this work.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer Journal for Clinicians. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Li-Ting C, Chung-Ho C, Yi-Hsin Y, Pei-Shan H. The development and validation of oral cancer staging using administrative health data. BMC Cancer. 2014;14(1, article 380) doi: 10.1186/1471-2407-14-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Y. C. Ko. Betel quid chewing, cigarette smoking and alcohol consumption related to oral cancer in Taiwan. Journal of Oral Pathology and Medicine. 1995;24(10):450–453. doi: 10.1111/j.1600-0714.1995.tb01132.x. [DOI] [PubMed] [Google Scholar]
- 4.Hsu WL, Chien YC, Chiang CJ, et al. Lifetime risk of distinct upper aerodigestive tract cancers and consumption of alcohol, betel and cigarette. International Journal of Cancer. 2014;135(6):1480–1486. doi: 10.1002/ijc.28791. [DOI] [PubMed] [Google Scholar]
- 5.Hung SH, Tsai MC, Liu TC, Lin HC, Chung SD. Routine endoscopy for esophageal cancer is suggestive for patients with oral, oropharyngeal and hypopharyngeal cancer. PLoS ONE. 2013;8(8) doi: 10.1371/journal.pone.0072097.e72097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen M, Huang W, Chan CH, Chen P, Lee K. Impact of second primary esophageal or lung cancer on survival of patients with head and neck cancer. Oral Oncology. 2010;46(4):249–254. doi: 10.1016/j.oraloncology.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Gupta CRPC. Ranks number three among ten leading causes of death. ANNALS Academy of Medicine Singapore. 2004;33:31S–36S. [Google Scholar]
- 8.Chiang S, Chen P, Lee C, et al. Up-regulation of inflammatory signalings by areca nut extract and role of cyclooxygenase-2 -1195G>A polymorphism reveal risk of oral cancer. Cancer Research. 2008;68(20):8489–8498. doi: 10.1158/0008-5472.CAN-08-0823. [DOI] [PubMed] [Google Scholar]
- 9.Rajalalitha P, Vali S. Molecular pathogenesis of oral submucous fibrosis—a collagen metabolic disorder. Journal of Oral Pathology and Medicine. 2005;34(6):321–328. doi: 10.1111/j.1600-0714.2005.00325.x. [DOI] [PubMed] [Google Scholar]
- 10.Ariyawardana A, Athukorala ADS, Arulanandam A. Effect of betel chewing, tobacco smoking and alcohol consumption on oral submucous fibrosis: a case-control study in Sri Lanka. Journal of Oral Pathology and Medicine. 2006;35(4):197–201. doi: 10.1111/j.1600-0714.2006.00400.x. [DOI] [PubMed] [Google Scholar]
- 11.Ramaesh T, Mendis BRRN, Ratnatunga N, Thattil RO. The effect of tobacco smoking and of betel chewing with tobacco on the buccal mucosa: a cytomorphometric analysis. Journal of Oral Pathology and Medicine. 1999;28(9):385–388. doi: 10.1111/j.1600-0714.1999.tb02108.x. [DOI] [PubMed] [Google Scholar]
- 12.Chung C, Yang Y, Wang T, Shieh T, Warnakulasuriya S. Oral precancerous disorders associated with areca quid chewing, smoking, and alcohol drinking in southern Taiwan. Journal of Oral Pathology and Medicine. 2005;34(8):460–466. doi: 10.1111/j.1600-0714.2005.00332.x. [DOI] [PubMed] [Google Scholar]
- 13.Tang X, Knudsen B, Bemis D, Tickoo S, Gudas LJ. Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clinical Cancer Research. 2004;10(1, part 1):301–313. doi: 10.1158/1078-0432.ccr-0999-3. [DOI] [PubMed] [Google Scholar]
- 14.Kitano M. Host genes controlling the susceptibility and resistance to squamous cell carcinoma of the tongue in a rat model. Pathology International. 2000;50(5):353–362. doi: 10.1046/j.1440-1827.2000.01058.x. [DOI] [PubMed] [Google Scholar]
- 15.Chang NW, Pei RJ, Tseng HC, et al. Co-treating with arecoline and 4-nitroquinoline 1-oxide to establish a mouse model mimicking oral tumorigenesis. Chemico-Biological Interactions. 2010;183(1):231–237. doi: 10.1016/j.cbi.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 16.Shaw RJ, Liloglou T, Rogers SN, et al. Promoter methylation of P16, RARβ, E-cadherin, cyclin A1 and cytoglobin in oral cancer: quantitative evaluation using pyrosequencing. The British Journal of Cancer. 2006;94(4):561–568. doi: 10.1038/sj.bjc.6602972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan CY. Epigenetic alterations in head and neck cancer: prevalence, clinical significance, and implications. Current Oncology Reports. 2004;6(2):152–161. doi: 10.1007/s11912-004-0027-0. [DOI] [PubMed] [Google Scholar]
- 18.Esteller M. Cancer epigenetics: DNA methylation and chromatin alterations in human cancer. Advances in Experimental Medicine and Biology. 2003;532:39–49. doi: 10.1007/978-1-4615-0081-0_5. [DOI] [PubMed] [Google Scholar]
- 19.Ha PK, Califano JA. Promoter methylation and inactivation of tumour-suppressor genes in oral squamous-cell carcinoma. The Lancet Oncology. 2006;7(1):77–82. doi: 10.1016/S1470-2045(05)70540-4. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki E, Imoto I, Pimkhaokham A, et al. PRTFDC1, a possible tumor-suppressor gene, is frequently silenced in oral squamous-cell carcinomas by aberrant promoter hypermethylation. Oncogene. 2007;26(57):7921–7932. doi: 10.1038/sj.onc.1210589. [DOI] [PubMed] [Google Scholar]
- 21.Scully C, Field JK, Tanzawa H. Genetic aberrations in oral or head and neck squamous cell carcinoma (SCCHN): 1. Carcinogen metabolism, DNA repair and cell cycle control. Oral Oncology. 2000;36(3):256–263. doi: 10.1016/s1368-8375(00)00007-5. [DOI] [PubMed] [Google Scholar]
- 22.Xiong Z, Laird PW. COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Research. 1997;25(12):2532–2534. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frommer M, McDonald LE, Millar DS, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(5):1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(18):9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang TH, Perry MR, Laux DE. Methylation profiling of CpG islands in human breast cancer cells. Human Molecular Genetics. 1999;8(3):459–470. doi: 10.1093/hmg/8.3.459. [DOI] [PubMed] [Google Scholar]
- 26.Adrien LR, Schlecht NF, Kawachi N, et al. Classification of DNA methylation patterns in tumor cell genomes using a CpG island microarray. Cytogenetic and Genome Research. 2006;114(1):16–23. doi: 10.1159/000091923. [DOI] [PubMed] [Google Scholar]
- 27.Wei SH, Chen CM, Strathdee G, et al. Methylation microarray analysis of late-stage ovarian carcinomas distinguishes progression-free survival in patients and identifies candidate epigenetic markers. Clinical Cancer Research. 2002;8(7):2246–2252. [PubMed] [Google Scholar]
- 28.Yan PS, Chen C, Shi H, Rahmatpanah F, Wei SH, Huang TH. Applications of CpG island microarrays for high-throughput analysis of DNA methylation. Journal of Nutrition. 2002;132(8):2430S–2434S. doi: 10.1093/jn/132.8.2430S. [DOI] [PubMed] [Google Scholar]
- 29.Chen CM, Chen HL, Hsiau TH, et al. Methylation target array for rapid analysis of CpG island hypermethylation in multiple tissue genomes. The American Journal of Pathology. 2003;163(1):37–45. doi: 10.1016/S0002-9440(10)63628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gitan RS, Shi H, Chen C, Yan PS, Huang TH. Methylation-specific oligonucleotide microarray: a new potential for high-throughput methylation analysis. Genome Research. 2002;12(1):158–164. doi: 10.1101/gr.202801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beier V, Mund C, Hoheisel JD. Monitoring methylation changes in cancer. Advances in biochemical engineering/biotechnology. 2007;104:1–11. doi: 10.1007/10_024. [DOI] [PubMed] [Google Scholar]
- 32.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA: A Cancer Journal for Clinicians. 2005;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 33.Neville BW, Day TA. Oral cancer and precancerous lesions. Ca: A Cancer Journal for Clinicians. 2002;52(4):195–215. doi: 10.3322/canjclin.52.4.195. [DOI] [PubMed] [Google Scholar]
- 34.Lotan R. Retinoids in cancer chemoprevention. The FASEB Journal. 1996;10(9):1031–1039. doi: 10.1096/fasebj.10.9.8801164. [DOI] [PubMed] [Google Scholar]
- 35.Shalinsky DR, Bischoff ED, Lamph WW, et al. A novel retinoic acid receptor-selective retinoid, ALRT1550, has potent antitumor activity against human oral squamous carcinoma xenografts in nude mice. Cancer Research. 1997;57(1):162–168. [PubMed] [Google Scholar]
- 36.McGregor F, Muntoni A, Fleming J, et al. Molecular changes associated with oral dysplasia progression and acquisition of immortality: potential for its reversal by 5-azacytidine. Cancer Research. 2002;62(16):4757–4766. [PubMed] [Google Scholar]
- 37.McGregor F, Wagner E, Felix D, Soutar D, Parkinson K, Harrison PR. Inappropriate retinoic acid receptor-β expression in oral dysplasias: correlation with acquisition of the immortal phenotype. Cancer Research. 1997;57(18):3886–3889. [PubMed] [Google Scholar]
- 38.Xu X, Ro JY, Lee JS, Shin DM, Hong WK, Lotan R. Differential expression of nuclear retinoid receptors in normal, premalignant, and malignant head and neck tissues. Cancer Research. 1994;54(13):3580–3587. [PubMed] [Google Scholar]
- 39.Hu L, Crowe DL, Rheinwald JG, Chambon P, Gudas LJ. Abnormal expression of retinoic acid receptors and keratin 19 by human oral and epidermal squamous cell carcinoma cell lines. Cancer Research. 1991;51(15):3972–3981. [PubMed] [Google Scholar]
- 40.Sheu JJ, Hua CH, Wan L, et al. Functional genomic analysis identified epidermal growth factor receptor activation as the most common genetic event in oral squamous cell carcinoma. Cancer Research. 2009;69(6):2568–2576. doi: 10.1158/0008-5472.CAN-08-3199. [DOI] [PubMed] [Google Scholar]
- 41.Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Research. 2008;68(7):2094–2105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
- 42.Chen CM, Wang CH, Wu SC, Lin C, Lin S, Cheng WTK. Temporal and spatial expression of biologically active human factor VIII in the milk of transgenic mice driven by mammary-specific bovine α-lactalbumin regulation sequences. Transgenic Research. 2002;11(3):257–268. doi: 10.1023/a:1015651302674. [DOI] [PubMed] [Google Scholar]
- 43.Ahluwalia A, Yan P, Hurteau JA, et al. Dna methylation and ovarian cancer: I. Analysis of CpG island hypermethylation in human ovarian cancer using differential methylation hybridization. Gynecologic Oncology. 2001;82(2):261–268. doi: 10.1006/gyno.2001.6291. [DOI] [PubMed] [Google Scholar]
- 44.Cross SH, Charlton JA, Nan X, Bird AP. Purification of CpG islands using a methylated DNA binding column. Nature Genetics. 1994;6(3):236–244. doi: 10.1038/ng0394-236. [DOI] [PubMed] [Google Scholar]
- 45.Nephew KP, Huang TH. Epigenetic gene silencing in cancer initiation and progression. Cancer Letters. 2003;190(2):125–133. doi: 10.1016/s0304-3835(02)00511-6. [DOI] [PubMed] [Google Scholar]
- 46.Yan PS, Perry MR, Laux DE, Asare AL, Caldwell CW, Huang TH. CpG island arrays: an application toward deciphering epigenetic signatures of breast cancer. Clinical Cancer Research. 2000;6(4):1432–1438. [PubMed] [Google Scholar]
- 47.Shi H, Yan PS, Chen C, et al. Expressed CpG island sequence tag microarray for dual screening of DNA hypermethylation and gene silencing in cancer cells. Cancer Research. 2002;62(11):3214–3220. [PubMed] [Google Scholar]
- 48.Yan PS, Chen C-, Shi H, et al. Dissecting complex epigenetic alterations in breast cancer using CpG island microarrays. Cancer Research. 2001;61(23):8375–8380. [PubMed] [Google Scholar]
- 49.Yan PS, Efferth T, Chen HL, et al. Use of CpG island microarrays to identify colorectal tumors with a high degree of concurrent methylation. Methods. 2002;27(2):162–169. doi: 10.1016/s1046-2023(02)00070-1. [DOI] [PubMed] [Google Scholar]
- 50.Chen YK, Huang HC, Lin LM, Lin CC. Primary oral squamous cell carcinoma: an analysis of 703 cases in southern Taiwan. Oral Oncology. 1999;35(2):173–179. doi: 10.1016/s1368-8375(98)00101-8. [DOI] [PubMed] [Google Scholar]
- 51.Jeng JH, Chang MC, Hahn LJ. Role of areca nut in betel quid-associated chemical carcinogenesis: current awareness and future perspectives. Oral Oncology. 2001;37(6):477–492. doi: 10.1016/s1368-8375(01)00003-3. [DOI] [PubMed] [Google Scholar]
- 52.Tanuma J, Hirano M, Hirayama Y, et al. Genetic predisposition to 4NQO-induced tongue carcinogenesis in the rat. Medical Principles and Practice. 2005;14(5):297–305. doi: 10.1159/000086926. [DOI] [PubMed] [Google Scholar]
- 53.Zhang X, Liu Y, Lee M, Pfahl M. A specific defect in the retinoic acid response associated with human lung cancer cell lines. Cancer Research. 1994;54(21):5663–5669. [PubMed] [Google Scholar]
- 54.Seeliger B, Wilop S, Osieka R, Galm O, Jost E. CpG island methylation patterns in chronic lymphocytic leukemia. Leukemia and Lymphoma. 2009;50(3):419–426. doi: 10.1080/10428190902756594. [DOI] [PubMed] [Google Scholar]
- 55.Wan H, Oridate N, Lotan D, Hong WK, Lotan R. Overexpression of retinoic acid receptor β in head and neck of squamous cell carcinoma cells increases their sensitivity to retinoid-induced suppression of squamous differentiation by retinoids. Cancer Research. 1999;59(14):3518–3526. [PubMed] [Google Scholar]
- 56.Sun S, Wan H, Yue P, Hong WK, Lotan R. Evidence that retinoic acid receptor β induction by retinoids is important for tumor cell growth inhibition. The Journal of Biological Chemistry. 2000;275(22):17149–17153. doi: 10.1074/jbc.M000527200. [DOI] [PubMed] [Google Scholar]
- 57.Widschwendter M, Daxenbichler G, Dapunt O, Marth C. Effects of retinoic acid and γ-interferon on expression of retinoic acid receptor and cellular retinoic acid-binding protein in breast cancer cells. Cancer Research. 1995;55(10):2135–2139. [PubMed] [Google Scholar]
- 58.Maruya S, Issa JJ, Weber RS, et al. Differential methylation status of tumor-associated genes in head and neck squamous carcinoma: incidence and potential implications. Clinical Cancer Research. 2004;10(11):3825–3830. doi: 10.1158/1078-0432.CCR-03-0370. [DOI] [PubMed] [Google Scholar]
- 59.Youssef EM, Lotan D, Issa JP, et al. Hypermethylation of the retinoic acid receptor-β2 gene in head and neck carcinogenesis. Clinical Cancer Research. 2004;10(5):1733–1742. doi: 10.1158/1078-0432.ccr-0989-3. [DOI] [PubMed] [Google Scholar]
- 60.Tischoff I, Tannapfel A. DNA methylation in hepatocellular carcinoma. World Journal of Gastroenterology. 2008;14(11):1741–1748. doi: 10.3748/wjg.14.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robertson KD, Uzvolgyi E, Liang G, et al. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Research. 1999;27(11):2291–2298. doi: 10.1093/nar/27.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xie S, Wang Z, Okano M, et al. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene. 1999;236(1):87–95. doi: 10.1016/s0378-1119(99)00252-8. [DOI] [PubMed] [Google Scholar]
- 63.Khuri FR, Lee JJ, Lippman SM, et al. Randomized phase III trial of low-dose isotretinoin for prevention of second primary tumors in stage I and II head and neck cancer patients. Journal of the National Cancer Institute. 2006;98(7):441–450. doi: 10.1093/jnci/djj091. [DOI] [PubMed] [Google Scholar]
- 64.Hong WK, Lippman SM, Itri LM, et al. Prevention of second primary tumors with isotretinoin in squamous-cell carcinoma of the head and neck. The New England Journal of Medicine. 1990;323(12):795–801. doi: 10.1056/NEJM199009203231205. [DOI] [PubMed] [Google Scholar]
- 65.Kao SY, Tu HF, Chang KW, Chang C, Yang C, Lin S. The retinoic acid receptor-β (RAR-β) mRNA expression in the oral squamous cell carcinoma associated with betel quid use. Journal of Oral Pathology and Medicine. 2002;31(4):220–226. doi: 10.1034/j.1600-0714.2002.310405.x. [DOI] [PubMed] [Google Scholar]
- 66.Mestre JR, Subbaramaiah K, Sacks PG, et al. Retinoids suppress epidermal growth factor-induced transcription of cyclooxygenase-2 in human oral squamous carcinoma cells. Cancer Research. 1997;57(14):2890–2895. [PubMed] [Google Scholar]