

Abstract
Benign familial neonatal seizures (BFNS) is an autosomal dominant disorder associated with heterozygous mutations of either the KCNQ2 or KCNQ3 gene. Most cases have mutations of the KCNQ2 gene. A handful of cases with KCNQ2 and CHRNA4 deletions have been identified with different phenotypic presentations. Only two cases presented with typical BFNS features. Benign familial neonatal seizures is associated with normal exam and work-up, and seizure remission is seen in the first month of life. We report three unrelated individuals with KCNQ2 and CHRNA4 deletions, presenting with neonatal seizures and developmental delay. Their seizures started within one week after birth; all required antiepileptic drugs. Each had normal brain magnetic resonance imaging and at least two electroencephalograms with either normal or abnormal findings. All were developmentally delayed. None presented with autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) phenotype associated with CHRNA4 mutation. This study supports reports of KCNQ2 and CHRNA4 deletions associated with phenotypes different from typical BFNS.
Keywords: Benign familial neonatal seizures, KCNQ2, CHRNA4, 20q13.33 deletion
1. Introduction
Benign familial neonatal seizures (BFNS) is characterized by a nonspecific type of seizure in healthy infants in their first few days of life, typically around postnatal day three. Affected individuals have normal neurological exam and studies, except for a small percentage of abnormal electroencephalograms (EEGs) with nonspecific findings. Spontaneous seizure remission is seen within one month, though about 10–15% of patients may have epilepsy later in life. Benign familial neonatal seizures is a rare autosomal dominant disorder caused by heterozygous mutations in either the KCNQ2 or KCNQ3 gene. About 80 families with BFNS have been reported to date [1]. Rare atypical features associated with heterozygous mutations in KCNQ2 are drug-resistant epilepsy and mental retardation [2] and epileptic encephalopathy [3]. A microdeletion on 20q13.33 including KCNQ2 and CHRNA4 has been associated with phenotypes indistinguishable from those with KCNQ2 deletion alone and without the autosomal dominant frontal lobe epilepsy (ADNFLE) phenotype [4]. On the other hand, several isolated cases with concomitant KCNQ2 and CHRNA4 deletions have been identified with different phenotypic presentations, including learning disability, hyperlaxity, and strabismus [5]; dysmorphic features, mild ataxia, developmental delay, and psychiatric problems [6]; and neurodevelopmental delay [7]. We report three unrelated individuals with deletions involving KCNQ2 and CHRNA4, presenting with neonatal seizures requiring antiepileptic drugs (AEDs) and developmental delay.
2. Methods
The affected individuals were evaluated for neonatal seizures. Each individual had full neurological work-up and genetic testing. Genetic testing was done using microarray analysis, with two patients diagnosed by 720K Agilent oligoarray and one diagnosed by Reveal SNP array containing 1.9M oligo and 750K SNP probes.
3. Case reports
3.1. Patient 1
A 10-month-old Mexican-American male was born at term, following induced labor due to oligohydramnios. Pregnancy was complicated by uterine fibroids, depression, and exposure to alcohol, marijuana, and cigarette smoking. He was a product of nonconsanguineous parents. At birth, he had normal APGAR scores and physical exam. On postnatal day seven, he started having several seizures daily, associated with head turning to the left, followed by secondary generalization for 15 to 30 s. Metabolic work-up was unrevealing, except for elevated thyroid stimulating hormone with normal free thyroxine. Brain magnetic resonance imaging (MRI) was normal. His first electroencephalogram (EEG) was normal. Repeat EEG a few days later showed excessive sharp transients with intermittent high voltage slowing. Monotherapy with levetiracetam failed to control his seizures. Oxcarbazepine was added, which stopped his seizures at age one month. At age three months, he had a seizure while off all AEDs. A subsequent EEG was normal. At six months, he was noted to have mild developmental delay. He had a seizure with fever after vaccination at six months. His mother had febrile seizures as a child, and a second-degree paternal uncle had seizures until age two years. No one else had seizures in the family (pedigree shown in Fig. 1-A). The patient's genetic testing showed a 521-kb deletion on 20q13.33 involving 22 genes (11 OMIM genes), including KCNQ2 and CHRNA4 (Fig. 2). Family members declined to undergo genetic testing.
Fig. 1.
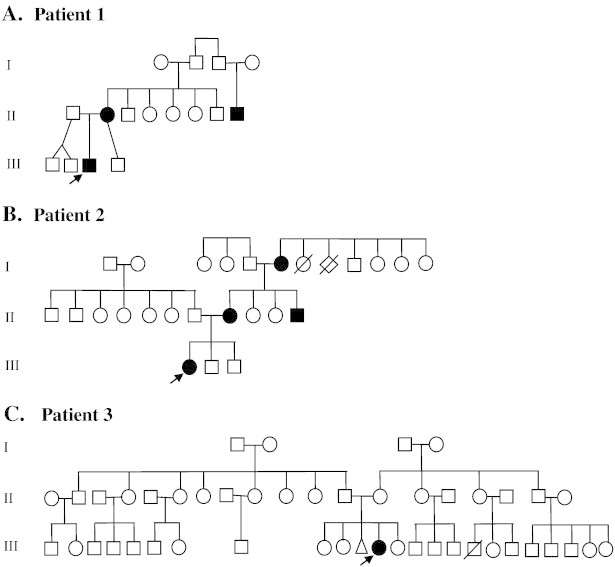
Three-generation pedigree. Patient 1's family has two generations affected by seizures in infancy. Patient 2's family is notable for seizures during infancy from each of the three generations. Patient 3 is the only one affected by seizures in three generations of her family. Arrows mark the affected patient. Cases of seizures in infancy are shown in solid symbols.
Fig. 2.
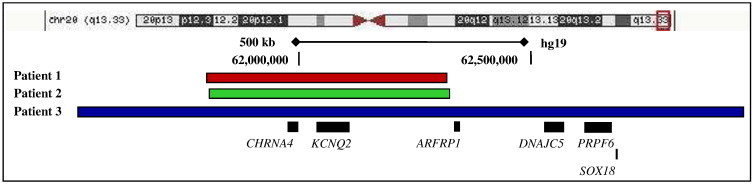
Chromosomal location of the microdeletions on 20q13.33. The size of contiguous chromosomal deletion for the three patients is depicted below the stylized chromosome. Further below is a cartoon of the genes mapping to the deleted region. The segment between the vertical dashed bars represents the contiguous deletion seen in patients 1 and 2, while the entire depicted segment represents the deletion seen in patient 3.
3.2. Patient 2
A 16-month-old Mexican/Native-American female was born at term, following normal pregnancy from a nonconsanguineous union. On postnatal day two, she started having convulsions until age six months, requiring levetiracetam, zonisamide, and topiramate. Her physical exam was normal, except for a dysconjugate gaze. The first EEG showed mild dysregulation, while the second one was normal. Her brain MRI was normal. She had global developmental delay. At 15 months, she was not yet talking or walking. She had a strong family history of seizures in infancy (pedigree shown in Fig. 1-B). Her mother, maternal uncle, and grandmother had seizures during infancy with spontaneous remission. The patient's genetic testing revealed a 273-kb duplication on 13q21.1 and a 520.7-kb deletion on 20q13.33 involving 22 genes (11 OMIM genes), which include KCNQ2 and CHRNA4 (Fig. 2). The 13q21.1 duplicated region contains one known gene, PCDH17, which has no known disease association. Family members declined to undergo genetic testing.
3.3. Patient 3
A 44-month-old Mexican-American term female was born via urgent Cesarean section due to failure to progress and fetal distress. She was the product of nonconsanguineous parents. The patient had normal exam at birth. She had two focal motor seizures on postnatal day two. Cerebrospinal fluid study was unremarkable. Initial EEG showed multifocal spikes and abnormal background. Her brain MRI was normal. She was started on phenobarbital, and she had no further seizures. A repeat EEG at four months showed rare left parietal spikes, while another EEG at 11 months was normal. She had global developmental delay. At 16 months, she was not walking yet. At 42 months, she could only say two words. A hearing test revealed mild to moderate hearing loss with poor localization. She was also diagnosed with idiopathic macrocytosis, severe eczema, asthma, benign systolic murmur, failure to thrive, short stature, and frequent otitis media. Microarray studies revealed an imbalance with a 127-kb duplication on 9p24.1 and 1.4-Mb deletion on 20q13.33-qter involving 64 genes (32 OMIM genes), including KCNQ2 and CHRNA4 (Fig. 2). There is no family history of seizures (pedigree shown in Fig. 1-C). Her 6-month-old sister has trisomy 21 and an Xq26 deletion. No other family members underwent genetic testing. A maternal cousin with suspected trisomy 21 passed away at 6 months.
4. Discussion
Benign familial neonatal seizures is an uncommon disorder, and its association with a microdeletion on 20q13.33, including KCNQ2 and CHRNA4, is also uncommon. Most cases have mutations in KCNQ2 with deletions, insertions, splice-site mutations, or missense mutations, while the minority has KCNQ3 mutation. Both genes encode voltage-gated potassium channel subunits. The major locus associated with KCNQ2 mutation is on chromosome 20q13.3, while the major locus associated with KCNQ3 mutation is on chromosome 8q24 [3]. Deletions in KCNQ2 have been identified in approximately 20% of families with BFNS [1]. Two cases with concomitant KCNQ2 and CHRNA4 deletions, ranging from 46.3 to 245.3 kb and presenting with typical BFNS features, have been reported [4]. The CHRNA4 gene encodes the α4 subunit of neuronal acetylcholine receptors, and its deletion is associated with ADNFLE [8]. The two cases had no ADNFLE phenotype. Kurahashi et al. [4] stated that cases with contiguous deletions of KCNQ2 and CHRNA4 only have a typical BFNS phenotype.
Interestingly, several cases highlighting atypical phenotypic presentations with contiguous deletion of KCNQ2 and CHRNA4 have been reported. A 1.1- to 1.6-Mb subtelomeric deletion of chromosome 20q13.33 involving KCNQ2 and CHRNA4 has been associated with learning disability, hyperlaxity, and strabismus [5]. A pericentric insertion of chromosome 20, causing deletion of KCNQ2 and CHRNA4, was seen in a female patient with generalized tonic seizure until 13 months, requiring phenobarbital; dysmorphic features; mild ataxia; developmental delay; and psychiatric problems. This gene mutation was caused by intrachromosomal insertion, resulting in a 4.7-Mb duplication of the terminal segment of 20p and a 1.6-Mb deletion of the terminal region of 20q [6]. Another case was initially thought to have pyridoxine-dependent epilepsy but was later confirmed to have a 1.5-Mb terminal deletion of chromosome 20, including KCNQ2 and CHRNA4. The patient stopped having seizures, but he continued to have neurodevelopmental delay [7].
We identified three cases with BFNS associated with atypical phenotype associated with contiguous KCNQ2 and CHRNA4 deletions. Unlike typical BFNS of which spontaneous remission is expected within one month, all cases required AEDs, with two needing more than one AED. Another striking, unusual feature was their varying degrees of developmental delay. None had the ADNFLE phenotype. Atypical BFNS features including mental retardation and drug resistance are rare [2,3]. In a report by Weckhuysen et al. [9], 10% of neonatal or early-infantile refractory epilepsy with developmental retardation have KCNQ2 mutations. Thus, atypical BFNS features may be more common than previously appreciated.
Our three cases had deletions of 20q13.3 with either 20q13.33 or 20q13.33-qter. Microdeletion of 20q13.33 with loss of gene group ARFGAP1, CHRNA4, and KCNQ2 has been associated with cognitive and language deficits, with or without behavior problems or seizures. There is no common physical feature [10]. The cases in the current study have loss of these three genes in addition to at least eight more OMIM (Online Mendelian Inheritance in Man) genes (Fig. 2). Five of these genes, including KCNQ2 and CHRNA4, are known to be associated with disorders. Three other deleted genes, DNAJC5, PRPF6, and SOX18, seen in patient 3 only have associations with neuronal ceroid lipofuscinosis 4B (CLN4B), retinitis pigmentosa, and hypotrichosis–lymphedema–telangiectasia syndrome, respectively. Patient 3 did not have any of these diseases. Most of these genes, including ARFGAP1, have no known associated disorder. It is possible that one or more common deleted genes, besides CHRNA4 and KCNQ2, have roles concerning these individuals' atypical features, possibly more likely in patient 3 who had a much larger 1.4-Mb deletion. When considering the previously reported cases, the size of gene deletions associated with atypical phenotypic features is larger, ranging from 1.1 to 1.6 Mb [5–7], compared to those with typical features [4]. It is conceivable that a shorter deletion may or may not present with unusual BFNS features. This could potentially explain why two other cases with concomitant CHRNA4 and KCNQ2 deletions had a phenotype similar to that of BFNS [4]. There is no clear explanation to the phenotypic variability of BFNS, but it is possible that a disorder with contiguous CHRNA4 and KCNQ2 deletions is a different entity from BFNS.
Acknowledgments
We acknowledge the contribution of Rachel Burnside, PhD (Laboratory Corporation of America) and Jiang Zhieje, PhD (University of Miami) in helping to create Fig. 2.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Pagon R.A., Bird T.D., Dolan C.R., Stephens K., Adam M.P., editors. Gene reviews [internet] University of Washington, Seattle; Seattle (WA): 1993. Benign neonatal seizures. [cited 2013 Jan 14. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1116/] [Google Scholar]
- 2.Borgatti R., Zucca C., Cavallini A. A novel mutation in KCNQ2 associated with BFNS, drug resistant epilepsy, and mental retardation. Neurology. 2004;63(1):57–65. doi: 10.1212/01.wnl.0000132979.08394.6d. [DOI] [PubMed] [Google Scholar]
- 3.Dedek K., Lucia F., Teloy N., Steinlein O. Neonatal convulsions and epileptic encephalopathy in an Italian family with missense mutation in the fifth transmembrane region of KCNQ2. Epilepsy Res. 2003;54(1):21–27. doi: 10.1016/s0920-1211(03)00037-8. [DOI] [PubMed] [Google Scholar]
- 4.Kurahashi H., Wang J.-w., Ishii A. Deletions involving both KCNQ2 and CHRNA4 present with benign familial neonatal seizures. Neurology. 2009;73(15):1214–1217. doi: 10.1212/WNL.0b013e3181bc0158. [DOI] [PubMed] [Google Scholar]
- 5.Bena F., Bottani A., Marcelli F. A de novo 1.1–1.6 Mb subteloremic deletion of chromosome 20q13.33 in a patient with learning difficulties but without obvious dysmorphic features. Am J Med Genet. 2007;143A(16):1894–1899. doi: 10.1002/ajmg.a.31789. [DOI] [PubMed] [Google Scholar]
- 6.Ardalan A., Prieur M., Choiset A. Intrachromosomal insertion mimicking a pericentric inversion: molecular cytogenetic characterization of three break rearrangement of chromosome 20. Am J Med Genet. 2005;138A(3):288–293. doi: 10.1002/ajmg.a.30966. [DOI] [PubMed] [Google Scholar]
- 7.Mefford H.C., Cook J., Gospe S.M. Epilepsy due to 20q13.33 subtelomere deletion masquerading as pyridoxine-dependent epilepsy. Am J Med Genet. 2012;158A(12):3190–3195. doi: 10.1002/ajmg.a.35633. [DOI] [PubMed] [Google Scholar]
- 8.Pagon R.A., Bird T.D., Dolan C.R., Stephens K., Adam M.P., editors. Gene reviews [internet] University of Washington, Seattle; Seattle (WA): 1993. Autosomal dominant nocturnal frontal lobe epilepsy. [cited 2013 Jan 14. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1116/] [Google Scholar]
- 9.Weckhuysen S., Mandelstam S., Suls A. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol. 2012;71(1):15–25. doi: 10.1002/ana.22644. [DOI] [PubMed] [Google Scholar]
- 10.Traylor R.N., Bruno D.L., Burgess T. A genotype-first approach for the molecular and clinical characterization of uncommon de novo microdeletion of 20q13.33. PLoS One. 2010;5(8):e12462. doi: 10.1371/journal.pone.0012462. [DOI] [PMC free article] [PubMed] [Google Scholar]