


Abstract
Despite systematic approaches to mapping networks of genetic interactions in Saccharomyces cerevisiae, exploration of genetic interactions on a genome-wide scale has been limited. The S. cerevisiae haploid genome has 110 regions that are longer than 10 kb but harbor only non-essential genes. Here, we attempted to delete these regions by PCR-mediated chromosomal deletion technology (PCD), which enables chromosomal segments to be deleted by a one-step transformation. Thirty-three of the 110 regions could be deleted, but the remaining 77 regions could not. To determine whether the 77 undeletable regions are essential, we successfully converted 67 of them to mini-chromosomes marked with URA3 using PCR-mediated chromosome splitting technology and conducted a mitotic loss assay of the mini-chromosomes. Fifty-six of the 67 regions were found to be essential for cell growth, and 49 of these carried co-lethal gene pair(s) that were not previously been detected by synthetic genetic array analysis. This result implies that regions harboring only non-essential genes contain unidentified synthetic lethal combinations at an unexpectedly high frequency, revealing a novel landscape of genetic interactions in the S. cerevisiae genome. Furthermore, this study indicates that segmental deletion might be exploited for not only revealing genome function but also breeding stress-tolerant strains.
INTRODUCTION
Despite systematic approaches to identifying networks of genetic interactions in Saccharomyces cerevisiae, mapping a complete genetic interaction network as a source both for discovering novel genes and pathways, and for predicting analogous networks in more complex systems is an ongoing problem in yeast systems biology. The gene deletion project has identified ∼5100 genes as non-essential for cell viability (1). Inactivation of each of the 5100 non-essential genes in the genome of S. cerevisiae has little discernible effect under laboratory conditions, indicating the functional robustness of the cell, which may arise from multiple redundancies (1). However, the inactivation of some non-essential genes in specific combinations has a lethal effect under exactly the same conditions (2,3); this makes the yeast genome resistant to engineering and could be problematic from the viewpoint of breeding new strains. Industrially preferable phenotypes, such as stress resistance during ethanol production, are controlled by multiple genes (4); as a result, the manipulation of a single gene is not sufficient for either strain breeding or functional characterization of the yeast genome. On the other hand, there are ∼6200 predicted genes, of which the function of nearly 1000 remains elusive (5). Thus, combining different mutations can give insight into gene function.
Synthetic genetic interactions are usually identified when a second site mutation or an increase in gene dosage suppresses or enhances the original mutant phenotype. Genetic interactions can be negative (more exaggerated) or positive (less exaggerated) in terms of phenotype with respect to the expected double mutant phenotype (6). Synthetic lethality represents an extreme example of a negative interaction where two mutations, each causing little to no fitness defect on their own, result in a lethal double-mutant phenotype (7). Systematic mapping of synthetic lethal genetic interactions was first facilitated by the development of an automated approach called ‘synthetic genetic array’ (SGA) analysis (8,9). SGA analysis facilitates the systematic construction of double disruptants as meiotic segregants by mating followed by sporulation, allowing large-scale mapping of synthetic genetic interactions (9,10).
Theoretically, pair-wise combinations of the ∼5100 non-essential genes in the yeast genome would generate ∼12.5 million double disruptants. In the first SGA screen using eight query genes, however, 291 synthetic lethal/sick interactions were identified among 204 genes (8). Three years later (9), the search was expanded to 132 query genes and 4000 synthetic lethal/sick interactions were identified among 1000 genes. In a recent large-scale endeavor, ∼5.4 million gene–gene pairs covering roughly 30% of the S. cerevisiae genome were examined to reveal ∼100 000 negative genetic interactions and to provide the first overall view of the genetic landscape of a cell (10).
The global set of quantitative negative genetic interactions included both synthetic lethal and synthetic sick (slow-growing) phenotypes. However, the number of the most extreme synthetic lethal phenotypes was ∼10 000 (10), which is roughly 10-fold higher than the number of essential yeast genes. Therefore, the results of SGA analysis in these genome-wide studies indicate that ∼0.2% (10 000/5 400 000) of gene combinations are synthetic lethal for the growth of S. cerevisiae. By contrast, it has been predicted that the complete genetic network of S. cerevisiae contains over 200 000 synthetic lethal combinations, which is ∼200-fold higher than the number of essential yeast genes (11).
Because in SGA analysis double mutants are created by meiotic recombination, the construction of a double disruptant is not possible if the two genes to be disrupted in the parental strains are tightly linked on the same chromosome. Consequently, double mutants involving linked gene-pairs tend to form smaller colonies, which are likely to be removed from genetic network analysis to prevent their misinterpretation as negative genetic interactions (10,12) because these combinations do not represent synthetic lethality. As a result, the functional relationship between linked gene-pairs remains largely unknown. In addition, the genetic interactions estimator criterion (S-score) of the SGA method does not explicitly measure single or double mutant fitness, which is critical for a detailed interpretation of genetic interactions (12). Furthermore, in SGA, genetic interaction screens are susceptible to systematic experimental effects such as differences in growth conditions from one array plate to the next, as well as subtle differences in local nutrient availability within the same plate that introduce noise in colony size measurements. Taking these facts into account, the proportion of false-negative interactions (true interactions that are not identified by detailed analysis) has been estimated to be ∼17–41% for a single SGA screen (9).
For all of the above-mentioned reasons, systematic screens of synthetic genetic interactions in yeast have failed to detect an enormous number of possible gene combinations, and complete mapping of synthetic lethal interactions remains some way off. The SGA method was devised to elucidate the complete global network structure of the S. cerevisiae genome, but it has been shown to be both time-consuming and costly, which in practice has subsequently limited its potential ability to provide a complete global synthetic lethal network.
Thus, one of the most important challenges in the post-genomic era is to create a more efficient approach to resolve the second aspect of the interaction map. To achieve this, we have used genome engineering technologies called PCD (PCR-mediated one-step chromosome deletion) and PCS (PCR-mediated chromosome splitting) (13–17). To facilitate an investigation of the lethal interactions of linked gene-pairs that escape detection by the SGA method and in order to provide a complete genetic interaction map of S. cerevisiae, in the present study, we focused our attention on the most extreme form of genetic interaction (synthetic lethality), where a double deletion mutant shows a no-growth phenotype that is not exhibited by either single deletion mutant. To do so, we perform the genome-wide mapping of the essential regions which conceive hidden lethal combinations between non-essential genes in S. cerevisiae genome by using PCD and PCS technologies. To this end, we first performed genome-wide mapping to identify large regions of non-essential genes flanked by essential genes in the S. cerevisiae genome. In total, 110 regions harboring only non-essential genes were mapped by surveying the Saccharomyces Genome Database (SGD) (http://www.yeastgenome.org/). These 110 regions were then subjected to deletion by using the PCD method, which surprisingly showed that 77 of them could not be deleted by PCD. Subsequently, 67 of the 77 regions were further verified for essentiality by converting each of them to artificial mini-chromosomes by PCS. Forty-nine of the 67 regions were identified to be indeed essential for cell growth. These findings suggest that the combined strategy of PCD and PCS is a unique approach to discover hidden synthetic lethal combinations of linked gene-pairs that are functionally informative and can provide additional insights into the functional organization of the yeast genome.
MATERIALS AND METHODS
Strains, plasmids, media and transformation
The yeast strains and all the plasmids used in this study are described in Table 1. S. cerevisiae strain BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) was used as the parental strain for the systematic genome-wide functional analysis. BY4741 (MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0) was used to make diploid cells. A collection of haploid strains in a MATα background carrying deletions in non-essential genes were used for phenotypic analyses (Invitrogen™, Carlsbad, CA, USA). Plasmid construction of p3009, p3121, p3122 and p3276 has been described previously (13,14). To generate the plasmid p3406, the 1-kb gene CEN4 was amplified with primer pairs CEN-f/CEN-r (Supplementary Table S2) and inserted next to the URA3 marker in plasmid p3276. Plasmid DNA was isolated from Escherichia coli according to the alkaline method (18).
Table 1. Yeast strains and plasmids used in this study.
| Designation | Description | Remarks |
|---|---|---|
| Strains | ||
| BY4741 (= SH6479) | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | |
| BY4742 (= SH6314) | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | |
| SH4233 | MATa pho3-1 leu1-58 | |
| Plasmids | ||
| p3009 | A derivative of pUG6 carrying loxP-CgHIS3-loxP | Sugiyama et al. (13) |
| p3121 | A derivative of pUG6 carrying CEN4 | Sugiyama et al. (13) |
| p3122 | A derivative of pUG6 carrying CEN4-loxP-CgLEU2-loxP | Sugiyama et al. (14) |
| p3406 | A derivative of pUG6 carrying CEN4-URA3 | This study |
| p3276 | A derivative of pUG6 carrying URA3 | Sugiyama et al. (14) |
Yeast strains were grown at 30°C in Yeast extract-Bacto peptone-Dextrose-Adenine (YPDA) medium containing 5% Difco Yeast extract-Bacto-peptone-Dextrose (YPD) broth supplemented with 0.004% of adenine (Wako), or selective media containing 0.67% yeast nitrogen base without amino acids (Difco) + 2% glucose (Wako). Synthetic complete glucose (SC) medium has been described (19). The amino-acid supplement powder mixture (drop-out mixture; DO) for making SC medium contained all possible supplements except leucine, histidine and uracil. 1.5 g of the DO powder mixture was used per liter of medium. When necessary for selection, leucine, histidine and uracil were omitted from the SC medium. 5-Fluoroorotic acid (5-FOA) was used to select strains containing the URA3 gene. URA3 encodes orotidine 5-phosphate decarboxylase, which can convert 5-FOA to 5-fluorouracil, which causes cell death; as a result, it is possible to determine whether a strain contains the URA3 gene based on cell viability (20). For solid media, 2% agar (Wako) was also added.
Transformation of yeast cells was carried out according to Gietz and Schiestl (21). BY4742 was transformed with a pair of fragments (called a module) consisting of CEN4-loxP-CgLEU2-loxP and loxP-CgHIS3-loxP cassettes flanked by two sequences, namely, the target sequence (∼400 bp) and six copies of a 5′-CCCCAA-3′ repeat sequence, 5′-(C4A2)6-3′, which can act as an artificial telomere. For each region, two pairs of primers (p1 and p2 for the left target and p3 and p4 for the right target) were designed using the SGD and used to amplify DNA segments flanking the region to be deleted by PCR using chromosomal DNA of the host strain (BY4742) as a template (14). For chromosome deletion or splitting, ∼5 μg of each PCR fragment was used in transformations. Deletion fragments were constructed by a two-step PCR with overlap extension as described previously (14).
Oligonucleotides, PCR and preparation of DNA fragments for PCD and PCS
Complete lists of the oligonucleotide primers used for segmental deletion, detection of the segmental deletion, mini-chromosome construction and mini-chromosome detection in this study are given in Supplementary Tables S1–S3, respectively. SGD (http://yeastgenome.org/) was used to select target regions for deletion, to identify the deletion and splitting points, and to design the oligonucleotides. As previously described, two DNA fragments generated by a two-step PCR are necessary for each deletion or splitting event in both PCS (13) and PCD (14). Briefly, in the first round of PCR, loxP and CA primers were used to amplify a DNA fragment containing the 5′-(C4A2)6-3′ telomere seed sequence and CgHIS3 marker from plasmid p3009; the 5′-(C4A2)6-3′ telomere seed sequence, CgLEU2 marker and CEN4 from plasmid p3122 for the PCD method; the 5′-(C4A2)6-3′ telomere seed sequence and CgURA3 marker from plasmid p3276; the 5′-(C4A2)6-3′ telomere seed sequence and CEN4 from p3121; and the 5′-(C4A2)6-3′ telomere seed sequence and CgHIS3 marker from p3009 for the PCS method. The PCR reaction was prepared in a final volume of 50 μl containing ∼50 ng of plasmid DNA and was performed as follows: 94°C for 5 min; followed by 35 cycles at 94°C for 30 s, 55°C for 30 s, 72°C for 3 min; and a final extension step at 72°C for 7 min. In parallel, 400 bp of upstream sequences and downstream sequences of the target region were amplified from the genomic DNA of strain BY4742. The PCR reaction mixture was prepared in a final volume of 50 μl containing ∼50 ng of genomic DNA and was performed as follows: 94°C for 5 min; followed by 35 cycles at 94°C for 30 s, 55°C for 30 s, 72°C for 30 s; and a final extension step at 72°C for 7 min. PCR products were gel-purified by using the Wizard SV Gel and PCR Clean-up System (Promega). DNA concentration was measured by using a Nanodrop 1000 spectrophotometer (Thermo Scientific). In the second-round PCR, used to overlap the two first PCR fragments, each target fragment was independently mixed with a fragment from the plasmid template, and then subjected to overlap extension PCR using the appropriate primers. The PCR reaction was carried out in a final volume of 100 μl, containing an equal amount of PCR product from the plasmid and genomic DNA, 1 μl of Ex Taq DNA Polymerase, 10 μl of each 10 μM primer and 10 μl of dNTP mixture, as follows: 94°C for 5 min; followed by 35 cycles at 94°C for 30 s, 55°C for 30 s, 72°C for 7 min; and a final extension step at 72°C for 7 min. All PCR amplifications were carried out on Gene Amp PCR System 9700 (Applied Biosystems). The PCR products were ethanol-precipitated and subsequently used to transform the yeast cells.
Pulsed-field gel electrophoresis and Southern hybridization
Chromosomal DNA from S. cerevisiae cultured in YPDA medium was prepared as agarose plugs according to Sheehan and Weiss (22). The DNA plugs were inserted into slots in a 1% pulsed-field agarose gel (Bio-Rad). The chromosomes were separated by electrophoresis using the CHEF MAPPER System (Bio-Rad Laboratories) in 0.5× TBE (Tris–borate–EDTA) buffer at 14°C and 6 V/cm using a 60-s pulse for 15 h, followed by a 90-s pulse for 9 h. After staining with ethidium bromide, the chromosomes were visualized and photographed under a UV trans-illuminator (UVP Bio Do-It Imaging System) and then transferred onto Hybond™-N+ membranes (GE Healthcare, Buckinghamshire, UK) by capillary blotting. The membranes were hybridized with probes generated by PCR using primers listed in Supplementary Table S3. Probe labeling, hybridization and hybridization signal detection were carried out by using an ECL Direct™ nucleic acid labeling and detection system (GE Healthcare).
Phenotypic assay
Yeast cells were pre-cultured overnight in selective medium supplemented with appropriate amino acids at 30°C. The cultures were then inoculated into fresh selective medium and incubated at 30°C for another 6 h. After that, the cells were collected, washed once with sterile water, and resuspended in sterile water. The concentration of cells was measured at OD660 nm. Then, the cell density was normalized to a concentration of 2.5 x 108 cells/ml. A ten-fold serial dilutions of this suspension was made, and 4μl of each dilution was spotted onto appropriate plates under different conditions including YPDA medium at 13, 30 and 41°C; or YDPA medium supplemented with 4, 5 and 6% lactic acid, 4, 6 and 8% ethanol and sulfuric acid (pH 2.5, pH 2.4 and pH 2.3). Cell growth was also measured on YPDA supplemented with 0.02% Sodium dodecyl sulfate (SDS), 80 mM acetic acid, 36 mM formic acid, 1.8 M NaCl, 150 mM CaCl2, 10 mM furfural and 2.0 M sorbitol. Growth differences were determined after 4 days of incubation. Experiments were carried out in triplicate.
RESULTS
Mapping of regions containing non-essential genes in the S. cerevisiae genome
First, we looked for large regions that contain only non-essential genes surrounded by essential genes along all 16 yeast chromosomes. By surveying SGD (http://www.yeastgenome.org/), we determined the location of all essential genes across the whole genome, and reasoned that theoretically it would be possible to delete regions located on the centromeric or telomeric sides of these genes. To avoid the possibility that the flanking essential genes might alter the efficiency of deletion, the position of the split points of the region to be deleted were designed to be at least 1 kb from the flanking essential genes. The region of the rDNA cluster on chromosome XII, and regions harboring known lethal combination(s) identified by SGA analysis (9,10) were not included in either the first or the second screening of the present study.
To present the results in a comprehensive way, we included 1 confirmed essential region (13) and 14 confirmed non-essential regions (13,14,17,23) in the genome-wide mapping and screening. We also included three regions that were confirmed to be deletable during a prior pilot study. Karyotype analysis and phenotypic characteristics of these 18 derivatives are not shown here. Across the whole S. cerevisiae genome, 110 regions varying in content from 9 non-essential genes in the smallest region to 47 non-essential genes in the largest region and an average of 20 non-essential genes were identified (Figure 1 and Table 2).
Figure 1.
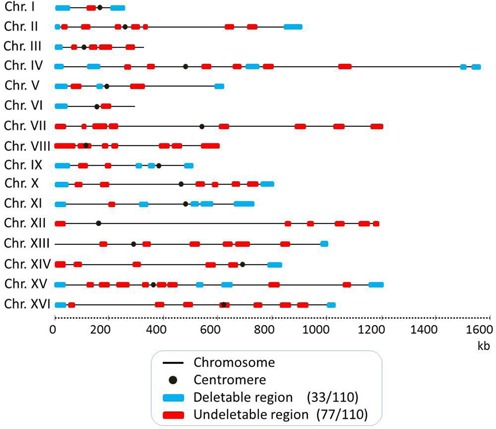
Genome-wide mapping of deletable and undeletable regions among 110 targeted chromosomal regions. Each chromosome is represented by a gray line with the centromere shown as a black solid circle. The chromosomal lengths are drawn to scale. Based on an SGD survey, these 110 regions were selected as having at least 9 non-essential genes. The distribution of 33 deletable and 77 undeletable regions among the 110 target regions, as identified by PCD, are indicated as blue and red horizontal filled bars, respectively.
Table 2. Characteristics of deletable and undeletable chromosomal regions.
| Chromosome (length) | Straina | Deleted regionb | Region namec | Deletion length (kb) | No. of genes (n) | No. of transformants obtained | No. of transformants examined for karyo-type | Transformants with expected deletion | Frequencyd | Deletable/ Undeletable | References/ remarks |
|---|---|---|---|---|---|---|---|---|---|---|---|
| I (230 kb) | ScΔ(Chr 1-1) | 1–51 910 | Chr 1-1 | 51.9 | 30 | 75 | 20 | 12 | 60.00% | Deletable | This study |
| 109 298–138 358 | Chr 1-2 | 29.1 | 19 | 55 | 29 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 1-3) | 178 545–230 218 | Chr 1-3 | 51.7 | 33 | 145 | 20 | 16 | 80.00% | Deletable | This study | |
| II (813 kb) | ScΔ(Chr 2-1) | 1–10 856 | Chr 2-1 | 10.8 | 18 | 118 | 28 | 26 | 92.85% | Deletable | This study |
| 21 866–37 346 | Chr 2-2 | 15.5 | 14 | 48 | 25 | 0 | 0.00% | Undeletable | This study | ||
| 92 578–121 720 | Chr 2-3 | 29.2 | 18 | 63 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 205 408–220 469 | Chr 2-4 | 15.0 | 9 | 64 | 28 | 0 | 0.00% | Undeletable | This study | ||
| 265 907–292 296 | Chr 2-5 | 26.3 | 15 | 75 | 28 | 0 | 0.00% | Undeletable | This study | ||
| 318 749–330 960 | Chr 2-6 | 12.2 | 9 | 71 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 577 219–602 500 | Chr 2-7 | 25.3 | 19 | 49 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 650 769–681 129 | Chr 2-8 | 30.4 | 22 | 56 | 20 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 2-9) | 744 846–813 184 | Chr 2-9 | 68.3 | 38 | 78 | 20 | 4 | 20.00% | Deletable | This study | |
| III (316 kb) | ScΔ(Chr 3-1) | 1–18 822 | Chr 3-1 | 18.8 | 20 | 163 | 20 | 18 | 90.00% | Deletable | This study |
| 68 950–83 624 | Chr 3-2 | 14.7 | 13 | 66 | 21 | 0 | 0.00% | Undeletable | This study | ||
| 116 424–133 170 | Chr 3-3 | 16.7 | 12 | 99 | 21 | 0 | 0.00% | Undeletable | This study | ||
| 145 265–186 097 | Chr 3-4 | 40.8 | 31 | 51 | 21 | 0 | 0.00% | Undeletable | This study | ||
| 246 961–268 191 | Chr 3-5 | 21.2 | 17 | 70 | 25 | 0 | 0.00% | Undeletable | This study | ||
| IV (1532 kb) | ScΔ(Chr 4-1) | 1–29 985 | Chr 4-1 | 29.9 | 17 | 198 | 20 | 20 | 100.00% | Deletable | This study |
| ScΔ(Chr 4-2) | 119 330–159 986 | Chr 4-2 | 40.6 | 24 | Deletable | Ueda et al. (17) | |||||
| 239 739–258 679 | Chr 4-3 | 18.9 | 13 | 59 | 21 | 0 | 0.00% | Undeletable | This study | ||
| 309 991–334 228 | Chr 4-4 | 24.2 | 16 | 52 | 21 | 0 | 0.00% | Undeletable | This study | ||
| 494 271–521 840 | Chr 4-5 | 27.6 | 24 | 87 | 25 | 0 | 0.00% | Undeletable | This study | ||
| 584 190–603 837 | Chr 4-6 | 19.7 | 14 | 54 | 21 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 4-7) | 632 193–676 133 | Chr 4-7 | 44.0 | 29 | Deletable | Ueda et al. (17) | |||||
| 691 278–730 916 | Chr 4-8 | 39.7 | 20 | 50 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 958 372–998 900 | Chr 4-9 | 40.6 | 31 | 88 | 20 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 4-10) | 1 473 549–1 487 620 | Chr 4-10 | 14.1 | 10 | 55 | 20 | 12 | 60.00% | Deletable | This study | |
| ScΔ(Chr 4-11) | 1 502 766–1 531 933 | Chr 4-11 | 29.2 | 19 | 106 | 20 | 20 | 100.00% | Deletable | This study | |
| V (577 kb) | ScΔ(Chr 5-1) | 1–38 703 | Chr 5-1 | 38.7 | 23 | 73 | 24 | 16 | 66.66% | Deletable | This study |
| 55 219–80 675 | Chr 5-2 | 25.4 | 22 | 90 | 20 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 5-3) | 126 605–141 203 | Chr 5-3 | 14.6 | 16 | 52 | 20 | 8 | 40.00% | Deletable | This study | |
| 260 306–303 330 | Chr 5-4 | 43.0 | 27 | 95 | 25 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 5-5) | 541 219–576 874 | Chr 5-5 | 35.6 | 28 | Deletable | Sugiyama et al. (14) | |||||
| VI (270 kb) | ScΔ(Chr 6-1) | 1–36 314 | Chr 6-1 | 36.3 | 24 | 95 | 20 | 14 | 70.00% | Deletable | This study |
| 161 023–196 416 | Chr 6-2 | 35.4 | 23 | 59 | 22 | 0 | 0.00% | Undeletable | This study | ||
| VII (1091 kb) | 1–32 647 | Chr 7-1 | 32.6 | 20 | 69 | 26 | 0 | 0.00% | Undeletable | This study | |
| 82 711–95 454 | Chr 7-2 | 12.7 | 11 | 73 | 25 | 0 | 0.00% | Undeletable | This study | ||
| 129 738–171 415 | Chr 7-3 | 41.7 | 23 | 56 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 190 867–210 017 | Chr 7-4 | 19.2 | 12 | 68 | 22 | 0 | 0.00% | Undeletable | This study | ||
| 555 493–579 150 | Chr 7-5 | 23.6 | 19 | 88 | 28 | 0 | 0.00% | Undeletable | This study | ||
| 809 906–835 553 | Chr 7-6 | 25.6 | 21 | 58 | 23 | 0 | 0.00% | Undeletable | This study | ||
| 939 364–974 120 | Chr 7-7 | 34.8 | 21 | 77 | 27 | 0 | 0.00% | Undeletable | This study | ||
| 1 055 720–1 090 940 | Chr 7-8 | 35.2 | 22 | 60 | 28 | 0 | 0.00% | Undeletable | This study | ||
| VIII (562 kb) | 1–71 481 | Chr 8-1 | 71.4 | 47 | 97 | 25 | 0 | 0.00% | Undeletable | This study | |
| 79 586–111 810 | Chr 8-2 | 32.3 | 28 | 86 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 162 591–176 986 | Chr 8-3 | 14.4 | 11 | 63 | 21 | 0 | 0.00% | Undeletable | This study | ||
| 196 188–214 563 | Chr 8-4 | 18.3 | 17 | 79 | 25 | 0 | 0.00% | Undeletable | This study | ||
| 358 040–383 752 | Chr 8-5 | 25.7 | 28 | 88 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 397 528–421 202 | Chr 8-6 | 23.7 | 13 | 49 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 502 800–562 643 | Chr 8-7 | 59.8 | 38 | 51 | 28 | 0 | 0.00% | Undeletable | This study | ||
| IX (440 kb) | ScΔ(Chr 9-1) | 1–56 480 | Chr 9-1 | 56.4 | 35 | 223 | 20 | 10 | 50.00% | Deletable | This study |
| 87 850–102 249 | Chr 9-2 | 14.4 | 12 | 55 | 21 | 0 | 0.00% | Undeletable | This study | ||
| 175 096–187 852 | Chr 9-3 | 12.7 | 11 | 60 | 21 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 9-4) | 273 992–288 932 | Chr 9-4 | 14.9 | 11 | 66 | 24 | 12 | 50.00% | Deletable | This study | |
| ScΔ(Chr 9-5) | 321 054–343 678 | Chr 9-5 | 22.6 | 20 | 67 | 20 | 16 | 80.00% | Deletable | This study | |
| ScΔ(Chr 9-6) | 402 831–439 888 | Chr 9-6 | 37.0 | 29 | 250 | 20 | 12 | 60.00% | Deletable | This study | |
| X (745 kb) | ScΔ(Chr 10-1) | 1–49 918 | Chr 10-1 | 49.9 | 28 | 88 | 29 | 4 | 13.79% | Deletable | This study |
| 74 248–91 729 | Chr 10-2 | 17.5 | 15 | 79 | 22 | 0 | 0.00% | Undeletable | This study | ||
| 150 919–181 823 | Chr 10-3 | 30.9 | 18 | 53 | 25 | 0 | 0.00% | Undeletable | This study | ||
| 472 448–506 851 | Chr 10-4 | 34.4 | 19 | 54 | 25 | 0 | 0.00% | Undeletable | This study | ||
| 526 499–540 234 | Chr 10-5 | 13.7 | 13 | 66 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 607 564–633 651 | Chr 10-6 | 26.1 | 18 | 82 | 21 | 0 | 0.00% | Undeletable | This study | ||
| 655 969–692 506 | Chr 10-7 | 36.6 | 19 | 87 | 25 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 10-8) | 700 629–745 751 | Chr 10-8 | 45.1 | 25 | Deletable | Sugiyama et al. (14) | |||||
| XI (667 kb) | ScΔ(Chr 11-1) | 1–35 760 | Chr 11-1 | 35.7 | 18 | 59 | 20 | 4 | 20.00% | Deletable | This study |
| 188 434–204 755 | Chr 11-2 | 16.3 | 11 | 51 | 20 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 11-3) | 294 480–323 059 | Chr 11-3 | 28.5 | 22 | Deletable | Sugiyama et al. (14) | |||||
| ScΔ(Chr 11-4) | 457 598–478 769 | Chr 11-4 | 21.1 | 13 | 55 | 24 | 8 | 33.33% | Deletable | This study | |
| ScΔ(Chr 11-5) | 516 847–556 301 | Chr 11-5 | 39.4 | 24 | Deletable | Ueda et al. (17) | |||||
| ScΔ(Chr 11-6) | 606 478–666 816 | Chr 11-6 | 60.3 | 24 | Deletable | Ueda et al. (17) | |||||
| XII (1078 kb) | 1–36 402 | Chr 12-1 | 36.4 | 27 | 62 | 24 | 0 | 0.00% | Undeletable | This study | |
| 782 360–796 254 | Chr 12-2 | 13.9 | 14 | 77 | 25 | 0 | 0.00% | Undeletable | This study | ||
| 849 154–872 370 | Chr 12-3 | 23.2 | 20 | 93 | 24 | 0 | 0.00% | Undeletable | This study | ||
| 940 622–969 960 | Chr 12-4 | 29.3 | 21 | 71 | 25 | 0 | 0.00% | Undeletable | This study | ||
| 1 021 075–1 051 682 | Chr 12-5 | 30.6 | 13 | 75 | 21 | 0 | 0.00% | Undeletable | This study | ||
| 1 061 445–1 078 177 | Chr 12-6 | 16.7 | 17 | 48 | 21 | 0 | 0.00% | Undeletable | This study | ||
| XIII (924 kb) | 149 349–170 801 | Chr 13-1 | 21.5 | 16 | 92 | 20 | 0 | 0.00% | Undeletable | This study | |
| 300 686–323 520 | Chr 13-2 | 22.9 | 13 | 55 | 24 | 0 | 0.00% | Undeletable | This study | ||
| 460 669–490 680 | Chr 13-3 | 30.0 | 20 | 110 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 565 257–593 930 | Chr 13-4 | 28.7 | 20 | 52 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 603 532–655 150 | Chr 13-5 | 51.6 | 35 | 46 | 21 | 0 | 0.00% | Undeletable | This study | ||
| 754 644–785 961 | Chr 13-6 | 31.3 | 23 | 66 | 27 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 13-7) | 906 127–924 431 | Chr 13-7 | 18.3 | 17 | Deletable | Murakami et al. (23) | |||||
| XIV (784 kb) | 1–37 073 | Chr 14-1 | 37.0 | 26 | 44 | 22 | 0 | 0.00% | Undeletable | This study | |
| 62 859–82 820 | Chr 14-2 | 20.0 | 11 | 83 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 260 029–280 867 | Chr 14-3 | 20.8 | 13 | 52 | 23 | 0 | 0.00% | Undeletable | This study | ||
| 515 793–551 621 | Chr 14-4 | 35.9 | 28 | 55 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 590 250–614 516 | Chr 14-5 | 24.3 | 15 | 88 | 20 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 14-6) | 728 119–784 333 | Chr 14-6 | 56.2 | 28 | Deletable | Murakami et al. (23) | |||||
| XV (1091 kb) | ScΔ(Chr 15-1) | 1–41 505 | Chr 15-1 | 41.5 | 26 | Deletable | Sugiyama et al. (13) | ||||
| 98 213–117 310 | Chr 15-2 | 19.1 | 17 | 58 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 146 179–178 230 | Chr 15-3 | 32.1 | 14 | 81 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 207 919–249 764 | Chr 15-4 | 41.8 | 29 | 105 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 288 798–304 553 | Chr 15-5 | 15.8 | 13 | 57 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 337 904–367 536 | Chr 15-6 | 29.6 | 21 | 58 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 374 378–411 086 | Chr 15-7 | 36.7 | 26 | 87 | 20 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 15-8) | 476 130–500 179 | Chr 15-8 | 24.0 | 14 | Deletable | Sugiyama et al. (14) | |||||
| ScΔ(Chr 15-9) | 556 270–593 762 | Chr 15-9 | 37.5 | 20 | Deletable | Sugiyama et al. (14) | |||||
| 672 254–702 299 | Chr 15-10 | 30.0 | 16 | Undeletable | Sugiyama et al., (13) | ||||||
| 969 010–995 011 | Chr 15-11 | 26.0 | 18 | 65 | 23 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 15-12) | 1 043 649–1 091 291 | Chr 15-12 | 47.6 | 29 | Deletable | Sugiyama et al. (13) | |||||
| XVI (948 kb) | ScΔ(Chr 16-1) | 1–35 668 | Chr 16-1 | 35.6 | 22 | Deletable | Murakami et al. (23) | ||||
| 43 629–64 215 | Chr 16-2 | 20.6 | 19 | 50 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 332 333–366 459 | Chr 16-3 | 34.1 | 21 | 63 | 23 | 0 | 0.00% | Undeletable | This study | ||
| 434 789–466 542 | Chr 16-4 | 31.8 | 23 | 49 | 20 | 0 | 0.00% | Undeletable | This study | ||
| 547 237–574 201 | Chr 16-5 | 26.9 | 19 | 66 | 25 | 0 | 0.00% | Undeletable | This study | ||
| 675 669–699 987 | Chr 16-6 | 24.3 | 22 | 58 | 25 | 0 | 0.00% | Undeletable | This study | ||
| 756 832–792 577 | Chr 16-7 | 35.7 | 21 | 93 | 24 | 0 | 0.00% | Undeletable | This study | ||
| 824 875–860 801 | Chr 16-8 | 36.0 | 30 | 60 | 20 | 0 | 0.00% | Undeletable | This study | ||
| ScΔ(Chr 16-9) | 922 998–948 066 | Chr 16-9 | 25.1 | 18 | 162 | 25 | 21 | 84.00% | Deletable | This study |
aScΔ(Chr x-y): ScΔ represents S. cerevisiae deletion mutant, Chr x represents chromosome number and -y represents region number.
bx-y: x represents first nucleotide number of chromosomal region and -y represents last nucleotide number of chromosomal region.
cChr x-y: Chr x represents chromosome number and -y represent region.
dFrequency for obtaining segmental chromosomal deletion (number of segmental deletions/number of candidate transformants that were analyzed in karyotype.
Systematic screen for deletable and undeletable regions by PCD
To systematically identify which regions could be deleted and which could not, each of the 110 regions was subjected to one-step deletion by the PCD method (14). Transformants containing the deletion module were selected as Leu+His+ colonies. The number of transformants obtained for each region is shown in Table 2. For each successful transformation, at least 20 transformants were subjected to karyotype analysis by Pulsed-Field Gel Electrophoresis (PFGE), followed by Southern hybridization. Because chromosomal deletion is accompanied by integration of the deletion module, the length of the split chromosomes generated is increased by the length of the loxP-CgHIS3-loxP (1.8 kb) and CEN4-loxP-CgLEU2-loxP (2.9 kb) cassettes that are integrated in place of the deleted region. Terminal regions were deleted by a module carrying a CgHIS3 marker.
We found that 33 (blue boxes in Figure 1) of the 110 regions harboring only non-essential genes could be deleted (Figure 1 and Table 2). Example results of the karyotype analysis of some transformants are shown in Figure 2. A deletion in the right terminal region of chromosome II (Chr 2-9) of 68 kb, in the left terminal region of chromosome III (Chr 3-1) of 19 kb, in the right terminal region of chromosome IV (Chr 4-11) of 29 kb, in the left terminal region of chromosome V (Chr 5-1) of 39 kb, in an internal region of chromosome IX (Chr 9-4) of 15 kb, and in the left terminal region of chromosome IX (Chr 9-1) of 56 kb are shown (Figure 2A). Figure 2B shows that the hybridization signal of all probes designed to hybridize with the deleted regions were detected, as expected, for the respective 813-, 316-, 1532-, 577-, 440- and 440-kb intact chromosomes. However, hybridization signals were absent for transformants harboring the expected deletions (lanes 2, 4, 6, 8, 10 and 12). These results indicate that a 68-kb region of chromosome II, 19-kb region of chromosome III, 29-kb region of chromosome IV, 39-kb region of chromosome V, 15-kb region of chromosome IX and 56-kb region of chromosome IX harboring, respectively, 38, 20, 19, 23, 11 and 35 genes were successfully deleted by a single transformation. Similar results were obtained for the other 27 regions identified as deletable by PCD (data not shown). The remaining 77 regions (red boxes in Figure 1) could not be deleted by PCD (Figure 1 and Table 2).
Figure 2.
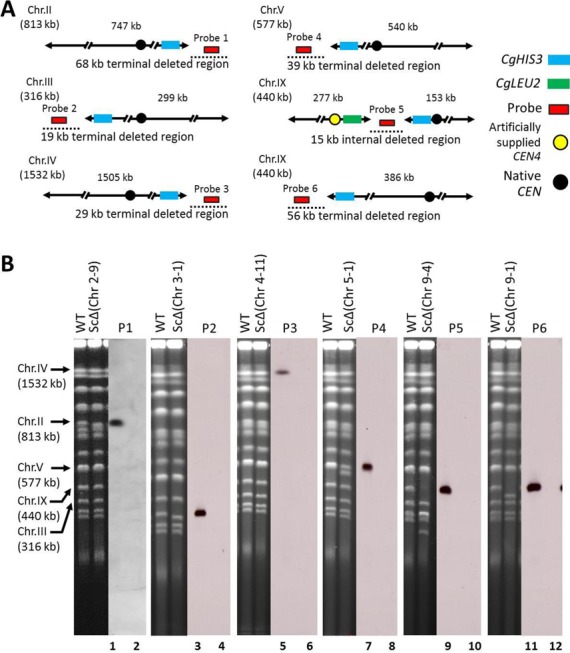
Construction of a segmental chromosomal deletion in chromosomes II, III, IV, V and IX. (A) Illustration of each target chromosome, deleted region and hybridization probe location. Deleted regions are shown as black dotted lines on the left, right and internal side of each chromosome. Probes 1, 2, 3, 4, 5 and 6 correspond to coordinate number between 768 247–768 628 of chromosomes II, 17 312–17 705 of chromosome III, 1 517 154–1 517 535 of chromosome IV, 26 785–27 159 of chromosome V, 277 726–278 122 and 37 397–37 878 of chromosome IX, respectively. The length of each deleted region is indicated. The blue box, green box, red box, yellow circle and black circle represent the CgHIS3 marker, CgLEU2 marker, probe, artificial CEN4 and natural CEN sequence, respectively. (B) PFGE and Southern hybridization showing karyotype analysis of the parental haploid and transformant clones carrying segmental deletion on chromosomes II, III, IV, V and IX. For all chromosomes, probes were prepared by PCR amplification of a 400-bp internal sequence of the target region.
Verification of essential regions harboring only non-essential genes by PCS
As described above, screening of the 110 regions by PCD identified 77 regions (red boxes in Figure 1) as undeletable (Table 3). To confirm whether these undeletable regions are in fact essential, we used the PCS method (13). PCS is a technology that can split a chromosome at any desired site. We converted 67 of the 77 potentially undeletable regions to mini-chromosomes marked with a URA3 gene by using PCS to split the chromosome at the left- and right-hand side of each region. Briefly, two modules consisting of the 400-bp upstream and downstream sequence of the target point, a CEN4/URA3 selective marker and six copies of the 5′-CCCCAA-3′ repeat sequence were prepared by PCR and introduced into cells by transformation. The target point then is thought to undergo homologous recombination at the right-hand end of the region to be deleted, resulting in splitting of the natural chromosome into two smaller chromosomes. Subsequently, a second splitting is conducted in a similar way at the left-hand end of the region. If splitting occurs at the correct site, a URA3-marked mini-chromosome of each region is created (Figure 3A). Figure 3B shows representative data from PFGE and Southern analysis demonstrating the expected generation of a mini-chromosome in strains generated to harbor a mini-chromosome of the 43-kb internal region of chromosome III, 30-kb internal region of chromosome IV, 39-kb internal region of chromosome X, 22-kb internal region of chromosome XV, 44-kb internal region of chromosome XV and 38-kb internal region of chromosome XVI.
Table 3. Characteristics of essential and dispensable chromosomal regions.
| Undeletable region by PCDa | Strainb | Undeletable regionc length (kb) | No. of genes (n) | First splitting transformants | Transformants with expected splitting/total no. of transformants examined for karyotype (%) | Second splitting transformants | Transformants with expected splitting/ total no. of transformants examined for karyotype (%) | Essential/ dispensable | References/ remarks |
|---|---|---|---|---|---|---|---|---|---|
| Chr 1-2 | 109.2–138.3 (29.1) | 19 | 164 | 5/5 (100) | 87 | 4/8 (50.0) | Essential | This study | |
| Chr 2-2 | 21.8–37.3 (15.5) | 14 | 83 | 3/4 (75.0) | 35 | 1/5 (20.0) | Essential | This study | |
| Chr 2-3 | 92.5–121.7 (29.2) | 18 | Essential | Harboring SL gene pair | |||||
| Chr 2-4 | 205.4–220.4 (15.0) | 9 | 190 | 4/4 (100) | 45 | 1/5 (20.0) | Essential | This study | |
| Chr 2-5 | 265.9–292.2 (26.3) | 15 | Essential | Harboring SL gene pair | |||||
| Chr 2-6 | 318.7–330.9 (12.2) | 9 | 47 | 13/15 (86.7) | 40 | 12/25 (48.0) | Essential | This study | |
| Chr 2-7 | 577.2–602.5 (25.3) | 19 | 76 | 8/10 (80.0) | 62 | 18/25 (72.0) | Essential | This study | |
| Chr 2-8 | 650.7–681.1 (30.4) | 22 | 111 | 3/5 (60.0) | 100 | 0.00% | ? | This study (not obtained) | |
| Chr 3-2 | ScΔ(Chr 3-2) | 68.9–83.6 (14.7) | 13 | 155 | 4/4 (100) | 86 | 1/4 (25.0) | Dispensable | This study |
| Chr 3-3 | 116.4–133.1 (16.7) | 12 | 35 | 4/4 (100) | 104 | 4/4 (100) | Essential | This study | |
| Chr 3-4 | 145.2–186.0 (40.8) | 31 | 65 | 4/4 (100) | 76 | 4/4 (100) | Essential | This study | |
| Chr 3-5 | 246.9–268.1 (21.2) | 17 | Essential | Harboring SL gene pair | |||||
| Chr 4-3 | 239.7–258.6 (18.9) | 13 | 91 | 4/4 (100) | 60 | 16/16 (100) | Essential | This study | |
| Chr 4-4 | ScΔ(Chr 4-4) | 309.9–334.2 (24.2) | 16 | 83 | 3/3 (100) | 65 | 2/4 (50.0) | Dispensable | This study |
| Chr 4-5 | 494.2–521.8 (27.6) | 24 | 150 | 4/4 (100) | 205 | 5/5 (100) | Essential | This study | |
| Chr 4-6 | 584.1–603.8 (19.7) | 14 | 43 | 3/3 (100) | 44 | 3/6 (50.0) | Essential | This study | |
| Chr 4-8 | 691.2–730.9 (39.7) | 20 | 112 | 5/5 (100) | 107 | 0.00% | ? | This study (not obtained) | |
| Chr 4-9 | 958.3–998.9 (40.6) | 31 | 146 | 3/3 (100) | 79 | 0.00% | ? | This study (not obtained) | |
| Chr 5-2 | 55.2–80.6 (25.4) | 22 | 167 | 6/6 (100) | 30 | 4/6 (67.0) | Essential | This study | |
| Chr 5-4 | 260.3–303.3 (43.0) | 27 | 65 | 3/3 (100) | 34 | 1/10 (10.0) | Essential | This study | |
| Chr 6-2 | 161.0–196.4 (35.4) | 23 | 58 | 5/6 (83.3) | 50 | 6/6 (100) | Essential | This study | |
| Chr 7-1 | 1–32.6 (32.6) | 20 | 47 | 4/8 (50.0) | 40 | 3/14 (21.0) | Essential | This study | |
| Chr 7-2 | 82.7–95.4 (12.7) | 11 | 108 | 4/4 (100) | 135 | 0.00% | ? | This study (not obtained) | |
| Chr 7-3 | ScΔ(Chr 7-3) | 129.7–171.4 (41.7) | 23 | 37 | 3/3 (100) | 115 | 5/5 (100) | Dispensable | This study |
| Chr 7-4 | ScΔ(Chr 7-4) | 190.8–210.0 (19.2) | 12 | 86 | 3/3 (100) | 82 | 4/6 (67.0) | Dispensable | This study |
| Chr 7-5 | 555.4–579.1 (23.6) | 19 | 143 | 3/3 (100) | 45 | 4/4 (100) | Essential | This study | |
| Chr 7-6 | 809.9–835.5 (25.6) | 21 | 121 | 3/3 (100) | 65 | 2/3 (66.7) | Essential | This study | |
| Chr 7-7 | 939.3–974.1 (34.8) | 21 | Essential | Harboring SL gene pair | |||||
| Chr 7-8 | 1055.7–1090.9 (35.2) | 22 | 205 | 5/5 (100) | 32 | 5/14 (35.7) | Essential | This study | |
| Chr 8-1 | 1–71.4 (71.4) | 47 | 210 | 3/4 (75.0) | 73 | 2/5 (40.0) | Essential | This study | |
| Chr 8-2 | 79.5–111.8 (32.3) | 28 | 66 | 3/3 (100) | 220 | 3/5 (60.0) | Essential | This study | |
| Chr 8-3 | 162.5–176.9 (14.4) | 11 | 97 | 3/4 (75.0) | 80 | 0.00% | ? | This study (not obtained) | |
| Chr 8-4 | 196.1–214.5 (18.3) | 17 | 274 | 5/5 (100) | 90 | 2/3 (66.7) | Essential | This study | |
| Chr 8-5 | 358.0–383.7 (25.7) | 28 | Essential | Harboring SL gene pair | |||||
| Chr 8-6 | 397.5–421.2 (23.7) | 13 | 54 | 5/5 (100) | 76 | 1/4 (25.0) | Essential | This study | |
| Chr 8-7 | 502.8–562.6 (59.8) | 38 | 83 | 5/5 (100) | 66 | 2/5 (40.0) | Essential | This study | |
| Chr 9-2 | 87.8–102.2 (14.4) | 12 | 60 | 4/4 (100) | 83 | 1/5 (20.0) | Essential | This study | |
| Chr 9-3 | 175.0–187.8 (12.7) | 11 | 123 | 4/4 (100) | 36 | 3/3 (100) | Essential | This study | |
| Chr 10-2 | 74.2–91.7 (17.5) | 15 | 245 | 5/5 (100) | 55 | 3/7 (42.8) | Essential | This study | |
| Chr 10-3 | 150.9–181.8 (30.9) | 18 | 183 | 4/4 (100) | 154 | 1/5 (20.0) | Essential | This study | |
| Chr 10-4 | ScΔ(Chr 10-4) | 472.4–506.8 (34.4) | 19 | 136 | 3/4 (75.0) | 33 | 1/5 (20.0) | Dispensable | This study |
| Chr 10-5 | 526.4–540.2 (13.7) | 13 | 104 | 4/4 (100) | 70 | 0.00% | ? | This study (not obtained) | |
| Chr 10-6 | 607.5–633.6 (26.1) | 18 | 49 | 5/5 (100) | 50 | 2/4 (50.0) | Essential | This study | |
| Chr 10-7 | 655.9–692.5 (36.6) | 19 | 88 | 3/3 (100) | 153 | 4/5 (80.0) | Essential | This study | |
| Chr 11-2 | 188.4–204.7 (16.3) | 11 | 57 | 4/4 (100) | 49 | 1/5 (20.0) | Essential | This study | |
| Chr 12-1 | 1–36.4 (36.4) | 27 | 145 | 4/6 (66.6) | 90 | 4/4 (100) | Essential | This study | |
| Chr 12-2 | ScΔ(Chr 12-2) | 782.3–796.2 (13.9) | 14 | 60 | 3/5 (60.0) | 87 | 3/5 (60.0) | Dispensable | This study |
| Chr 12-3 | 849.1–872.3 (23.2) | 20 | 44 | 4/5 (80.0) | 65 | 4/5 (80.0) | Essential | This study | |
| Chr 12-4 | 940.6–969.9 (29.3) | 21 | 172 | 3/4 (75.0) | 68 | 5/5 (100) | Essential | This study | |
| Chr 12-5 | 1021.0–1051.6 (30.6) | 13 | 123 | 3/3 (100) | 160 | 5/5 (100) | Essential | This study | |
| Chr 12-6 | 1061.4–1078.1 (16.7) | 17 | 249 | 6/10 (60.0) | 43 | 2/14 (14.2) | Essential | This study | |
| Chr 13-1 | ScΔ(Chr 13-1) | 149.3–170.8 (21.5) | 16 | 114 | 5/5 (100) | 55 | 6/9 (66.6) | Dispensable | This study |
| Chr 13-2 | 300.6–323.5 (22.9) | 13 | 182 | 5/5 (100) | 80 | 0.00% | ? | This study (not obtained) | |
| Chr 13-3 | 460.6–490.6 (30.0) | 20 | 96 | 6/6 (100) | 53 | 13/15 (86.7) | Essential | This study | |
| Chr 13-4 | 565.2–593.9 (28.7) | 20 | 83 | 5/5 (100) | 110 | 4/5 (80.0) | Essential | This study | |
| Chr 13-5 | 603.5–655.1 (51.6) | 35 | 118 | 3/3 (100) | 200 | 5/5 (100) | Essential | This study | |
| Chr 13-6 | 754.6–785.9 (31.3) | 23 | 78 | 5/7 (71.4) | 57 | 2/6 (33.3) | Essential | This study | |
| Chr 14-1 | 1–37.0 (37.0) | 26 | 66 | 1/9 (11.1) | 80 | 2/6 (33.3) | Essential | This study | |
| Chr 14-2 | 62.8–82.8 (20.0) | 11 | 120 | 5/5 (100) | 42 | 1/4 (25.0) | Essential | This study | |
| Chr 14-3 | 260.0–280.8 (20.8) | 13 | Essential | Harboring SL gene pair | |||||
| Chr 14-4 | 515.7–551.6 (35.9) | 28 | 135 | 3/3 (100) | 185 | 4/5 (80.0) | Essential | This study | |
| Chr 14-5 | 590.2–614.5 (24.3) | 15 | Essential | Harboring SL gene pair | |||||
| Chr 15-2 | 98.2–117.3 (19.1) | 17 | 37 | 2/5 (40.0) | 271 | 6/6 (100) | Essential | This study | |
| Chr 15-3 | 146.1–178.2 (32.1) | 14 | 59 | 5/5 (100) | 60 | 3/8 (37.5) | Essential | This study | |
| Chr 15-4 | 207.9–249.7 (41.8) | 29 | 86 | 3/3 (100) | 150 | 5/6 (83.3) | Essential | This study | |
| Chr 15-5 | 288.7–304.5 (15.8) | 13 | 160 | 5/5 (100) | 30 | 3/3 (100) | Essential | This study | |
| Chr 15-6 | 337.9–367.5 (29.6) | 21 | 297 | 4/4 (100) | 152 | 5/5 (100) | Essential | This study | |
| Chr 15-7 | 374.3–411.0 (36.7) | 26 | 76 | 7/7 (100) | 45 | 0.00% | ? | This study (not obtained) | |
| Chr 15-10 | 672.2–702.2 (30.0) | 16 | Essential | Sugiyama et al. (13) | |||||
| Chr 15-11 | ScΔ(Chr 15-11) | 969.0–995.0 (26.0) | 18 | 42 | 4/4 (100) | 63 | 5/5 (100) | Dispensable | This study |
| Chr 16-2 | 43.6–64.2 (20.6) | 19 | 265 | 4/4 (100) | 66 | 0.00% | ? | This study (not obtained) | |
| Chr 16-3 | 332.3–366.4 (34.1) | 21 | 164 | 3/3 (100) | 49 | 0.00% | ? | This study (not obtained) | |
| Chr 16-4 | 434.7–466.5 (31.8) | 23 | 66 | 5/5 (100) | 50 | 3/5 (60.0) | Essential | This study | |
| Chr 16-5 | ScΔ(Chr 16-5) | 547.2–574.2 (26.9) | 19 | 140 | 5/6 (83.3) | 95 | 5/5 (100) | Dispensable | This study |
| Chr 16-6 | 675.6–699.9 (24.3) | 22 | 95 | 5/5 (100) | 70 | 4/4 (100) | Essential | This study | |
| Chr 16-7 | ScΔ(Chr 16-7) | 756.8–792.5 (35.7) | 21 | 77 | 2/3 (66.7) | 80 | 6/6 (100) | Dispensable | This study |
| Chr 16-8 | ScΔ(Chr 16-8) | 824.8–860.8 (36.0) | 30 | 119 | 11/14 (78.6) | 50 | 5/6 (83.3) | Dispensable | This study |
aChr x-y: Chr x represents chromosome number and -y represents region.
bScΔ(Chr x-y): ScΔ represents S. cerevisiae deletion mutant, Chr x represents chromosome number and -y represents region number.
cx-y: x represents the left-hand side chromosomal region and -y represents the right-hand side of chromosomal region.
Figure 3.
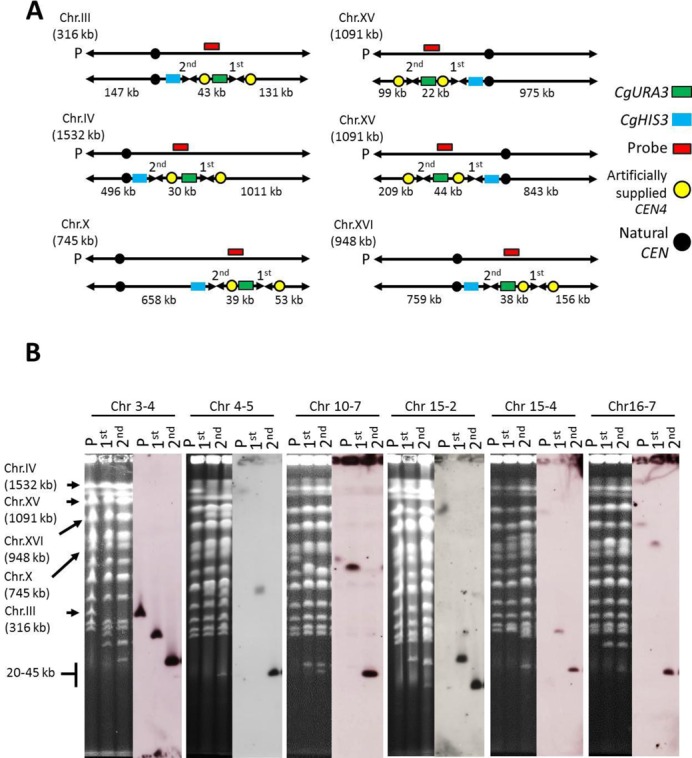
Construction of mini-chromosome in chromosomes III, IV, X, XV and XVI. (A) Illustration of the target chromosome, target region, length of artificial chromosomes and hybridization probe location. Mini-chromosomes were constructed by splitting chromosomes at the left- and right-hand side of each region using PCS technology. Each mini-chromosome harbored the CgURA3 marker (dark green box) and artificial CEN4 (yellow circle). The length of each mini-chromosome and other fragments is indicated. Red boxes represent probes corresponding to an internal sequence of the mini-chromosome. Blue box and black circle represent the CgHIS3 marker and natural CEN, respectively. (B) PFGE and Southern analysis showing the karyotype analysis of the parental haploid (P), strain after first splitting (designated 1st), and strain after second splitting (designated 2nd) harboring mini-chromosomes of regions Chr 3-4, Chr 4-5, Chr 10-7, Chr 15-2, Chr 15-4 and Chr 16-7. For all chromosomes, probes were prepared by PCR amplification of a 400-bp internal sequence of the constructed mini-chromosome.
Next, the essentiality of these regions was examined by using diploids constructed by crossing each of these strains with a wild-type strain (ura3) with no split chromosomes on 5-FOA selection medium on which only Ura− clones can grow (20). In brief, haploid strains harboring the URA3-marked mini-chromosome, called ‘H-mini’, were mated with wild-type strain BY4741 to produce a resultant diploid, called D-mini. Cells of the haploid parental strain (BY4742), H-mini, and D-mini were then spotted on YPDA, SC-Ura and 5-FOA selection medium and observed for growth (Figure 4). 5-FOA is toxic to yeast cells that can synthesize the enzyme orotidine-5′-phosphate decarboxylase encoded by URA3; as a result, Ura+ cells die when plated on 5-FOA-containing media, whereas ura3 cells survive. Thus, if the lost chromosomal region in uracil auxotroph strains (H-mini) is non-essential, the H-mini strain will grow on medium containing 5-FOA, whereas it will not grow on 5-FOA selection medium if the lost region is essential. By contrast, a D-mini strain harboring a mini-chromosome comprising an essential region is expected to grow on 5-FOA selection medium even if the mini-chromosome marked with URA3 is lost, owing to the presence of an intact chromosome from the wild-type parent (Figure 4).
Figure 4.

Comparative growth of the Ura− strain (BY4742), Ura+ strain (SH4233), H-mini and D-mini on YPDA, SC-Ura and 5-FOA media. Each strain was first grown overnight at 30°C on SC medium, and then washed twice with water. Aliquots of 105 cells were spotted onto YPDA, SC-Ura and 5-FOA media and incubated at 30°C for 2 days. In total, 67 H-mini strains harboring a URA3-marked mini-chromosome of either an essential or non-essential region were observed to grow on 5-FOA medium. The strains are indicated above their respective lanes. H-mini strains showing growth on 5-FOA contain deletions of non-essential regions; those showing no growth on 5-FOA contain mini-chromosomes of essential regions.
We found that strains harboring mini-chromosomes of regions Chr 1-2, Chr 2-2, Chr 2-4, Chr 2-6, Chr 2-7, Chr 3-3, Chr 3-4, Chr 4-3, Chr 4-5, Chr 4-6, Chr 5-2, Chr 5-4, Chr 6-2, Chr 7-1, Chr 7-5, Chr 7-6, Chr 7-8, Chr 8-1, Chr 8-2, Chr 8-4, Chr 8-6, Chr 8-7, Chr 9-2, Chr 9-3, Chr 10-2, Chr 10-3, Chr 10-6, Chr 10-7, Chr 11-2, Chr 12-1, Chr 12-3, Chr 12-4, Chr 12-5, Chr 12-6, Chr 13-3, Chr 13-4, Chr 13-5, Chr 13-6, Chr 14-1, Chr 14-2, Chr 14-4, Chr 15-2, Chr 15-3, Chr 15-4, Chr 15-5, Chr 15-6, Chr 16-4, Chr 16-6 were sensitive to growth on 5-FOA medium (Figure 4 and Table 3), suggesting that these regions are essential for cell growth. On the other hand, 11 strains with mini-chromosomes containing the regions Chr 3-2, Chr 4-4, Chr 7-3, Chr 7-4, Chr 10-4, Chr 12-2, Chr 13-1, Chr 15-11, Chr 16-5, Chr 16-7 and Chr 16-8 grew on both 5-FOA and Sc-Ura media (Figure 4 and Table 3), suggesting that these regions have no role in cell viability at least under nutrient-rich conditions.
To construct strains lacking each of the 11 non-essential regions identified by PCS, namely, ScΔ(Chr 3-2), ScΔ(Chr 4-4), ScΔ(Chr 7-3), ScΔ(Chr 7-4), ScΔ(Chr 10-4), ScΔ(Chr 12-2), ScΔ(Chr 13-1), ScΔ(Chr 15-11), ScΔ(Chr 16-5), ScΔ(Chr 16-7) and ScΔ(Chr 16-8) (Table 3), we used 5-FOA selection medium to select for loss of the URA3-containing mini-chromosome corresponding to each of these regions.
Because 49 of the 77 undeletable regions identified by PCD were not found to carry a pair of genes previously shown to cause a synthetic lethal phenotype in double knockout screens during SGA analysis (9,10), the present results strongly suggest that these undeletable regions contain unidentified synthetic lethal combinations of two or more non-essential genes. Overall, these findings indicate that most of the essential regions discovered in this study contain lethal interactions of linked gene-pairs. These linked gene-pairs are biologically informative and their identification will contribute towards comprehensive coverage of all double mutant combinations in yeast.
Characterization of strains lacking deletable regions
Among the 44 deletable regions (33 identified by PCD and 11 identified by PCS), phenotypic information for 17 regions could be derived from previous studies (13,14,17,23). To evaluate the consequences of deleting the non-essential chromosomal regions on cell growth, 27 deletion strains, each containing one of the remaining deletable regions, i.e. ScΔ(Chr 1-1), ScΔ(Chr 1-3), ScΔ(Chr 2-1), ScΔ(Chr 2-9), ScΔ(Chr 3-1), ScΔ(Chr 3-2), ScΔ(Chr 4-1), ScΔ(Chr 4-4), ScΔ(Chr 4-10), ScΔ(Chr 4-11), ScΔ(Chr 5-1), ScΔ(Chr 6-1), ScΔ(Chr 7-3), ScΔ(Chr 7-4), ScΔ(Chr 9-1), ScΔ(Chr 9-4), ScΔ(Chr 9-5), ScΔ(Chr 9-6), ScΔ(Chr 10-4), ScΔ(Chr 11-1), ScΔ(Chr 12-2), ScΔ(Chr 13-1), ScΔ(Chr 15-11), ScΔ(Chr 16-5), ScΔ(Chr 16-7), ScΔ(Chr 16-8) and ScΔ(Chr 16-9), were constructed for the first time subjected to phenotypic analysis. Different culture conditions were used for phenotypic analysis including YPDA (incubated at 13, 30 and 41°C) and YPDA supplemented with 4, 6 and 8% ethanol, 4, 5 and 6% lactic acid, sulfuric acid (pH 2.5, pH 2.4 and pH 2.3), 0.02% SDS, 80 mM acetic acid, 36 mM formic acid, 1.8 M NaCl, 150 mM CaCl2, 10 mM furfural and 2.0 M sorbitol. In addition, to investigate the effect of deleting the non-essential chromosomal region on cell growth in liquid culture, deletion strains were observed to grow on YPDA broth (Figure 6).
Figure 6.
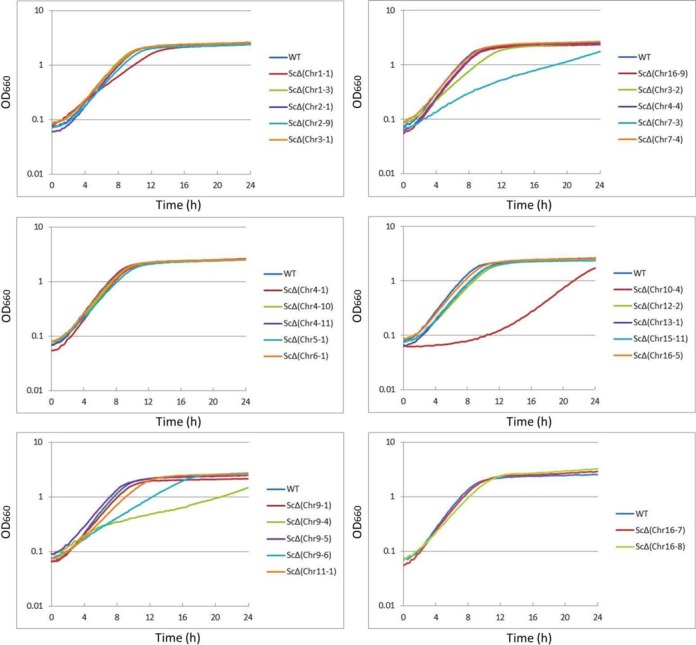
Growth profiles of the wild-type strain (WT) and deletion strains. Growth of the 27 deletion strains at 30°C in liquid YPDA culture, starting at an initial OD660of 0.1 were examined by monitoring OD660 at the indicated times.
Some of the strains displayed a sensitive or resistant phenotype under the stressful environment (Figure 5A, B and C and Supplementary Table S4) including a low-temperature-sensitive phenotype, ScΔ(Chr 10-4), ScΔ(Chr 11-1) and ScΔ(Chr 16-8); a high-temperature-sensitive phenotype, ScΔ(Chr 5-1), ScΔ(Chr 9-1), ScΔ(Chr 10-4), ScΔ(Chr 11-1), ScΔ(Chr 13-1), ScΔ(Chr 16-5), ScΔ(Chr 16-7) and ScΔ(Chr 16-8) (Figure 5A); a high-temperature-resistant phenotype, ScΔ(Chr 4-10) and ScΔ(Chr 4-11) (Figure 5A); a ethanol-sensitive phenotype, ScΔ(Chr 5-1), ScΔ(Chr 9-4), ScΔ(Chr 9-6), ScΔ(Chr 10-4), ScΔ(Chr 11-1), ScΔ(Chr 13-1) and ScΔ(Chr 16-8); a lactic-acid-resistant phenotype, ScΔ(Chr 3-1), ScΔ(Chr 4-1) and ScΔ(Chr 16-9) (Figure 5B) and a sulfuric acid-sensitive phenotype (all strains except for ScΔ(Chr 1-1), ScΔ(Chr 6-1) and ScΔ(Chr 7-4)) (Figure 5B). We did not find strains that were resistant to the ethanol or the sulfuric acid. Based on information in the SGD, neither of the regions deleted in the high-temperature-resistant strains, ScΔ(Chr 4-10) and ScΔ(Chr 4-11), contains the genes whose single deletion is known to cause high-temperature resistance; instead, each region contain a gene (FPR2 and STL1, respectively) whose single deletion cause heat sensitivity. This result suggests that the deletion of either of these genes in combination with another in the respective chromosomal regions caused the observed tolerant phenotype of those strains. We found that all regions deleted in the ethanol-sensitive strains, ScΔ(Chr 5-1), ScΔ(Chr 9-4), ScΔ(Chr 9-6), ScΔ(Chr 10-4), ScΔ(Chr 11-1), ScΔ(Chr 13-1) and ScΔ(Chr 16-8), contain the genes, (CIN8, GVP36 and TED1, YVH1, RAV1, URA1 and SAC1, PIF1 and TDA6 and KRE6, respectively), whose single deletion cause ethanol sensitivity (SGD)(http://www.yeastgenome.org/). Therefore, observed sensitive phenotypes due to deletion of those regions might be conferred by deletion of respective genes. Lack of resistance to ethanol in deletion strains implies that neither of deleted regions nor genes have important role in conferring ethanol-tolerance phenotype.
Figure 5.

Phenotypic characteristics of deletion mutants under various stress conditions. Deletion strains as well as the wild-type strain were tested for their ability to growth on YPDA medium incubated at 13°C, 30°C, and 41°C, and YPDA medium supplemented with 4%, 6% and 8% ethanol (A), 4%, 5% and 6% lactic acid, and sulfuric acid (pH2.5, pH2.4 and pH2.3) (B),0.02% SDS, 80 mM acetic acid, 36 mM formic acid, 1.8 M NaCl, 150 mM CaCl2,10 mM furfural,and 2.0 M sorbitol (C). For panel (A) and (B), each strain was plated in serial ten-fold dilutions from 2.5 × 108 cells/ml to 2.5 × 101 cells/ml, from left to right, as indicated. For panel (C), 4μl from cell suspension with concentration of 2.5 × 108 cells/ml was spotted on plate. Plates were incubated for 3 days. White and black boxes represent tolerant and sensitive phenotypes, respectively.
We also found that strains ScΔ(Chr 1-1), ScΔ(Chr 5-1), ScΔ(Chr 9-1), ScΔ(Chr 9-4), ScΔ(Chr 9-5), ScΔ(Chr 9-6), ScΔ(Chr 10-4), ScΔ(Chr 11-1) and ScΔ(Chr 13-1) were sensitive to 0.02% SDS; strains ScΔ(Chr 1-1), ScΔ(Chr 3-2), ScΔ(Chr 4-4), ScΔ(Chr 7-3), ScΔ(Chr 7-4), ScΔ(Chr 9-1), ScΔ(Chr 9-4), ScΔ(Chr 9-6), ScΔ(Chr 10-4), ScΔ(Chr 11-1), ScΔ(Chr 13-1), ScΔ(Chr 15-11), ScΔ(Chr 16-5), ScΔ(Chr 16-8) and ScΔ(Chr 16-9) were sensitive to acetic and formic acid; strains ScΔ(Chr 1-1), ScΔ(Chr 5-1), ScΔ(Chr 9-1), ScΔ(Chr 9-4), ScΔ(Chr 9-6), ScΔ(Chr 10-4), ScΔ(Chr 11-1), ScΔ(Chr 13-1) and ScΔ(Chr 16-8) were sensitive to 1.8 M NaCl; strain ScΔ(Chr 9-1) was sensitive to 150 mM CaCl2; strain ScΔ(Chr 9-6) was sensitive to 10 mM furfural; and strain ScΔ(Chr 5-1) was sensitive to 2.0 M sorbitol (Figure 5C and Supplementary Table S4). However, we did not find strains that were resistant to these stress conditions.
Strains lacking the left terminal region of chromosome III, ScΔ(Chr 3-1), the left terminal region of chromosome IV, ScΔ(Chr 4-1), and the right terminal region of chromosome XVI, ScΔ(Chr 16-9) showed a resistant phenotype to lactic acid (Figure 5B). Because the region deleted in strain ScΔ(Chr 4-1) contains the gene LRG1, whose single deletion causes lactic acid resistance (24), the resistant phenotype that ScΔ(Chr 4-1) displays may be due to the deletion of LRG1 in this region. Many of the regions deleted in the sulfuric acid-sensitive strains do not have a gene whose single deletion cause sulfuric acid-sensitivity (http://www.yeastgenome.org/). Therefore, observed sensitive phenotypes suggest two possibilities. First is that particular combinations of genes present in those regions are important for sulfuric acid stress sensitivity. Second is that adaptation to sulfuric acid stress condition requires a complex organization of genetic network beyond those regions and modulation of sulfuric acid related pathways is partially collapsed in deletion strains, which could drastically affect the adaptation. In addition, absence of deletion strains showing tolerant phenotype to sulfuric acid suggests that none of genes in deleted regions are involved in mechanisms underlying sulfuric acid stress tolerance. Notably, the region deleted in ScΔ(Chr 16-9) does not contain any genes whose single deletion causes lactic acid tolerance. We found that strains ScΔ(Chr 7-3), ScΔ(Chr 9-4), ScΔ(Chr 9-6) and ScΔ(Chr 10-4) are susceptible to grow on YPDA broth compared to the wild-type (Figure 6). Because in both YPDA agar and broth at 30°C many of the deletion strains exhibited parent-like phenotype, this observation suggests that gene of deleted regions in those strains are not involved in molecular mechanism underlying cell growth in both media (Figures 5A and 6). Our result also suggests that genes deleted in strains ScΔ(Chr 9-6) and ScΔ(Chr 10-4) are important for yeast cell to be viable at normal condition of both media. Altogether, these results indicate that phenotypic alterations in strains displaying an unexpected tolerant phenotype might be conferred by the synergistic effects of two or more genes on the deleted region. Hence, segmental chromosomal deletion technology provides a new tool for both analyzing the functions and interactions of genes and screening for target genes associated with a desired phenotype. Even more valuable, this strategy can be applied to the breeding of multi-stress tolerant yeast strains by generating an optimal combination of beneficial segmental chromosomal deletions in a single strain.
DISCUSSION
Our data have revealed that there are many unexplored lethal combinations into the genome of S. cerevisiae. Because the efficiency of double mutant formation is often reduced for gene-pairs that are located in close proximity on the same chromosome, these lethal interactions among non-essential genes were not previously detected by SGA analysis, which is the standard approach for the global analysis of synthetic lethal genetic interactions (8–10). We found that 56 of the 110 regions investigated were essential, even though they harbor only non-essential genes. Furthermore, 49 out of the 56 (87.5%) regions did not carry any of the co-lethal gene pair(s) previously identified by SGA analysis. This result implies that regions found to harbor only non-essential genes by single knockout experiments in fact contain unidentified synthetic lethal combinations at unexpectedly high frequency.
A recent large-scale genetic interaction mapping study on S. cerevisiae (10) has revealed that, among 5.4 million gene-pairs tested so far, ∼10 000 (0.2%) cause a synthetic lethal phenotype. Therefore, probability of a gene-pair being a non-synthetic lethal combination, P(non-SL), would be 0.998, i.e. [P(non-SL) = 1 – P(SL) = 1 – 0.002 = 0.998]. Given a region consisting of 20 non-essential genes (the average number of non-essential genes in the 110 regions), the number of possible gene-pair combinations among those genes would be (20 × 20/2 = 200). Thus, under assumption that all combinations are equally likely to occur, the probability (defined as non-SLno. of combinations) of obtaining a successful deletion of this region would be (0.998)200 = 0.67. On the basis of these calculations, the expected number of deletable regions among 110 regions harboring only non-essential genes would be 110 × 0.67 = 74 regions, and correspondingly the expected number of undeletable regions would be 110 – 74 = 36 (33%). However, the present findings showed that 56 out of 100 regions (56%) were undeletable. This result predicts that regions that harbor only non-essential genes according to single knockout experiments contain unidentified synthetic lethal combinations at a significantly high frequency; moreover, it emphasizes the need to validate screens for synthetic lethal interactions by novel genome engineering technologies, such as those used in this study, in addition to SGA methods.
The high frequency of unidentified synthetic lethal combinations observed in the present study (i.e. 87.5% of the undeletable regions) is consistent with the view of previous studies (9,10) that the number of real synthetic lethal gene-pairs across the complete network is likely to be much higher than the number observed so far. In theory, to test all possible pair-wise combinations of the ∼5000 non-essential genes in the yeast genome would require the construction of ∼12.5 million double disruptants (10). In a double-knockout test, however, only 3% of the theoretical combinations (170 000 interactions among 5 400 000 gene-pairs) were achieved using SGA analysis (10). In addition, by extrapolating the results of that study (10) for extreme synthetic lethal pairs (∼10 000 interactions among 5 400 000 gene-pairs), the number of synthetic lethal pairs across the complete network would be 25 000 pairs (12.5 million × 0.002). However, the quantitative estimate of synthetic lethality for the global network of non-essential genes is more than 100 000 among 12.5 million combinations (9,10). Because the formation of double mutants in SGA analysis depends on meiotic recombination, the construction of double disruptants is not possible if the two genes to be combined are tightly linked on the same chromosome. Thus, numerous linked gene-pairs throughout the genome form small colonies of double mutants that have been overlooked in analysis to prevent them from being mistaken for true negative genetic interactions. Although the incomplete network of 10 000 lethal interactions has so far provided valuable insight into the general principles of network connectivity, the results of the present study indicate that the application of a more sensitive method for detecting genetic interactions would have a significant impact on increasing the network size toward the estimated number (100 000 combinations), and would provide a more explicit evolutionarily-connected picture of essential interactions between non-essential genes.
None of the essential regions discovered for the first time in this study carries a pair of genes with lethal combinations revealed by SGA assay. Therefore, this result indicates that some regions, especially those containing many genes, may harbor lethal interactions among more than two genes. On the basis of our current results, we could not conclude which gene-pairs, gene-triplets or gene sets were responsible for the essentiality of the 49 identified regions; however, synthetic lethality often involves genes with overlapping functions. For example, we noted that genes FCY2, FCY21 and FCY22 belonging to the same gene family mediate the active transport of purines and cytosine, and these genes were simultaneously deleted from the region Chr 5-4. Because deletion mutants for all three genes must lead to defects in active transport of purine and cytosine, it is likely that simultaneous deletion of these genes caused the lethality. In another region (Chr 10-2), genes RPS22A, RPL39 and RPL17B encoding ribosomal subunit protein were deleted at once. Because lethal interaction between these three physically linked genes very likely failed to be detected from SGA screening on lethal interaction, simultaneous deletion of these three genes may be responsible for lethal phenotype observed for ScΔ(Chr 10-2). If we continue to narrow down these regions, we should be able to pinpoint shorter chromosomal segments including the essential gene sets. Accordingly, from such regions, we may reveal how and in what way essentiality is extended all over the genome. In addition to possible lethal interaction of gene pairs, deletion of genes encoding non-coding RNAs and other non-coding elements such as tRNAs, tmRNAs, snoRNAs and miRNAs could be responsible for the essentiality to the 49 essential regions as well. For instance, essential regions harbored in the strains ScΔ(Chr 6-2) and ScΔ(Chr 8-4), were found to contain non-coding RNAs (RUF22, RUF5-1 and RUF5-2, respectively) (http://www.yeastgenome.org/). Although the function of these non-coding RNAs is unknown, there is a possibility that the essentiality of those regions is caused by possible essential function of non-coding RNA genes, individually or in combination.
We found that, in many mutant strains, the deleted regions do not play an important role in cell viability under the stress-free and stressful conditions tested because those strains exhibited parent-like phenotypes. Among 44 constructed mutant strains, however, strains ScΔ(Chr 4-10) and ScΔ(Chr 4-11) and strains ScΔ(Chr 3-1), ScΔ(Chr 4-1) and ScΔ(Chr 16-9) displayed more a tolerant phenotype than the parent strain under, respectively, heat and lactic acid stress conditions (Figure 5A and B and Supplementary Table S4). On the basis of a recent study that used fitness profiling on synthetic complete medium (25), strain ScΔ(Chr 16-9) harbors six haplo-insufficient (AQY1, HPA2, YPR195C, YPR196W, YPR197C and ARR1) genes that cause slower growth rate of the diploid, and one haplo-proficient (ARR3) gene that causes faster growth rate of the diploid. Thus, we propose that deletion of these haplo-insufficient and haplo-proficient genes simultaneously in a haploid background might give the observed tolerant phenotype. This can be explained in terms of the dosage of these seven genes in both backgrounds. Having a single copy of the genes can be easily tolerated in the diploid because robustness in the gene network is better in the diploid than in the haploid background, and the consequence of a single change in gene dosage (i.e. one copy of the gene in the diploid state) would be to increase or decrease the fitness. In the haploid background, however, deleting a gene, especially in a cluster form, clearly collapses the genetic network, which is usually detrimental. The tolerant phenotype that was observed might be a type of drastic response to the segmental deletion of the chromosome in the haploid background (Figure 5B and Table 4). It is predicted that under conditions of stress, the cell might rely on very different genetic pathways to promote viability, and may rewire its biological networks to deal with the altered conditions. Thus, the stress resistant or sensitive phenotypes might be conferred by synergistic effects of the deletion of two or more genes in a genetic network that operates across the genes normally present in those deleted regions. Another possibility is that, because segmental deletion might lead to an imbalance in gene expression, the altered phenotypes of the deletion strains might result from a change in gene expression induced by the deletion of multiple genes. Finally, because transcription of genes located near telomeres might be repressed in the deletion mutants' strains (26), observed phenotypes by those strains could somewhat be explained by gene silencing effect by artificially added telomere. Although the phenotypic consequences of segmental deletion might generally be detrimental, the results of the present study suggest that this type of deletion occasionally confers a growth advantage on S. cerevisae, which can apparently be explained by the change in copy number of many genes at once. Altogether, these observations indicate that segmental chromosomal deletion may be a powerful approach not only for better understanding of genome function but also for obtaining knowledge for breeding.
Table 4. Genes in deleted regions associated with heat and lactic acid tolerance.
| Region names | Classification (no. of genes) | Gene names |
|---|---|---|
| Chr 4-10 | RNA binding protein (1) | SLF1 |
| Isomerase (2) | EUG1, FPR2 | |
| Transcription (1) | URC2 | |
| Protein kinase (1) | SPS1 | |
| GTPase (1) | AGE1 | |
| Unknown (4) | EMI2, GRH1, YDR521W, SPS2 | |
| Chr 4-11 | Transport (2) | STL1 |
| Acid decarboxylase (1) | PAD1 | |
| DNA helicase (1) | YRF1-1 | |
| Unknown (10) | FIT1, YDR535C, YDR537C, FDC1, IRC4, YDR541C, PAU10, YDR543C, YDR544C, YDR545C-A | |
| Chr 3-1 | Protein anchor (1) | VAC17 |
| Deaminase (1) | CHA1 | |
| Transcription (2) | HMLALPHA1, HMLALPHA2 | |
| Transport (2) | VBA3, GEX1 | |
| Unknown (3) | YCL065W, YCL068C, YCL076W | |
| Chr 4-1 | Dehydrogenase (1) | AAD4 |
| Deaminase (1) | GUD1 | |
| GTPase (1) | LRG1 | |
| Transport (2) | HXT15, MPH2 | |
| Unknown (8) | ADY3, YDL240C-A, YDL241W, YDL242W, THI13, SOR2, YDL247W-A, COS7 | |
| Chr 16-9 | Acetyltransferase (1) | HPA2 |
| Arsenate resistance (1) | ARR2 | |
| Transcription (1) | ARR1 | |
| Transport (4) | AQY1, OPT2, SGE1, ARR3 | |
| Unknown (7) | YPR195C, YPR196W, YPR197C, YPR202W, YPR203W, YPR204W, YPR204C-A |
Screening for genetic interactions that occur when a genetic perturbation leads to an extreme phenotype provides a simple but powerful approach for revealing gene function. Here we validated the utility of our strategies by revealing many hidden lethal combinations between two physically linked genes that would not be detected by SGA array analysis. The ability of this approach to discover novel lethal combinations has enabled us to demonstrate a new view of the genetic landscape for the yeast gene interaction network.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
One of the authors (S.K.) expresses his special thanks to the Global COE (center of excellence) Program ‘Global Education and Research Center for Bio-Environmental Chemistry’ of Osaka University. S.K. is a Ph.D. student supported by the Iran Ministry of Science, Research and Technology. The authors are thankful to Deasty Imara for technical assistance.
FUNDING
Grant-in-Aid for Challenging Exploratory Research [24658081, 19658132 to S.H.]; Grant-in-Aid for Scientific Research (B) [24380048 to S.H.]; Grant-in-Aid for Young Scientists (B) [23780080 to M.S.] from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. Funding for open access charge: Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
Conflict of interest statement. None declared.
REFERENCES
- 1.Giaever G., Chu A.M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 2.Novick P., Osmond B.C., Botstein D. Suppressors of yeast actin mutations. Genetics. 1989;121:659–674. doi: 10.1093/genetics/121.4.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guarente L. Synthetic enhancement in gene interaction: a genetic tool come of age. Trends Genet. 1993;9:362–366. doi: 10.1016/0168-9525(93)90042-g. [DOI] [PubMed] [Google Scholar]
- 4.He X., Qian W., Wang Z., Li Y., Zhang J. Prevalent positive epistasis in Escherichia coli and Saccharomyces cerevisiae metabolic networks. Nat. Genet. 2010;42:272–276. doi: 10.1038/ng.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peña-Castillo L., Hughes T.R. Why are there still over 1000 uncharacterized yeast genes. Genetics. 2007;176:7–14. doi: 10.1534/genetics.107.074468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mani R., St Onge R.P., Hartman J.L., Giaever G., Roth F.P. Defining genetic interaction. Proc. Natl. Acad. Sci. U.S.A. 2008;105:3461–3466. doi: 10.1073/pnas.0712255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixon S.J., Costanzo M., Baryshnikova A., Andrews B., Boone C. Systematic mapping of genetic interaction networks. Annu. Rev. Genet. 2009;43:601–625. doi: 10.1146/annurev.genet.39.073003.114751. [DOI] [PubMed] [Google Scholar]
- 8.Tong A.H.Y., Evangelista M., Parsons A.B., Xu H., Bader G.D., Pagé N., Robinson M., Raghibizadeh S., Hogue C.W., Bussey H., et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 9.Tong A.H.Y., Lesage G., Bader G.D., Ding H., Xu H., Xin X., Young J., Berriz G.F., Brost R.L., Chang M., et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- 10.Costanzo M., Baryshnikova A., Bellay J., Kim Y., Spear E.D., Sevier C.S., Ding H., Koh J.L.Y., Toufighi K., Mostafavi S., et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baryshnikova A., Costanzo M., Myers C.L., Andrews B., Boone C. Genetic interaction networks: toward an understanding of heritability. Annu. Rev. Genomics Hum. Genet. 2013;14:111–133. doi: 10.1146/annurev-genom-082509-141730. [DOI] [PubMed] [Google Scholar]
- 12.Collins S.R., Schuldiner M., Krogan N.J., Weissman J.S. A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol. 2006;7:R63. doi: 10.1186/gb-2006-7-7-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugiyama M., Ikushima S., Nakazawa T., Kaneko Y., Harashima S. PCR-mediated repeated chromosome splitting in Saccharomyces cerevisiae. Biotechniques. 2005;38:909–914. doi: 10.2144/05386RR01. [DOI] [PubMed] [Google Scholar]
- 14.Sugiyama M., Nakazawa T., Murakami K., Sumiya T., Nakamura A., Kaneko Y., Nishizawa M., Harashima S. PCR-mediated one-step deletion of targeted chromosomal regions in haploid Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2008;80:545–553. doi: 10.1007/s00253-008-1609-9. [DOI] [PubMed] [Google Scholar]
- 15.Sugiyama M., Yamagishi K., Kim Y.-H., Kaneko Y., Nishizawa M., Harashima S. Advances in molecular methods to alter chromosomes and genome in the yeast Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2009;84:1045–1052. doi: 10.1007/s00253-009-2144-z. [DOI] [PubMed] [Google Scholar]
- 16.Park A.-H., Sugiyama M., Harashima S., Kim Y.-H. Creation of an ethanol-tolerant yeast strain by genome reconstruction based on chromosome splitting technology. J. Microbiol. Biotechnol. 2012;22:184–189. doi: 10.4014/jmb.1109.09046. [DOI] [PubMed] [Google Scholar]
- 17.Ueda Y., Ikushima S., Sugiyama M., Matoba R., Kaneko Y., Matsubara K., Harashima S. Large-scale genome reorganization in Saccharomyces cerevisiae through combinatorial loss of mini-chromosomes. J. Biosci. Bioeng. 2012;113:675–682. doi: 10.1016/j.jbiosc.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 18.Sambrook J., Fritsch E.F., Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 19.Amberg D.C., Burke D.J., Strathern J.N. Methods in Yeast Genetics. Cold Spring Harbor, New York: Cold Harbor Spring Laboratory; 2005. [Google Scholar]
- 20.Green S.R., Moehle C.M. Current Protocols in Cell Biology. John Wiley and Sons, Inc; 2003. [Google Scholar]
- 21.Gietz R.D., Schiestl R.H. Transforming yeast with DNA. Methods Mol. Cell Biol. 1995;5:255–269. [Google Scholar]
- 22.Sheehan C., Weiss A.S. Yeast artificial chromosomes: rapid extraction for high resolution analysis. Nucleic Acids Res. 1990;18:2193. doi: 10.1093/nar/18.8.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murakami K., Tao E., Ito Y., Sugiyama M., Kaneko Y., Harashima S., Sumiya T., Nakamura A., Nishizawa M. Large scale deletions in the Saccharomyces cerevisiae genome create strains with altered regulation of carbon metabolism. Appl. Microbiol. Biotechnol. 2007;75:589–597. doi: 10.1007/s00253-007-0859-2. [DOI] [PubMed] [Google Scholar]
- 24.Kawahata M., Masaki K., Fujii T., Iefuji H. Yeast genes involved in response to lactic acid and acetic acid: acidic conditions caused by the organic acids in Saccharomyces cerevisiae cultures induce expression of intracellular metal metabolism genes regulated by Aft1p. FEMS Yeast Res. 2006;6:924–936. doi: 10.1111/j.1567-1364.2006.00089.x. [DOI] [PubMed] [Google Scholar]
- 25.Pir P., Gutteridge A., Wu J., Rash B., Kell D.B., Zhang N., Oliver S.G. The genetic control of growth rate: a systems biology study in yeast. BMC Syst. Biol. 2012;6:4. doi: 10.1186/1752-0509-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tham W.H., Zakian V.A. Transcriptional silencing at Saccharomyces telomeres: implications for other organisms. Oncogene. 2002;21:512–521. doi: 10.1038/sj.onc.1205078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.