

Abstract
Certain environmental factors including drugs exacerbate or precipitate psoriasis. Lithium is the commonest cause of drug-induced psoriasis but underlying mechanisms are currently unknown. Lithium inhibits glycogen synthase kinase 3 (GSK-3). As lithium does not exacerbate other T-cell-mediated chronic inflammatory diseases, we investigated whether lithium may be acting directly on epidermal keratinocytes by inhibiting GSK-3. We report that lithium-induced keratinocyte proliferation at therapeutically relevant doses (1–2 mM) and increased the proportion of cells in S phase of the cell cycle. Inhibition of GSK-3 in keratinocytes by retroviral transduction of GSK-binding protein (an endogenous inhibitory protein) or through a highly selective pharmacological inhibitor also resulted in increased keratinocyte proliferation. Nuclear factor of activated T cells (NFAT) is an important substrate for GSK-3 and for cyclosporin, an effective treatment for psoriasis that inhibits NFAT activation in keratinocytes as well as in lymphocytes. Both lithium and genetic/pharmacological inhibition of GSK-3 resulted in increased nuclear localization of NFAT2 (NFATc1) and increased NFAT transcriptional activation. Finally, retroviral transduction of NFAT2 increased keratinocyte proliferation whereas siRNA-mediated knockdown of NFAT2 reduced keratinocyte proliferation and decreased epidermal thickness in an organotypic skin equivalent model. Taken together, these data identify GSK-3 and NFAT2 as key regulators of keratinocyte proliferation and as potential molecular targets relevant to lithium-provoked psoriasis. J. Cell. Physiol. 227: 1529–1537, 2012. © 2011 Wiley Periodicals, Inc.
Psoriasis is a common inflammatory skin disease characterized by abnormal cellular regulation involving the immune system, dermal vasculature, and the epidermis. The pathogenesis of psoriasis is recognized to be complex with interplay between innate and adaptive immune responses. Although psoriasis has a major genetic component there is thought to be significant interplay between genetic predisposition and external environmental triggers such as streptococcal infection and drugs. Provocation or exacerbation of psoriasis by lithium is well described and re-challenge studies have confirmed lithium as a pharmacological trigger for psoriasis (Skoven and Thormann, 1979).
Proliferation of resident skin T lymphocytes has been shown to be an early initiating event in psoriasis (Boyman et al., 2004). More recent studies have highlighted the role of innate immunity and cytokine signals from resident plasmocytoid dermal dendritic cells in regulating Th-17/Th-22 T cells which appear critical in initiating epidermal remodeling and the formation of psoriatic plaques (Zheng et al., 2007; Di Cesare et al., 2009; Nestle et al., 2009). Lithium has been previously shown to induce T-cell proliferation and it has been suggested that this may explain the action of lithium in psoriasis (Ohteki et al., 2000). However, lithium has not been shown to trigger or exacerbate other T-cell-mediated inflammatory diseases such as rheumatoid arthritis (Oliver and Silman, 2009) or multiple sclerosis (Ramagopalan et al., 2010). Moreover, lithium has been shown to down-regulate an inflammatory mRNA signature identified in monocytes from bipolar patients (Padmos et al., 2008), protect against cytokine-mediated damage to cartilage (Hui et al., 2010) and be an effective treatment for experimental autoimmune encephalomyelitis which models some features of multiple sclerosis in mice (Beurel et al., 2010). Together, these studies provide no support for a direct pro-inflammatory effect of lithium in psoriasis. In this study, we have examined the effect of lithium on keratinocytes as there is good evidence to support an intrinsic defect of keratinocytes in psoriasis. For example, linear psoriasis has been reported in a pattern following Blashko's lines, the routes of embryological keratinocyte migration (Happle, 1991). Also, uninvolved psoriatic skin shows increased keratinocyte proliferation under basal conditions (Hell and Hodgson, 1966; Goodwin et al., 1973) and is hyperresponsive to proliferation stimuli (Goodwin et al., 1973; Hatta et al., 1997). Recently, a large-scale genetic association study has identified polymorphisms within the IL-23 receptor gene as a risk factor for psoriasis (Cargill et al., 2007). The overexpression of IL-23 by dermal dendritic cells has been identified as an important component in the psoriasis inflammatory cascade (Di Cesare et al., 2009). Keratinocytes have also been shown to express IL-23 (Piskin et al., 2006), in sufficient quantities to activate memory T cells. Moreover, gene expression profile studies on uninvolved psoriatic skin emphasize distinct differences to normal skin, particularly with respect to lipid processing (Gudjonsson et al., 2009) and recent genetic studies have shown that PSORS4 and loss of LCE3B and LCE3C within the epidermal differentiation complex are linked to an increased risk of psoriasis (de Cid et al., 2009; Huffmeier et al., 2010). Taken in conjunction with a psoriasis-like phenotype observed following expression of activated signal transducer and activator of transcription 3 (STAT3) within mouse epidermis (Sano et al., 2005), these data emphasize that keratinocytes may play a key role in psoriasis, may be genetically predisposed to be hyperresponsive to cytokine signals and may modulate inflammatory responses.
The molecular mechanisms underlying lithium-provoked psoriasis are not well understood. Lithium is known to inhibit a variety of enzymes including inositol monophosphatase (IMPase) and glycogen synthase kinase-3 (GSK-3; Hedgepeth et al., 1997; Manji et al., 1999). The consequences of inhibition of IMPase have been mainly described in the brain and are thought to be due to inositol depletion, a phenomenon perhaps unique to the brain due to the blood–brain barrier. On the other hand, GSK-3 is ubiquitously expressed (Jope and Johnson, 2004) and inhibition of GSK-3 is not believed to be tissue specific in the way that has been suggested for IMPase.
GSK-3 is a serine threonine kinase which was first characterized for its role in glycogen metabolism (Welsh and Proud, 1993). Two isoforms α and β have been described. Lithium and other pharmacological GSK-3 inhibitors, inhibit the activity of both isoforms. Inhibition of GSK-3 has been reported to both reduce and promote inflammation via different downstream pathways (Martin et al., 2005; Vines et al., 2006; Shen et al., 2009). GSK-3 is also known to influence a multitude of other intracellular signaling pathways that regulate differentiation, cell survival, and cell proliferation (Beurel et al., 2010). Previous reports of the role of GSK-3 in cell proliferation have focused on the GSK-3β isoform (Cui et al., 1998; Park et al., 2003). GSK-3 has been demonstrated to regulate a wide variety of metabolic and structural proteins and also a number of transcription factors including the nuclear factor of activated T cells (NFAT; Jope and Johnson, 2004). GSK-3 activity is also negatively regulated by binding proteins such as GSK-binding protein (GSKBP; Jonkers et al., 1997; Yost et al., 1998). GSKBP has been shown to inhibit GSK-3 activity and to elevate levels of the GSK-3 substrate, β-catenin, in NIH3T3 cells (Farr et al., 2000).
Lithium is known to inhibit the activity of GSK-3 at therapeutic concentrations. Ryves and Harwood (2001) calculated that lithium inhibits GSK-3β with an IC50 of 2.0 mM and GSK-3α with an IC50 of 3.5 mM. Lithium has also been shown to mediate PI3 kinase-dependent protein kinase C-α dependent phosphorylation of GSK-3 (Kirshenboim et al., 2004).
We investigated possible downstream mechanisms following inhibition of GSK-3 in keratinocytes. NFAT is a GSK-3 substrate that has been implicated in proliferative responses. Expression of calcineurin and NFAT 1–4 has been previously demonstrated in human epidermal keratinocytes (Santini et al., 2001; Al-Daraji et al., 2002). NFAT has been implicated in regulating proliferation of a wide variety of cells including T cells, mast cells, endothelial cells, preadipocytes, and pancreatic cells, and is also the target of the highly effective anti-psoriatic therapy cyclosporin (Murphy and Hughes, 2002; Lipskaia and Lompre, 2004; Buchholz and Ellenrieder, 2007).
We reasoned that inhibition of GSK-3 by lithium in keratinocytes may lead to increased nuclear NFAT with subsequent effects on keratinocyte proliferative responses. NFAT2 transcripts are overexpressed in mouse epidermal stem cells (Horsley et al., 2008) and we therefore studied the effect of lithium and GSK-3 on NFAT and focused specifically on NFAT2.
In this study, we show that lithium inhibits GSK-3 in keratinocytes and that inhibition of GSK-3 induces keratinocyte proliferation. The downstream signaling events include activation of NFAT, specifically NFAT2, overexpression of which induces proliferation of keratinocytes, consistent with its pro-proliferative role described in other cell types.
Materials and Methods
Materials
The GSKBP vector (Xenopus origin in CS2-Flag vector) was a kind gift from Dr. D. Kimelman (Washington). The GSK vectors wild-type (WT) and constitutively active GSK-3β (CaGSK-3β-S9A) were a gift from Dr. B. Eldar Finkelman. pEGFP-NFAT2 (NFATc1) was a kind gift from Dr. Dorothy Cantrell (ICFR, London, UK; Turner et al., 1998). The anti-NFAT 2 antibody was a kind gift from Dr. Nancy Rice, (NCI, Frederick, MD; Lyakh et al., 1997). The GSK-3β antibodies were from Cell Signalling Technologies, Beverly, MA. The NFAT luciferase vector was a gift from Dr. D.J. McKean (Mayo Foundation, Rochester, MN). The GSK-3 inhibitor, 6-bromo-indirubin-3'-oxime (BIO) was purchased from Calbiochem, La Jolla, CA.
Cells
After obtaining informed consent, redundant skin from circumcisions and pinoplasty operations was used to culture keratinocytes as previously described (Todd and Reynolds, 1998). An SV-40 transformed human foreskin keratinocyte cell line, SV-K14 obtained from J Taylor (Cell Services, Cancer Research UK, Clare Hall Labs, Herts, UK) and HaCaT keratinocytes, a non-tumorgenic cell line, a kind gift from Dr. NE Fusenig (German Cancer Research Center, Heidelberg, Germany), were grown in DMEM supplemented with antibiotics and 5% and 10% fetal calf serum (FCS) respectively.
Retroviral transduction
The GSKBP segment was digested from the CS2 vector using HindIII and XbaI. After gel extraction, the DNA fragment was inserted into the pBluescriptIISK(+) cloning vector. Primers were designed with new restriction sites incorporated to amplify the fragment and enable insertion into the retroviral GFP vector pLEGFPc1 (Clontec, BD Biosciences, San Jose, CA) at 5′ HindIII and 3′ BamHI sites. Forward primer was ggtccgaagctttgccgtgtcgcaaggagagtttcctg and reverse primer (5′–3′) was gcttctggatccggttgtctcagtgcactcggacac. Full-length human NFATc1 cDNA was cloned from pEGFPc1-NFAT2 into the retroviral vector pLEGFPc1 using BspEI and HindIII restriction sites. Vectors were sequenced to confirm that the inserts were in frame with the GFP. The pLEGFP-GSKBP, pLEGFP-NFAT2, and pLEGFP empty vector were then transiently transfected into the Phoenix Ampho retroviral packaging cell line, a kind gift from Dr. G. Nolan (Stanford University, Stanford, CA). These cells were grown in DMEM and selected using G418 (Invitrogen, Foster City, CA) until over 90% were GFP positive. They were then transferred to an incubator at 32°C for production of virus which was collected from confluent cells after 48 h. Retroviral medium was either used immediately or frozen at −80°C until needed. Retroviral transductions of human keratinocytes were performed essentially as previously described (Regl et al., 2002). Human keratinocytes were seeded into 96-well plates and left over night. The medium was then substituted for growth factor free medium for 2 days before retroviral transduction. Following washing with PBS, retroviral medium with polybrene (5 µg/ml) was pipetted onto cells and then spun in a Sorvall plate spinning centrifuge at 1,500 rpm for 2 h. Following washing with PBS normal medium was replaced onto the cells which were returned to the incubator. Similarly, for SRB assays with K14 keratinocytes, cells were seeded into 96-well plates and cultured for 2 days in DMEM without FCS prior to retroviral transduction in the 96-well plates. HaCaTs were retrovirally transduced in six-well plates and incubated for 2 days after transduction, then transferred to T75 culture flasks. Culture for 3 days in DMEM without FCS resulted in an almost synchronous population with around 90% of cells transiently arresting in G1 phase of the cell cycle. Cells were then seeded for SRB assays or flow cytometry. For all cell types the transduction efficiency was greater than 75%.
Sulphorhodamine B assays
Sulphorhodamine B (SRB) assays provide a sensitive measure of total cellular protein, are linear with cell number and cellular protein at densities from 1% to 200% and perform similarly compared with other proliferation assays such as MTT assay or clonogenic assays (Skehan et al., 1990; Voigt, 2005). Cells were seeded into flat bottomed 96-well plates with 5 × 103 cells/well (keratinocytes) or 3 × 103 cells/well (K14 keratinocytes) or in 24-well plates with 2.5 × 104 cells/well (HaCaT cells), incubated in growth factor free medium (keratinocytes) or serum-free medium for 48 h (K14 keratinocytes) or DMEM plus 10% FCS (HaCaTs, following synchronization) before transduction with indicated plasmids or treatment with lithium or 6-bromoindirubin-3′-oxime (BIO) in complete medium for 4 or 7 days, and SRB assay performed (McGill et al., 2005). After removing the medium the cells were fixed in 10% trichloroacetic acid for 1 h at 4° and then washed with water five times. SRB 0.4% dissolved in 1% glacial acetic acid was added for 30 min after which the cells were washed with 1% glacial acetic acid. The cells were left to dry overnight and then the protein bound SRB was solubilized by adding 200 µl 10 mM Tris per well. The absorbance was measured at wavelengths 490 nM using a plate reader (Spectra MAX 250; Molecular Devices, Sunnyvale, CA).
Flow cytometry
Synchronized HaCaT cells were seeded in six-well plates at a density of 1.5 × 105 cells/well in DMEM plus 10% FCS. Cells were harvested at various time points post-seeding, fixed in ice-cold methanol, and stored at 4°C until required for analysis. For cell cycle analysis, cells were stained with 100 µg/ml propidium iodide (Sigma, Dorset, UK) plus 0.2% Triton-X (Sigma), and 0.15 mg/ml ribonuclease A (Sigma) for 20 min at 37°C in the dark. Keratinocytes were seeded into flasks and treated as indicated. At the desired time points cells were fixed and labeled with propidium iodide as described above. The DNA content of the cells was measured using a gated amplifier for FL2 (λ 625 nm) and 10,000 cells per sample were counted. Cell cycle analysis was performed using Multicycle software (University of Washington, Verity, WA).
Western blotting
Following treatment with agonists for designated times, medium was removed and keratinocytes lysed in buffer containing 15 mM KCl, 3.75 mM NaCl, 37.5 µM Spermine, 125 µM Spermidine, 500 µM EDTA, and 3.75 mM Tris–HCl and 0.1% (w/v) digitonin, harvested with a cell scraper and sonicated. Lysis buffer was mixed with sample buffer and protein separation performed on 10% Bis-Tris gels from Novex (Invitrogen). Primary antibodies were incubated in 5% dried milk in PBS/0.05% Tween-20 for 1 h. The antibodies used were: rabbit anti-NFAT2 antibody (1:200; Lyakh et al., 1997) and rabbit anti-Phospho-GSK-3β (Ser 9) antibody (Cell Signalling Technologies, Catalog. No.# 9336). Secondary biotinylated antibodies at 1 in 2,000 were incubated for 1 h (Vector Labs, Burlingame, CA). Following washing, membranes were incubated in ABC (avidin and biotinylated horseradish peroxidase macromolecular complex) reagents (Vector Labs) for 1 h at room temperature. Membranes were washed again before incubation with ECL superadvance™ reagent. Blots were imaged using a FluroChem™ CCD camera (Alpha Innotech, Staffordshire, UK).
Immunohistochemistry of cells
Keratinocytes were seeded onto 16 mm glass cover slips and treated as specified when approximately 70% confluent. Cells were fixed in 4% paraformaldehye for 15 min and permeabilized with 0.01% Triton X-100, and immunostained as previously described with rabbit anti-NFAT2 antibodies (1:200; Lyakh et al., 1997) as previously described (Al-Daraji et al., 2002). Cells were imaged using a Leica TCS SP II laser scanningconfocal microscope (Leica Microsystems, Heidelberg, Germany) as previously described (Flockhart, 2008). Line by line scanning mode was utilized to minimize cross-talk between channels. Images were exported and subsequently processed in Photoshop™ (Adobe, San Jose, CA).
Luciferase assays
The following vectors were used: NFAT luciferase vector (pNF-AT-luc) containing three tandem repeats of the NF-AT/AP-1-binding site at approximately −287 bp of the murine IL-2 promoter a gift from Dr. D.J. McKean, Mayo Foundation (Hedin et al., 1997), USA: pRLtk Renilla luciferase (Promega, Southampton, UK) was used as an internal control for transfection efficiency. Approximately 50,000 keratinocytes were seeded into each well of a 12-well plate. One microgram of reporter vector, 0.05 µg of renilla and 1 µg of vector were transfected per well using Lipofectamine Plus (Invitrogen). The transfection reagents were incubated with the cells for 3 h before being replaced with normal medium. For vector only experiments, cell lysis was performed 24 h after replacement of normal medium unless stated otherwise. For experiments using lithium, cells were incubated in normal medium for 16 h before treatments were added. Treatments were for 24 h unless stated otherwise. Luciferase activity was measured using the Promega Dual Luciferase assay (Promega) and luminescence was measured with a Wallac 96-well plate luminescence detecting Beta counter.
RNA interference to knock down NFAT2 expression
Chemically modified (to prevent off-target effects) scrambled siRNA and NFAT2-specific siRNA were obtained from Qiagen (Crawley, UK). siRNA were transfected into keratinocytes using the Human Keratinocyte Nucleofector kit and device from Amaxa/Lonza (Cologne, Germany) according to the manufacturer's protocol (2 µg siRNA/106 cells). Knockdown of NFAT2 was verified by real-time PCR using standard procedure. Poly-A RNA was isolated from 3 × 106 transfected cells using a Direct mRNA isolation kit (Sigma), retrotranscribed using a High-Capacity cDNA RT kit (Applied Biosystems, Foster City, CA) and subjected to real-time PCR using iQ SYBR Green Supermix and a Chromo4 cycler (Bio-Rad, Hemel Hempstead, UK). Primers used to detect NFAT2 were 5′-CTTCTCCAACACCAAAGTCC-3′ (forward) and 5′-CGTACCCGTGTGTTCTTCCT-3′ (reverse). NFAT2 expression was normalized for GAPDH expression (forward primer: 5′-GTCAGTGGTGGACCTGACCT-3′, reverse primer: 5′-AGGGGTCTACATGGCAACTG-3′). Annealing temperature was 56°C for both NFAT2 and GAPDH.
Assessment of effect of NFAT2 knockdown on keratinocyte growth by manual cell counting
Keratinocytes were transfected using scrambled or NFAT2-specific siRNA and seeded into T25 flasks at 106 cells per flask. Three days later, the cells were trypsinized and passaged at a 1:3 ratio into new T25 flasks to prevent them from becoming confluent and growth arrested. Two days later, the cells were trypsinized again and counted using a hemocytometer.
Epidermal equivalents
Epidermal equivalents were established using keratinocytes transfected with scrambled or NFAT2-specific siRNA (Poumay et al., 2004; Jans et al., 2008). Briefly, 500,000 keratinocytes were seeded onto polycarbonate filters (0.4 µm pore size, Millipore, Durham, UK) and allowed to stratify for 13 days following lifting to air–liquid interface culture. The resulting epidermal equivalents were then fixed with 4% (w/v) paraformaldehyde for 20 min and washed with PBS. The filters were cut out of the inserts and split into half-moons using a scalpel blade and were processed for routine histology and H&E staining.
Statistics and Graphical illustrations
Data were analyzed using either un-paired t-tests, one-way ANOVA, ANOVA using a generalized linear model or two-way ANOVA to detect differences at a level of significance of P < 0.05, with two-tailed tests. Subset ANOVA analysis was also performed where appropriate. Minitab software was used. For comparisons of cell growth over time following retroviral transduction with different vectors, two-way ANOVA was used (GraphPad Prism version 4.00 for Windows (GraphPad, San Diego CA)).
Results
Lithium induces increased keratinocyte proliferation
Lithium at 1 or 2 mM induced increased proliferation of primary human keratinocytes (Fig. 1A), as measured by SRB assay maximal at 7 days. Stimulation of primary human keratinocyte growth appeared maximal at 1–2 mM lithium (Fig. 1A), consistent with the therapeutic range for lithium in patients treated for bipolar disorder of 0.4–1.2 mM (1 mM, P < 0.05, 2 mM P = 0.05, ANOVA, four independent experiments). The activity of GSK-3 is inhibited by N-terminal phosphorylation. Western analysis using an antibody specific to GSK-3β phosphorylated at N-terminal serine 9 showed that lithium (2, 5, or 10 mM lithium for 7 days) induced N-terminal phosphorylation in human keratinocytes (Fig. 1B) but did not increase total GSK-3. Keratinocytes treated with 1, 2, and 5 mM lithium were assessed by flow cytometry and an increase in the percentage of cells in S phase was seen with 2 mM lithium (Fig. 1C) and 5 mM lithium (data not shown) at 7 days. These data show that lithium promotes keratinocyte proliferation at therapeutic doses.
Fig. 1.

Lithium inhibits GSK3β in keratinocytes and induces proliferation of keratinocytes. A: SRB assays were performed on human keratinocytes following treatment for 7 days with lithium or vehicle. Lithium 1 mM (P < 0.05) and 2 mM (P = 0.05) significantly induced cell growth at 7 days (ANOVA, 24 replicates per group in four independent experiments). B: Western blotting of human keratinocyte lysates using anti-pGSK(Ser 9)3β antibody and anti-total GSK antibody. As expected an increase in phosphoGSK3β was seen but no increase in total GSK following treatment with lithium. C: Flow cytometry of human keratinocytes treated with lithium for 4 days. Cell cycle analysis showed an increase in S phase with 2 mM lithium (50.4%) compared with control (34.8%). One representative of three independent experiments is shown.
Pharmacological and genetic inhibition of GSK-3 induced proliferation of keratinocytes
Having confirmed that lithium-induced keratinocyte proliferation and inhibited GSK-3β, we further investigated the role of GSK-3 in regulating keratinocyte proliferation. To investigate whether inhibition of GSK-3 by lithium accounted for the effect of lithium on keratinocyte proliferation, we overexpressed GSKBP. GSKBP is a specific GSK-3 inhibitor, which we have shown, using Tcf/LEF-dependent TOPFLASH luciferase assays, to be functionally active in human keratinocytes (data not shown). We transduced SV-k14 keratinocytes with the empty vector, pLEGFP empty vector (Fig. 2A) or with a retroviral GFP–GSKBP fusion protein, pLEGFP-GSKBP (Fig. 2B), and showed that pLEGFP-GSKBP induced significantly greater proliferation at 7 days compared to pLEGFP empty vector (Fig. 2C, P < 0.005). Transduction of human keratinocytes with pLEGFP-GSKBP for 4 days induced increased proliferation compared to empty vector control pLEGFP empty (Fig. 2D, P = 0.02). To further confirm the role of GSK-3 in regulating keratinocyte proliferation, we next investigated pharmacological inhibition of GSK-3 using a specific GSK-3 inhibitor BIO (Meijer et al., 2003) and showed that BIO induced increased proliferation of SV-k14 keratinocytes at 7 days (Fig. 2E, P < 0.005 at 5, 50, and 500 nM).
Fig. 2.
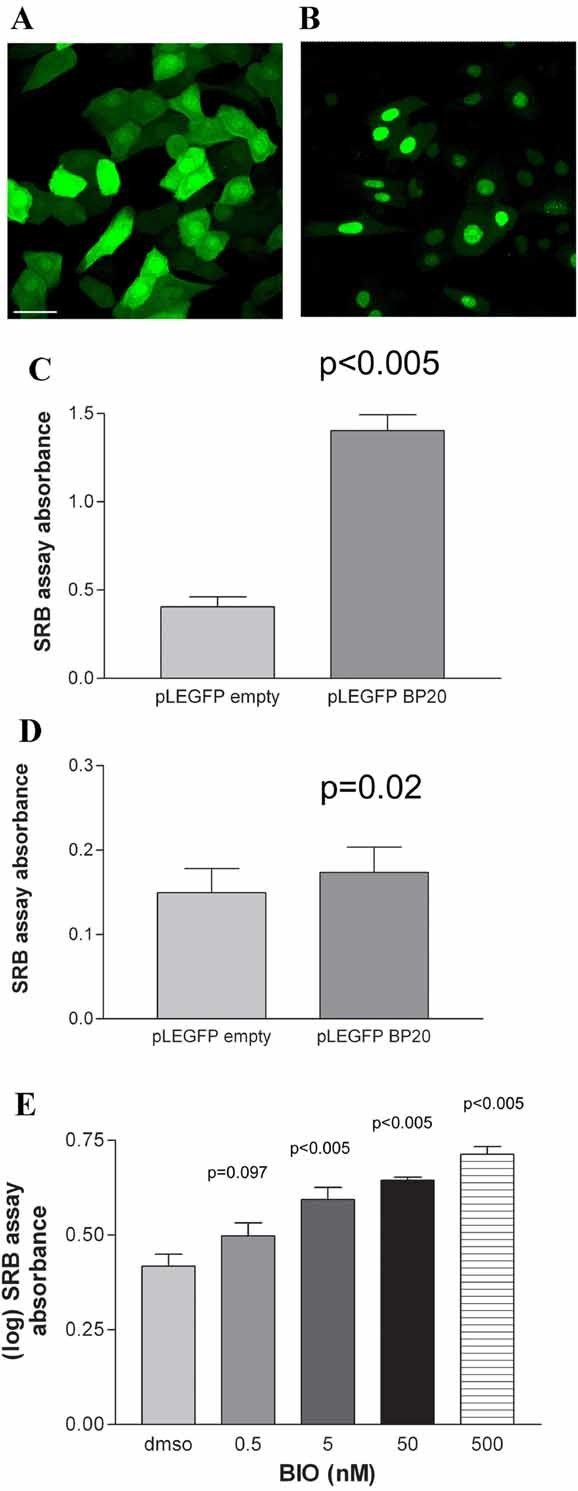
Inhibition of GSK3 by both pharmacological and genetic methods induces proliferation of keratinocytes. Keratinocytes were retrovirally transduced with pLEGFP empty vector (A) or pLEGFP GSKBP (B) and imaged by confocal microscopy. Scale bar represents 40 µm. SRB assays of K14 keratinocytes (C) or human keratinocytes (D). Transduction with pLEGFP GSKBP (BP20) induced significant proliferation compared to pLEGFP empty vector in K14 keratinocytes at 7 days (C: P < 0.005, t-test, three independent experiments, 30 wells per group per experiment) and in human keratinocytes at 4 days (D: P = 0.02, t-test, three independent experiments, 30 wells per group per experiment). E: SRB assays of K14 keratinocytes following treatment with BIO for 7 days induced significant proliferation at 5, 50, and 500 nM concentrations (P < 0.005, P < 0.006, P < 0.005, respectively, ANOVA, three independent experiments, 12 wells per group per experiment).
Inhibition of GSK-3 increased nuclear NFAT2 and induced transcriptional activation of an NFAT-luciferase reporter
Having demonstrated that inhibition of GSK-3 induced keratinocyte proliferation, we next investigated the role of the GSK substrate NFAT in keratinocyte proliferation. Since NFAT is a negatively regulated substrate of GSK-3, we investigated the effect of inhibition of GSK-3 on NFAT nuclear localization and transcriptional activity. Primary keratinocytes were transiently transfected with an NFAT-luciferase reporter and either the GSKBP or empty vector. As expected, GSKBP induced significantly greater activation of the NFAT-luciferase reporter than empty vector (Fig. 3A, P < 0.005). A similar effect on the NFAT-luciferase reporter was seen following retroviral transduction with the fusion protein pLEGFP-GSKBP compared to empty vector, further validating that this construct was functionally active in this system (data not shown). Consistent with the effect of GSKBP, overexpression of a constitutively active GSK mutant (CaGSK-3β-S9A), reduced activation of the NFAT-luciferase reporter (Fig. 3B, P = 0.002). Retroviral transduction of pLEGFP GSKBP induced increased nuclear NFAT2 compared with transduction of pLEGFP empty (Fig. 3C).
Fig. 3.

NFAT nuclear localization and transcriptional activation in human keratinocytes is regulated by GSK3. A: Transfection of the GSKBP (BP20) induced a fourfold increase of NFAT-luciferase activity (P < 0.005, unpaired t-test, n = 3 independent experiments, three replicates per experiment) at 24 h. B: Transfection of caGSK-3β-S9A inhibited NFAT-luciferase activity at 24 h compared with an empty vector control (P = 0.002, t-test, n = 2 independent experiments, three replicates per experiment). C: Human keratinocytes were transduced with either pLEGFP GSKBP or pLEGFP empty and were then subsequently immunostained 48 h later with anti-NFAT2 antibodies and the nuclear dye Toto-3. The rabbit anti-NFAT2 antibody was labeled with an Alexa Fluor 568 anti-rabbit secondary antibody. Increased nuclear NFAT2 was seen in the GSKBP positive cells (arrow heads), compared to the empty vector GFP positive cells. Mid z images are shown taken in sequential scanning mode to minimize cross talk. The following excitation wavelengths were used; GFP 488 nM, Alexa Fluor 543 nM, and Toto-3 633 nM. Scale bar represents 40 µm.
Lithium induced nuclear translocation of NFAT2, transcriptional activation of NFAT-luciferase, and increased expression of NFAT2 protein
The level of NFAT gene activation is determined in part by the duration that NFAT remains in the nucleus. Nuclear export of NFAT is therefore a crucial component in the regulation of NFAT-dependent gene transcription. GSK-3 has been shown to inhibit the DNA-binding activity of NFAT2 (Beals et al., 1997a; Neal and Clipstone, 2001).
Primary keratinocytes were treated with lithium (10 mM) for 48 h and NFAT2 localization was assessed by immunostaining. Increased nuclear localization of NFAT2 was seen following lithium treatment (Fig. 4A). Western blotting of lysates from primary human keratinocytes treated with lithium (10 mM) showed an increase in expression of NFAT2 isoforms at 48 h (Fig. 4B). Significantly increased transcriptional activation by therapeutically relevant concentrations of lithium (2 mM) was shown between 24 and 168 h, two-way ANOVA P = 0.039 (Fig. 4C). Thus, 2 mM lithium induced prolonged NFAT transcriptional activation in keratinocytes up to 7 days and at time points where we observed lithium-induced keratinocyte proliferation (Fig. 1).
Fig. 4.
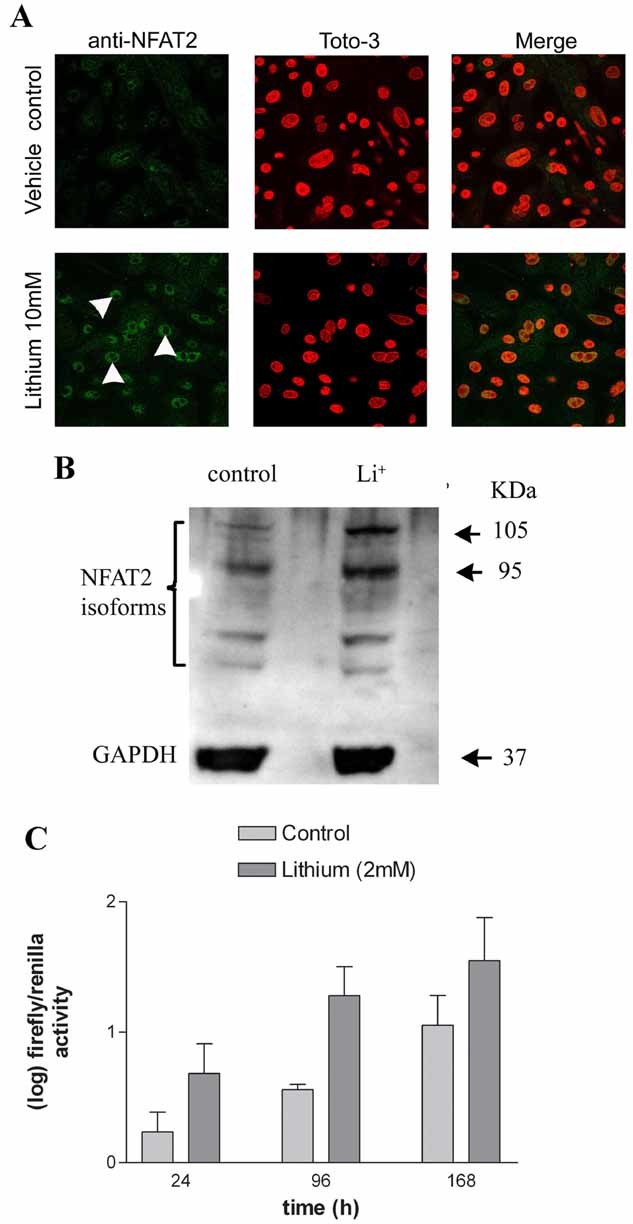
Lithium induces nuclear translocation of NFAT2, NFAT transcriptional activation and NFAT2 protein expression in keratinocytes. A: Human keratinocytes were treated with lithium (10 mM) for 48 h, immunostained with an anti-NFAT2 antibody and visualized by confocal microscopy. One representative of three experiments is shown. Increased nuclear NFAT2 (arrow heads) was seen in the lithium group compared with vehicle control. B: Western blotting of lysates of human keratinocytes treated with lithium (10 mM) for 48 h. Protein levels were calculated by band densitometry and adjusted for GAPDH expression. All of the NFAT2 isoforms were increased by all the treatments. Band densitometry showed the following percentage increases over control: 105 kDa band, Li+ (293%), 95 kDa band, Li+ (148%). C: Lithium (2 mM) activated NFAT-luciferase at 24, 96, and 168 h. (Two-way ANOVA P = 0.039 for lithium vs. control, three replicates per treatment in 2–3 independent experiments.)
Retroviral transduction of NFAT2 induces keratinocyte proliferation
NFAT signaling regulates proliferation in a number of cellular systems as previously indicated. We hypothesized that inhibition of GSK-3 by lithium leading to increased NFAT activity could result in increased proliferation of keratinocytes and that this may be relevant to the actions of lithium in provoking psoriasis. Overexpression of pLEGFP-GSKBP induced increased nuclear NFAT2 (Fig. 3C). To investigate the role of NFAT2 in regulating keratinocyte proliferation, we synchronized HaCaTs in G1 by serum starvation and then transduced cells with pLEGFP-NFAT2. A time-dependent increase in proliferation was seen up to 120 h (Fig. 5A, P < 0.001) following transduction with pLEGFP-NFAT2 compared to empty vector. In addition, transduction with pLEGFP-NFAT2 resulted in an increase in the proportion of cells in S phase compared to empty vector at 24 h (Fig. 5B). Also, following retroviral transduction of K14 keratinocytes with pLEGFP-NFAT2, increased proliferation was seen at 7 days (P = 0.01) compared to empty vector (data not shown).
Fig. 5.
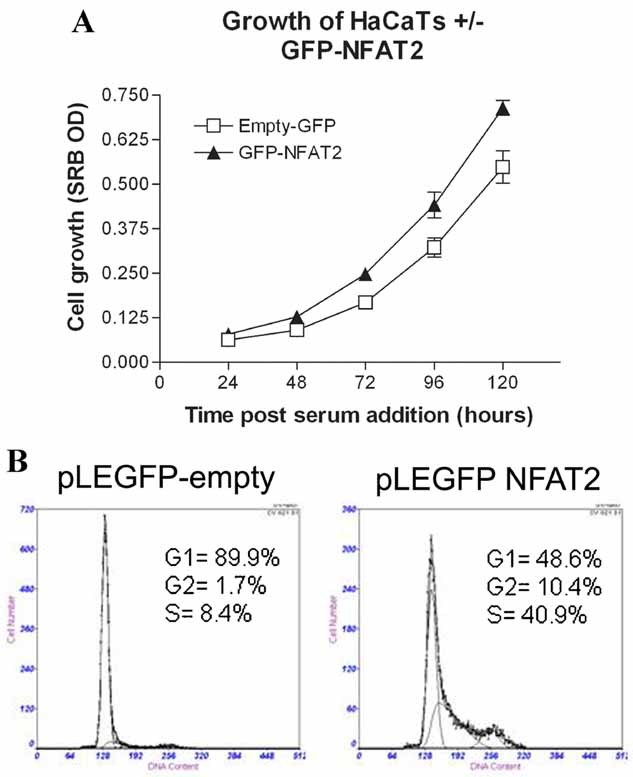
Overexpression of NFAT2 induces keratinocyte proliferation. A: HaCaT keratinocytes synchronized in G1 were retrovirally transduced with either pLEGFP empty or pLEGFP-NFAT2 and proliferation assessed by SRB assay. Two-way ANOVA analysis showed a significant difference in proliferation between the pLEGFP empty and the pLEGFP-NFAT2 groups (P = 0.008) and a significant difference in proliferation over time (P < 0.001), two independent experiments with 12 replicates. B: Synchronized HaCaT keratinocytes were retrovirally transduced with either pLEGFP empty or pLEGFP-NFAT2 and cell cycle progression was assessed by flow cytometry. At 24 h there was an increase in the percentage of cells in S phase in the pLEGFP-NFAT2 group.
Finally, we assessed the effect of knocking down NFAT2 using NFAT siRNA in an epidermal equivalent model and in monolayer cultured human keratinocytes. Knockdown of NFAT2 in primary keratinocytes was confirmed by Western blotting (Jans et al. (manuscript in preparation)). Keratinocytes transfected with NFAT2 siRNA were grown in an epidermal skin equivalent model. Knockdown of NFAT2 resulted in reduced epidermal thickness (Fig. 6A). siRNA-mediated knockdown of NFAT2 reduced keratinocyte growth as assessed by cell counting (Fig. 6B) and SRB assay (data not shown). Together, these data indicate a role of NFAT2 in the regulation of keratinocyte proliferation and cell cycle progression and suggest that lithium induced keratinocyte proliferation may be regulated via GSK-3 and NFAT2.
Fig. 6.

NFAT2 knockdown by siRNA reduces cell number and reduces epidermal thickness in an in vitro epidermal equivalent model. A: Keratinocytes were transfected with either scrambled RNA or with NFAT2 siRNA and then grown as an in vitro epidermal skin equivalent. The NFAT2 siRNA group resulted in a thinner epidermis than control (P < 0.05). B: Cell counts confirmed that NFAT2 siRNA reduced keratinocyte proliferation (P < 0.001).
Discussion
Our results have identified GSK-3 as a molecular target for lithium in human epidermal keratinocytes and as a negative regulator of keratinocyte proliferation. Thus, lithium at therapeutic doses resulted in inhibitory phosphorylation of GSK-3β and also induced proliferation of human keratinocytes. Using a genetic and pharmacological approach, we have confirmed that GSK3 is a key regulator of human interfollicular keratinocyte cell growth. Inhibition of GSK-3, using retroviral transduction of GSKBP (Franca-Koh et al., 2002), or through using the GSK-3 inhibitor BIO (Sato et al., 2004), increased keratinocyte proliferation. Furthermore, our data identify NFAT, and NFAT2 in particular, as an important down-stream target for GSK3 in keratinocytes. Inhibition of GSK-3 by lithium or overexpression of GSKBP increased NFAT transcriptional activity and increased NFAT2 nuclear localization. Importantly, overexpression of NFAT2 increased growth of human keratinocytes and HaCaT cells, consistent with the inhibitory action of cyclosporin on keratinocyte growth (Fisher et al., 1988).
We observed proliferation of keratinocytes in response to (1) lithium which inhibits both GSK-3 isoforms, (2) the specific GSK-3 inhibitor BIO, and (3) overexpression of GSKBP which inhibits GSK-3 activity, underscoring the role of GSK-3 in regulating keratinocyte growth (Ferkey and Kimelman, 2002; Bhat et al., 2004). These results are consistent with evidence from a variety of cell types which indicates that genetic inhibition of GSK-3β induces cell proliferation (Hamilton et al., 1995; Ohteki et al., 2000; Park et al., 2003). Moreover, we observed an increase in pGSK-3β (Ser 9) in keratinocytes following lithium treatment, suggesting that GSK-3β is a likely target for lithium-induced keratinocyte proliferation. Finally, epidermal hyperproliferation in response to lithium has been demonstrated in an in vitro skin explant model (Wolf et al., 2000).
Our data showing that lithium and inhibition of GSK-3 induced transcriptional activation of NFAT and increased translation of NFAT2 isoforms in keratinocytes are consistent with the known role of GSK-3 in regulating NFAT phosphorylation in vitro and NFAT nuclear export (Beals et al., 1997b). Regulation of NFAT2 (NFATc1) is complex, controlled by at least two promoters, as well as alternative splicing and two polyadenylation sites and may result in the transcription and translation of up to 6 isoforms (Serfling et al., 2006). Moreover, two NFAT-binding sites have been identified in the P1 promoter and binding of NFAT may therefore result in auto-induction particularly of the shorter NFAT2/αA isoform but also NFAT2/αB and NFAT2/αC (Serfling et al., 2006). Induction of 4 NFAT2 isoforms in keratinocytes following treatment with lithium is consistent with this model which appears to operate in a variety of cellular systems including T cells and bone (Lyakh et al., 1997; Serfling et al., 2006). In addition, lithium treatment resulted in nuclear NFAT2 as did overexpression of pLEGFP GSKBP. Importantly and potentially relevant to lithium-provoked psoriasis, we demonstrated that transduction of NFAT2 induced keratinocyte proliferation and that NFAT2 knockdown caused a reduction in thickness in an epidermal skin equivalent model.
The effect of lithium alone is unlikely to be sufficient to induce the psoriatic phenotype. A predisposition to psoriasis seems to be regulated by a complex interplay between keratinocytes, T cells, and the innate immune system (Gaspari, 2006; Nestle et al., 2009). We hypothesize that lithium is likely to trigger psoriasis in genetically predisposed individuals whose skin innate immune system is in an activated state. Our results suggest that lithium-induced psoriasis may involve a GSK3-mediated effect on NFAT2. By inhibiting GSK3, lithium may reduce the nuclear export of NFAT2 thereby maintaining NFAT2 in the nucleus and moreover this may lead to NFAT2 auto-induction and a positive feedback loop (Chuvpilo et al., 2002). Thus, lithium may reduce the threshold required to induce a significant NFAT-mediated effect on keratinocyte proliferation. However, in the clinical situation lithium may not itself be sufficient to induce clinically overt psoriasis in the absence of a further, yet to be identified, co-stimuli, or the relevant cytokine milieu. In other words lithium, by inhibiting GSK3 and inducing NFAT2, may reduce the threshold for induction of epidermal proliferation and remodeling required for the formation of the psoriatic plaques in predisposed individuals. This hypothesis may also explain the widely varying times between commencement of lithium and onset of psoriasis.
Acknowledgments
We are grateful to Dr. B. Eldar-Finkelman for the kind gift of the GSKBP vector, to Dr. D.J. McKean (Mayo Foundation, USA) for the NFAT-luciferase reporter, and to Dr. Nancy Rice (NCI, Frederick, USA) for the NFAT2 antibody. We thank Prof. John Matthews (Newcastle University, UK) for statistical advice and Carole Todd for expert technical assistance. We thank the Departments of Paediatric Surgery, Plastic Surgery and Urology, Newcastle upon Tyne Hospitals NHS Foundation Trust for their help in recruiting patients.
Literature Cited
- Al-Daraji WI, Grant KR, Ryan K, Saxton A, Reynolds NJ. Localization of calcineurin/NFAT in human skin and psoriasis and inhibition of calcineurin/NFAT activation in human keratinocytes by cyclosporin A. J Invest Dermatol. 2002;118:779–788. doi: 10.1046/j.1523-1747.2002.01709.x. [DOI] [PubMed] [Google Scholar]
- Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997a;275:1930–1934. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997b;275:1930–1934. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- Beurel E, Michalek SM, Jope RS. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3) Trends Immunol. 2010;31:24–31. doi: 10.1016/j.it.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Budd Haeberlein SL, Avila J. Glycogen synthase kinase 3: A drug target for CNS therapies. J Neurochem. 2004;89:1313–1317. doi: 10.1111/j.1471-4159.2004.02422.x. [DOI] [PubMed] [Google Scholar]
- Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med. 2004;199:731–736. doi: 10.1084/jem.20031482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz M, Ellenrieder V. An emerging role for Ca2+/calcineurin/NFAT signaling in cancerogenesis. Cell Cycle. 2007;6:16–19. doi: 10.4161/cc.6.1.3650. [DOI] [PubMed] [Google Scholar]
- Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, Matsunami N, Ardlie KG, Civello D, Catanese JJ, Leong DU, Panko JM, McAllister LB, Hansen CB, Papenfuss J, Prescott SM, White TJ, Leppert MF, Krueger GG, Begovich AB. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuvpilo S, Jankevics E, Tyrsin D, Akimzhanov A, Moroz D, Jha MK, Schulze-Luehrmann J, Santner-Nanan B, Feoktistova E, Konig T, Avots A, Schmitt E, Berberich-Siebelt F, Schimpl A, Serfling E. Autoregulation of NFATc1/A expression facilitates effector T cells to escape from rapid apoptosis. Immunity. 2002;16:881–895. doi: 10.1016/s1074-7613(02)00329-1. [DOI] [PubMed] [Google Scholar]
- Cui H, Meng Y, Bulleit RF. Inhibition of glycogen synthase kinase 3beta activity regulates proliferation of cultured cerebellar granule cells. Brain Res Dev Brain Res. 1998;111:177–188. doi: 10.1016/s0165-3806(98)00136-9. [DOI] [PubMed] [Google Scholar]
- de Cid R, Riveira-Munoz E, Zeeuwen PL, Robarge J, Liao W, Dannhauser EN, Giardina E, Stuart PE, Nair R, Helms C, Escaramis G, Ballana E, Martin-Ezquerra G, den Heijer M, Kamsteeg M, Joosten I, Eichler EE, Lazaro C, Pujol RM, Armengol L, Abecasis G, Elder JT, Novelli G, Armour JA, Kwok PY, Bowcock A, Schalkwijk J, Estivill X. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat Genet. 2009;41:211–215. doi: 10.1038/ng.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129:1339–1350. doi: 10.1038/jid.2009.59. [DOI] [PubMed] [Google Scholar]
- Farr GH, III, Ferkey DM, Yost C, Pierce SB, Weaver C, Kimelman D. Interaction among GSK-3, GBP, axin, and APC in Xenopus axis specification. J Cell Biol. 2000;148:691–702. doi: 10.1083/jcb.148.4.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferkey DM, Kimelman D. Glycogen synthase kinase-3 beta mutagenesis identifies a common binding domain for GBP and Axin. J Biol Chem. 2002;277:16147–16152. doi: 10.1074/jbc.M112363200. [DOI] [PubMed] [Google Scholar]
- Fisher GJ, Duell EA, Nickoloff BJ, Annesley TM, Kowalke JK, Ellis CN, Voorhees JJ. Levels of cyclosporin in epidermis of treated psoriasis patients differentially inhibit growth of keratinocytes cultured in serum free versus serum containing media. J Invest Dermatol. 1988;91:142–146. doi: 10.1111/1523-1747.ep12464387. [DOI] [PubMed] [Google Scholar]
- Franca-Koh J, Yeo M, Fraser E, Young N, Dale TC. The regulation of glycogen synthase kinase-3 nuclear export by Frat/GBP. J Biol Chem. 2002;277:43844–43848. doi: 10.1074/jbc.M207265200. [DOI] [PubMed] [Google Scholar]
- Gaspari AA. Innate and adaptive immunity and the pathophysiology of psoriasis. J Am Acad Dermatol. 2006;54:S67–S80. doi: 10.1016/j.jaad.2005.10.057. [DOI] [PubMed] [Google Scholar]
- Goodwin P, Hamilton S, Fry L. A comparison between DNA synthesis and mitosis in uninvolved and involved psoriatic epidermis and normal epidermis. Br J Dermatol. 1973;89:613–618. doi: 10.1111/j.1365-2133.1973.tb07587.x. [DOI] [PubMed] [Google Scholar]
- Gudjonsson JE, Ding J, Li X, Nair RP, Tejasvi T, Qin ZS, Ghosh D, Aphale A, Gumucio DL, Voorhees JJ, Abecasis GR, Elder JT. Global gene expression analysis reveals evidence for decreased lipid biosynthesis and increased innate immunity in uninvolved psoriatic skin. J Invest Dermatol. 2009;129:2795–2804. doi: 10.1038/jid.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B, Burger PC, Wood PA, Taqi F, Booker SV, Petersen GM, Offerhaus JA, Tersmette AC. Giardiello FM, Volgelstein B, Kinzler KW. The molecular basis of Turcot's syndrome. N Engl J Med. 1995;332:839–847. doi: 10.1056/NEJM199503303321302. [DOI] [PubMed] [Google Scholar]
- Happle R. Somatic recombination may explain linear psoriasis. J Med Genet. 1991;28:337. doi: 10.1136/jmg.28.5.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatta N, Takata M, Kawara S, Hirone T, Takehara K. Tape stripping induces marked epidermal proliferation and altered TGF-alpha expression in non-lesional psoriatic skin. J Dermatol Sci. 1997;14:154–161. doi: 10.1016/s0923-1811(96)00567-1. [DOI] [PubMed] [Google Scholar]
- Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VM, Klein PS. Activation of the Wnt signaling pathway: A molecular mechanism for lithium action. Dev Biol. 1997;185:82–91. doi: 10.1006/dbio.1997.8552. [DOI] [PubMed] [Google Scholar]
- Hedin KE, Bell MP, Kalli KR, Huntoon CJ, Sharp BM, McKean DJ. Delta-opioid receptors expressed by Jurkat T cells enhance IL-2 secretion by increasing AP-1 complexes and activity of the NF-AT/AP-1-binding promoter element. J Immunol. 1997;159:5431–5440. [PubMed] [Google Scholar]
- Hell E, Hodgson C. The uptake of 3H-thymidine by epidermal cells in normal and psoriatic subjects. DNA synthesis in psoriasis. Br J Dermatol. 1966;78:262–268. doi: 10.1111/j.1365-2133.1966.tb12220.x. [DOI] [PubMed] [Google Scholar]
- Horsley V, Aliprantis AO, Polak L, Glimcher LH, Fuchs E. NFATc1 balances quiescence and proliferation of skin stem cells. Cell. 2008;132:299–310. doi: 10.1016/j.cell.2007.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffmeier U, Bergboer JG, Becker T, Armour JA, Traupe H, Estivill X, Riveira-Munoz E, Mossner R, Reich K, Kurrat W, Wienker TF, Schalkwijk J, Zeeuwen PL, Reis A. Replication of LCE3C-LCE3B CNV as a risk factor for psoriasis and analysis of interaction with other genetic risk factors. J Invest Dermatol. 2010;130:979–984. doi: 10.1038/jid.2009.385. [DOI] [PubMed] [Google Scholar]
- Hui W, Litherland GJ, Jefferson M, Barter MJ, Elias MS, Cawston TE, Rowan AD, Young DA. Lithium protects cartilage from cytokine-mediated degradation by reducing collagen-degrading MMP production via inhibition of the P38 mitogen-activated protein kinase pathway. Rheumatology (Oxford) 2010;49:2043–2053. doi: 10.1093/rheumatology/keq217. [DOI] [PubMed] [Google Scholar]
- Jans R, Sturniolo MT, Eckert RL. Localization of the TIG3 transglutaminase interaction domain and demonstration that the amino-terminal region is required for TIG3 function as a keratinocyte differentiation regulator. J Invest Dermatol. 2008;128:517–529. doi: 10.1038/sj.jid.5701035. [DOI] [PubMed] [Google Scholar]
- Jonkers J, Korswagen HC, Acton D, Breuer M, Berns A. Activation of a novel proto-oncogene, Frat1, contributes to progression of mouse T-cell lymphomas. EMBO J. 1997;16:441–450. doi: 10.1093/emboj/16.3.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kirshenboim N, Plotkin B, Shlomo SB, Kaidanovich-Beilin O, Eldar-Finkelman H. Lithium-mediated phosphorylation of glycogen synthase kinase-3b involves PI3 kinase-dependent activation of protein kinase C-alpha. J Mol Neurosci. 2004;24:237–245. doi: 10.1385/JMN:24:2:237. [DOI] [PubMed] [Google Scholar]
- Lipskaia L, Lompre AM. Alteration in temporal kinetics of Ca2+ signaling and control of growth and proliferation. Biol Cell. 2004;96:55–68. doi: 10.1016/j.biolcel.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Lyakh L, Ghosh P, Rice NR. Expression of NFAT-family proteins in normal human T cells. Mol Cell Biol. 1997;17:2475–2484. doi: 10.1128/mcb.17.5.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji HK, Bebchuk JM, Moore GJ, Glitz D, Hasanat KA, Chen G. Modulation of CNS signal transduction pathways and gene expression by mood-stabilizing agents: Therapeutic implications. J Clin Psychiatry. 1999;60:27–39. Discussion 40–21, 113–116. [PubMed] [Google Scholar]
- Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill A, Frank A, Emmett N, Turnbull DM, Birch-Machin MA, Reynolds NJ. The anti-psoriatic drug anthralin accumulates in keratinocyte mitochondria, dissipates mitochondrial membrane potential, and induces apoptosis through a pathway dependent on respiratory competent mitochondria. FASEB J. 2005;19:1012–1014. doi: 10.1096/fj.04-2664fje. [DOI] [PubMed] [Google Scholar]
- Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, Crovace C, Tarricone C, Musacchio A, Roe SM, Pearl L, Greengard P. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Murphy LL, Hughes CC. Endothelial cells stimulate T cell NFAT nuclear translocation in the presence of cyclosporin A: Involvement of the wnt/glycogen synthase kinase-3 beta pathway. J Immunol. 2002;169:3717–3725. doi: 10.4049/jimmunol.169.7.3717. [DOI] [PubMed] [Google Scholar]
- Neal JW, Clipstone NA. Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. J Biol Chem. 2001;276:3666–3673. doi: 10.1074/jbc.M004888200. [DOI] [PubMed] [Google Scholar]
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496–509. doi: 10.1056/NEJMra0804595. [DOI] [PubMed] [Google Scholar]
- Ohteki T, Parsons M, Zakarian A, Jones RG, Nguyen LT, Woodgett JR, Ohashi PS. Negative regulation of T cell proliferation and interleukin 2 production by the serine threonine kinase GSK-3. J Exp Med. 2000;192:99–104. doi: 10.1084/jem.192.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver JE, Silman AJ. What epidemiology has told us about risk factors and aetiopathogenesis in rheumatic diseases. Arthritis Res Ther. 2009;11:223. doi: 10.1186/ar2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmos RC, Hillegers MH, Knijff EM, Vonk R, Bouvy A, Staal FJ, de Ridder D, Kupka RW, Nolen WA, Drexhage HA. A discriminating messenger RNA signature for bipolar disorder formed by an aberrant expression of inflammatory genes in monocytes. Arch Gen Psychiatry. 2008;65:395–407. doi: 10.1001/archpsyc.65.4.395. [DOI] [PubMed] [Google Scholar]
- Park KW, Yang HM, Youn SW, Yang HJ, Chae IH, Oh BH, Lee MM, Park YB, Choi YS, Kim HS, Walsh K. Constitutively active glycogen synthase kinase-3beta gene transfer sustains apoptosis, inhibits proliferation of vascular smooth muscle cells, and reduces neointima formation after balloon injury in rats. Arterioscler Thromb Vasc Biol. 2003;23:1364–1369. doi: 10.1161/01.ATV.0000081633.53390.B4. [DOI] [PubMed] [Google Scholar]
- Piskin G, Sylva-Steenland RM, Bos JD, Teunissen MB. In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: Enhanced expression in psoriatic skin. J Immunol. 2006;176:1908–1915. doi: 10.4049/jimmunol.176.3.1908. [DOI] [PubMed] [Google Scholar]
- Poumay Y, Dupont F, Marcoux S, Leclercq-Smekens M, Herin M, Coquette A. A simple reconstructed human epidermis: Preparation of the culture model and utilization in in vitro studies. Arch Dermatol Res. 2004;296:203–211. doi: 10.1007/s00403-004-0507-y. [DOI] [PubMed] [Google Scholar]
- Ramagopalan SV, Dobson R, Meier UC, Giovannoni G. Multiple sclerosis: Risk factors, prodromes, and potential causal pathways. Lancet Neurol. 2010;9:727–739. doi: 10.1016/S1474-4422(10)70094-6. [DOI] [PubMed] [Google Scholar]
- Regl G, Neill GW, Eichberger T, Kasper M, Ikram MS, Koller J, Hintner H, Quinn AG, Frischauf AM, Aberger F. Human GLI2 and GLI1 are part of a positive feedback mechanism in basal cell carcinoma. Oncogene. 2002;21:5529–5539. doi: 10.1038/sj.onc.1205748. [DOI] [PubMed] [Google Scholar]
- Ryves WJ, Harwood AJ. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem Biophys Res Commun. 2001;280:720–725. doi: 10.1006/bbrc.2000.4169. [DOI] [PubMed] [Google Scholar]
- Sano S, Chan KS, Carbajal S, Clifford J, Peavey M, Kiguchi K, Itami S, Nickoloff BJ, DiGiovanni J. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat Med. 2005;11:43–49. doi: 10.1038/nm1162. [DOI] [PubMed] [Google Scholar]
- Santini MP, Talora C, Seki T, Bolgan L, Dotto GP. Cross talk among calcineurin, Sp1/Sp3, and NFAT in control of p21(WAF1/CIP1) expression in keratinocyte differentiation. Proc Natl Acad Sci USA. 2001;98:9575–9580. doi: 10.1073/pnas.161299698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med. 2004;10:55–63. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- Serfling E, Chuvpilo S, Liu J, Hofer T, Palmetshofer A. NFATc1 autoregulation: A crucial step for cell-fate determination. Trends Immunol. 2006;27:461–469. doi: 10.1016/j.it.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Shen F, Li N, Gade P, Kalvakolanu DV, Weibley T, Doble B, Woodgett JR, Wood TD, Gaffen SL. IL-17 receptor signaling inhibits C/EBPbeta by sequential phosphorylation of the regulatory 2 domain. Sci Signal. 2009;2:ra8. doi: 10.1126/scisignal.2000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- Skoven I, Thormann J. Lithium compound treatment and psoriasis. Arch Dermatol. 1979;115:1185–1187. [PubMed] [Google Scholar]
- Todd C, Reynolds NJ. Up-regulation of p21WAF1 by phorbol ester and calcium in human keratinocytes through a protein kinase C-dependent pathway. Am J Pathol. 1998;153:39–45. doi: 10.1016/S0002-9440(10)65543-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner H, Gomez M, McKenzie E, Kirchem A, Lennard A, Cantrell DA. Rac-1 regulates nuclear factor of activated T cells (NFAT) C1 nuclear translocation in response to Fcepsilon receptor type 1 stimulation of mast cells. J Exp Med. 1998;188:527–537. doi: 10.1084/jem.188.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vines A, Cahoon S, Goldberg I, Saxena U, Pillarisetti S. Novel anti-inflammatory role for glycogen synthase kinase-3beta in the inhibition of tumor necrosis factor-alpha- and interleukin-1beta-induced inflammatory gene expression. J Biol Chem. 2006;281:16985–16990. doi: 10.1074/jbc.M602446200. [DOI] [PubMed] [Google Scholar]
- Voigt W. Sulforhodamine B assay and chemosensitivity. Methods Mol Med. 2005;110:39–48. doi: 10.1385/1-59259-869-2:039. [DOI] [PubMed] [Google Scholar]
- Welsh GI, Proud CG. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem J. 1993;294:625–629. doi: 10.1042/bj2940625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf R, D'Avino M, De Angelis F, Ruocco E, Lombardi ML. Effects of lithium carbonate (Li2CO3) on in-vitro-cultured normal human skin explants. J Eur Acad Dermatol Venereol. 2000;14:97–99. doi: 10.1046/j.1468-3083.2000.00031.x. [DOI] [PubMed] [Google Scholar]
- Yost C, Farr GH, III, Pierce SB, Ferkey DM, Chen MM, Kimelman D. GBP, an inhibitor of GSK-3, is implicated in Xenopus development and oncogenesis. Cell. 1998;93:1031–1041. doi: 10.1016/s0092-8674(00)81208-8. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]